
Structural and spectroscopic studies of iodine dimer radical anion hydrated clusters: an approach using a combination of stochastic and quantum chemical methods†
Abstract
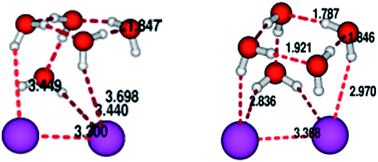
In this communication we would like to explore the idea of obtaining structures and IR spectral features of I2−(H2O)n cluster with n = 2 to 6. The approach taken up by us is a two tire one with initial good quality pre-optimized structures being found out by exploring a suitably constructed empirical potential energy surface and using the stochastic optimizer simulated annealing to explore the surface. These structures are then further refined using quantum chemical calculation to obtain the final structure and the IR spectra. We clearly show that the initial pre-optimization can indeed pave the way for quick and better convergence on subsequent quantum chemical refinement. To give credence to our effort we have also shown the evolutionary trend in vertical detachment energy (VDE) as a function of cluster size.
 Please wait while we load your content...
Please wait while we load your content...

