
A structure guided drug-discovery approach towards identification of Plasmodium inhibitors
Abstract
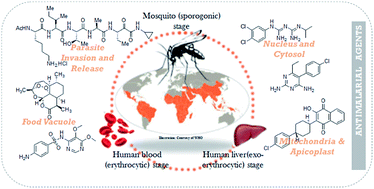
Rapidly increasing resistance to the currently available antimalarial drugs has drawn attention globally for the search for more potent novel drugs with a high therapeutic index. The genome sequencing of the human malarial parasite Plasmodium falciparum has provided extensive information to understand potential target pathways and efforts are being made to develop lead inhibitors with a hope to eliminate the disease. This review is focused on a brief description of key biochemical targets identified from the genome sequence of P. falciparum. This review also summarizes the work undertaken by different scientific groups over the last five years to develop inhibitors from natural, semisynthetic or synthetic sources, which will be valuable to medicinal chemists to develop novel antimalarial agents.
 Please wait while we load your content...
Please wait while we load your content...

