
One-pot protocol for J-aggregated anthraimidazolediones catalyzed by phosphotungstic acid in PEG-400 under aerobic condition†
Bhaswati Bhattacharyyaa,
Arijit Kundub,
Aniruddha Dasc,
Kaliprasanna Dhara*c and
Nikhil Guchhaitc
aDepartment of Chemistry, A. P. C. Roy Govt. College, Darjeeling- 734010, India
bDepartment of Chemistry, Maulana Azad College, Kolkata-700013, India
cDepartment of Chemistry, University College of Science & Technology, University of Calcutta, 92, A.P.C. Road, Kolkata-700009, India. E-mail: chemkpd@gmail.com
First published on 18th February 2016
Abstract
A precise and productive one pot green protocol has been developed for the chemoselective synthesis of anthra[1,2-d]imidazole-6,11-diones using phosphotungstic acid (PTA) as a reusable catalyst on polyethylene glycol-400 (PEG-400) support under aerobic condition. It involves a simple synthetic procedure for a wide range of anthraimidazolediones from 1,2-diaminoanthraquinone and various aldehydes possessing π-enriched aromatic/heteroaromatic rings and lipophilic alkyl chains. In inert atmosphere, the reaction followed a different pathway, yielding a unique naphtho-quinoxaline derivative. Synthesized anthraimidazoledione derivatives had shown different self-aggregation morphologies influenced by the substitution pattern. A nice agreement between crystal packing pattern and photophysical properties with J-aggregation morphology was also observed. Representative derivatives exhibited contrasting fluorescence behaviors in transition from solution state to nanostructured solid state.
Introduction
In the new era of research, low-dimensional self-aggregated organic materials (LMSOM) have gained much attention for their broad spectrum of electrical, magnetic and optical properties.1–4 Performances of these organo-derived devices are correlated with the associative patterns present in their nanoarchitectures.5 Therefore, designing of self-assembled structures with preferred shape and size by tailoring the structural features of the building blocks, i.e. modulating the π–π stacking, hydrogen bonding and van der Waal's interactions etc., is the real challenge towards scientists.6 Literature study revealed that some typical glycal-based chiral anthraimidazoledione derivatives had exhibited self-association properties.7 Motivated by this observation we herein fabricated anthraimidazoledione scaffolds with a long lipophilic aliphatic chain or π-enriched aromatic/heteroaromatic substituents at the 2-position of the imidazole ring which might find applications in the field of organic nano-materials.The regular synthetic methodology for imidazole derivatives is cyclocondensation-cum-oxidation (CCO) reaction between 1,2-diamino compounds with aldehydes.8 However, 1,2-diaminoanthraquinone is exceptionally unreactive owing to the presence of an intramolecular hydrogen bond between N–H proton of the –NH2 group and the neighboring quinone C9-carbonyl oxygen. Reported methodologies for the syntheses of anthraimidazolediones involved harsh reaction conditions viz. refluxing in ethanol in presence of CF3CO2H,9 or Na2S2O5 at 130 °C,10 boiling in glacial CH3CO2H with sodium acetate,11a or with two equivalents of cupric acetate,11b in nitrobenzene at 120–140 °C for 8–24 hours,12 in PEG-400 at 120 °C for 18 hours13 and a two step process which required refluxing overnight with formic acid in ethanol followed by in situ oxidation with lead tetraacetate in acetic acid.14 Huang and Lin et.al.15 and Maiti et.al.7 had achieved syntheses of certain types of anthraimidazoledione derivatives at room temperature using concentrated sulphuric acid in DMF and a combo catalyst VO(acac)2–Ti(OBu)4–CeCl3 with molecular O2 as oxidant, respectively. It is pertinent to mention here that several of these methodologies were explored with some selected substrates only. So the search for an easier general protocol with environmentally benign catalytic system for the preparation of anthraimidazoledione derivatives was the major thrust of our work.
Commercially available heteropolyacids (HPAs) is very much significant for their “value adding properties”.16 HPAs with their Keggin structure, manifest very high Brönsted acidic character comparable to super-acids and show unique redox properties under aerobic condition.16 Of them, phosphotungstic acid (PTA) and phosphomolybdic acid (PMA) are already in use for several types of organic transformations.17–21 In this methodology preference was given to PTA over PMA for its higher acidity and greater thermal stability.22
The difficulties with solid HPAs as heterogeneous catalysts are their inherent nature to absorb polar and especially basic compounds by forming a “pseudoliquid” phase.23 The extent of absorption of those molecules depend on their molecular sizes.23 Additionally the compounds bearing alcoholic functionality are easily desorbed from HPAs whereas, for the basic amino compounds, elevated temperature is required for desorption.23 So, to maintain the virtues of hydroxylic environment and to avoid the “pseudoliquid” formation, poly(ethylene glycol) (PEG-400) was chosen as a medium because of its high thermal stability (up to 150–250 °C) towards oxidative environment of HPAs.24 PEGs are also known to act as phase-transfer catalyst since they possess both hydrophobic crown ether framework along with polar hydroxyl groups. These substances are cheap, easily available, biodegradable and may stand in comparison to other recently much publicized media like micellar systems or costly ionic liquids.25
Considering the above facts we have used PTA as solid heterogeneous catalyst on PEG-400 support to prepare anthra[1,2-d]imidazole-6,11-dione derivatives in presence of air. A comparative study was performed with the crystal structure, morphology and photo-physical property to correlate the substituent effect of the anthraimidazoledione derivatives.
Results and discussion
In the hunt for optimum reaction condition we carried out extensive screening tests employing a representative reaction between 1,2-diaminoanthraquinone (1) (1 mmol) and p-chlorobenzaldehyde (2c) (1 mmol) to furnish the product 3c (Scheme 1) in presence of different oxidants and acid catalysts. The other parameters such as solvent, temperature and reaction time etc. were also varied and the results have been summarized in Table 1. It is evident from Table 1 that the oxidant ammonium persulfate [(NH4)2S2O8] in ethanol solvent, used successfully for the synthesis of benzimidazoles,26 had negligible efficacy in this specific case even after 10 hours (Table 1, entry 1). N-Iodosuccinimide (NIS) in acetonitrile (Table 1, entries 2 and 3), which was initially employed in our laboratory for simple CCO type reaction at room temperature with good yield,27 led to moderate yield of 3c even at 60 °C. Both L-proline, as an organocatalyst in ethanol and polyphosphoric acid in dichloromethane with its dual nature as acid and dehydrating agent (Table 1, entries 4 and 5) failed to prove as suitable reagents after six and ten hours respectively. At this stage we turned our attention towards HPAs as more proficient catalytic system. We had chosen phosphotungstic acid (PTA) in dichloromethane at room temperature which increased the yield of 3c (35%) a bit though the reaction was slow and incomplete even after 20 h (Table 1, entry 6). But, employing PMA as catalyst in similar solvent system diminished the yield of 3c to 20% (Table 1, entry 7). It may be mentioned here that the general limitations of HPAs as solid heterogeneous catalysts are their low specific surface area (approx. 5 m2 g−1).23 To overcome this predicament, we had dispersed PTA on solid surface such as silica gel to obtain better surface catalytic activity (Table 1, entries 8 and 9) but no significant change in the yield of the product was obtained. This phenomenon could be interpreted as the loss of Keggin structure due to interaction between the surface silanol groups of silica and surface P atoms of PTA.23 Since HPAs absorb polar molecules which affect the yields of the corresponding products, we had explored the effects of polar protic and polar aprotic solvents to raise the yield of the products. It provided yields of the product in the order 55%, 60%, 63% and 70% in ethanol, acetonitrile, dimethylformamide and PEG-400 respectively (Table 1, entries 10–13). So PEG-400 was favored for its better activity as supportive medium. Henceforth, additional screening tests were performed with PTA in PEG-400 in order to find out the best reaction condition by varying temperature and time (Table 1, entries 14–18). Finally the optimum condition was achieved with the reaction being carried out at 80 °C for 2 hours by using PTA (10 mol%) in PEG-400 (3 mL) when 85% yield of the desired product (Table 1, entry 16) was obtained. | ||
| Scheme 1 Chemoselective CCO protocol for the synthesis of anthraimidazoledione derivatives. | ||
| Sl. no. | Oxidant/catalyst | Conditions | Time | Temp | Yieldb (%) |
|---|---|---|---|---|---|
| a Reaction: aldehyde (1 mmol), 1,2-diaminoanthraquinone (1 mmol).b Isolated and optimised yield, r.t. = room temperature. | |||||
| 1 | (NH4)2S2O8 (1.1 mmol) | EtOH | 10 h | r.t. | 0 |
| 2 | NIS (1.2 mmol) | ACN | 4 h | r.t. | 22 |
| 3 | NIS (1.2 mmol) | ACN | 6 h | 60 °C | 32 |
| 4 | L-Proline (30 mol%) | EtOH | 6 h | 60 °C | 15 |
| 5 | Polyphosphoric acid (10 mol%) | CH2Cl2 | 10 h | r.t. | 10 |
| 6 | Phosphotungstic acid (10 mol%) | CH2Cl2 | 20 h | r.t. | 35 |
| 7 | Phosphomolybdic acid (10 mol%) | CH2Cl2 | 20 h | r.t. | 20 |
| 8 | Phosphotungstic acid–SiO2 | ACN | 10 h | r.t. | 10 |
| 9 | Phosphotungstic acid–SiO2 | ACN | 6 h | Reflux | 20 |
| 10 | Phosphotungstic acid (10 mol%) | EtOH | 6 h | Reflux | 55 |
| 11 | Phosphotungstic acid (10 mol%) | ACN | 6 h | Reflux | 60 |
| 12 | Phosphotungstic acid (10 mol%) | DMF | 6 h | 60 °C | 63 |
| 13 | Phosphotungstic acid (10 mol%) | PEG-400 | 6 h | 50 °C | 70 |
| 14 | Phosphotungstic acid (20 mol%) | PEG-400 | 6 h | 50 °C | 75 |
| 15 | Phosphotungstic acid (10 mol%) | PEG-400 | 3 h | 70 °C | 80 |
| 16 | Phosphotungstic acid (10 mol%) | PEG-400 | 2 h | 80 °C | 85 |
| 17 | Phosphotungstic acid (10 mol%) | PEG-400 | 2 h | 100 °C | 85 |
| 18 | Phosphotungstic acid (10 mol%) | PEG-400 | 3 h | 80 °C | 85 |
To study the extent and limitations of this CCO protocol we explored the new methodology with variety of aldehydes possessing π-enriched aromatic/heteroaromatic rings and lipophilic alkyl chains reacting with 1,2-diaminoanthraquinone 1 (Scheme 1, Table 2). In all cases reaction progressed smoothly with good to excellent yields of the products. The reactions were consistently executed at the 1 mmol scale and no change of the product yield was observed when scaled up to the 10 mmol. High chemoselectivity was a rewarding aspect of this CCO protocol as no undesired 1,2-disubstituted derivative was obtained. It was pertinent to mention here that among the three types of aldehydes used, in terms of reaction times and isolated yields, aliphatic aldehydes with a chain length of C4–C16 (2m–2q, Table 2) showed highest reactivity followed by heteroaromatic aldehydes (2s–2w, Table 2) and then aromatic aldehydes (2a–2l, Table 2). Metallocene aldehyde like ferrocene carboxaldehyde, 2r was equally reactive and yielded 85% of 3r (Table 2) within 2 hours. All the new products were well characterized by spectral (IR, 1H and 13C NMR) and elemental analyses. The known products were identified by comparing the literature reports with their respective 1H-NMR spectra and melting points.
| a Reactions: aldehyde (1 mmol), 1,2-diaminoanthraquinone (1 mmol); PTA (10 mol%); solvent PEG 400; 80 °C. |
|---|
 |
In order to comprehend the reaction pathway for the formation of anthraimidazoledione derivatives involving PTA and PEG-400, an experimental strategy was developed with aldehyde 2o since 3o was formed within a short period of time. A mixture of 2o and 1 was stirred in PEG-400 at 80 °C for 45 minutes in absence of PTA and then quenched with water and worked up. Compound 1 was returned back in almost quantitative amount without isolation of imine or cyclo-condensed product. In a successive experiment, PTA (10 mol%) was added to the reaction mixture after 45 minutes and the reaction was complete in next 1 hour with the formation of the product, 3o. This result demonstrated the importance of both PTA and PEG-400 for cyclocondensation step which involved the aldehydic functional group of 2o with amino groups of 1. PEG-400 formed strong inter-molecular H-bonds with its free hydroxyl groups and C9-carbonyl oxygen of 1 thus reducing the strength of intra-molecular H-bond existing in compound 1 as discussed earlier. Both of the amino groups would now be available on the catalyst surface to react in tandem with aldehyde group in presence of PTA. At this stage it became imperative to understand the oxidation step of this CCO reaction which was carried out under aerobic condition. To investigate the role of aerial oxygen, if any, we repeated the experiment under dry nitrogen atmosphere when an altogether different reaction pathway was identified after the isolation of a novel compound, 2,3-dipentylnaphtho[2,3-f]quinoxaline-7,12(1H,4H)-dione, 3x along with unreacted 1,2-diaminoanthraquinone 1 approximately in the ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 (Scheme 2).
:2 (Scheme 2).
 | ||
| Scheme 2 1,2-Diaminoanthraquinone 1 reacts with hexanal 2o in presence of PTA under nitrogen atmosphere forming 3x. | ||
A plausible mechanistic pathway was presented (Scheme 3) to explain the formation of 3x under oxygen-free condition. Initially 1,2-diaminoanthraquinone 1 would form the diimine derivative 4 with hexanal 2o followed by a cascade pathway involving 6π electrocyclisation and two consecutive 1,5- and 1,3-prototropic shifts to produce 3x. Aromatization of the compound 3x did not occur in absence of oxidative environment probably due to the formation of intra-molecular hydrogen bond between N–H proton and the C-12 carbonyl group of the quinone ring which was reflected in the 1H-NMR spectrum of 3x. A broad singlet at δ 11.04 was assigned to H-bonded –NH– whereas the singlet at δ 6.89 was attributed to free –NH– function. This type of 6π electrocyclisation of diimine derived from aromatic 1,2-diamine and aldehyde in inert atmosphere was not reported earlier in the literature.
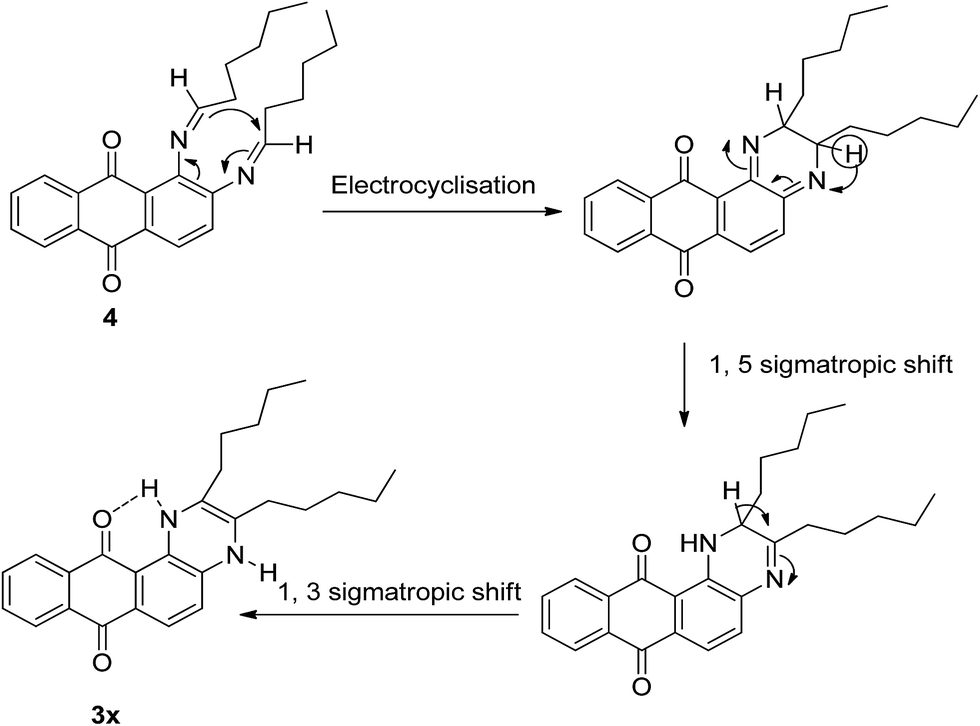 | ||
| Scheme 3 A plausible mechanistic pathway to the formation of 3x. | ||
Therefore from the above study it may be concluded that the aerial oxygen was required for oxidative dehydrogenation of the dihydroimidazole intermediate leading to the formation of heteroaromatic imidazole moiety and/or regeneration of PTA as oxidant from low valence states of tungsten.16e
A study regarding recycling of PTA was also performed and it was successfully reused four times without any pretreatment (Fig. 1). To verify the property of reused PTA, field emission scanning electron microscopy (FESEM) images of fresh and reused PTA (after first cycle) was taken which indicated no obvious change in the morphology of the catalyst (see ESI, Fig. S1†).
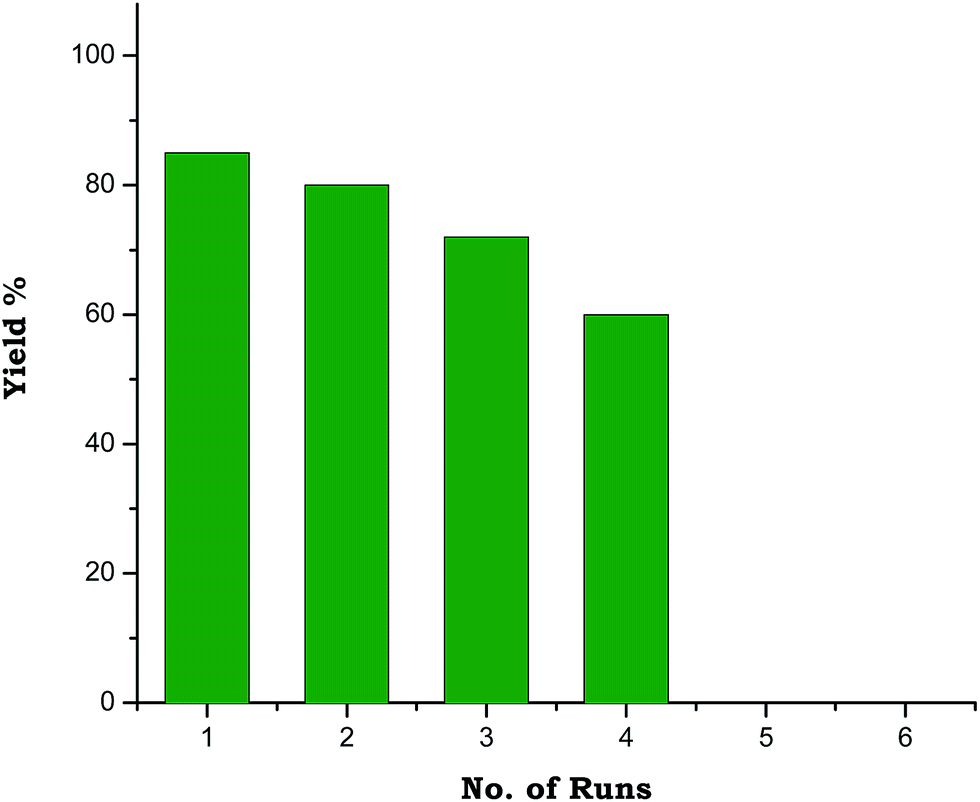 | ||
| Fig. 1 Reusability graph of PTA. | ||
The intrinsic non-covalent interactions of low molecular mass self-aggregated organic materials (LMSOM) are identifiable from its stacking pattern in the crystal lattice.4b,28,29 We could obtain a single crystal of the compound 3t (Fig. 2) (more information see ESI†).
 | ||
| Fig. 2 X-ray crystallographic structure of compound 3t. | ||
To investigate the effect of the crystal structure on molecular aggregation, we had performed analysis of the X-ray data of 3t using SHELXS 97,30 SHELXL 97,31 PLATON 99,32 ORTEP-32 (ref. 33) and WINGX system ver-1.64.34 (Fig. 3a–d). It has been observed from the analysis that several types of intermolecular hydrogen bonding interactions (Fig. 3a and b) are present in the crystal lattice of 3t (see ESI, Table S2†). Besides these intermolecular H-bonding interactions, an inter-molecular π–π stacking interaction is also present between two phenyl rings of anthraquinone unit of the two neighboring molecules with a centroid Cg⋯Cg distance of 3.596(4) Å to generate a 1D chain (Fig. 3c) along the crystallographic ‘c’ axis. Both H-bonding and π–π stacking interactions present in the crystal lattice of 3t formed a 3D network (Fig. 3d) with a brickwork type arrangement containing a knot of water molecule. A bent and head-to-tail array of the molecules in the crystal lattice indicated the J-aggregation property of the compound.4b
 | ||
| Fig. 3 (a) Hydrogen bonding pattern between water molecule and anthraimidazoledione moiety; (b) H-bonding between carbonyl oxygen and the aromatic hydrogen atom of neighboring molecule; (c) intermolecular π–π stacking between the two phenyl rings of neighboring molecule; (d) the 3D network of compound 3t having brickwork-like arrangement with a water molecule like a knot. | ||
A powder X-ray diffraction (PXRD) study was performed for 3t by using X-ray diffraction (XRD) analysis applying Sherrer's formula Dp = 0.941l/bcosq; where X-ray wave length (l) = 1.5406 Å, q = Bragg's diffraction angle for the planes (010), (110), (111), (200), (211), (220), (222), (311) and (440) and b as the corresponding full width at half maximum (FWHM) value.35 A comparison between the simulated PXRD derived from single crystal diffraction data of 3t with the experimental one clearly showed that it was pure and entirely crystalline in the bulk state (see ESI, Fig. S2†). Similar comparative PXRD studies between 3t and 3p (see ESI, Fig. S3†) using the planes (010), (110), (111), (200), (210) and (310) for 3p also demonstrated the existence of micro-crystallinity of 3p in its bulk state.35,36
It is relevant to mention here that 3t and 3p varies in their substituent pattern at 2-position of the imidazole ring with a thiophene ring for the former and a long aliphatic chain of ten carbon atoms for the latter. Non-covalent interactions observed in the crystal lattice of 3t and the micro-crystalline nature of both 3p and 3t as evinced from their respective PXRD data prompted us to Scanning Electron Microscopy (SEM) study. Compound 3t in toluene formed well-defined bundles of needle-shaped nanowires (width 129 nm) with a knot (Fig. 4a and b), whereas 3p spontaneously aggregates as ordered hemispherical particle assemblies with several holes on the surface ranging from 148–410 nm (Fig. 4c and d) in toluene. Presence of several hydrogen bonding and π–π stacking interactions within the framework of the π-extended planar molecule 3t would be the main driving force for the formation of the aforesaid morphology. On the other hand, molecule 3p assumes a unique amphiphilic nature bearing a polar head (anthraimidazoledione part) and an elongated flexible hydrophobic tail (ten carbon alkyl chain) which might compel the molecule to aggregate as an ordered particle assembly with holes on the surface.37 Thus in case of 3p, morphology is dominated by the van der Waals interactions involving the alkyl chains.37,38
 | ||
| Fig. 4 (a and b) SEM images of compound 3t showing bundle of needle-shaped nanowire with a knot; scale bar represent 1 μm and 200 nm respectively; (c and d) SEM images of compound 3p showing an ordered hemispherical particle assembly with several holes on surface; scale bar represents 1 μm and 200 nm respectively. | ||
As LMSOMs exhibit unique optical and optoelectronic properties both in their solid and solution state28 we took UV-VIS absorption spectra of the representative compounds (1 × 10−6 M) in polar aprotic and non-polar solvents and in the solid state (Fig. 5). In case of 3p, a π–π* transition band of the aromatic ring appears at ∼390 nm in toluene (1 × 10−6 M) and it shows slight solvent dependency in acetonitrile solvent (λmax = ∼380 nm, 1 × 10−6 M). The absorption spectrum of 3p in the solid state is found to be comparatively broad and flat at the peak position (Fig. 5a) may be due to the self-aggregation in the solid state.39 The same π–π* transition of 3t (421 nm in toluene and 417 nm in acetonitrile, both concentrations are 1 × 10−6 M) exhibited red shift due to extended conjugation (Fig. 5b). Here also absorption band in solid is broad and flat at the peak position (J-aggregation as per X-ray study). Systematic concentration dependant UV-VIS spectroscopy experiments for both 3p and 3t revealed that with increase in concentrations (from 10−6 to 10−4 M), the peak saturated at the OD value which were almost equivalent to the spectra obtained in their solid state.
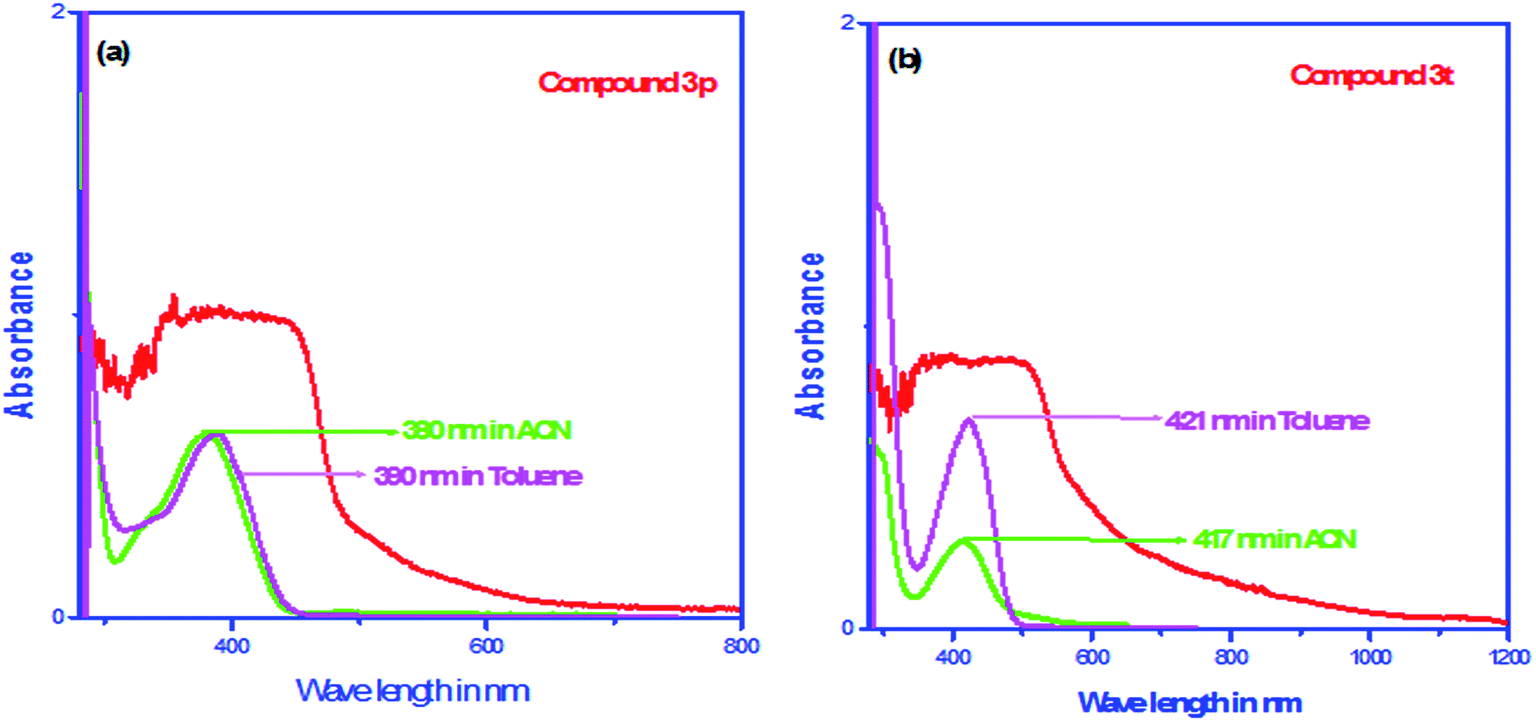 | ||
| Fig. 5 (a) Comparison of UV-VIS spectra of the compound 3p in solution (1 × 10−6 M) and solid phase. (b) Comparison of UV-VIS spectra of the compound 3t in solution (1 × 10−6 M) and solid phase. | ||
However 3p and 3t showed contrasting behavior in their emission spectroscopy. Compound 3t exhibited emission maxima at 527 nm (λex = 421 nm) in toluene solution (1 × 10−6 M) and showed higher solvent dependency (557 nm in ACN solvent, λex = 417 nm, 1 × 10−6 M Fig. 6a). In solid form, it furnished slightly different spectral pattern with peak at 607 nm (irradiated at ∼502 nm) having higher fluorescence intensity (Fig. 6b). In comparison, compound 3p was found to be non-emissive in acetonitrile (λex = 380 nm) as well as in toluene solution (λex = 390 nm) (both concentrations are 1 × 10−6 M), but showed high fluorescence intensity (nearly 400 times) in the aggregated solid state (λem = 530 nm, irradiated at ∼439 nm) as indicated in Fig. 6a. Presence of long tail with high flexibility may be the cause of non-emission property of 3p in solution (1 × 10−6 M) as flexible bonds usually open up non-radiative channels.
 | ||
| Fig. 6 (a) Comparison of fluorescence spectra of the compound 3p in acetonitrile (λex = 380 nm, 1 × 10−6 M) and toluene solution (λex = 390 nm, 1 × 10−6 M) and solid phase (λex = 439 nm). (b) Comparison of fluorescence spectra of the compound 3t in ACN (λex = 417 nm, 1 × 10−6 M) and in toluene solution (λex = 421 nm, 1 × 10−6 M) and solid phase (λex = 502 nm). | ||
Citations of some particular organic entities having enhanced emission in their solid state are correlated with the intra- and intermolecular forces present in the molecules.39 Intra-molecular effects like conformational aspects of the chromophore influence the radiation process in solid state or in solution phase.40 Inter-molecular interactions such as molecular aggregation (H-type or J-type) are reported to control the fluorescence efficiency of the molecule.41 Generally J-aggregated molecules showed comparatively higher fluorescence intensity due to their bent or head-to-tail arrangement with a red shift in UV absorption maxima.41 But the unusual strong fluorescence efficiency of the compound 3p in J-aggregated form compared to non-emissive nature of its acetonitrile or toluene solution can be explained in terms of non-radiative channels operated through flexible bonds. In 3p, intermolecular forces cause aggregation induced rigidity resulting in high solid-state emission. But in solution, both in polar or non-polar medium, conformational aspects of the long flexible aliphatic chain is dominating, thus favoring energy dissipative paths and it becomes non-emissive. As a consequence, 3p imparts a fluorescence “ON–OFF” mechanism from solid to solution state. In case of 3t, structural flexibility is less and it shows emission in the solution phase and enhances its emission intensity in the solid by the formation of J-aggregated state.
The results obtained from emission spectrum prompted us to carry out DFT calculations for 3p and 3t. The HOMO–LUMO pictures obtained by using B3LYP/6-311++G as the functional and basis set showed that HOMOs for both molecules are localized on their respective substituent arms whereas the LUMOs are contributed by the anthraquinone units (see ESI, Fig. S4†). For this reason, photophysical properties of these molecules would be guided by the nature of the substituent present at the 2-position of the imidazole ring as observed in this case.42
Considering all the morphological and photophysical phenomena we may anticipate that anthraimidazolediones containing a lipophilic long aliphatic chain and polar anthraquinone moiety could find application in the field of organogelators.37 Furthermore, the holes present in the SEM structure of 3p are very unusual which might find use as a receptor site or host or a transporter.43 Both 3p and 3t, due to their remarkable fluorescent nature, are eligible candidates for application as chemical sensors.
Conclusions
In conclusion, a straightforward tandem green protocol was developed for the chemoselective synthesis of anthra[1,2-d]imidazole-6,11-dione derivatives via CCO reaction employing PTA as reusable catalyst on PEG-400 support in open air. In inert atmosphere, methodology adopted a complete different pathway leading to the formation of unique naphtho-quinoxaline derivative. Wide range of applicability of the reaction procedure was proved by using an array of aldehydes which furnish good to excellent yields of the anthraimidazoledione derivatives. Single crystal data of compound 3t reveals distinct crystal packing pattern with π–π stacking and hydrogen bonding interactions. 3p and 3t, bearing different substituents at 2-position of the anthra[1,2-d]imidazole-6,11-dione moieties, showed micro-crystalline nature in bulk state from PXRD studies and different morphologies in their corresponding SEM images which justified the effect of substituents. DFT studies of aforesaid compounds revealed similarities in their HOMO–LUMO characteristics. Both molecules show comparable J-aggregation behavior and form LMSOMs in their solid state as evidenced from photophysical studies. Further studies related to the generation of novel nanomaterials based on this scaffold possessing interesting optoelectronic and gelation properties and also as chemosensing agents are in progress in our laboratory.Experimental
General reaction procedure for the formation of anthra [1,2-d]imidazole-6,11-diones (3a–w)
A mixture of 1,2-diaminoanthraquinone 1 (1 mmol), aldehydes 2 (1 mmol) were stirred at 80 °C in an oil bath in 3 mL PEG-400 support in open air. To this stirring mixture, phosphotungstic acid (10 mol%) was added and stirred for stipulated time. After completion (as monitored by TLC), the reaction mixture was cooled to 0–5 °C and extracted with ethyl acetate. The reaction mixture was filtered and the filtered out solid PTA was washed with acetone and dried under vacuum for reuse. Ethyl acetate part was washed with water thrice to remove PEG-400 and evaporated under vacuum. Pure product was isolated by column chromatography over silica gel using different mixtures of petroleum ether: ethyl acetate.Known products were compared with their mp and spectral data as obtained from concerned literature. Spectral data of representative compounds, 3p and 3t are listed below;
General reaction procedure for synthesis of 2,3-dipentylnaphtho[2,3-f]quinoxaline-7,12(1H,4H)-dione (3x)
A mixture of 1,2-diaminoanthraquinone 1 (1 mmol), aldehyde 2o (1.2 mmol) were stirred at 80 °C in an oil bath in 3 mL PEG-400 support under N2-atmosphere. To this stirring mixture phosphotungstic acid (10 mol%) was added and stirred for one hour. Then the reaction mixture was cooled to 0–5 °C and extracted with ethyl acetate. Ethyl acetate part was washed with water thrice to remove PEG-400 and evaporated under vacuum. Pure product was isolated by column chromatography over silica gel using different mixtures of petroleum ether:ethyl acetate.
Acknowledgements
We gratefully acknowledge the financial support received from the University of Calcutta. Crystallography was performed at the DST-FIST, India-funded Single Crystal Diffractometer facility at the Department of Chemistry, University of Calcutta. We would like to thank the Centre for Research in Nanoscience and Nanotechnology, University of Calcutta for providing SEM and FESEM facilities. We express our gratitude to Lakshmi Kanta Das and Aniruddha Ganguly for their valuable suggestions. We further express our gratefulness to Dr Dipankar Chakraborty for providing powder XRD facilities.References
- (a) M.-B. Madec, D. Crouch, G. Llorente, T. J. Whittle, M. Geogheganc and S. G. Yeates, J. Mater. Chem., 2008, 18, 3230 RSC; (b) T.-T. Bui and F. Goubard, EPJ Photovoltaics, 2013, 4, 40402 CrossRef; (c) V. Coropceanu, H. Li, P. Winget, L. Zhu and J.-L. Brédas, Annu. Rev. Mater. Res., 2013, 43, 63 CrossRef CAS.
- (a) C. Reese and Z. Bao, Mater. Today, 2007, 10, 20 CrossRef CAS; (b) C. D. Dimitrakopoulos and D. J. Mascaro, IBM J. Res. Dev., 2001, 45, 11 CrossRef CAS; (c) A. Facchetti, Mater. Today, 2007, 10, 28 CrossRef CAS.
- D. Volz, M. Wallesch, C. Fléchon, M. Danz, A. Verma, J. M. Navarro, D. M. Zink, S. Bräse and T. Baumann, Green Chem., 2015, 17, 1988 RSC.
- (a) T. W. Bell and N. M. Hext, Chem. Soc. Rev., 2004, 33, 589 CAS; (b) Y. S. Zhao, H. Fu, A. Peng, Y. Ma, D. Xiao and J. Yao, Adv. Mater., 2008, 20, 2859 CrossRef CAS; (c) M. Mille, J.-F. Lamère, F. Rodrigues and S. Fery-Forgues, Langmuir, 2008, 24, 2671 CrossRef CAS PubMed.
- (a) Q. H. Cui, Y. S. Zhao and J. Yao, Adv. Mater., 2014, 26, 6852 CrossRef CAS PubMed; (b) M.-S. Schiedel, C. A. Briehn and P. Bäuerle, Angew. Chem., Int. Ed., 2001, 40, 4677 CrossRef CAS.
- (a) L. Zang, Y. Che and J. S. Moore, Acc. Chem. Res., 2008, 41, 1596 CrossRef CAS PubMed; (b) Y. S. Zhao, W. Yang, D. Xiao, X. Sheng, X. Yang, Z. Shuai, Y. Luo and J. Yao, Chem. Mater., 2005, 17, 6430 CrossRef CAS.
- D. K. Maiti, S. Halder, P. Pandit, N. Chatterjee, D. De Joarder, N. Pramanik, Y. Saima, A. Patra and P. K. Maiti, J. Org. Chem., 2009, 74, 8086 CrossRef CAS PubMed.
- S. S. Panda, R. Malik and S. C. Jain, Curr. Org. Chem., 2012, 16, 1905 CrossRef CAS.
- S. Saha, A. Ghosh, P. Mahato, S. Mishra, S. K. Mishra, E. Suresh, S. Das and A. Das, Org. Lett., 2010, 12, 3406 CrossRef CAS PubMed.
- S. M. Sondhi, J. Singh, P. Roy, S. K. Agrawal and A. K. Saxena, Med. Chem. Res., 2011, 20, 887 CrossRef CAS.
- (a) E. N. da Silva Jr, B. C. Cavalcanti, T. T. Guimarães, M. do C. F. R. Pinto, I. O. Cabral, C. Pessoa, L. V. Costa-Lotufo, M. O. de Moraes, C. K. Z. de Andrade, M. R. dos Santos, C. A. de Simone, M. O. F. Goulart and A. V. Pinto, Eur. J. Med. Chem., 2011, 46, 399 CrossRef PubMed; (b) Y. Ooyama, T. Nakamura and K. Yoshida, New J. Chem., 2005, 29, 447 RSC.
- N. Kumari, S. Jha and S. Bhattacharya, J. Org. Chem., 2011, 76, 8215 CrossRef CAS PubMed.
- V. Luxami and S. Kumar, Dalton Trans., 2012, 41, 4588 RSC.
- (a) R. M. F. Batista, E. Oliveira, S. P. G. Costa, C. Lodeiro and M. M. M. Raposo, Org. Lett., 2007, 9, 3201 CrossRef CAS PubMed; (b) R. M. F. Batista, S. P. G. Costa and M. M. M. Raposo, J. Photochem. Photobiol., A, 2013, 259, 33 CrossRef CAS; (c) R. M. F. Batista, E. Oliveira, S. P. G. Costa, C. Lodeiro and M. M. M. Raposo, Supramol. Chem., 2014, 26, 71 CrossRef CAS; (d) R. M. F. Batista, S. P. G. Costa and M. M. M. Raposo, Sens. Actuators, B, 2014, 191, 791 CrossRef CAS.
- H.-S. Huang, T.-C. Chen, R.-H. Chen, K.-F. Huang, F.-C. Huang, J.-R. Jhan, C.-L. Chen, C.-C. Lee, Y. Lo and J.-J. Lin, Bioorg. Med. Chem., 2009, 17, 7418 CrossRef CAS PubMed.
- (a) G. Li, Y. Ding, J. Wang, X. Wang and J. Suo, J. Mol. Catal. A: Chem., 2007, 262, 67 CrossRef CAS; (b) A. Haimov and R. Neumann, Chem. Commun., 2002, 876 RSC; (c) D. C. Duncan and C. L. Hill, J. Am. Chem. Soc., 1997, 119, 243 CrossRef CAS; (d) R. Neumann and M. Levin, J. Am. Chem. Soc., 1992, 114, 7278 CrossRef CAS; (e) A. Hiskia and E. Papaconstantinou, Inorg. Chem., 1992, 31, 167 CrossRef; (f) R. Neumann and M. Levin, J. Org. Chem., 1991, 56, 5707 CrossRef CAS.
- (a) S.-S. Wang and G.-Y. Yang, Chem. Rev., 2015, 115, 4893 CrossRef CAS PubMed; (b) Y. Zhou, G. Chen, Z. Long and J. Wang, RSC Adv., 2014, 4, 42092 RSC; (c) M. Misono, I. Ono, G. Koyano and A. Aoshima, Pure Appl. Chem., 2000, 72, 1305 CrossRef CAS.
- M. Dabiri and S. Bashribod, Molecules, 2009, 14, 1126 CrossRef CAS PubMed.
- R. S. Keri and K. M. Hosamani, Catal. Lett., 2009, 131, 321 CrossRef CAS.
- K. Vanlaldinpuia and G. Bez, Tetrahedron Lett., 2011, 52, 3759 CrossRef CAS.
- K. C. S. Achar, K. M. Hosamani and H. R. Seetharamareddy, Synth. Commun., 2011, 41, 33 CrossRef CAS.
- I. V. Kozhevnikov, J. Mol. Catal. A: Chem., 2009, 305, 104 CrossRef CAS.
- N. Mizuno and M. Misono, Chem. Rev., 1998, 98, 199 CrossRef CAS PubMed.
- E. Colacino, J. Martinez, F. Lamaty, L. S. Patrikeeva, L. L. Khemchyan, V. P. Ananikov and I. P. Beletskaya, Coord. Chem. Rev., 2012, 256, 2893 CrossRef CAS.
- J. Chen, S. K. Spear, J. G. Huddleston and R. D. Rogers, Green Chem., 2005, 7, 64 RSC.
- K. Bahrami, M. M. Khodaei and A. Nejati, Green Chem., 2010, 12, 1237 RSC.
- B. Bhattacharyya and K. P. Dhara, J. Indian Chem. Soc., 2013, 90, 1749 CAS.
- Y. Yan and Y. S. Zhao, Chem. Soc. Rev., 2014, 43, 4325 RSC.
- L. J. Prins, D. N. Reinhoudt and P. Timmerman, Angew. Chem., Int. Ed., 2001, 40, 2382 CrossRef CAS.
- G. M. Sheldrick, SHELXS 97, Program for Structure Solution, University of Göttingen, Germany, 1997 Search PubMed.
- G. M. Sheldrick, SHELXL 97, Program for Crystal Structure Refinement, University of Göttingen, Germany, 1997 Search PubMed.
- A. L. Spek, PLATON. Molecular Geometry Program, J. Appl. Crystallogr., 2003, 36, 7 CrossRef CAS.
- L. J. Farrugia, J. Appl. Crystallogr., 1997, 30, 565 CrossRef CAS.
- L. J. Farrugia, J. Appl. Crystallogr., 1999, 32, 837 CrossRef CAS.
- Y. Waseda, E. Matsubara and K. Shinoda, X- Ray Diffraction Crystallography Introduction, Examples and Solved Problems, Springer-Verlag Berlin Heidelberg, 2011 Search PubMed.
- Y. Wang, H. Fu, A. Peng, Y. Zhao, J. Ma, Y. Ma and J. Yao, Chem. Commun., 2007, 1623 RSC.
- X. Shen, T. Jiao, Q. Zhang, H. Guo, Y. Lv, J. Zhou and F. Gao, J. Nanomater., 2013, 409087 Search PubMed.
- T. Jiao, Q. Huang, Q. Zhang, D. Xiao, J. Zhou and F. Gao, Nanoscale Res. Lett., 2013, 8, 278 CrossRef PubMed.
- B.-K. An, S.-K. Kwon, S.-D. Jung and S. Y. Park, J. Am. Chem. Soc., 2002, 124, 14410 CrossRef CAS PubMed.
- M. M. Souza, G. Rumbles, I. R. Gould, H. Amer, I. D. W. Samuel, S. C. Moratti and A. B. Holmes, Synth. Met., 2000, 111, 539 CrossRef.
- W. I. Gruszecki, J. Biol. Phys., 1991, 18, 99 CrossRef CAS.
- (a) Y. Yang, H. Wang, F. Liu, D. Yang, S. Bo, L. Qui, Z. Zhen and X. Liu, Phys. Chem. Chem. Phys., 2015, 17, 5776 RSC; (b) G.-Y. Li, G.-J. Zhao, Y.-H. Liu, K.-L. Han and G.-Z. He, J. Comput. Chem., 2010, 31, 1759 CrossRef CAS PubMed.
- T.-T. Bui and F. Goubard, EPJ Photovoltaics, 2013, 4, 40402 CrossRef.
- X. J. Peng, Y. K. Wu, J. L. Fan, M. Z. Tian and K.-L. Han, J. Org. Chem., 2005, 170, 10524 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1053279. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ra19190d |
| This journal is © The Royal Society of Chemistry 2016 |