
Effect of anion type on the synthesis of mesoporous nanostructured MgO, and its excellent adsorption capacity for the removal of toxic heavy metal ions from water†
Abstract
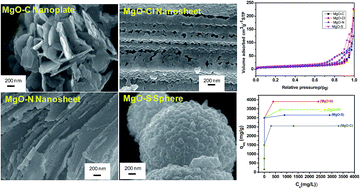
Mesoporous MgO nanostructures of different morphologies like nanoplates, nanosheets and nanoparticle assembled microspheres were prepared by a simple hydrothermal method at 180 °C/5 h in the absence of any organic templates. The products were characterized by X-ray diffraction (XRD), differential thermal analysis (DTA), thermogravimetry (TG), Fourier transform infrared spectroscopy (FTIR), N2 adsorption–desorption, field emission scanning electron microscopy (FESEM), and transmission electron microscopy (TEM). The role of anions of different magnesium precursors on the morphology, textural properties, and heavy metal ion (Pb(II) and Cd(II)) adsorption capacity of the products was studied. The adsorption data were interpreted with Langmuir and Freundlich models. The effect of contact time, adsorbate concentration, pH and temperature on the adsorption capacity of the products was investigated. Nanosheet-like MgO obtained from chloride and nitrate salts of magnesium having higher pore size rendered better adsorption capacity than that prepared from sulphate and carbonate sources of magnesium. The prepared MgO nanostructures showed maximum adsorption capacities up to 3900 mg g−1 and 2980 mg g−1 for Pb(II) and Cd(II) ions, respectively.
 Please wait while we load your content...
Please wait while we load your content...

