
Magnetized property effect of a non-aqueous solvent upon complex formation between kryptofix 22DD with lanthanum(iii) cation: experimental aspects and molecular dynamics simulation
Abstract
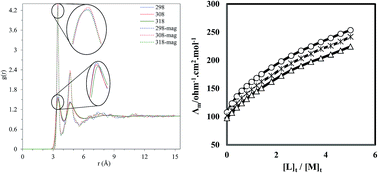
The complexation reaction between the La3+ cation and a macrocyclic ligand (kryptofix 22DD) in ordinary methanol (MeOH) and extraordinary methanol solvent (magnetized methanol) exposed to a magnetic field at different temperatures has been investigated using the conductometric method, in order to determine the effect of magnetic solvents on the thermodynamics of the complexation process between the metal cation and the ligand. The stoichiometry of the complex formed between the La3+ cation and the ligand is 1 : 1 [M : L] in both solvent systems. Comparison of the corresponding molar conductivity versus [L]t/[M]t for (kryptofix 22DD·La)3+ complex shows that the complex formed between the metal cation and kryptofix 22DD in the magnetized methanol is weaker compared to the ordinary methanol. The results of the density functional theory shows that addition of the metal ion to the ligand completely deforms the ligand from its planar shape and the orientation of two carbon chains attached to the ring changes in comparison with the free ligand. In addition, the molecular dynamics simulation can shed light on the influence of the magnetic field, from the molecular point of view, on the properties of solvents and is able to explain the variation of hydrogen bond and transport properties. These changes may be the reason for the interaction of the solvent and the solute as well as the stability constant of the complex formed between the La3+ cation and kryptofix 22DD in solution. As the strength of the magnetic field increases, the number of the hydrogen bonds formed between the methanol molecules increases and the methanol self-diffusion coefficient decreases. These changes restrict the movement of the molecules which results in increasing the viscosity of the methanol and therefore the stability constant of the (kryptofix 22DD·La)3+ complex decreases in magnetized methanol with respect to the ordinary methanol.
 Please wait while we load your content...
Please wait while we load your content...

