
Enzymatically crosslinked epsilon-poly-l-lysine hydrogels with inherent antibacterial properties for wound infection prevention†
Abstract
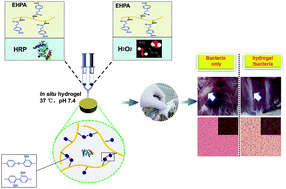
In this study, in situ forming epsilon-poly-L-lysine (EPL) bioadhesive hydrogels were fabricated for wound infection prevention. The hydrogel precursor polymer consisted of EPL backbone conjugated with phenol groups of hydroxyphenylpropionic acid (HPA). The chemical modification was characterized by 1H NMR and UV spectroscopies. An enzymatic crosslinking method was employed to copolymerize EPL-HPA (EHPA) in the presence of horseradish peroxidase (HRP) and hydrogen peroxide (H2O2). The HRP crosslinking system allows rapid gelation within several seconds. The hydrogel properties, including the gelation rate, the mechanical strength, and the degradation behavior, were adjustable by controlling the concentration of HRP, H2O2, and the polymer. The adhesiveness ranged from 10 kPa to 35 kPa, which is higher than that of fibrin glue. Cytotoxicity assay with L929 fibroblasts revealed that the hydrogels possessed good in vitro biocompatibility. In addition, in vitro antibacterial studies showed that these bioadhesive hydrogels exhibited an impressively wide spectrum of antimicrobial activity against both Gram-negative and Gram-positive bacteria. Furthermore, evaluations of in vivo subcutaneous injection revealed that the hydrogels have a significant in vivo anti-infection effect. These results suggest that the in situ forming EPL-based hydrogels are interesting and promising bioadhesive materials with inherent antibacterial properties for wound infection prevention.
 Please wait while we load your content...
Please wait while we load your content...

