
Theoretical and experimental investigation of the polyeletrophilic β-enamino diketone: straightforward and highly regioselective synthesis of 1,4,5-trisubstituted pyrazoles and pyrazolo[3,4-d]pyridazinones†
Michael J. V. da Silvaa,
Raí G. M. Silvaa,
Ulisses Z. Meloa,
Davana S. Gonçalvesa,
Davi F. Backb,
Sidnei Mourac,
Rodrigo M. Pontesa,
Ernani A. Bassoa,
Gisele F. Gauzea and
Fernanda A. Rosa*a
aEstereoquímica de Compostos Orgânicos e Docking Molecular, Departamento de Química, Universidade Estadual de Maringá (UEM), 87030-900, Maringá PR, Brazil. Web: http://www.dqi.uem.br/professores/eabasso/geco/E-mail: farosa@uem.br
bDepartamento de Química, Universidade Federal de Santa Maria (UFSM), 97110-970 – Santa Maria, RS, Brazil
cLaboratório de Produtos Naturais e Sintéticos, Instituto de Biotecnologia, Universidade de Caxias do Sul (UCS), 295070-560, Caxias do Sul, RS, Brazil
First published on 22nd December 2015
Abstract
Obtaining a new precursor enamino diketone with five electrophilic centers is reported, along with theoretical and experimental studies of its reactivity against mono- or dinucleophiles. The Fukui function showed that the β-carbon is the most electrophilic center, followed by the carbonyl ketone and the carbonyl ester, respectively. The reaction of enamino diketone with aniline and hydrazines allowed for the synthesis of a new enamino diketone and 1,4-disubstituted pyrazoles-5-carboxylates, respectively. The regiochemistry and mechanism of syntheses of 1,4-disubstituted pyrazoles-5-carboxylates were determined from reaction monitoring by ESI-MS, NMR analysis and crystallographic data, and fully agreed with the theoretical results. The versatility and efficiency of the enamino diketone was demonstrated by the reaction with hydrazines furnishing multi-functionalized pyrazoles and pyrazolo[3,4-d]pyridazinone derivatives with high regioselectivity.
Introduction
The synthesis of nitrogen-containing heterocycles is a very useful and attractive area of research in organic chemistry because of the wide pharmacological and agrochemical applications of such compounds.1 Multi-functionalized pyrazoles constitute the core structure of several commercial drugs such as celecoxib,2 sildenafil3 and the insecticide fipronil.4 Moreover, compounds containing a pyrazole scaffold have been shown to exhibit a wide spectrum of biological activities2,3,5–11 such as anti-hyperglycemic,6 anti-bacterial,7 sedative-hypnotic,8 anti-inflammatory,9 anti-cancer10 and analgesic11 properties. In addition, multi-functionalized pyrazoles represent useful synthetic building blocks in organic chemistry, for example to obtain pyrazole–pyridazinones.12 Thus, there is continuing interest in the development of efficient methods to obtain multi-functionalized pyrazoles. One of the most popular, oldest, and most explored methods for the synthesis of pyrazoles is classical cyclocondensation 3 + 2 atom fragments, where β-diketones or analogous compounds are used as three-atom building blocks and monosubstituted hydrazines as the two-atom fragment (Knorr synthesis).13 However, the application of this method using unsymmetrical β-diketones leads to the frequent formation of a regioisomeric mixture of pyrazoles with generally poor selectivity.14 A variation of this method by using β-enamino diketones instead β-diketones generally allows better control of regioselectivity.In recent years, the β-enamino diketones15 have been employed as building blocks in multi-functionalized heterocyclic compound synthesis due to their excellent 1,3-dieletrophilic system. The chemical structure of these compounds has more than two electrophilic centers, so cyclocondensation reactions with nucleophiles such as arylamines (Ar-NH2), hydrazines (R–NHNH2) and amidines (NH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C(R)–NH2) in general, have shown high regioselectivity.16 These reactions have proven to be attractive because it permitted the attainment of a variety of multi-functionalized heterocyclic structures such as 1,4-disubstituted pyrazole-5-carboxylates,16h 2,5-disubstituted pyrimidine-4-carboxylates,16i pyrimide-pyridazinones,16i pyrazole-pyridazinones12f and 1,4-disubstituted-pyrrolidine-2,3,5-triones16j (Scheme 1). In relation to regiochemistry, the studies revealed that when using an R–NHNH2 dinucleophile such as tert-butylhydrazine hydrochloride or carboxymethylhydrazine, the reactions were highly regioselective with the first nucleophilic attack on the β-carbon followed by a second attack on the carbonylic carbon neighbor of the ester group. On the other hand, preliminary results have shown that the reaction with phenylhydrazine and with hydrazine monohydrate leads to a regioisomeric mixture of pyrazoles.16h The reaction with NHC(R)–NH2 dinucleophiles such as benzamidine hydrochloride and 1H-pyrazole-1-carboxamidine hydrochloride also has been invastigted.16i When benzamidine hydrochloride was used, the regiochemistry of the reaction depended on the structure of the enamino diketone, while the reaction with 1H-pyrazole-1-carboxamidine hydrochloride was highly selective. Another study showed that the reaction of enamino diketones (R = CCl3) with arylamines was also highly selective. It should be noted that the first nucleophilic attack occurred at the carbonylic carbon neighbor of the CCl3 group and the second attack was at the carbonylic carbon neighbor of the ester group16j (Scheme 1).
C(R)–NH2) in general, have shown high regioselectivity.16 These reactions have proven to be attractive because it permitted the attainment of a variety of multi-functionalized heterocyclic structures such as 1,4-disubstituted pyrazole-5-carboxylates,16h 2,5-disubstituted pyrimidine-4-carboxylates,16i pyrimide-pyridazinones,16i pyrazole-pyridazinones12f and 1,4-disubstituted-pyrrolidine-2,3,5-triones16j (Scheme 1). In relation to regiochemistry, the studies revealed that when using an R–NHNH2 dinucleophile such as tert-butylhydrazine hydrochloride or carboxymethylhydrazine, the reactions were highly regioselective with the first nucleophilic attack on the β-carbon followed by a second attack on the carbonylic carbon neighbor of the ester group. On the other hand, preliminary results have shown that the reaction with phenylhydrazine and with hydrazine monohydrate leads to a regioisomeric mixture of pyrazoles.16h The reaction with NHC(R)–NH2 dinucleophiles such as benzamidine hydrochloride and 1H-pyrazole-1-carboxamidine hydrochloride also has been invastigted.16i When benzamidine hydrochloride was used, the regiochemistry of the reaction depended on the structure of the enamino diketone, while the reaction with 1H-pyrazole-1-carboxamidine hydrochloride was highly selective. Another study showed that the reaction of enamino diketones (R = CCl3) with arylamines was also highly selective. It should be noted that the first nucleophilic attack occurred at the carbonylic carbon neighbor of the CCl3 group and the second attack was at the carbonylic carbon neighbor of the ester group16j (Scheme 1).
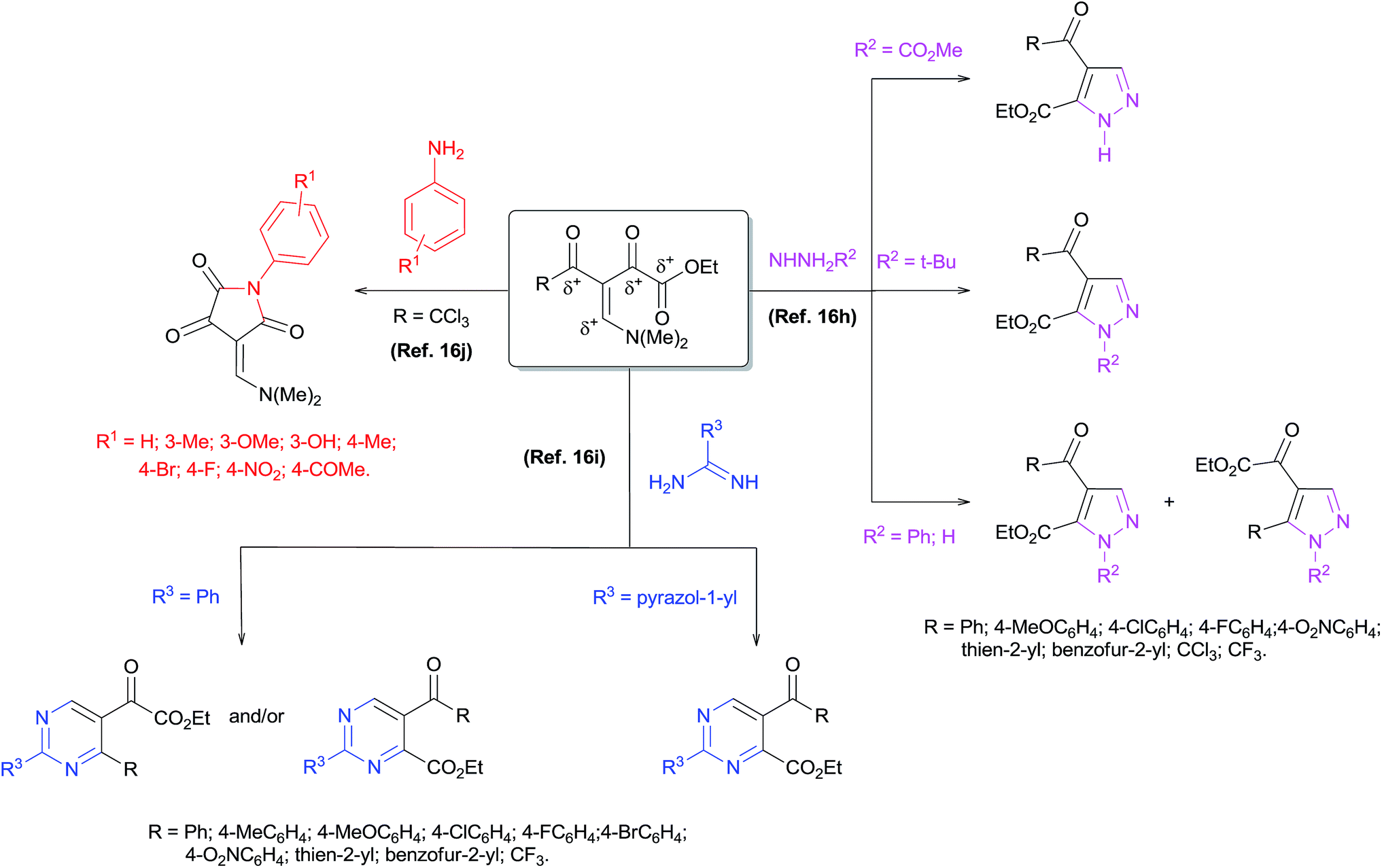 | ||
| Scheme 1 Syntheses of azo-heterocycles from β-enamino diketones. | ||
Therefore, the enamino diketones have become potentially useful 1,3-dieletrophilic synthons for broad regioselective construction of five- and six-membered multi-functionalized heterocycles. Whereas in Knorr pyrazole synthesis the regioselectivity of the reaction depends on the reactivity of both carbonyl carbons of β-diketone or the reactivity difference of the carbonyl group and the β-carbon in the case of enamino ketones, a detailed study of the reactivity of a dieletrophilic system of enamino diketones would be fundamental because of the significant differences in reactivity and electronic properties of the carbonyl groups and β-carbons present in these compounds. Furthermore, to our knowledge, a study of the quantum mechanics of enamino diketones in order to evaluate global and local reactivity descriptors of this polyeletrophilic system has not been reported in the literature. Considering that the development of efficient protocols for the regioselective synthesis of polysubstituted pyrazoles is highly desirable, we conducted a theoretical and experimental investigation of the structure of a new β-enamino diketone 2 (Scheme 2). This allowed us to analyze the reactive potential of this precursor against nucleophiles/dinucleophiles. Furthermore, we have developed an efficient synthesis of 1,4,5-trisubstituted pyrazoles with high regioselectivity by the cyclocondensation reaction of β-enamino diketone 2 with hydrazines. We have also demonstrated the effective synthesis of pyrazolo[3,4-d]pyridazinones by the reaction of polysubstituted pyrazoles with hydrazines or directly from precursor 2 in a one-pot reaction. This paper reports the results of these studies.
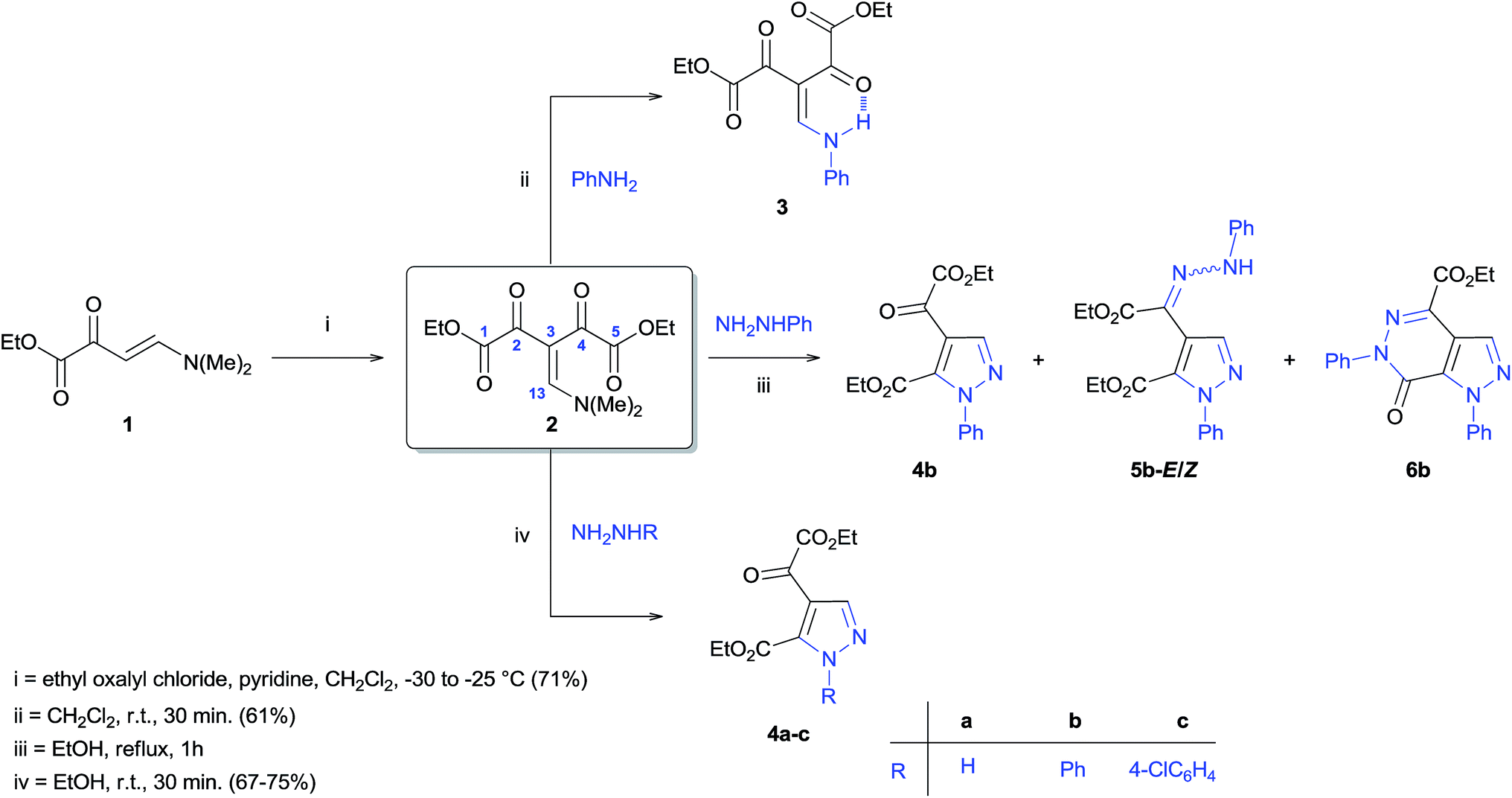 | ||
| Scheme 2 Synthesis of β-enamino diketones 2 and 3, reaction of 2 with unsymmetrical dinucleophiles R–NHNH2. | ||
Results and discussion
Theoretical investigation of β-enamino diketone 2
C1–C2O), d2 (OC2–C3C13), d3 (C13C3–C4O) and d4 (OC4–C5O). From these combinations, 16 different conformations (Fig. 1 and Table 1, ESI†) were generated and subsequently optimized at M06-2X/6-31+G(d,p) level.17 The dipole moment, relative energies and populations for the most stable optimized conformations are presented in Table 1, along with the numbered structures, from which the relations between the energy, structural and reactivity characteristics of the molecule can be clarified. Solvation effects in dichlorometane and ethanol were added with the integral equation formalism polarizable continuum model (IEF-PCM)18 that not include explicit hydrogen bond.
|
|||||||
|---|---|---|---|---|---|---|---|
| Conformation | μ | Gas phase | Dichloromethane | Ethanol | |||
| Erel | Populationc | Erel | Populationc | Erel | Populationc | ||
| a In M06-2X/6-31+G(d,p) at T = 298 K.b In IEF-PCM-M06-2X/6-31+G(d,p) at T = 298 K.c Calculated from x2/x1 = e−Erel/RT were x1, x2 = mole fraction and R = gas constant. | |||||||
| ASSA | 3.00 | 0.00 | 71 | 1.85 | 23 | 3.05 | 16 |
| ASSS | 4.60 | 3.60 | 17 | 0.00 | 47 | 0.00 | 56 |
| SSSA | 6.02 | 4.56 | 11 | → SSSS | → ASSA | ||
| SSSS | 4.57 | 11.01 | 1 | 1.14 | 30 | 1.81 | 27 |
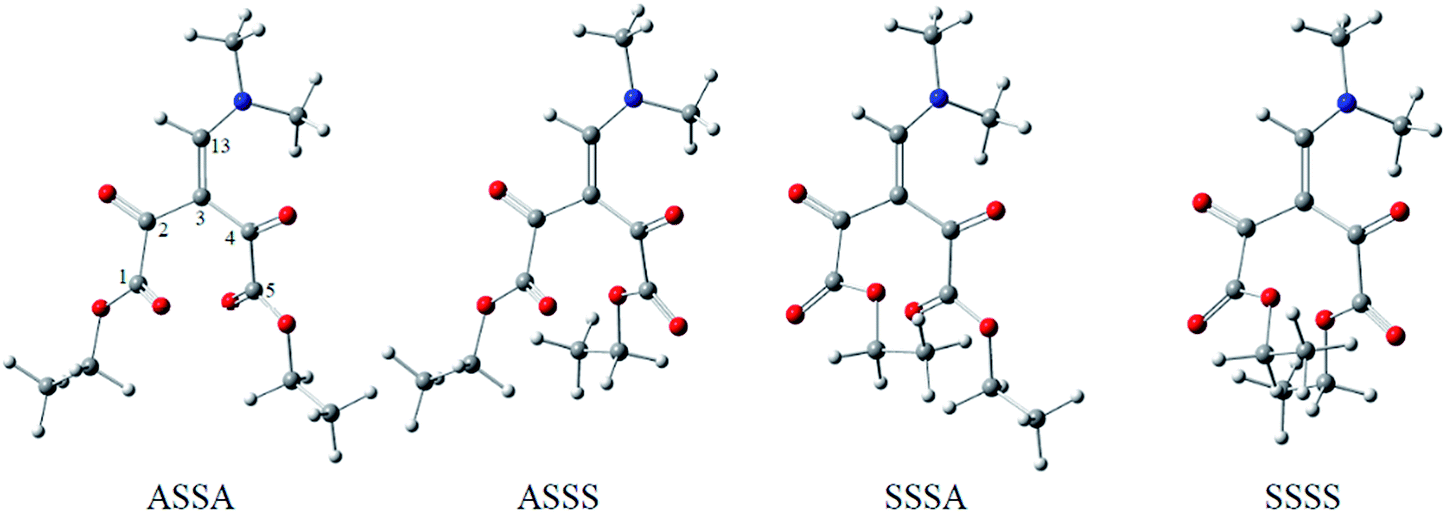
We observe that the four selected conformations, i.e. that have the lowest energy among the 16 possible conformations, have ketone carbonyls (C2O and C4O) with syn orientation to the double bond (C3C13). On the other hand, the carbonyl of the ester (C1O and C5O) showed different orientations. Thus, the orientation of the carbonyl of the ester groups plays a fundamental role in the dipole moment of each conformation and thus influences the population in each of the studied media, whereas the quantity most easily associated to the solvation energy is the dipole moment. According to the gas phase calculations (Table 1), the ASSA conformer that has the smallest dipole moment is the most stable. However, in the solvent phase, the ASSS and SSSS conformations are preferred because their greater dipole moment favors interactions that stabilize these conformations in solvents with intermediate and high relative permittivity. The SSSA conformer was not found to be stable in dichloromethane and ethanol, converging to SSSS and ASSA, respectively.
| fk+ = qk(N + 1) − qk(N) |
The condensed-to-atom variant for local electrophilicity index (ωk+) is defined as ωk+ = fk+ω where ω = μ2/2η. In our study, local reactivity parameters were calculated using the M06-2X/6-31+G(d,p) theory level; some atomic Fukui function (fk+) and local electrophilicity (ωk+) values are listed in Table 2.
| Gas phase | Solvent phasea | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ASSA | ASSS | SSSA | ASSA | ASSS | SSSS | |||||||
| f+ | ω+ | f+ | ω+ | f+ | ω+ | f+ | ω+ | f+ | ω+ | f+ | ω+ | |
| a In dichloromethane at IEF-PCM-M06-2X/6-31+G(d,p). The data in ethanol are very similar, thus were not presented. | ||||||||||||
| C1 | 0.016 | 0.013 | 0.031 | 0.026 | −0.005 | −0.004 | 0.009 | 0.008 | 0.024 | 0.020 | −0.004 | −0.003 |
| C2 | 0.031 | 0.026 | 0.058 | 0.048 | 0.016 | 0.013 | 0.028 | 0.023 | 0.060 | 0.050 | 0.024 | 0.020 |
| C3 | −0.045 | −0.037 | −0.040 | −0.033 | −0.050 | −0.041 | −0.045 | −0.038 | −0.040 | −0.033 | −0.046 | −0.038 |
| C4 | 0.116 | 0.096 | 0.080 | 0.067 | 0.153 | 0.128 | 0.115 | 0.096 | 0.068 | 0.056 | 0.132 | 0.110 |
| C5 | 0.040 | 0.033 | 0.022 | 0.018 | 0.050 | 0.042 | 0.034 | 0.028 | 0.012 | 0.010 | 0.030 | 0.025 |
| C13 | 0.200 | 0.167 | 0.206 | 0.172 | 0.182 | 0.152 | 0.219 | 0.182 | 0.235 | 0.196 | 0.214 | 0.179 |
Although the results of the calculations for the more stable conformations in the gas phase and the solvent phase are presented in Table 2, the following discussion is based on the solvent phase to ASSS conformation that has the lowest energy. Let us first look at the regioselectivity, in regards to the first nucleophilic attack on enamino diketone. Local indices have the highest values in C13, which implies that this position is the most preferable site for the first nucleophilic attack in enamino diketone. Furthermore, we analyzed the regioselectivity with regards to the second nucleophilic attack, as in reactions with dinucleophiles. The C2 and C4 carbons (CO of keto group) have much higher values of Fukui function (fk+) and local electrophilicity (ωk+) than the C1 and C5 carbons (CO of ester group), correctly reproducing the reactivity order of these functional groups. Thus, for the reaction with a dinucleophile, it was expected that the nucleophilic attack would take place first at the C13 with the second nucleophilic attack at the ketone carbonyl.
Experimental investigation
The β-enamino diketones has been prepared by a C-acylation reaction of enaminones with ethyl oxalyl chloride in CH2Cl2 under reflux for 15 hours.15 However, the desired β-enamino diketone 2 was obtained by a C-acylation of enaminone 120 with ethyl oxalyl chloride in CH2Cl2 under low temperature (−30 to −25 °C) for 15 minutes, with satisfactory yield (71%) (Scheme 2). The structure of precursor 2 was established by spectroscopic methods (1H NMR and 13C NMR) and mass spectrometry (Fig. S10, S30 and S31, ESI†). The 1H NMR spectrum shows two singlets at δ = 2.97 (3H) and 3.43 ppm (3H) corresponding to the methyl hydrogens of N(Me)2 group. The splitting of signals for N(Me)2 group is due electron delocalization in the (Me)2N–C(13)–C(3)–C(O) system, that prevents rotation around the C13–N(Me)2 bond. Also was observe a triplet at δ = 1.37 (6H) and a quartet at δ = 4.29 ppm (4H) equivalents to the hydrogens of the two carboethoxy groups (CO2Et), evidencing symmetry in the compound 2. This symmetry exists due electronic delocalization reported above, and the rotation around the C(13)–C(3) bond, which is presumably rapid on the NMR timescale. Furthermore, the 1H NMR spectrum show at 7.97 ppm (1H) a singlet corresponding to vinylic hydrogen (H13). The 13C NMR spectrum shows the same observations evidenced in the 1H spectrum, so the number of observed signals relates only to a single 2-ethoxy-2-oxoacetyl group (C(O)CO2Et), confirming symmetry in the precursor 2, two signals corresponding to the methyl carbons of N(Me)2 group, showing that both are different chemically, and also were observed signals corresponding to the vinylic carbons (C3 and C13).In order to verify if the data obtained from theoretical calculations agrees with the experimental data, the reactivity of enamino diketone 2 was first examined with the mononucleophile aniline. The reaction was carried out in dichloromethane at room temperature for 30 minutes. The reaction was highly regioselective and yielded only one product which resulted in aniline nitrogen attacks on the β-carbon (C13) atom of enamino diketone 2 and the elimination of N,N-dimethylamine to form product 3 (61%) (Scheme 2). This result is fully consistent with the theoretical study described above. The structure of product 3 was characterized on the basis of its spectral and analytical data (Fig. S16, S32 and S33, ESI†). The spectroscopic data of compound 3, in contrast to the data of the compound 2, show that this structure of 3 does not has symmetry, since signals shown in the 1H and 13C NMR spectra corresponds to two 2-ethoxy-2-oxoacetyl (C(O)CO2Et) groups chemically inequivalence. The non-symmetry observed for compound 3 can be explained presumable by intramolecular hydrogen bond between the aminic hydrogen (NH) and carbonyl oxygen (C2O or C4O) resulting in the formation of a six-membered pseudo-ring, avoiding rotation around the C3–C13 bond (Scheme 2). Also, it is noteworthy that the signals correspondents to aminic hydrogen (NH) and vinylic hydrogen (13H), at 12.5 and at 8.8 ppm, respectively, appears as doublets, indicating coupling between those hydrogens (J = 13.8 Hz).
Our next step was to investigate the reactivity of precursor 2 against unsymmetrical dinucleophiles R–NHNH2, through the reaction with phenylhydrazine, under ethanol reflux, according to methodology described in the literature.16h After one hour of reaction (monitored by TLC), the formation of four different products was observed which were characterized as: pyrazole 4b and the by-products hydrazonyl-pyrazole 5b, in the stereoisomeric forms E and Z, and pyrazolo[3,4-d]pyridazinone 6b (Scheme 2). The reaction conditions were optimized in order to develop general protocols for the regiocontrolled synthesis those aza-heterocycles.
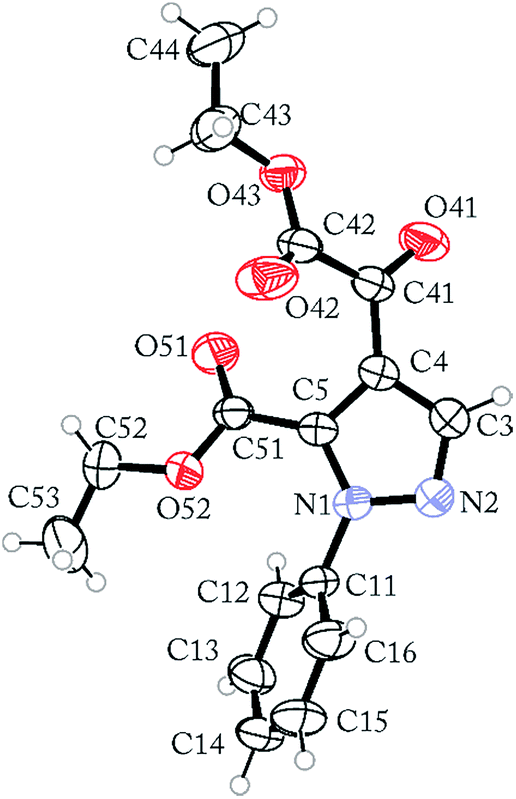
The regioselective synthesis of pyrazole 4b was obtained from the reaction of the enamino diketone 2 with phenylhydrazine in ethanol at room temperature for 30 minutes (monitored by TLC). On the basis of its 1H, 13C NMR and mass spectra (Fig. S18, S36 and S37, ESI†), the pyrazole 4b was isolated in good yield (71%) as a single regioisomer, 1,4-disubstituted pyrazole-5-carboxylate (Scheme 2). Moreover, X-ray crystallography data confirmed the structure of compound 4b (Fig. 1)21 The formation of 1,4-disubstituted pyrazole-5-carboxylate regioisomer, indicating that the regiochemistry of the cyclocondensation reaction of 2 with phenylhydrazine proceeds via 1,4-addition, where the NH2 group of hydrazine first attacks at the β-carbon atom (C13) of 2 (intermediate I – Fig. 2) with subsequent heterocyclization from the attack of the second amine group of hydrazine at the carbonyl group C2 or C4 (intermediate III – Fig. 2).
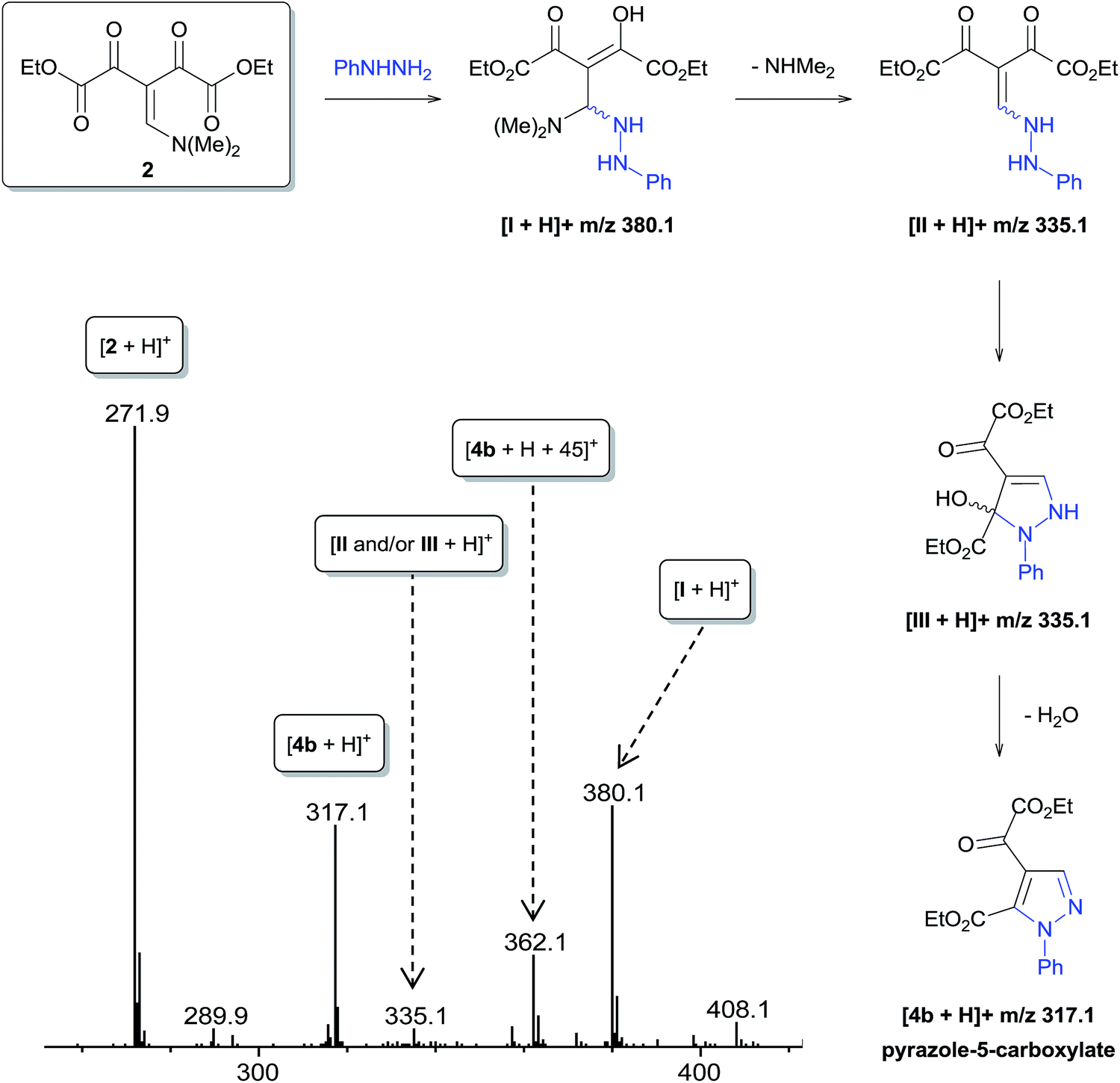 | ||
| Fig. 2 Intermediates proposed for the formation of regioisomers of 1,4-disubstituted pyrazole-5-carboxylate (4b) and ESI-MS for the reaction of 2 with phenylhydrazine in ethanol at 0 °C after 3 min of reaction. | ||
To perform a mechanistic study of the reaction of 2 with phenylhydrazine, we used direct infusion electrospray ionization mass and tandem mass spectrometry (ESI-MS/MS).22 Initially, the reaction solution of 2 and phenylhydrazine in ethanol at 0 °C was immediately injected into the ESI source operating in positive ion mode. Approximately three minutes later, ESI-MS (Fig. 2) detected some protonated forms of neutral species that were consistent with the intermediaries proposed for the possible reaction mechanisms (Fig. 2): the protonated reactant [2 + H]+ of m/z 271.9; [I + H]+ of m/z 380.1; [II and/or III + H]+ of m/z 335.1; and protonated final product [4b + H]+ of m/z 317.1. The characterization of these key species was carried out through collision-induced dissociation (CID) via high resolution ESI(+)-MS/MS experiments (Fig. S12 and S14, ESI†). However, the ion of m/z 335.1 showed a fragmentation pattern equivalent to intermediates II and III involved in the proposed mechanism (Fig. 2). A specie of m/z 362.1 (Fig. 2) also was detected, which was characterized via ESI(+)-MS/MS (Fig. S13, ESI†) as the protonated specie [4b + H + 45]+.
After 6 hours of reaction at room temperature, ESI-MS showed that most of the intermediates I and II and/or III had been consumed and the specie [4b + H + 45]+ was now detected as the most abundant specie (Fig. S11, ESI†).
To gain further insight into the specie [4b + H + 45]+, we performed a series of NMR experiments with the intent of investigating the chemical nature this ion, since this species demonstrated stability in the reaction medium for a long period of time. For this purpose, 1H, 13C NMR and 1D NOESY experiments were performed for the crude product of the reaction of 2 with phenylhydrazine carried out in CDCl3 for 6 hours. The results show the conversion of reagent 2 to the product pyrazole 4b and N,N-dimethylamine by-product (Fig. 3A and B and ESI S15†). The Fig. 3B demonstrates the use of the 1D NOESY experiment for the crude product obtained after 6 hours of reaction, where the phenyl ring hydrogens were selectively irradiated and the nOes were observed on the ethoxyl, N,N-dimethylamine groups and H3 on the pyrazole ring, which suggests that these groups are spatially close to the phenyl ring. From this behavior, we propose that N,N-dimethylamine is attached to the pyrazole 4b through a hydrogen bond. When the crude product 4b was washed with water and analyzed again by 1D NOESY, nOes was not observed on the N,N-dimethylamine group, which corroborates with our suggestion (Fig. 3C). Thus, the specie of m/z 362.1 was characterized as [4b + NHMe2 + H]+. ESI-MS/MS analysis (Fig. S13, ESI†) of this ion showed that dissociation occurred mainly by the loss of neutral N,N-dimethylamine to form a ion m/z 317.1 [4b + H]+.
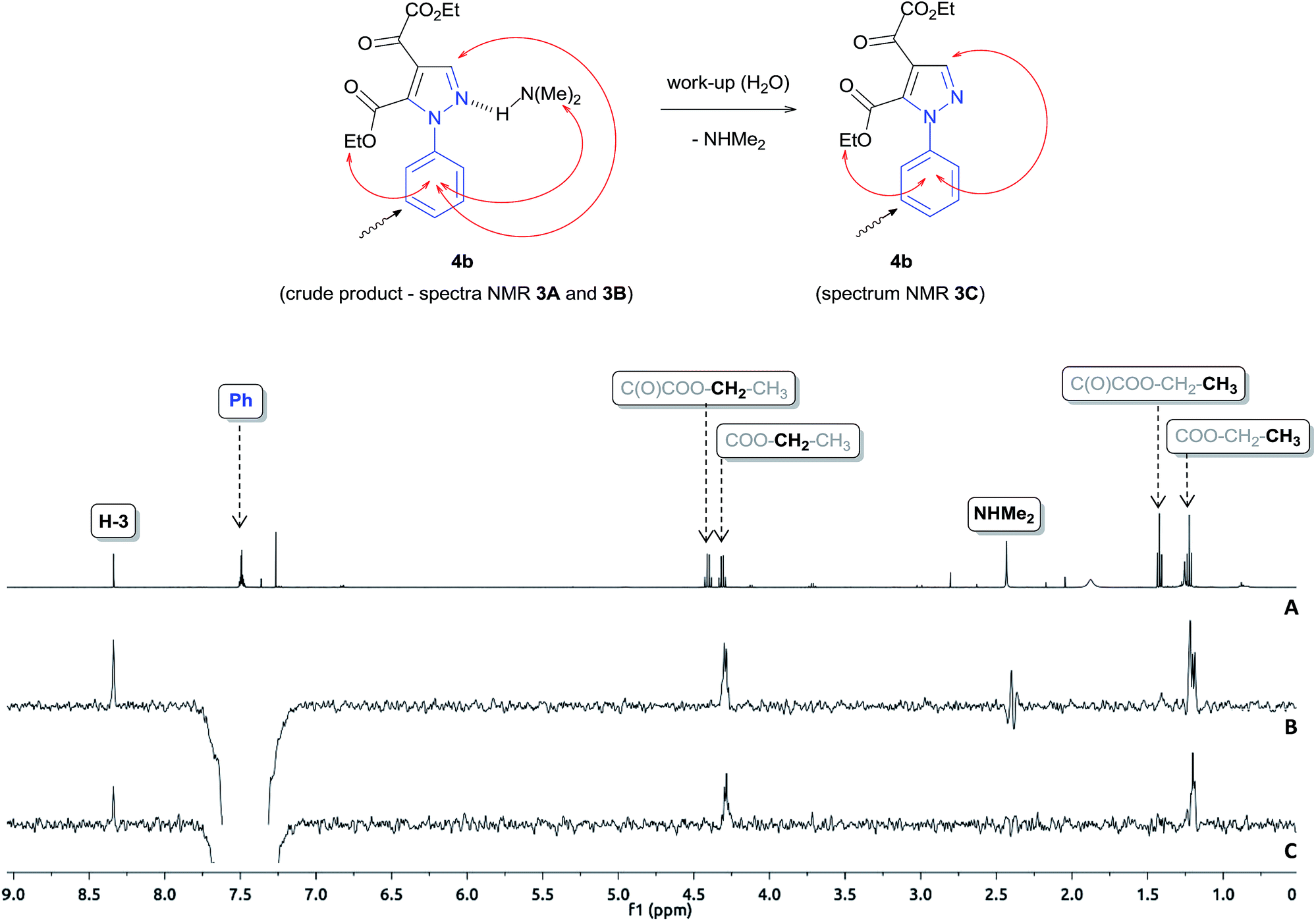 | ||
| Fig. 3 A) – 1H MNR spectrum of crude product after 6 hours of reaction. (B) – 1D NOESY spectrum of crude product after 6 hours of reaction. (C) – 1D NOESY spectrum of the reaction after washing with water. | ||
The synthesis regioselective of 1,4-disubstituted pyrazole-5-carboxylate also was possible from the reaction of 2 with hydrazine monohydrate and 4-chlorophenylhydrazine, using identical condition, furnishing the pyrazoles 4a and 4c, respectively, with good yields (67–75%) (Scheme 2).
These results reveal that the reaction of 2 against hydrazines dinucleophiles (R–NHNH2, R = H, Ph, 4-ClC6H4) it presents a well-defined regiochemistry, fact that is also fully in accordance with the theoretical study described before.
The versatility and the synthetic potential of the building block 2 were demonstrated by the synthesis of the intermediate hydrazonyl-pyrazole 5b and the pyrazole[3,4-d]pyridazinone 6b–g (Scheme 3). Our studies revealed that the reaction of 2 with two phenylhydrazine equivalents, Lewis acid BF3·MeOH catalysis, in dichloromethane at room temperature for 90 minutes led to the formation of the intermediate hydrazonyl-pyrazole (5b), which was found to be a mixture of E and Z stereoisomers (Protocol B, Scheme 3) based on their 1H NMR spectra (Fig. S40 and S45, ESI†). The formation of the E stereoisomer was favored; however, the exact quantification of the stereoisomers was not possible, since over time it was observed (by 1H NMR) slow isomerization between the E and Z forms. This isomerization is provided by the hydrazone–azoenol tautomerization23 and, it is justified by the intramolecular hydrogen bond between the aminic hydrogen (NH) and carbonyl oxygen resulting in the formation of a six-membered pseudo-ring in the Z stereoisomer, providing greater stability for molecule (Scheme 3).
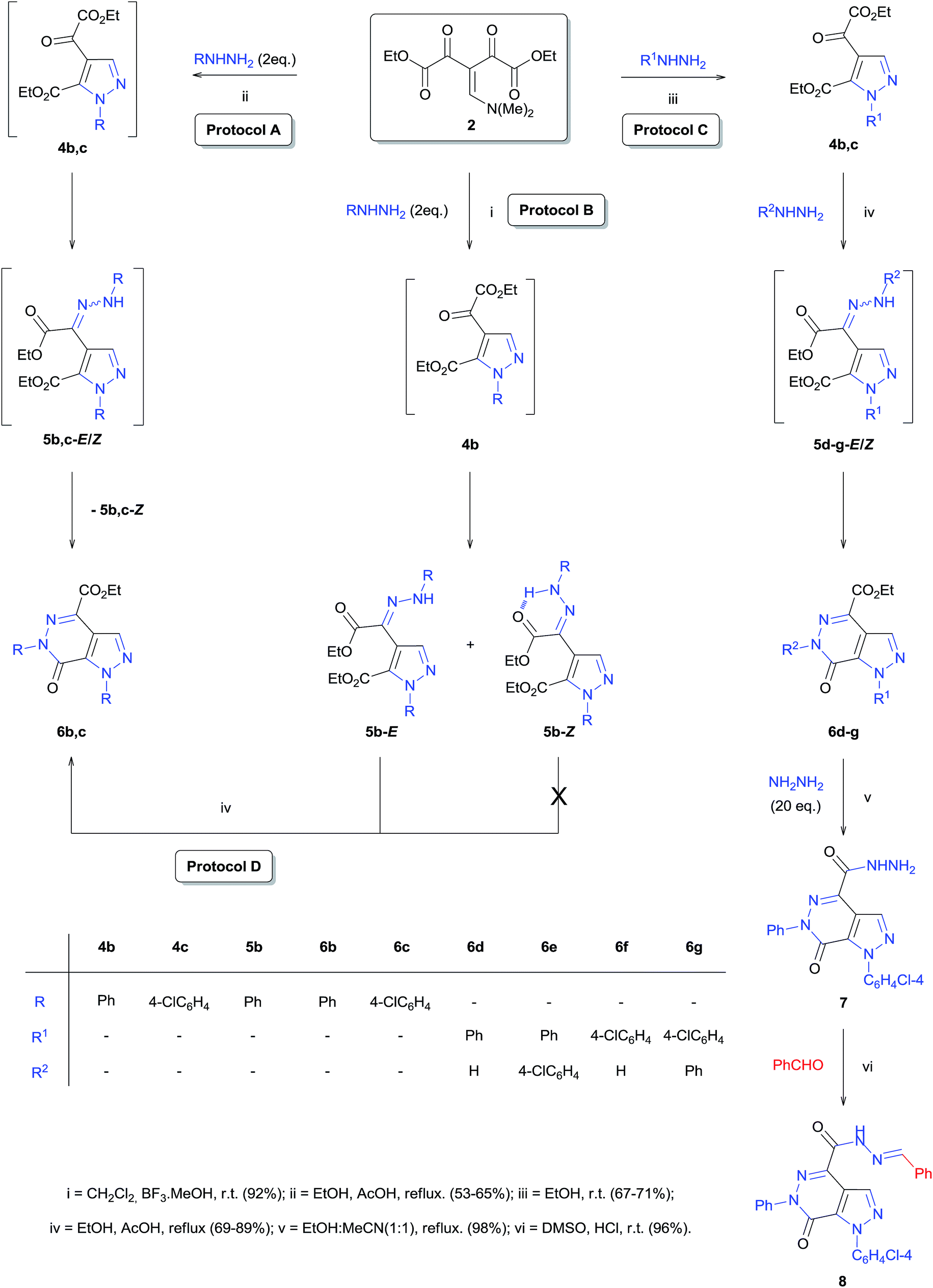 | ||
| Scheme 3 Synthesis of hydrazonyl-pyrazole 5b and pyrazolo[3,4-d]pyridazinone 6b,c from enamino diketone 2 and pyrazolo[3,4-d]pyridazinones 6d–g and the corresponding N-acylhydrazone 8 from pyrazole 4. | ||
On the other hand, when 2 was reacted with two hydrazine equivalents (R = Ph or Cl-4-Ph) in the presence of a protic acid (AcOH, 4 equivalents) in refluxing ethanol for 1 hour, the product was identified by 1H and 13C NMR and ESI-MS spectra (Fig. S22, S23, S50–S53, ESI†) as pyrazole[3,4-d]pyridazinones 6b,c (Protocol A, Scheme 3).
After successfully isolating the intermediate hydrazonyl-pyrazole 5b in the form of E and Z stereoisomers, we performed a preliminary study of the reactivity of both stereoisomers in the formation of pyrazolo[3,4-d]pyridazinone 6 under the conditions of refluxing ethanol and 4 equivalents of AcOH. Interestingly, only the E stereoisomer led to the formation of the fused heterocycle, while the Z stereoisomer proved to be inactive against the intramolecular heterocyclization reaction. This can be explained by formation of the intramolecular hydrogen bond between the aminic hydrogen (NH) and carbonyl oxygen (Protocol D, Scheme 3).
Furthermore, we explored the reaction of pyrazoles 4 with hydrazines, with the view of expanding the scope of the reaction for the synthesis of pyrazole[3,4-d]pyridazinones 6d–g. The best result was obtained when the reaction was performed in refluxing ethanol with 4 equivalents of AcOH for 12 h (Protocol C, Scheme 3).
Finally, the versatility of precursor 2 was demonstrated by the synthesis of the derivative N-acylhydrazone 8 from the carbohydrazide 7 (Scheme 3). The N-acylhydrazone class has been shown great relevance in organic synthesis24 and exhibits a wide spectrum of biological activities25 such as anti-microbial,25a anti-convulsant,25b analgesic,25c anti-inflammatory,25d anti-viral,25e anti-tumor,25f anti-malarial,25g anti-depressant,25h and vasodilatory.25i
Conclusions
In summary, we have synthesized a new building block enamino diketone 2 and described its reactivity by theoretical and experimental studies. Fukui function showed that the β-carbon is the most electrophilic center, followed by the carbonyl ketone and carbonyl ester, respectively. The reactivity of 2 could be verified experimentally by reaction with aniline and hydrazines. These results demonstrate the complementarity and efficiency of the utilized methods. Furthermore, we developed an efficient method for the synthesis of 1,4-disubstituted pyrazole-5-carboxylates as the sole regioisomer. The regiochemistry was confirmed by X-ray crystallography data and a concise mechanism of reaction of 2 with phenylhydrazine were determined via ESI-MS followed by characterization via ESI-MS/MS and NMR. Obtaining a single building block (2) and simple variation of the reaction conditions allowed for efficient and rapid access to a variety multi-functionalized pyrazoles and their hydrazonyl-pyrazole and pyrazolo-pyridazinone derivatives with high regioselectivity, which resulted in the synthesis of 13 new aza-heterocycles. This makes enamino diketone 2 a useful and powerful precursor of heterocyclic compounds, especially for the regioselective synthesis of five- and six-membered multi-functionalized heterocycles.Experimental section
General methods and materials
Reagents were used as obtained from commercial suppliers without further purification. The solvents were dried and purified according to recommended procedures.26 The reactions were monitored by thin-layer chromatography using Merck TLC silica gel plates and visualized with UV light. All melting points were measured using a MQAPF-307-Microquímica apparatus using benzoic acid as an internal standard. 1H NMR (300.06 MHz) and 13C NMR (75.46 MHz) spectra were acquired on a Mercury Plus VARIAN spectrometer, at 300 K, 5 mm samples tubes with CDCl3 or DMSO-d6 as the solvent. Chemical shifts are reported in ppm using residual solvent protons as the internal standard for DMSO-d6 and TMS as the internal standard for CDCl3. Monitoring reactions were performed by maintaining the probe temperature approximately equivalent to the reaction temperature. The samples were analyzed in MS and MS/MS mode by quadrupole-time-of-flight (Q-TOF) and ion trap (IT) mass spectrometry, respectively, with an electrospray ionization (ESI) source for both (MicrOTOF-QII and Amazon Bruker®). Data were acquired in positive mode under the following conditions: nitrogen as collision gas (in Q-TOF) and helium (in IT) and the energy for the collision induced dissociations (CID) was optimized for each component. The flow of drying gas was 10 L min−1 at 200 °C. In both, the data acquisition and processing were carried out using Hystar software (Bruker Scientific). The data were collected in the m/z range of 50–1000 at the speed of two scans per second, providing the resolution of 50.000 (FWHM) at m/z 200 in Q-TOF and 5.000 (FWHM) at m/z 200 in IT. X-Ray crystallography data were collected with a Bruker APEX II CCD area-detector diffractometer and graphite-monochromatized Mo-Ka radiation. The structure was solved by direct methods using SHELXS.27 Subsequent Fourier-difference map analyses yielded the positions of the non-hydrogen atoms. Refinements were carried out with the SHELXL package.27 All refinements were made by full-matrix least squares on F2 with anisotropic displacement parameters for all non-hydrogen atoms. Hydrogen atoms were included in the refinement in calculated positions but the atoms (of hydrogens) that are commenting performing special bond were located in the Fourier map. Illustration of complexes was done using ORTEP-3 for Windows.28Procedure for the synthesis of diethyl 3-((dimethylamine)methylene)-2,4-dioxopentanedioate (2)
A solution of β-enamino ketone 1 (0.514 g, 3.0 mmol), pyridine (0.6 mL, 7.26 mmol) and dry dichloromethane (10 mL) was added dropwise to a stirred solution of ethyl oxalyl chloride (0.37 mL, 3.3 mmol) in dry dichloromethane (5 mL), at −30 to −25 °C under a nitrogen atmosphere. After the addition was completed, the reaction was maintained for 15 minutes at −30 to −25 °C under agitation. The organic layer was then washed with an aqueous solution 10% HCl (1 × 20 mL) and with distilled water (3 × 20 mL). The organic phase was dried over anhydrous sodium sulfate, then filtered and the solvent was evaporated under reduced pressure. The residue was purified on a silica gel chromatography column, utilizing a mixture of 70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :30 of ethyl acetate:hexane as the eluent, affording the product as a viscous yellow oil: yield 0.578 g, 71%; 1H NMR (300 MHz; CDCl3; δ, ppm) 1.37 (t, J = 7.1 Hz, 6H), 2.97 (s, 3H), 3.43 (s, 3H), 4.29 (q, J = 7.1 Hz, 4H), 7.97 (s, 1H); 13C NMR (75 MHz; CDCl3; δ, ppm) 14.2, 44.1, 48.9, 62.5, 106.1, 162.6, 164.0, 181.6; HRMS (ESI) m/z calcd for C12H18NO6 [M + H]+ 272.1134, found 272.1128.
:30 of ethyl acetate:hexane as the eluent, affording the product as a viscous yellow oil: yield 0.578 g, 71%; 1H NMR (300 MHz; CDCl3; δ, ppm) 1.37 (t, J = 7.1 Hz, 6H), 2.97 (s, 3H), 3.43 (s, 3H), 4.29 (q, J = 7.1 Hz, 4H), 7.97 (s, 1H); 13C NMR (75 MHz; CDCl3; δ, ppm) 14.2, 44.1, 48.9, 62.5, 106.1, 162.6, 164.0, 181.6; HRMS (ESI) m/z calcd for C12H18NO6 [M + H]+ 272.1134, found 272.1128.
Procedure for the synthesis of diethyl 3-(anilinomethylene)-2,4-dioxopentanedioate (3)
A mixture of β-enamino diketone (2) (0.271 g, 1 mmol) and aniline (0.094 g, 1 mmol) in dichloromethane (2 mL) was stirred at room temperature for 30 min. Then the solvent was evaporated under vacuum. The residue was then purified by column chromatography using ethyl acetate:hexane 70:30, to give the pure product as a yellow solid: yield 0.195 g, 61%; mp 110.3–112.1 °C; 1H NMR (300 MHz; CDCl3; δ, ppm) 1.40 (t, J = 7.1 Hz, 3H), 1.41 (t, J = 7.1 Hz, 3H), 4.32–4.41 (m, 4H), 7.25–7.36 (m, 3H), 7.45–7.50 (m, 2H), 8.88 (d, J = 13.8 Hz, 1H), 12.45 (d, J = 13.8 Hz, 1H); 13C NMR (75 MHz; CDCl3; δ, ppm) 14.0, 14.1, 62.3, 62.7, 106.5, 118.7, 127.6, 130.3, 137.8, 155.5, 163.0, 164.2, 180.7, 186.8. HRMS (ESI) m/z calcd for C16H18NO6 [M + H]+ 320.1134, found 320.1135.
General procedure for the synthesis of 5-carboxyethyl-4-[ethoxy(oxo)acetyl]-1H-pyrazol and 1-substituted (4a–c)
A mixture of the compound 2 (0.271 g, 1 mmol) and hydrazine (hydrazine monohydrate: 0.036 g; phenylhydrazine: 0.099 g; 4-chlorophenylhydrazine: 0.131 g, 0.9 mmol) in ethanol (2 mL), was stirred at room temperature for 30 minutes. After, the solvent was evaporated under reduced pressure and the residue was washed with distilled water (25 mL), extracted with ethyl acetate (3 × 20 mL) and dried with anhydrous sodium sulfate. The solvent was evaporated once again under reduced pressure and the obtained residue was purified on a silica gel chromatography column, utilizing a 90:10 mixture of hexane:ethyl acetate as the eluent.
5-Carboxyethyl-4-[ethoxy(oxo)acetyl]-1H-pyrazol (4a)
White solid: 0.180 g, yield 75%; mp 107.7–109.8 °C; 1H NMR (300 MHz; CDCl3; δ, ppm) 1.41 (t, J = 7.1 Hz, 3H), 1.41 (t, J = 7.1 Hz, 3H), 4.40 (q, J = 7.1 Hz, 2H), 4.45 (q, J = 7.1 Hz, 2H), 8.51 (s, 1H), 14.01 (bs, 1H); 13C NMR (75 MHz; CDCl3; δ, ppm) 14.2, 14.3, 62.5, 62.8, 119.3, 136.5, 143.2, 161.7, 162.3, 180.0; HRMS (ESI) m/z calcd for C10H13N2O5 [M + H]+ 241.0824, found 241.0823.5-Carboxyethyl-4-[ethoxy(oxo)acetyl]-1-phenyl-1H-pyrazol (4b)
Yellow solid: 0.225 g, yield 71%; mp 38.4–39.1 °C; 1H NMR (300 MHz; CDCl3; δ, ppm) 1.23 (t, J = 7.1 Hz, 3H), 1.42 (t, J = 7.1 Hz, 3H), 4.31 (q, J = 7.1 Hz, 2H), 4.41 (q, J = 7.1 Hz, 2H), 7.49 (bs, 5H), 8.34 (s, 1H); 13C NMR (75 MHz; CDCl3; δ, ppm) 13.8, 14.2, 62.9, 63.1, 120.5, 124.7, 129.5, 129.7, 138.8, 138.0, 142.6, 160.2, 161.7, 178.2; HRMS (ESI) m/z calcd for C16H17N2O5 [M + H]+ 317.1137, found 317.1125.5-Carboxyethyl-1-(4-chlorophenyl)-4-[ethoxy(oxo)acetyl]-1H-pyrazol (4c)
Yellow solid: 0.235 g, yield 67%; mp 57.2–59.0 °C; 1H NMR (300 MHz; CDCl3; δ, ppm) 1.26 (t, J = 7.1 Hz, 3H), 1.42 (t, J = 7.1 Hz, 3H), 4.33 (q, J = 7.1 Hz, 2H), 4.40 (q, J = 7.1 Hz, 2H), 7.46 (bs, 4H), 8.33 (s, 1H); 13C NMR (75 MHz; CDCl3; δ, ppm) 13.9, 14.2, 63.0, 63.3, 120.8, 126.0, 129.6, 135.6, 137.3, 137.9, 142.7, 160.0, 161.5, 178.1; HRMS (ESI) m/z calcd for C16H16ClN2O5 [M + H]+ 351.0748, found 351.0754.General procedure for the synthesis of 5-carboxyethyl-4-[(1E/Z)-2-ethoxy-2-oxo-N-phenylethanehydrazonoyl]-1-phenyl-1H-pyrazol stereoisomers (5b-E/5b-Z)
A mixture of compound 2 (0.271 g, 1 mmol) and phenylhydrazine (0.122 g, 1.1 mmol) in dichloromethane (2 mL) was stirred at room temperature for 30 min. Next, 0.03 mL (5 mol%) of a boron trifluoride–methanol solution 20% and a solution of phenylhydrazine (0.122 g, 1.1 mmol) in dichloromethane (1 mL) were added to the mixture. After 1 h, the solvent was evaporated under reduced pressure. The residue was washed with distilled water (25 mL), extracted with dichloromethane (3 × 20 mL) and dried with anhydrous sodium sulfate. The solvent was evaporated under reduced pressure and the products were isolated on a silica gel chromatography column using a 90:10 mixture of hexane:ethyl acetate as the eluent.
5-Carboxyethyl-4-[(1E)-2-ethoxy-2-oxo-N-phenylethanehydrazonoyl]-1-phenyl-1H-pyrazol (5b-E)
Yellow solid: 0.260 g, yield 64%; mp 124.0–126.5 °C; 1H NMR (300 MHz; CDCl3; δ, ppm) 1.12 (t, J = 7.1 Hz, 3H), 1.35 (t, J = 7.1 Hz, 3H), 4.15 (q, J = 7.1 Hz, 2H), 4.32 (q, J = 7.1 Hz, 2H), 6.97–7.02 (m, 1H), 7.20–7.34 (m, 4H), 7.49 (sl, 5H), 7.79 (s, 1H), 8.25 (s, 1H); 13C NMR (75 MHz; CDCl3; δ, ppm) 13.8, 14.5, 61.5, 62.0, 114.4, 122.6, 125.8, 129.0, 129.3, 129.5, 140.0, 142.8, 114.8, 126.6, 132.8, 139.8, 158.8, 164.4; HRMS (ESI) m/z calcd for C22H23N4O4 [M + H]+ 407.1719, found 407.1710.5-Carboxyethyl-4-[(1Z)-2-ethoxy-2-oxo-N-phenylethanehydrazonoyl]-1-phenyl-1H-pyrazol (5b-Z)
Yellow solid: 0.114 g, yield 28%; mp 101.2–104.1 °C; 1H NMR (300 MHz; CDCl3; δ, ppm) 1.11 (t, J = 7.1 Hz, 3H), 1.32 (t, J = 7.1 Hz, 3H), 4.16 (q, J = 7.1 Hz, 2H), 4.29 (q, J = 7.1 Hz, 2H), 6.97–7.02 (m, 1H), 7.21–7.34 (m, 4H), 7.41–7.50 (m, 5H) 7.87 (s, 1H), 12.42 (s, 1H); 13C NMR (75 MHz; CDCl3; δ, ppm) 14.0, 14.2, 61.3, 61.7, 114.3, 122.8, 125.5, 128.7, 128.9, 129.5, 140.3, 143.1, 120.8, 122.5, 132.2, 140.5, 160.7, 163.5; HRMS (ESI) m/z calcd for C22H23N4O4 [M + H]+ 407.1719, found 407.1720.General procedure for the synthesis of 1,6-disubstituted 4-carboxyethyl-1H-pyrazolo[3,4-d]pyridazin-7-one (6b,c)
A mixture of compound 2 (0.271 g, 1 mmol), hydrazine (phenylhydrazine: 0.243 g; 4-chlorophenylhydrazine: 0.319 g, 2.2 mmol) and acetic acid (4 mmol, 0.2 mL) was stirred under reflux in ethanol (5 mL) for 1 hour. Then, the mixture was cooled to 0 °C and the solid was filtered, washed with cold ethanol (10 mL) and dried under vacuum.4-Carboxyethyl-1,6-diphenyl-1H-pyrazolo[3,4-d]pyridazin-7-one (6b)
White solid: 0.191 g, yield 53%; mp 179.3–181.9 °C; 1H NMR (300 MHz; CDCl3; δ, ppm) 1.48 (t, J = 7.1 Hz, 3H), 4.54 (q, J = 7.1 Hz, 2H), 7.37–7.71 (m, 10H), 8.60 (s, 1H); 13C NMR (75 MHz; CDCl3; δ, ppm) 14.5, 62.6, 120.7, 125.8, 126.5, 128.7, 128.8, 129.1, 138.7, 141.0, 131.9, 132.9, 136.8, 152.8, 162.5; HRMS (ESI) m/z calcd for C20H17N4O3 [M + H]+ 361.1301, found 361.1283.4-Carboxyethyl-1,6-bis(4-chlorophenyl)-1H-pyrazolo[3,4-d]pyridazin-7-one (6c)
White solid: 0.279 g, yield 65%; mp 239.8–241.4 °C; 1H NMR (300 MHz; CDCl3; δ, ppm) 1.48 (t, J = 7.1 Hz, 3H), 4.55 (q, J = 7.1 Hz, 2H), 7.43–7.49 (m, 4H), 7.54–7.58 (m, 2H), 7.63–7.67 (m, 2H), 8.61 (s, 1H); 13C NMR (75 MHz; CDCl3; δ, ppm) 14.5, 62.8, 120.9, 127.0, 127.8, 129.1, 129.3, 134.7, 135.2, 137.0, 139.3, 131.8, 133.3, 137.2, 152.7, 162.3; HRMS (ESI) m/z calcd for C20H15Cl2N4O3 [M + H]+ 429.0521, found 429.0515.General procedure for the synthesis of 1,6-disubstituted 4-carboxyethyl-1H-pyrazolo[3,4-d]pyridazin-7-one (6d–g)
A mixture of compound 4 (4b: 0.158 g; 4c: 0.175 g, 0.50 mmol), hydrazine (phenylhydrazine: 0.61 g; 4-chlorophenylhydrazine: 0.80 g, 0.55 mmol) and acetic acid (2 mmol, 0.1 mL), was stirred under reflux in ethanol (5 mL) for 12 h. Then, the mixture was cooled to 0 °C and the solid was filtered, washed with cold ethanol (10 mL) and dried under vacuum.4-Carboxyethyl-6,7-dihydro-1-phenyl-1H-pyrazolo[3,4-d]pyridazin-7-one (6d)
White solid: 0.239 g, yield 84%; mp 214.6–217.5 °C; 1H NMR (300 MHz; DMSO-d6; δ, ppm) 1.39 (t, J = 7.1 Hz, 3H), 4.43 (q, J = 7.1 Hz, 2H), 7.48–7.61 (m, 3H), 7.68–7.75 (m, 2H), 8.54 (s, 1H), 13.38 (s, 1H); 13C NMR (75 MHz; DMSO-d6; δ, ppm) 14.1, 61.7, 120.2, 125.4, 128.6, 138.3, 131.4, 132.2, 136.3, 153.1, 162.0; HRMS (ESI) m/z calcd for C14H13N4O3 [M + H]+ 285.0988, found 285.0991.4-Carboxyethyl-6-(4-chlorophenyl)-1-phenyl-1H-pyrazolo[3,4-d]pyridazin-7-one (6e)
White solid: 0.273 g, yield 69%; mp 220.1–223.0 °C; 1H NMR (300 MHz; CDCl3; δ, ppm) 1.48 (t, J = 7.1 Hz, 3H), 4.54 (q, J = 7.1 Hz, 2H), 7.42–7.59 (m, 7H), 7.65–7.70 (m, 2H), 8.60 (s, 1H); 13C NMR (75 MHz; CDCl3; δ, ppm) 14.5, 62.8, 120.6, 125.7, 127.8, 128.8, 129.2, 134.5, 138.5, 139.4, 131.8, 133.2, 136.9, 152.7, 162.3; HRMS (ESI) m/z calcd for C20H16ClN4O3 [M + H]+ 395.0911, found 395.0905.4-Carboxyethyl-1-(4-chlorophenyl)-6,7-dihydro-1H-pyrazolo[3,4-d]pyridazin-7-one (6f)
White solid: 0.284 g, yield 89%; mp 265.3–267.8 °C; 1H NMR (300 MHz; DMSO-d6; δ, ppm) 1.39 (t, J = 7.1 Hz, 3H), 4.43 (q, J = 7.1 Hz, 2H), 7.63 (d, J = 8.7 Hz, 2H), 7.76 (d, J = 8.7 Hz, 2H), 8.55 (s, 1H), 13.41 (s, 1H); 13C NMR (75 MHz; DMSO-d6; δ, ppm) 14.0, 61.7, 120.3, 127.1, 128.6, 133.1, 137.1, 132.2, 131.6, 136.5, 153.1, 162.0; HRMS (ESI) m/z calcd for C14H12ClN4O3 [M + H]+ 319.0598, found 319.0593.4-Carboxyetil-1-(4-chlorophenyl)-6-phenyl-1H-pyrazolo[3,4-d]pyridazin-7-one (6g)
White solid: 0.273 g, yield 69%; mp 197.3–200.1 °C; 1H NMR (300 MHz; CDCl3; δ, ppm) 1.48 (t, J = 7.1 Hz, 3H), 4.54 (q, J = 7.1 Hz, 2H), 7.39–7.69 (m, 9H), 8.60 (s, 1H); 13C NMR (75 MHz; CDCl3; δ, ppm) 14.5, 62.7, 120.9, 126.5, 126.9, 128.9, 129.0, 129.1, 135.0, 137.1, 140.9, 131.9, 132.9, 137.1, 152.8, 162.4; HRMS (ESI) m/z calcd for C20H16ClN4O3 [M + H]+ 395.0911, found 395.0905.Procedure for the synthesis of 1-(4-chlorophenyl)-4-(hydrazinecarbonyl)-6-phenyl-1H-pyrazolo[3,4-d]pyridazin-7-one (7)
A mixture of compound 6g (0.118 g, 0.3 mmol) and hydrazine monohydrate (0.25 mL, 6 mmol) was stirred under reflux in ethanol:acetonitrile (1:1, 3 mL) for 24 h. Then, the mixture was cooled to room temperature and the solvent evaporated under vacuum, affording a white solid: yield 0.112 g, 98%; mp 283.6–286.0 °C; 1H NMR (300 MHz; DMSO-d6; δ, ppm) 4.65 (sl, 2H), 7.40–7.81 (m, 9H), 8.63 (s, 1H), 9.94 (s, 1H); 13C NMR (75 MHz; DMSO-d6; δ, ppm) 119.6, 126.5, 127.4, 128.0, 128.4, 128.5, 133.2, 137.2, 140.9, 132.5, 134.8, 136.6, 152.0, 160.7; HRMS (ESI) m/z calcd for C18H14ClN6O2 [M + H]+ 381.0867, found 381.0861.
Procedure for the synthesis of 4-{[(2E)-2-benzylidenehydrazine]carbonyl}-1-(4-chlorophenyl)-6-phenyl-1H-pyrazolo[3,4-d]pyridazin-7-one (8)
A mixture of compound 7 (0.3 mmol, 0.114 g), benzaldehyde (0.36 mmol, 0.039 g) and HCl 37% (two drops) in DMSO was stirred at room temperature for 1 h. Then, cold distilled water (20 mL) was added to the mixture and the product was obtained as a white solid. The product was washed with cold water and dried under vacuum: yield 0.135 g, 96%; mp 265.2–267.4 °C; 1H NMR (300 MHz; DMSO-d6; δ, ppm) 7.43–7.84 (m, 14H), 8.59 (s, 1H), 8.72 (s, 1H), 11.95 (s, 1H); 13C NMR (75 MHz; DMSO-d6; δ, ppm) 119.7, 126.8, 127.2, 127.4, 128.3, 128.5, 128.6, 128.9, 130.4, 133.2, 134.2, 137.2, 140.8, 132.5, 134.4, 136.8, 149.6, 152.1, 158.4; HRMS (ESI) m/z calcd for C25H18ClN6O2 [M + H]+ 469.1180, found 469.1174.Computational details
All electronic structure calculations were performed with the GAUSSIAN 09 (ref. 29) suite of programs. The geometric optimization of the possible conformations for the compounds was performed by DFT (density functional theory) calculations with a hybrid functional, M06-2X/6-31+G(d,p). Vibrational frequencies were calculated using the same basis set with optimization; the absence of an imaginary frequency indicated the structures were at the global minimum. The reactivity descriptors used here, namely the condensed Fukui function for nucleophilic attack (fk+) and local electrophilicity index (ωk+), were calculated according to the literature.30 Condensed Fukui functions were obtained using NPA populations obtained from the single point energy calculations performed at the M06-2X/6-31+G(d,p) level for the N-electron species as well as the N + 1 electron species. All energy calculations were done at the optimized geometry of the N-electron species. Solvation effects were included with the IEF-PCM model31 using dielectric constants stored in the program database. The solvents employed were dichloromethane and ethanol, which were chosen on the basis of experimental studies. The NBO calculations32 were performed on all optimized structures at the M06-2X/6-31+G(d,p) level.Acknowledgements
The authors are grateful for the financial support from CNPq-Brazil (CNPq/Universal Process No. 454920/2014-8). Fellowships from CAPES are also acknowledged.Notes and references
- (a) O. Meth-Cohn, Comprehensive Heterocyclic Chemistry, Pergamon press, Oxford, 1984, p. 665 Search PubMed; (b) A. W. Czarnik, Acc. Chem. Res., 1996, 29, 112 CrossRef CAS.
- (a) T. D. Penning, J. J. Talley, S. R. Bertenshaw, J. S. Carter, P. W. Collins, S. Docter, M. J. Graneto, L. F. Lee, J. W. Malecha, J. M. Miyashiro, R. S. Rogers, D. J. Rogier, S. S. Yu, G. D. Anderson, E. G. Burton, J. N. Cogburn, S. A. Gregory, C. M. Koboldt, W. E. Perkins, K. Seibert, A. W. Veenhuizen, Y. Y. Zhang and P. C. Isakson, J. Med. Chem., 1997, 40, 1347 CrossRef CAS PubMed; (b) A. G. Habeeb, P. N. Praveen Rao and E. E. Knaus, J. Med. Chem., 2001, 44, 3039 CrossRef CAS PubMed; (c) T. D. Penning, S. W. Kramer, L. F. Lee, P. W. Collins, C. M. Koboldt, K. Seibert, A. W. Veenhuizen, Y. Y. Zhang and P. C. Isakson, Bioorg. Med. Chem. Lett., 1997, 7, 2121 CrossRef CAS; (d) S. H. Hwang, K. M. Wagner, C. Morisseau, J.-Y. Liu, H. Dong, A. T. Wecksler and B. D. Hammock, J. Med. Chem., 2011, 54, 3037 CrossRef CAS PubMed , and references therein.
- N. K. Terrett, A. S. Bell, D. Brown and P. Ellis, Bioorg. Med. Chem. Lett., 1996, 6, 1819 CrossRef.
- J. E. Casida, D. Hainzl and L. M. Cole, Chem. Res. Toxicol., 1998, 11, 1529 CrossRef PubMed.
- (a) A. Jamwal, A. Javed and V. Bhardwaj, J. Pharm. BioSci., 2013, 1, 114 CAS; (b) D. Dias, B. S. Pacheco, W. Cunico, L. Pizzuti and C. M. P. Pereira, Mini-Rev. Med. Chem., 2014, 14, 1078 CrossRef CAS; (c) S. G. Kuçukguzel and S. Senkardes, Eur. J. Med. Chem., 2015, 97, 786 CrossRef PubMed; (d) R. S. Keri, K. Chand, T. Ramakrishnappa and B. M. Nagaraja, Arch. Pharm. Chem. Life Sci., 2015, 348, 299 CrossRef CAS PubMed.
- K. L. Kees, J. J. Fitzgerald Jr, K. E. Steiner, J. F. Mattes, B. Mihan, T. Tosi, D. Mondoro and M. L. McCaleb, J. Med. Chem., 1996, 39, 3920 CrossRef CAS PubMed.
- (a) L. N. Jungheim, Tetrahedron Lett., 1992, 30, 1889 CrossRef; (b) S. Manfredini, R. Bazzanini, P. G. Baralgi, M. Guarneri, D. Simoni, M. E. Morongiu, A. Pani, E. Tramontano and P. L. Colla, J. Med. Chem., 1992, 35, 917 CrossRef CAS PubMed; (c) A. Tanitame, Y. Oyamada, K. Ofuji, M. Fujimoto, K. Suzuki, T. Ueda, H. Terauchi, M. Kawasaki, K. Nagai, M. Wachi and J. I. Yamagishi, Bioorg. Med. Chem., 2004, 12, 5515 CrossRef CAS PubMed.
- (a) K. W. Weitzel, J. M. Wickman, S. G. Augustin and J. G. Strom, Clin. Ther., 2000, 22, 1254 CrossRef CAS PubMed; (b) C. F. P. George, Lancet, 2001, 357, 1623 CrossRef.
- (a) Z. Sui, J. Guan, M. P. Ferro, K. McCoy, M. P. Wachter, W. V. Murray, M. Singer, M. Steber, D. M. Ritchie and D. C. Argentieri, Bioorg. Med. Chem. Lett., 2000, 10, 601 CrossRef CAS PubMed; (b) A. A. Bekhit and T. AbdelAziem, Bioorg. Med. Chem., 2004, 12, 1935 CrossRef CAS PubMed; (c) C. Selvam, S. M. Jachak, R. Thilagavathi and A. K. Chakraborti, Bioorg. Med. Chem. Lett., 2005, 15, 1793 CrossRef CAS PubMed.
- (a) S. R. Stauffer, C. J. Coletta, R. Tedesco, G. Nishiguchi, K. Carlson, J. Sun, B. S. Katzenellenbogen and J. A. Katzenellenbogen, J. Med. Chem., 2000, 43, 4934 CrossRef CAS PubMed; (b) S. R. Stauffer, Y. Huang, C. J. Coletta, R. Tedesco and J. A. Katzenellenbogen, Bioorg. Med. Chem., 2001, 9, 141 CrossRef CAS PubMed.
- R. Katoch-Rouse, L. A. Pavlova, T. Caulder, A. F. Hoffman, A. G. Mukhin and A. G. Horti, J. Med. Chem., 2003, 46, 642 CrossRef CAS PubMed.
- (a) S. Gelin and D. Hatmam, J. Heterocycl. Chem., 1978, 15, 813 CrossRef CAS; (b) T. Kurihara, T. Uno and Y. Sakomoto, J. Heterocycl. Chem., 1980, 17, 231 CrossRef CAS; (c) Y. Akaçamur, A. Sener, A. M. Ipekoglu and G. Kollenz, J. Heterocycl. Chem., 1997, 34, 221 CrossRef; (d) A. Sener, R. Kasimogullari, M. K. Sener, I. Bildirici and Y. Akaçamur, J. Heterocycl. Chem., 2002, 39, 869 CrossRef CAS; (e) P. Biagini, C. Biancalani, A. Graziano, N. Cesari, M. P. Giovannoni, A. Cilibrizzi, V. D. Piaz, C. Vergelli, L. Crocetti, M. Delcanale, E. Armani, A. Rizzi, P. Puccini, P. M. Gallo, D. Spinabelli and P. Caruso, Bioorg. Med. Chem., 2010, 18, 3506 CrossRef CAS PubMed; (f) C. P. Frizzo, M. A. Villetti, A. Z. Tier, I. M. Gindri, L. Buriol, F. A. Rosa, R. M. Claramunt, D. Sanz and M. A. P. Martins, Thermochim. Acta, 2013, 574, 63–72 CrossRef CAS.
- (a) L. Knorr, BER: Economic Prospects, 1883, 16, 2587 Search PubMed; (b) M. V. Patel, R. Bell, S. Majest, R. Henry and T. Kolasa, J. Org. Chem., 2004, 69, 7058 CrossRef CAS PubMed; (c) S. Peruncheralathan, T. A. Khan, H. Ila and H. Junjappa, J. Org. Chem., 2005, 70, 10030 CrossRef CAS PubMed; (d) B. A. Donohue, E. L. Michelotti, J. C. Reader, V. Reader, M. Stirling and C. M. Tice, J. Comb. Chem., 2002, 4, 23 CrossRef CAS PubMed; (e) S. M. Sakya and B. Rast, Tetrahedron Lett., 2003, 44, 7629 CrossRef CAS; (f) E. Vickerstaffe, B. H. Warrington, M. Ladlow and S. V. Ley, J. Comb. Chem., 2004, 6, 332 CrossRef CAS PubMed; (g) S. K. Singh, M. S. Reddy, S. Shivaramakrishna, D. Kavitha, R. Vasudev, J. Moses Babu, A. Sivalakshmidevi and Y. Koteswar Rao, Tetrahedron Lett., 2004, 45, 7679 CrossRef CAS; (h) V. Polshettiwar and R. S. Varma, Tetrahedron, 2010, 66, 1091 CrossRef CAS; (i) A. DeAngelis, D.-H. Wang and S. L. Buchwald, Angew. Chem., Int. Ed., 2013, 52, 1 CrossRef PubMed.
- (a) C. Kashima, H. Harada, I. Kita, I. Fukuchi and A. Hosomi, Synthesis, 1994, 61 CrossRef CAS; (b) A. C. Spivey, C. M. Diaper, H. Adams and A. J. Rudge, J. Org. Chem., 2000, 65, 5253 CrossRef CAS PubMed; (c) Y. R. Huang and J. A. Katzenellenbogen, Org. Lett., 2000, 2, 2833 CrossRef CAS PubMed; (d) V. L. M. Silva, A. M. S. Silva, D. C. G. A. Pinto, J. A. S. Cavaleiro and J. Elguero, Eur. J. Org. Chem., 2004, 4348 CrossRef CAS; (e) S. T. Heller and S. R. Natarajan, Org. Lett., 2006, 8, 2675 CrossRef CAS PubMed; (f) F. Gosselin, P. D. O'Shea, R. A. Webster, R. A. Reamer, R. D. Tillyer and E. J. J. Grabowski, Synlett, 2006, 3267 CrossRef CAS; (g) S. Fustero, R. Roman, J. F. Sanz-Cervera, A. Simon-Fuentes, S. Villanova and J. Bueno, J. Org. Chem., 2008, 73, 8545 CrossRef CAS PubMed; (h) S. Fustero, R. Roman, J. F. Sanz-Cervera, A. Simon-Fuentes, A. C. Cunat, S. Villanova and M. Murguia, J. Org. Chem., 2008, 73, 3523 CrossRef CAS PubMed , and references therein.
- F. A. Rosa, P. Machado, M. Rossatto, S. P. Vargas, H. G. Bonacorso, N. Zanatta and M. A. P. Martins, Synlett, 2007, 3165 CAS.
- (a) E. Okada, R. Masuda and M. Hojo, Heterocycles, 1992, 34, 791 CrossRef CAS; (b) M. Soufyane, C. Mirand and J. Lévy, Tetrahedron Lett., 1993, 34, 7737 CrossRef CAS; (c) M. Soufyane, S. van den Brock, L. Khamlich and C. Mirand, J. Heterocycl. Chem., 1999, 51, 2445 CrossRef CAS; (d) G. Negri and C. Kascheres, J. Heterocycl. Chem., 2001, 38, 109 CrossRef CAS; (e) H. Beber, M. Soufyane, C. Mirand, S. Schmidt and A. M. Aubertin, Tetrahedron, 2001, 57, 7369 CrossRef; (f) H. Beber, M. Soufyane, M. Santillana-Hayat and C. Mirand, Tetrahedron Lett., 2002, 43, 9233 CrossRef; (g) A. Touzo, M. Soufyane, H. Berber, L. Toupet and C. Mirand, J. Fluorine Chem., 2004, 125, 1299 CrossRef; (h) F. A. Rosa, P. Machado, P. S. Vargas, H. G. Bonacorso, N. Zanatta and M. A. P. Martins, Synlett, 2008, 1673 CAS; (i) F. A. Rosa, P. Machado, G. F. Fiss, P. S. Vargas, T. S. Fernandes, H. G. Bonacorso, N. Zanatta and M. A. P. Martins, Synthesis, 2008, 3639 CAS; (j) P. S. Vargas, F. A. Rosa, L. Buriol, M. Rotta, D. N. Moreira, C. P. Frizzo, H. G. Bonacorso, N. Zanatta and M. A. P. Martins, Tetrahedron Lett., 2012, 53, 3131 CrossRef CAS.
- T. C. Rozada, M. J. V. Silva, D. S. Gonçalves, M. A. P. Martins, R. M. Pontes, G. F. Gauze, E. A. Basso and F. A. Rosa, Struct. Chem., 2015, 26, 1007 CrossRef CAS.
- E. Cances, B. Mennucci and J. Tomasi, J. Chem. Phys., 1997, 107, 3032 CrossRef CAS.
- (a) P. Geerlings, F. de Proft and W. Langenaeker, Chem. Rev., 2003, 103, 1793 CrossRef CAS PubMed; (b) R. G. Parr and W. Yang, in Density Functional Theory of Atoms and Molecules, Oxford University Press, Oxford, 1989 Search PubMed; (c) R. G. Pearson, in Chemical Hardness – Applications from Molecules to Solids, VCH Wiley, Weinheim, 1997 Search PubMed; (d) P. Kolandaivel, G. Praveena and P. Selvarengan, J. Chem. Sci., 2005, 117, 591 CrossRef CAS.
- A. Hanzlowsky, B. Jelencic, S. Recnick, J. Svete, A. Golobic and B. Stanovnik, J. Heterocycl. Chem., 2003, 40, 487 CrossRef CAS.
- Crystallographic data for compound 4b (CCDC-1425933)†.
- (a) C. A. M. Abella, M. Benassi, L. S. Santos, M. N. Eberlin and F. Coelho, J. Org. Chem., 2007, 72, 4048 CrossRef CAS PubMed; (b) G. W. Amarante, H. M. S. Milagre, B. G. Vaz, B. R. V. Ferreira, M. N. Eberlin and F. Coelho, J. Org. Chem., 2009, 74, 3031 CrossRef CAS PubMed; (c) R. C. Barcelos, L. A. Zeoly, M. T. Rodrigues Jr, B. R. V. Ferreira, M. N. Eberlin and F. Coelho, Monatsh. Chem., 2015, 146, 1557 CrossRef CAS.
- (a) K. T. Mahmudov, A. M. Maharramov, R. A. Aliyeva, F. M. Chyragov, R. K. Askerov, P. Q. Hasanov, M. N. Kopylovich and A. J. L. Pombeiro, J. Mol. Struct., 2011, 1006, 576 CrossRef CAS; (b) C. D. a. L. Demange, Chem. Rev., 2003, 103, 2475 CrossRef PubMed.
- M. S. a. S. Kobayashi, Angew. Chem., Int. Ed., 2005, 44, 5176 CrossRef PubMed.
- (a) P. Vicini, F. Zani, P. Cozzini and I. Doytchinova, Eur. J. Med. Chem., 2002, 37, 553 CrossRef CAS PubMed; (b) J. R. Dimmock, S. C. Vashishtha and J. P. Stables, Eur. J. Med. Chem., 2000, 35, 241 CrossRef CAS PubMed; (c) G. A. Silva, L. M. M. Costa, F. C. F. Brito, A. L. P. Miranda, E. J. Barreiro and C. A. M. Fraga, Bioorg. Med. Chem., 2004, 12, 3149 CrossRef CAS PubMed; (d) C. D. Duarte, J. L. M. Tributino, D. I. Lacerda, M. V. Martins, M. S. Alexandre-Moreira, F. Dutra, E. J. H. Bechara, F. S. de-Paula, M. O. F. Goulart, J. Ferreira, J. B. Calixto, M. P. Nunes, A. L. Bertho, A. L. P. Miranda, E. J. Barreiro and C. A. M. Fraga, Bioorg. Med. Chem., 2007, 15, 2421 CrossRef CAS PubMed; (e) M. T. Abdel-Aal, W. A. El-Sayed and E. H. El-Ashry, Arch. Pharm. Chem. Life Sci., 2006, 339, 656 CrossRef CAS PubMed; (f) N. Terzioğlu and A. Gürsoy, Eur. J. Med. Chem., 2003, 38, 781 CrossRef; (g) A. Bernardino, A. C. K. Gomes, A. Freitas, G. Machado, M. Canto-Cavalheiro, L. Leon and V. Amaral, Eur. J. Med. Chem., 2006, 41, 80 CrossRef CAS PubMed; (h) N. Ergenç and N. S. Günay, Eur. J. Med. Chem., 1998, 33, 143 CrossRef; (i) A. G. Silva, G. Zapata-Suto, A. E. Kummerle, C. A. M. Fraga, E. J. Barreiro and R. T. Sudo, Bioorg. Med. Chem., 2005, 13, 3431 CrossRef CAS PubMed.
- D. D. Perrin and L. F. Armarego, in Purification of Laboratory Chemicals, Pergamon Press, New York, 3rd edn, 1996 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 1997, 30, 565 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi,C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision B.01, Gaussian Inc., Wallingford, CT, 2010 Search PubMed.
- A. Reyes, P. A. Cuervo, F. Orozco, R. Abonia, M. Duque-Noreña, P. Pérez and E. Chamorro, J. Mol. Model., 2013, 19, 3611 CrossRef CAS PubMed.
- M. T. Cancès, B. Menucci and J. Tomasi, J. Chem. Phys., 1997, 107, 3032 CrossRef.
- (a) F. Weinhold and C. Landis, in Valency and Bonding: A Natural Bond Orbital Donor-Acceptor Perspective, Cambridge University Press, Cambridge, 2005 Search PubMed; (b) E. D. Glendening, J. K. Badenhoop, A. E. Reed, J. E. Carpenter, J. A. Bohmann, C. M. Morales and F. Weinhold, in NBO 5.G, ed. Theoretical Chemistry Institute, University of Wisconsin, Madison, WI, 2001 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1425933. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ra12968k |
| This journal is © The Royal Society of Chemistry 2016 |