
Precisely designed rattle-type mTiO2@P(NIPAM-co-MBA) microspheres with screening gel network for highly selective extraction of phosphopeptidome
Wanfu Ma†
a,
Ying Zhang†b,
Weili Miaob,
Cheng Zhangb,
Meng Yua,
Yuting Zhanga,
Jia Guoa,
Haojie Lu*b and
Changchun Wang*a
aState Key Laboratory of Molecular Engineering of Polymers, Department of Macromolecular Science, Laboratory of Advanced Materials, Fudan University, Shanghai 200433, China. E-mail: ccwang@fudan.edu.cn,; Fax: +86-21-65640293; Tel: +86-21-65642385
bDepartment of Chemistry and Institutes of Biomedical Sciences, Fudan University, Shanghai 200032, People's Republic of China. E-mail: luhaojie@fudan.edu.cn; Fax: +86-21-5423-7618; Tel: +86-21-54237618
First published on 18th August 2014
Abstract
The selective extraction of phosphopeptidome from complicated biological samples is of great importance for the development of diagnostic and prognostic biomarkers, but still remains a challenge. In this work, rattle-type mTiO2@P(NIPAM-co-MBA) composite microspheres comprising a mesoporous crystalline mTiO2 core, an intermediate hollow space and a crosslinked P(NIPAM-co-MBA) network shell were elaborately designed and fabricated via two-step reflux-precipitation polymerization followed by a hydrothermal process. First, a non-crosslinked PMAA layer was directly coated onto the surface of the TiO2 core without any pretreatment. Then, the formed TiO2@PMAA was encapsulated with another crosslinked P(NIPAM-co-MBA) layer with the aid of the strong hydrogen-bonding interaction between the two polymer layers. Finally, a hydrothermal process was adopted to convert the TiO2 core into a crystalline and mesoporous counterpart. At the same time, a non-crosslinked PMAA layer was selectively removed to form a rattle-type structure. The crosslinked P(NIPAM-co-MBA) shell makes the rattle-type mTiO2@P(NIPAM-co-MBA) possess great size-exclusion effect against both high-molecular-weight nonphosphoproteins and high-molecular-weight phosphoproteins, while the mTiO2 core was responsible of the selective enrichment of the low-molecular-weight phosphopeptides. With the help of these unique properties, the rattle-type mTiO2@P(NIPAM-co-MBA) microspheres show excellent potential for the one-step selective extraction of the phosphopeptidome.
Introduction
Peptidome, the naturally existing peptides and low-molecular-weight (LMW) proteins (with molecular weight less than 20 kDa) in the biological samples, plays pivotal roles in the regulation of diverse biological functions and activities of the proteins. The comprehensive analysis of the peptidome can contribute to a better elucidation of the biochemical functions, as well as to the development of diagnostic and prognostic biomarkers.1–5 To reveal the peptidome in complicated samples, several steps are usually involved, including sample collection, selective extraction of the peptidome, mass spectrometric (MS) detection and data mining. Of these four key steps, the selective extraction step has been recognized as the major obstacle for the successful identification of the peptidome.6 There is a fundamental difference between proteome and peptidome extraction. In the proteome research, all the proteins are first digested into the peptides mixture, and then the peptides are enriched by functional materials to avoid the interference of lipids and salts. In contrast, because extra peptides will be produced from the digestion of proteins, the digestion process could not be executed for the peptidome research. For this reason, a large amount of high-molecular-weight (HMW) proteins (with molecular weight greater than 20 kDa) will exist in the test samples, and their presence seriously hampers the mass spectrometric analysis of the peptidome.Toward this end, three main methodologies are currently used for the selective extraction of the peptidome. Organic solvent precipitation is a simple approach for discarding highly abundant proteins.7–9 However, the proteins may act as carriers for the less abundant peptides during the precipitation process. As a result, the depletion of the HMW proteins would lead to a concomitant loss of the physiologically important peptides. Centrifugal ultrafiltration (UF) with an accurate molecular cutoff is considered to be a useful technology for the separation of proteins with different masses.10–13 Nevertheless, when a relatively large amount of the sample is applied, the ultrafiltration time will sharply increase. Moreover, other contaminants with low molecular weights, such as small molecules and salts, will also be concentrated and result in severe interference in the MS detection. Therefore, additional peptide enrichment and salt removal achieved by solid-phase extraction, particularly using C18 sorbents, need to be employed before MS analysis. To simplify the extraction procedures, mesoporous nanomaterials endowed with a size-exclusion effect have shed new light on the peptidome research. Ordered mesoporous nanomaterials, including mesoporous silica, mesoporous carbon and other materials with high porous surface areas, highly ordered channel and adjustable pore sizes facilitate the selective extraction of the peptidome, while effectively excluding the HMW proteins.14–25 In addition, for modification-specific peptidome, such as glycopeptidome26 and phosphopeptidome,27–29 except for the size-exclusion effect, enrichment of the specific glycopeptidome or phosphopeptidome from the entire peptidome is also indispensable. Further, manipulating the surface chemistry of the pore channel has nicely solved this issue. Although this simple approach could block most proteins both in peptidome and modification-specific peptidome research, the outer surface of the mesoporous nanomaterials will also adsorb a few of the specific proteins. As a result, this brings unfavorable influence to the optimal identification of the peptidome and modification-specific peptidome.
To overcome the limitations of the state-of-the-art methods, we herein proposed a new technique to implement the facile one-step extraction of peptidome and an efficient exclusion of the HMW proteins using a composite microsphere with a rattle-type structure. The composite microspheres were deliberately designed to be constructed with a freely moveable core, an intermediate hollow space and a polymeric gel network shell. When this type of microarchitecture was applied, the HMW component, such as HMW proteins, could be effectively blocked by the gel network, whereas the peptidome and other compositions with smaller sizes could pass through to get to the intermediate hollow space. Then, the peptidome could be enriched by the core, and the other LMW contaminants are eliminated. As a typical example, in this article, we designed and fabricated rattle-type mTiO2@P(NIPAM-co-MBA) microspheres for highly specific and facile extraction of the phosphopeptidome. The rattle-type mTiO2@P(NIPAM-co-MBA) microspheres were designed to possess the following features: (1) a poly(N-isopropyl acrylamide-co-N,N′-methylenebisacrylamide) (P(NIPAM-co-MBA)) gel network shell showing the effective exclusion of the HMW constitutes, including both HMW nonphosphoproteins and HMW phosphoproteins; (2) a mesoporous and highly crystalline mTiO2 core possessing a remarkable selectivity and effectiveness toward the enrichment of the phosphopeptides and LMW phosphoproteins; and (3) an intermediate hollow space offering interspace for the storage of the LMW components and allowing the easy removal of the nonphosphopeptides and the LMW nonphosphoproteins through washing. These rattle-type mTiO2@P(NIPAM-co-MBA) microspheres with the aforementioned unique properties are anticipated to have high performance in the selective extraction of the phosphopeptidome from complicated biological samples.
Experimental methods
Materials
Tetrabutyl orthotitanate (TBOT) was purchased from Jiangsu Qiang Sheng Chemical Reagent Co., Ltd. Anhydrous ethanol, potassium chloride, methylacrylic acid (MAA), iron(III) chloride hexahydrate (FeCl3·6H2O), ammonium acetate (NH4Ac), ethylene glycol (EG), trisodium citrate dehydrate and aqueous ammonia solution (25%) were purchased from Shanghai Chemical Reagents Company and used as received. N,N′-Methylenebisacrylamide (MBA) was purchased from Fluka and recrystallized using acetone. 2,2-Azobisisobutyronitrile (AIBN) was supplied by Sinopharm Chemical Reagents Company. N-Isopropyl acrylamide (NIPAM, 97%) was obtained from Sigma-Aldrich and recrystallized using hexane. β-Casein, asialofetuin (ASF), bovine serum albumin (BSA, 95%), cytochrome c from horse heart (Cyto C), 2,5-dihydroxybenzoic acid (2,5-DHB, 98%), ammonium bicarbonate (ABC, 99.5%) and 1-1-(tosylamido)-2-phenyl-ethyl chloromethyl ketone (TPCK)-treated trypsin (E.G 2.4.21.4) were purchased from Sigma-Aldrich (St. Louis, Mo., USA). Acetonitrile (ACN, 99.9%) and trifluoroacetic acid (TFA, 99.8%) were purchased from Merck (Darmstadt, Germany). Phosphoric acid (85%) was purchased from Shanghai Feida Chemical Reagents Ltd. (Shanghai, China). Matrix DHB was dissolved in acetonitrile (ACN)–water (50/50, v/v) solution containing 1% H3PO4 by maintaining DHB at 10 mg mL−1. Deionized water (18.4 MΩ cm) used for all experiments was obtained from a Milli-Q system (Millipore, Bedford, Mass., USA).Preparation of monodisperse titania microspheres
Monodisperse spherical titania (TiO2) microspheres were prepared via controlled hydrolysis of TBOT in ethanol–acetonitrile mixed solvent (ethanol–acetonitrile = 1/1, v/v). For a typical experiment, an acetonitrile–ethanol volume of 100 mL was mixed with 0.50 mL of KCl aqueous solution (0.40 mol L−1, containing 0.2 mmol KCl), followed by adding 4.0 mL of TBOT at ambient temperature with vigorous mechanical stirring. Reagents were completely mixed for the formation of the uniform nucleation throughout the entire solution. The initially homogeneous reaction mixtures became milky white after the addition of TBOT, which indicated the formation of visible titania particles as a uniform suspension. The controlled hydrolysis was ended after stirring the reaction system for 2 h at room temperature. The resultant titania microspheres were purified by three cycles of centrifugation, decantation and resuspension in acetonitrile. The resultant titania microspheres were dispersed in acetonitrile for further use.Preparation of TiO2@PMAA core–shell microspheres
The TiO2@PMAA core–shell microspheres were synthesized by directly coating uncrosslinked polymethylacrylic acid (PMAA) onto the surface of the titania microspheres without any modification. Coating the PMAA layer onto TiO2 microspheres was executed by the reflux-precipitation polymerization30 of MAA with AIBN as the initiator in acetonitrile. Typically, about 50 mg of TiO2 seed nanoparticles were dispersed in 50 mL acetonitrile in a dried 100 mL single-necked flask with the aid of ultrasonication. Then, a mixture of 0.5 mL of MAA and 12.5 mg of AIBN was added to the flask to initiate the polymerization. The flask submerged in a heating oil bath was attached with a Liebig condenser. The reaction mixture was heated from ambient temperature to the boiling state within 30 min and the reaction was ended after 3 h. The obtained TiO2@PMAA microspheres were collected by centrifugation and washed with acetonitrile in order to eliminate excess reactants. The final product was dispersed in acetonitrile for further use.Preparation of TiO2@PMAA@P(NIPAM-co-MBA) core–shell–shell microspheres
TiO2@PMAA@P(NIPAM-co-MBA) core–shell–shell microspheres were synthesized by coating a crosslinked P(NIPAM-co-MBA) shell onto the surface of TiO2@PMAA via the second-step reflux-precipitation polymerization of NIPAM, with MBA as the crosslinker and AIBN as the initiator, in acetonitrile. Typically, about 100 mg of TiO2@PMAA seed nanoparticles were dispersed in 80 mL acetonitrile in a dried 150 mL single-necked flask with the aid of ultrasonication. Then, a mixture of 600 mg of NIPAM, 150 mg of MBA and 15 mg of AIBN was added to the flask to initiate the polymerization. The reaction mixture was heated from ambient temperature to the boiling state within 30 min, and the reaction was ended after 3 h. The obtained TiO2@PMAA@P(NIPAM-co-MBA) microspheres were collected by centrifugation and washed with acetonitrile in order to eliminate excess reactants.Preparation of rattle-type mTiO2@P(NIPAM-co-MBA) microspheres
The rattle-type mTiO2@P(NIPAM-co-MBA) microspheres were achieved by hydrothermally treating the obtained TiO2@PMAA@P(NIPAM-co-MBA) core–shell–shell microspheres. Typically, about 200 mg of the as-synthesized TiO2@PMAA@P(NIPAM-co-MBA) microspheres was dispersed in a 60 mL mixed solvent containing 40 mL of ethanol and 20 mL of deionized water, and 3 mL of NH3·H2O was then added to aforementioned suspension. Then, the mixture was transferred to a Teflon-lined stainless-steel autoclave (100 mL capacity). The autoclave was heated at 160 °C and maintained for 24 h. Then, it was cooled to room temperature, the obtained rattle-type mTiO2@P(NIPAM-co-MBA) microspheres were collected by centrifugation and washed with ethanol and deionized water to remove the uncrosslinked PMAA chains.Preparation of tryptic digest of standard proteins
β-casein, BSA were each dissolved in 25 mM ABC at pH 8.0 (1 mg mL−1 for each protein) and denatured by boiling for 10 min. Protein solutions were then incubated with trypsin at an enzyme–substrate ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :40 (w/w) for 12 h at 37 °C to produce proteolytic digests, respectively. The tryptic peptide mixtures were stored at −20 °C until further use.
:40 (w/w) for 12 h at 37 °C to produce proteolytic digests, respectively. The tryptic peptide mixtures were stored at −20 °C until further use.
Selective enrichment of phosphopeptides with mTiO2@P(NIPAM-co-MBA)
The obtained rattle-type mTiO2@P(NIPAM-co-MBA) was first washed three times with ethanol and then suspended in deionized water with a concentration of 10 mg mL−1. Tryptic digests of β-casein and BSA were dissolved in 100 μL of loading buffer (50% ACN containing 5% TFA), then 2 μL of the mTiO2@P(NIPAM-co-MBA) was added and incubated at room temperature. After that, the mTiO2@P(NIPAM-co-MBA) with captured phosphopeptides was separated from the mixed solutions by centrifugation. After washing with 200 μL of washing buffer (50% ethanol containing 5% TFA) to remove the nonspecifically adsorbed peptides, the trapped phosphopeptides were eluted with NH3·H2O (5%, 10 μL) for further MS analysis. Enrichment of phosphopeptides from the protein mixture was the same as previously described; the protein mixture contains cyto-c (protein), BSA (protein), β-casein (protein) and β-casein digest with a mass ratio of 100:100:10:1.
MALDI mass spectrometry
1 μL of the eluate was deposited on the MALDI probe, and then the matrix solution DHB (1 μL) was deposited for MS analysis. MALDI-TOF mass spectrometry analysis was performed in the positive reflection mode on a 5800 Proteomic Analyzer (Applied Biosystems, Framingham, Mass., USA) with an Nd:YAG laser at 355 nm, a repetition rate of 400 Hz and an acceleration voltage of 20 kV. The range of the laser energy was optimized to obtain good resolution and signal-to-noise ratio (S/N) and kept constant for further analysis. External mass calibration was performed by using standard peptides from myoglobin digests.Characterization
Field-emission transmission electron microscopy (FE-TEM) images were taken on a JEM-2100F transmission electron microscope at an accelerating voltage of 200 kV. Samples dispersed at an appropriate concentration were cast onto a carbon-coated copper grid. Fourier transform infrared spectra (FTIR) were obtained on a NEXUS-470 FTIR spectrometer using a potassium bromide pellet, and the diffuse reflectance spectra were scanned in the range of 400–4000 cm−1. Thermogravimetric analysis (TGA) was performed on a Pyris 1 TGA instrument. All data were obtained under a constant flow of nitrogen at a rate of 40 mL min−1. The temperature was first increased from room temperature to 100 °C and held until constant weight, and then increased from 100 °C to 800 °C at a rate of 20 °C min−1. XRD patterns were collected on an X'Pert Pro (Panalytical, The Netherlands) diffraction meter with Cu Kα radiation at λ = 0.154 nm operating at 40 kV and 40 mA. Nitrogen adsorption–desorption measurements were performed on an ASAP2020 (Micromeritics, USA) accelerated surface area analyzer at 77 K. Before measuring, the samples were degassed in a vacuum at 120 °C for at least 6 h.Results and discussion
The overall synthetic route employed for the preparation of the rattle-type mTiO2@P(NIPAM-co-MBA) microspheres is schematically illustrated in Scheme 1. In brief, monodisperse spherical titania (TiO2) microspheres were first synthesized via controlled hydrolysis of TBOT in a mixed solvent containing ethanol and acetonitrile with a volume ratio of 1:1. Then, the as-prepared TiO2 was directly coated with a non-crosslinked polymethylacrylic acid (PMAA) interim layer by reflux-precipitation polymerization (RPP) of the monomer MAA in the solvent of acetonitrile without any pretreatment of the titania seed microspheres. This interim layer is crucial for the formation of the ultimate rattle-type structure. On one hand, the strong hydrogen-bonding interaction between the carboxyl group and the amide groups of N-isopropyl acrylamide (NIPAM) facilitated the direct coating of the P(NIPAM-co-MBA) layer on the PMAA layer via the second-step RPP process. On the other hand, the PMAA layer could be selectively removed to form the rattle-type structure in order to make the surface of the titania core available for anchoring phosphopeptidome. Subsequently, the TiO2@PMAA microspheres were encapsulated with another crosslinked P(NIPAM-co-MBA) gel network shell via the second step RPP of monomer NIPAM and crosslinker MBA in acetonitrile solvent. Finally, the obtained TiO2@PMAA@P(NIPAM-co-MBA) was subjected to a hydrothermal process for following reason: hydrothermal crystallization of the TiO2 to form mesoporous crystalline mTiO2 core and removal of the PMAA interim layer to shape into a rattle-type structure.
 | ||
| Scheme 1 Schematic illustration of the synthetic procedure for the preparation of rattle-type mTiO2@P(NIPAM-co-MBA) microspheres. | ||
Representative TEM images of TiO2 and TiO2@PMAA core–shell microspheres are shown in Fig. 1a and b. The spherical TiO2 have an average diameter of ca. 385 nm and were uniform in shape and size. After coating with a non-crosslinked PMAA layer, the obtained TiO2@PMAA microspheres possessed a distinctive core–shell structure and the size of the composite microspheres increased to around 455 nm, indicating the shell thickness to be about 35 nm. It is necessary to note that the noncrosslinked PMAA layer is stable only in acetonitrile solvent. When the TiO2@PMAA is transferred to the solvent of PMAA, such as ethanol and deionized water, the PMAA layer is removed. Therefore, the TiO2@PMAA must be stored in acetonitrile before further use and could not come in contact with the solvent of PMAA. As seen in Fig. 1c, when the TiO2@PMAA was encapsulated with the P(NIPAM-co-MBA) gel network shell, the boundary between the two polymer layers is not clearly observed because of the low contrast between the PMAA and the P(NIPAM-co-MBA) layer. Nevertheless, the shell thickness was visibly increased from about 35 nm to around 95 nm, which suggested that the thickness of the P(NIPAM-co-MBA) shell is about 60 nm. With a further hydrothermal treatment at a temperature of 160 °C for 24 h, the noncrosslinked PMAA interim layer could be completely removed to form the intermediate hollow space (Fig. 1d). At the same time, the TiO2 core was no longer solid, but was porous instead and constructed with many small nanocrystals (Fig. 1e). The diameter of the final rattle-type product is about 950 nm.
 | ||
| Fig. 1 Representative TEM images of (a) TiO2, (b) TiO2@PMAA, (c) TiO2@PMAA@P(NIPAM-co-MBA), (d and e) rattle-type mTiO2@P(NIPAM-co-MBA) and (f) mTiO2. (The scale bars are 300 nm for a, b, c, e, f and 1 μm for d.) | ||
The entire fabrication process was continued to be investigated by FTIR spectroscopy. As shown in Fig. 2a, in addition to the characteristic peaks of TiO2, the new peak appearing at 1705 cm−1 attributed to the stretching vibration of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O of the carboxyl groups confirms the presence of the PMAA layer in TiO2@PMAA. After coating with another layer of P(NIPAM-co-MBA), in addition to the peak at 1705 cm−1, the peaks corresponding to amide I (CO stretching) and amide II bands (N–H bending) emerged at 1649 cm−1 and 1527 cm−1, respectively. Because the PMAA layer was almost completely removed after the hydrothermal process, the signal of the characteristic peak of the carboxyl group (1705 cm−1) was not detected.
O of the carboxyl groups confirms the presence of the PMAA layer in TiO2@PMAA. After coating with another layer of P(NIPAM-co-MBA), in addition to the peak at 1705 cm−1, the peaks corresponding to amide I (CO stretching) and amide II bands (N–H bending) emerged at 1649 cm−1 and 1527 cm−1, respectively. Because the PMAA layer was almost completely removed after the hydrothermal process, the signal of the characteristic peak of the carboxyl group (1705 cm−1) was not detected.
 | ||
| Fig. 2 (a) FT-IR spectra and (b) TGA curves of (i) TiO2, (ii) TiO2@PMAA, (iii) TiO2@PMAA@P(NIPAM-co-MBA), (iv) rattle-type mTiO2@P(NIPAM-co-MBA) and (v) mTiO2. | ||
To quantitatively determine the composition of these composite microspheres, thermogravimetric analysis (TGA) was executed (Fig. 2b). The 15.1 wt% weight loss of TiO2 is attributed to the dehydration reaction of the hydroxyls, indicating that the TiO2 contains a considerable amount of hydroxyls. The uncommon segment between 400 °C and 500 °C is caused by the phase transition.31 After coating by the PMAA layer, the weight loss dramatically increased to 67.2 wt%. Through calculation, the weight percentage of the PMAA layer is about 61.4% of the total weight of TiO2@PMAA. When the P(NIPAM-co-MBA) shell was introduced to the system, the weight loss continuously increased to 82.2 wt%, which suggests that the TiO2@PMAA@P(NIPAM-co-MBA) is composed of 21 wt% of TiO2, 33.3 wt% of PMAA and 45.7 wt% of P(NIPAM-co-MBA). In addition, the PMAA layer was eliminated and another important transition for rattle-type mTiO2@P(NIPAM-co-MBA) is that the hydroxyls of TiO2 have reacted with each other during the hydrothermal process. This fact could be effectively proven by the TGA curve of mTiO2, which was prepared by directly treating the TiO2 microspheres under the same hydrothermal condition and used as a reference. The morphology and structure of mTiO2 (Fig. 1f) is same as the mTiO2 core in rattle-type mTiO2@P(NIPAM-co-MBA). Because the mTiO2 core nearly has no weight loss, the 45.9 wt% weight loss of rattle-type mTiO2@P(NIPAM-co-MBA) is ascribed to the weight percentage of the P(NIPAM-co-MBA) shell. By comparing the weight ratio of the P(NIPAM-co-MBA) shell to the TiO2 core in rattle-type mTiO2@P(NIPAM-co-MBA) and that in TiO2@PMAA@P(NIPAM-co-MBA), it could be found that some noncrosslinked PNIPAM or PMBA chains were also simultaneously removed.
The crystallization transition before and after the hydrothermal process was further studied by powder X-ray diffraction. As shown in Fig. 3a, prior to the hydrothermal process, TiO2, TiO2@PMAA and TiO2@PMAA@P(NIPAM-co-MBA) all have no signal peak, which is indicative of the amorphous TiO2 in all the three types of microspheres. However, after hydrothermal processing for 24 h, the PXRD pattern for the synthesized rattle-type mTiO2@P(NIPAM-co-MBA) microspheres was noticeably different from the former patterns. In addition, distinct XRD peaks were clearly observed at 2θ values of 25.2°, 37.8°, 48.0°, 53.9°, 55.0°, 62.7°, 68.8°, 70.2°, and 75.1°, which are well assigned to the (101), (004), (200), (105), (211), (204), (116), (220), and (215) crystallographic planes of an anatase-phase TiO2 (JCPDS card no. 21-1272), respectively. The rattle-type mTiO2@P(NIPAM-co-MBA) microspheres were subjected to further study of the porosity of the mTiO2 core by means of a nitrogen sorption analysis performed at 77 K. As shown in Fig. 3b, the specific surface area and the pore volume of the rattle-type mTiO2@P(NIPAM-co-MBA) are all very low (only about 5 m2 g−1 and 0.009 cm3 g−1, respectively). The reason is that the polymer shell collapsed when the sample was dried, thus the pore of the mTiO2 core was blocked by the polymer shell. However, the mTiO2 exhibited type-IV gas sorption isotherms, which were indicative of the mesoporous character. According to the calculations made using the BET model, the mTiO2 resulted in a specific surface area of 77.3 m2 g−1 and pore volumes of 0.43 cm3 g−1. The corresponding pore-size distribution was evaluated using the Barrett–Joyner–Halenda (BJH) model, and the average pore diameter is calculated to be about 19 nm (Fig. 3b inset). Considering the result together with the TEM image, as well as the PXRD patterns, the creation of the mesopore should be attributed to the tiny slit between the neighboring nanocrystals in both the mTiO2 and mTiO2 cores. The crystalline and mesoporous structures will lead to a considerably better selectivity and capacity for the mTiO2 in the enrichment of the phosphopeptidome than its solid counterpart.32
 | ||
| Fig. 3 (a) X-ray diffraction patterns of (i) TiO2, (ii) TiO2@PMAA, (iii) TiO2@PMAA@P(NIPAM-co-MBA) and (iv) rattle-type mTiO2@P(NIPAM-co-MBA); (b) nitrogen adsorption–desorption isotherm (● = adsorption, ○ = desorption) curves of (iv) rattle-type mTiO2@P(NIPAM-co-MBA) and (v) mTiO2 (the inset is the BJH pore-size distribution of mTiO2). | ||
The requirements of the material used for the extraction of phosphopeptidome can be summarized by the following two points:27 one is that the material should have high selectivity toward phosphopeptides and LMW phosphoproteins enrichment, and the other is that the material should have an excellent capability to achieve the size-exclusion effect. The procedures of phosphopeptidome studies using rattle-type mTiO2@P(NIPAM-co-MBA) are illustrated in Scheme 2. When both proteins and peptides were mixed with rattle-type mTiO2@P(NIPAM-co-MBA), HMW proteins were excluded by the crosslinking gel network because of the size-exclusion effect, whereas peptidome was allowed to pass through the P(NIPAM-co-MBA) shell and reach the surface of the mTiO2 core. With the aid of strong interaction between the phosphoric acid groups and TiO2, the phosphopeptidome was anchored onto the surface of the TiO2 nanocrystals in the mTiO2 core and other nonphosphopeptidome could then be washed away. Through centrifugal separation, the phosphopeptidome-captured microspheres could be well isolated from the mixture and then the adsorbed phosphopeptidome was desorbed for further mass spectrometric analysis.
 | ||
| Scheme 2 Schematic illustration of the typical process for the selective extraction of the phosphopeptidome by using rattle-type mTiO2@P(NIPAM-co-MBA) microspheres. | ||
To test the specificity of the rattle-type mTiO2@P(NIPAM-co-MBA) in phosphopeptidome enrichment, a tryptic digest of standard phosphoprotein β-casein was mixed with a digest of standard non-phosphoprotein BSA at a molar ratio of 1:100. The standard phosphoprotein β-casein harbors three phosphorylated sites, and it would generate three phosphopeptides after trypsin digestion, including m/z at 2061.83, 2556.09, and 3122.27 in the MALDI spectrum. In a typical enrichment procedure, the β-casein digest and BSA digest were first dissolved in a 100 μL loading buffer consisting of 50% acetonitrile containing 5% trifluoroacetic acid (TFA). The solution was then mixed with rattle-type mTiO2@P(NIPAM-co-MBA), which, with captured phosphopeptides, were separated from the mixed solution by centrifugation and washed several times with the washing buffer (50% ethanol containing 5% TFA) to remove nonspecifically adsorbed nonphosphopeptides. Finally, the phosphopeptides were eluted from the rattle-type mTiO2@P(NIPAM-co-MBA) with 10 μL of 5% NH3·H2O, and 4 μL of this solution were used for MALDI-TOF MS analysis. Before enrichment, the spectrum is dominated by nonphosphopeptides without the detection of any phosphopeptides (Fig. 4a). After selective enrichment, signals of the three phosphopeptides were easily detected with a very clean background, shown in Fig. 4b. This result clearly proves that the peptides could pass through the P(NIPAM-co-MBA) gel network shell, and the rattle-type mTiO2@P(NIPAM-co-MBA) has high enrichment selectivity toward phosphopeptides. As we know, the concentration of biologically active peptides is always at an extremely low level. Therefore, the enrichment sensitivity of the rattle-type mTiO2@P(NIPAM-co-MBA) was investigated, as illustrated in the MALDI mass spectrum shown in Fig. 4c. The three targeted phosphopeptides could be easily enriched and detected at a signal-to-noise ratio of 55, 164 and 42 for the phosphopeptide with m/z of 2061.83, 2556.09, and 3122.27, respectively, even when the total amount of β-casein was decreased to only 1 fmol μL−1.
 | ||
| Fig. 4 MALDI mass spectra of the tryptic digest mixture of β-casein and BSA (with a molar ratio of β-casein to BSA of 1:100): (a) direct analysis, (b) analysis after enrichment using rattle-type mTiO2@P(NIPAM-co-MBA). (c) MALDI mass spectrum of the tryptic digests of β-casein (1 nM, 2 mL), after enrichment. “*” indicates phosphorylated peptides, “#” indicates their dephosphorylated counterparts and “●” indicates their doubly charged phosphorylated peptides. | ||
To investigate the size-exclusion capability of the P(NIPAM-co-MBA) gel network shell, a tryptic digest of β-casein was mixed into a protein mixture, including a standard phosphoprotein of β-casein (the molecular weight is 24 kDa), a standard nonphosphoprotein of BSA (the molecular weight is 66 kDa) and a standard nonphosphoprotein of cytochrome c (the molecular weight is 11 kDa) with a mass ratio of 1:10:100:100. Before enrichment, the mixture was directly analyzed with MALDI-MS, and the signals of the peptides were too weak to be detected because of the interference of a large amount of proteins (Fig. 5a). After enrichment, the phosphopeptides were eluted from rattle-type mTiO2@P(NIPAM-co-MBA) and subjected to mass spectrometric analysis. As the mass spectrum (Fig. 5b) revealed, low-abundant phosphopeptides were efficiently removed from the mixture with a prominent signal-to-noise ratio. The size-exclusion effect definitely helped the sensitive detection of the phosphopeptides from the complex sample.
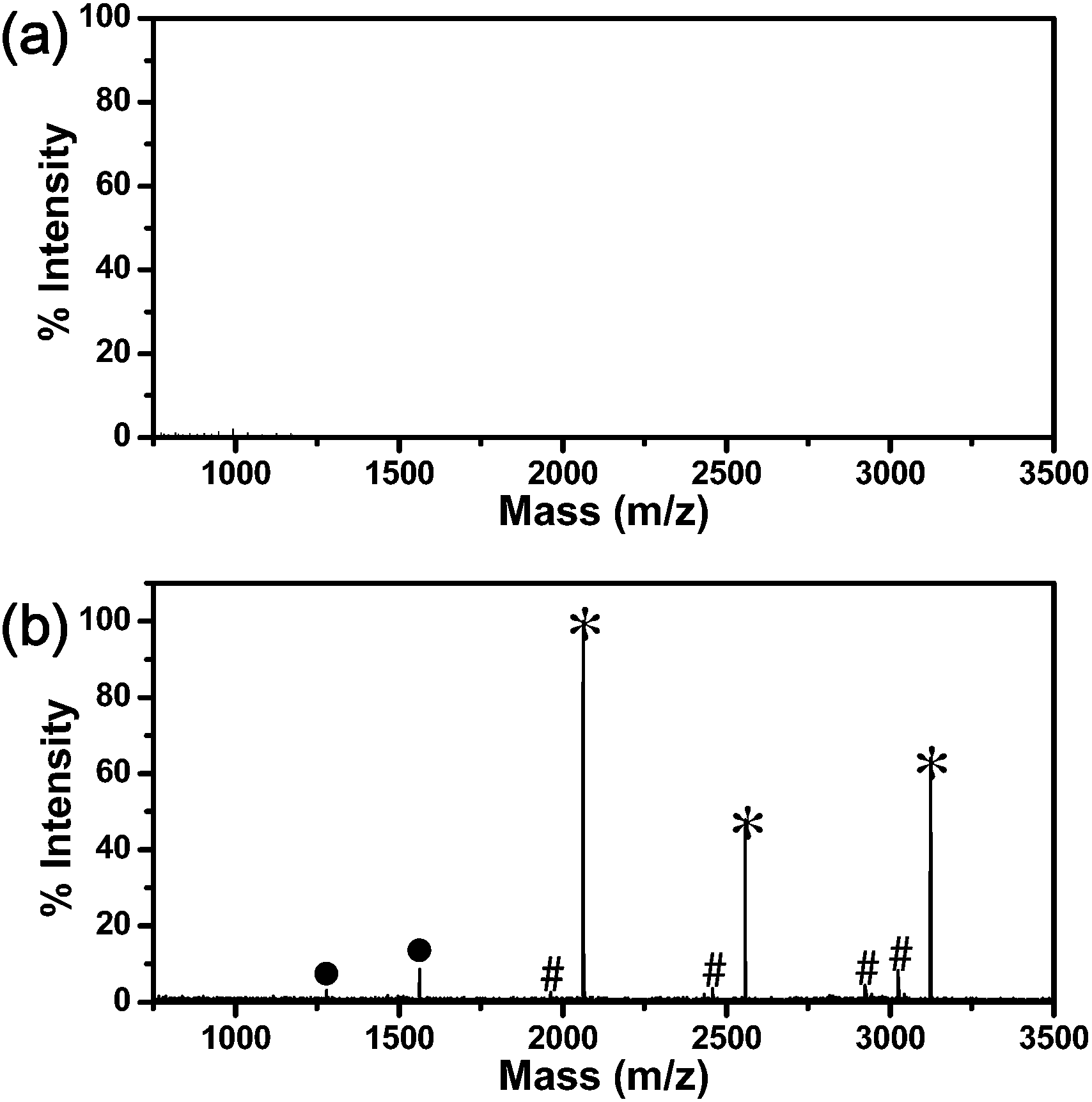 | ||
| Fig. 5 MALDI mass spectra of a tryptic digest of β-casein in the mixture of proteins containing β-casein, BSA and Cyto C (mass ratio of 1:10:100:100): (a) before and (b) after enrichment with rattle-type mTiO2@P(NIPAM-co-MBA). “*”, “#” and “●” indicate phosphorylated peptides, their dephosphorylated counterparts and double-charged counterparts, respectively. | ||
In order to further verify the size-exclusion effect against proteins and identify the molecular range in which the analyte could freely pass through the P(NIPAM-co-MBA) gel network shell, we measured the adsorption capability of rattle-type mTiO2@P(NIPAM-co-MBA) toward both nonphosphoproteins and phosphoproteins with different molecular weights by SDS-PAGE analysis. Before adding the rattle-type mTiO2@P(NIPAM-co-MBA), BSA, Cyto C, β-casein and ASF (phosphoprotein with a molecular weight of 38 kDa) were mixed with a mass ratio of nearly 1:1:1:1. After enrichment by rattle-type mTiO2@P(NIPAM-co-MBA), the stock solution, supernatant, eluate, as well as the residue on the material were collected and lyophilized for SDS-PAGE analysis. The protein adsorption capability of rattle-type mTiO2@P(NIPAM-co-MBA) was evaluated by comparing the difference in densities of each protein collected from the previously mentioned four steps. As shown in Fig. 6, BSA, Cyto C and ASF were effectively excluded by rattle-type mTiO2@P(NIPAM-co-MBA). Although the molecular weight of Cyto C is smaller than β-casein, rattle-type mTiO2@P(NIPAM-co-MBA) did not adsorb Cyto C because the mTiO2 core only selectively captures phosphoproteins. It is worth noting that a small amount of β-casein was found in the eluate, which indicates that these β-caseins could partially pass through the P(NIPAM-co-MBA) shell. The partial adsorption of β-casein suggests that the critical exclusion molecular weight of the P(NIPAM-co-MBA) network shell is around 24 kDa, which perfectly meets the requirement of the molecular weight range in the peptidome research (<20 kDa). All these abovementioned results clearly demonstrate that the rattle-type mTiO2@P(NIPAM-co-MBA) possesses the desired great size-exclusion effect against both HMW nonphosphoproteins and HMW phosphoproteins.
 | ||
| Fig. 6 SDS-PAGE analysis of standard protein mixtures (BSA + ASF + β-casein + Cyto C) before and after treatment with rattle-type mTiO2@P(NIPAM-co-MBA). Lane 1: marker; Lane 2: protein mixture (BSA + ASF + β-casein + Cyto C) before treatment; Lane 3: the supernatant of protein mixture (BSA + ASF + β-casein + Cyto C) after treatment by rattle-type mTiO2@P(NIPAM-co-MBA); Lane 4: the eluate from rattle-type mTiO2@P(NIPAM-co-MBA); Lane 5: the residual on the rattle-type mTiO2@P(NIPAM-co-MBA). | ||
Conclusions
In summary, we have presented a facile and repeatable synthetic route for the preparation of rattle-type mTiO2@P(NIPAM-co-MBA) with well-defined core–void–shell structure and desired functionality. The non-crosslinked PMAA sacrificial layer plays a key role in the successful preparation of the target material. Taking advantage of the excellent size-exclusion effect against high-molecular-weight proteins offered by the polymer gel network shell, as well as the remarkable enrichment capability toward phosphopeptides of the mTiO2 core, the rattle-type mTiO2@P(NIPAM-co-MBA) microspheres exhibit great capabilities for the one-step selective extraction of phosphopeptidome from complicated samples containing large amounts of high-molecular-weight proteins and nonphosphopeptides. In addition, we believe that our strategy can also be applied in the selective extraction of peptidome and other modification-specific peptidome by simply alternating the mTiO2 core with a mesoporous carbon core or some other specific cores.Acknowledgements
This work was supported by the National Science and Technology Key Project of China (2012AA020204 and 2012CB910602), the National Science Foundation of China (Grant no. 21025519 and 21034003) and Shanghai Projects (Grants Eastern Scholar, 13520720200, 13JC1400500 and B109).Notes and references
- L. A. Liotta, M. Ferrari and E. Petricoin, Nature, 2003, 425, 905 CrossRef CAS PubMed.
- E. F. Petricoin, C. Belluco, R. P. Araujo and L. A. Liotta, Nat. Rev. Cancer, 2006, 6, 961–967 CrossRef CAS PubMed.
- E. P. Diamandis, J. Proteome Res., 2006, 5, 2079–2082 CrossRef CAS PubMed.
- G. Menschaert, T. T. M. Vandekerckhove, G. Baggerman, L. Schoofs, W. Luyten and W. Van Criekinge, J. Proteome Res., 2010, 9, 2051–2061 CrossRef CAS PubMed.
- W. H. Robinson and L. Steinman, Nat. Biotechnol., 2011, 29, 500–502 CrossRef CAS PubMed.
- L. Zhao, H. Q. Qin, R. A. Wu and H. F. Zou, J. Chromatogr. A, 2012, 1228, 193–204 CrossRef CAS PubMed.
- J. Z. Chen, M. Anderson, D. E. Misek, D. M. Simeone and D. M. Lubman, J. Chromatogr. A, 2007, 1162, 117–125 CrossRef CAS PubMed.
- R. Vitorino, A. S. Barros, A. Caseiro, R. Ferreira and F. Amado, Talanta, 2012, 94, 209–215 CrossRef CAS PubMed.
- T. C. Li, S. P. Dai, Z. M. Wang and H. Q. Zhang, Electrophoresis, 2010, 31, 1090–1096 CAS.
- R. S. Tirumalai, K. C. Chan, D. A. Prieto, H. J. Issaq, T. P. Conrads and T. D. Veenstra, Mol. Cell. Proteomics, 2003, 2, 1096–1103 CAS.
- K. L. Johnson, C. J. Mason, D. C. Muddiman and J. E. Eckel, Anal. Chem., 2004, 76, 5097–5103 CrossRef CAS PubMed.
- X. Y. Zheng, H. Baker and W. S. Hancock, J. Chromatogr. A, 2006, 1120, 173–184 CrossRef CAS PubMed.
- D. W. Greening and R. J. Simpson, J. Proteomics, 2010, 73, 637–648 CrossRef CAS PubMed.
- R. J. Tian, H. Zhang, M. L. Ye, X. G. Jiang, L. H. Hu, X. Li, X. H. Bao and H. F. Zou, Angew. Chem., Int. Ed., 2007, 46, 962–965 CrossRef CAS PubMed.
- R. J. Tian, L. B. Ren, H. J. Ma, X. Li, L. H. Hu, M. L. Ye, R. Wu, Z. J. Tian, Z. Liu and H. F. Zou, J. Chromatogr. A, 2009, 1216, 1270–1278 CrossRef CAS PubMed.
- A. Bouamrani, Y. Hu, E. Tasciotti, L. Li, C. Chiappini, X. W. Liu and M. Ferrari, Proteomics, 2010, 10, 496–505 CrossRef CAS PubMed.
- S. S. Liu, Y. Li, C. H. Deng, Y. Mao, X. M. Zhang and P. Y. Yang, Proteomics, 2011, 11, 4503–4513 CrossRef CAS PubMed.
- H. Q. Qin, P. Gao, F. J. Wang, L. Zhao, J. Zhu, A. Q. Wang, T. Zhang, R. Wu and H. F. Zou, Angew. Chem., Int. Ed., 2011, 50, 12218–12221 CrossRef CAS PubMed.
- F. Li, B. Dever, H. Q. Zhang, X. F. Li and X. C. Le, Angew. Chem., Int. Ed., 2012, 51, 3518–3519 CrossRef CAS PubMed.
- K. Qian, W. Y. Gu, P. Yuan, F. Liu, Y. H. Wang, M. Monteiro and C. Z. Yu, Small, 2012, 8, 231–236 CrossRef CAS PubMed.
- P. Yin, Y. H. Wang, Y. Li, C. H. Deng, X. M. Zhang and P. Y. Yang, Proteomics, 2012, 12, 2784–2791 CrossRef CAS PubMed.
- L. Zhang, S. B. Wu, C. Lia and Q. H. Yang, Chem. Commun., 2012, 48, 4190–4192 RSC.
- G. T. Zhu, X. S. Li, Q. Gao, N. W. Zhao, B. F. Yuan and Y. Q. Feng, J. Chromatogr. A, 2012, 1224, 11–18 CrossRef CAS PubMed.
- H. Wan, H. Q. Qin, Z. C. Xiong, W. B. Zhang and H. F. Zou, Nanoscale, 2013, 5, 10936–10944 RSC.
- G. T. Zhu, X. M. He, X. S. Li, S. T. Wang, Y. B. Luo, B. F. Yuan and Y. Q. Feng, J. Chromatogr. A, 2013, 1316, 23–28 CrossRef CAS PubMed.
- L. T. Liu, Y. Zhang, L. Zhang, G. Q. Yan, J. Yao, P. Y. Yang and H. J. Lu, Anal. Chim. Acta, 2012, 753, 64–72 CrossRef CAS PubMed.
- L. H. Hu, H. J. Zhou, Y. H. Li, S. T. Sun, L. H. Guo, M. L. Ye, X. F. Tian, J. R. Gu, S. L. Yang and H. F. Zou, Anal. Chem., 2009, 81, 94–104 CrossRef CAS PubMed.
- H. Q. Qin, F. J. Wang, P. Y. Wang, L. Zhao, J. Zhu, Q. H. Yang, R. A. Wu, M. L. Ye and H. F. Zou, Chem. Commun., 2012, 48, 961–963 RSC.
- X. S. Li, Y. N. Pan, Y. Zhao, B. F. Yuan, L. Guo and Y. Q. Feng, J. Chromatogr. A, 2013, 1315, 61–69 CrossRef CAS PubMed.
- S. Jin, Y. J. Pan and C. C. Wang, Acta Chim. Sin., 2013, 71, 1500–1504 CrossRef CAS.
- S. Eiden-Assmann, J. Widoniak and G. Maret, Chem. Mater., 2004, 16, 6–11 CrossRef CAS.
- W. F. Ma, Y. Zhang, L. L. Li, L. J. You, P. Zhang, Y. T. Zhang, J. M. Li, M. Yu, J. Guo, H. J. Lu and C. C. Wang, ACS Nano, 2012, 6, 3179–3188 CrossRef CAS PubMed.
Footnote |
| † Wanfu Ma and Ying Zhang contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2014 |