
[3 + 2] Cycloaddition of aza-oxyallyl cations with aldehydes†
Qianfa
Jia
a,
Zhiyun
Du
a,
Kun
Zhang
a and
Jian
Wang
*b
aAllan H. Conney Laboratory for Anticancer Research. Guangdong University of Technology, Guang Dong, 510006, China
bSchool of Pharmaceutical Sciences, Tsinghua University, Beijing, 100084, China. E-mail: wangjian2012@tsinghua.edu.cn
First published on 24th October 2016
Abstract
We report here the [3 + 2] cycloaddition reactions of in situ-generated aza-oxyallyl cations with commercially available α,β-unsaturated aldehydes or aldehydes, providing a rapid and efficient synthetic approach to access a variety of oxazolidin-4-ones in high yields.
Introduction
The scaffold of oxazolidinones is widely spread in natural products and bioactive molecules.1 As outlined in Fig. 1, molecule I is an oxazolidinone calcitonin gene-related peptide (CGRP) receptor antagonist for the treatment of migraine headaches.2 Synoxazolidinone C (compound II) is isolated from the crude S. pulmonaria extract which exhibited promising antibacterial and anticancer activities.3 In recent years, the preparation of 4-oxazolidinone derivatives has received much attention from the synthetic community due to their significant applications. In this context, oxaziridines have been identified as one of the practical and useful synthons in the construction of oxazolidin-4-ones. To date, Ye,4 Feng5 and Smith6 groups have successfully utilized oxaziridines to react with ketenes, azlactones or anhydrides to assemble oxazolin-4-ones respectively. In these cases, good yields and high diastereo- and enantioselectivities were achieved regularly (Scheme 1a). Later on, Comesse and co-workers reported a domino reaction to generate oxazolidin-4-ones by using enol ethers and hydroxyl α-bromoamides7 as starting materials (Scheme 1b). Despite these advances, a more practical and efficient protocol for the rapid construction of diversified oxazolidin-4-ones is still in high demand.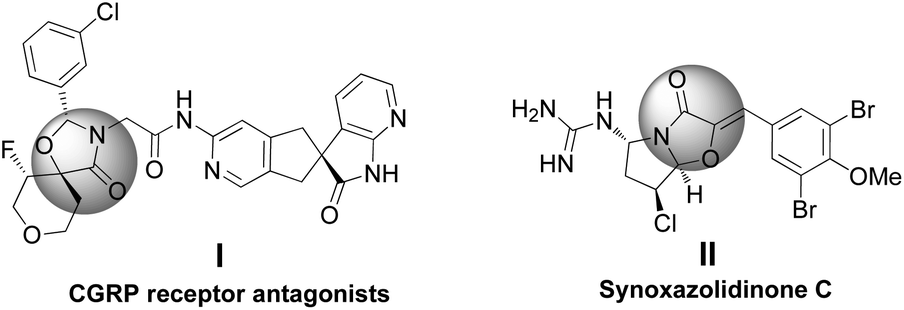 | ||
| Fig. 1 Biologically important oxazolidin-4-ones. | ||
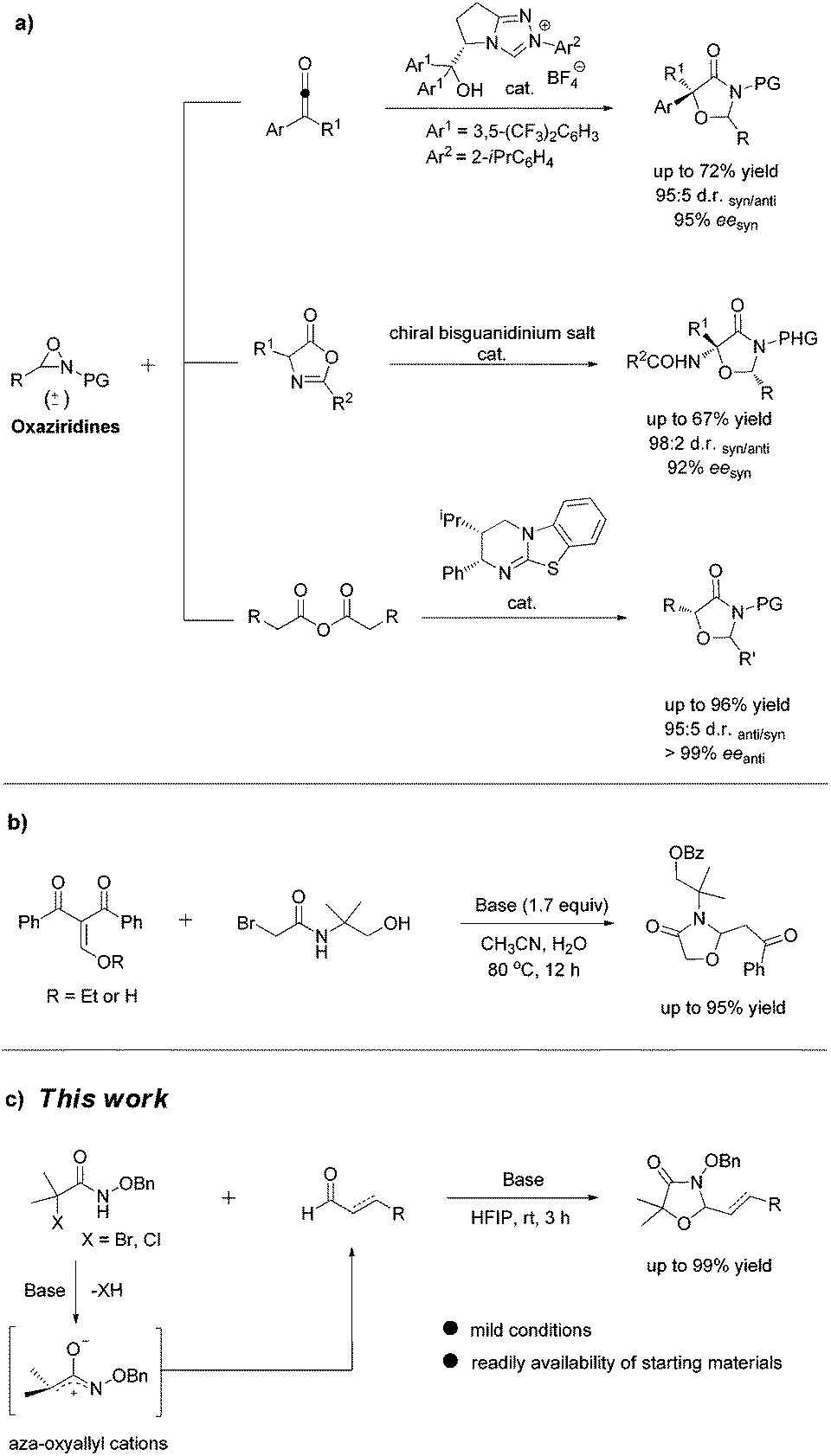 | ||
| Scheme 1 Synthetic strategies for the synthesis of oxazolidin-4-ones via [3 + 2] cycloadditions. | ||
Although aza-oxyallylic cation intermediates had long been speculated, they remained elusive due to the failure in trapping the key active species.8 Until recently, the Jeffrey group has found definitive evidence to approve the existence of in situ-generated aza-oxyallylic cations.9 Subsequently, Jeffrey, Wu and other groups described a formal [3 + 2] cycloaddition reaction of substituted indoles with aza-oxyallylic cation intermediates to prepare pyrrolindoline scaffolds.10 Based on our own research interest on heterocycle synthesis,11 we envisioned that readily available unsaturated aldehydes or simple aldehydes may also react with aza-oxyallylic cations in a one-pot fashion to generate five-membered oxazolidinones with diversified substituents. Herein, we wish to report a practical procedure for the synthesis of styryloxazolidin-4-ones under ambient conditions (Scheme 1c).12
Results and discussion
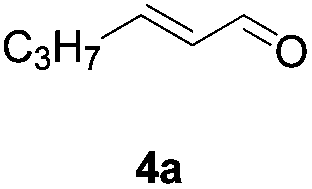
N-(Benzyloxy)-2-bromo-2-methylpropanamide 1a and cinnamaldehyde 2a were selected as model substrates in reaction optimization (Table 1). Several inorganic and organic bases were screened firstly (entries 1–8). Na2CO3 was found to be efficient and afforded (E)-3-(benzyloxy)-5,5-dimethyl-2-styryloxazolidin-4-one 3a in 94% yield at room temperature (entry 3). Further parameter screening showed that the solvent significantly affected the reaction conversion. When TFE (2,2,2-trifluoroethanol), trifluoromethylbenzene or 1,2,3,4,5-pentafluoro-6-methylbenzene was used instead of HFIP (hexafluoroisopropanol)13 as a solvent, the chemical yield of 3a dropped dramatically (entries 9, 11 and 12). We deduce that TFE might be a suitable solvent in stabilizing the in situ-generated aza-oxyallyl cations. When reactions were carried out in nonpolar solvents (e.g. toluene and Et2O), low chemical yields were obtained (26% and 32%, respectively, entries 10 and 13). Surprisingly, no product was obtained when the reaction was carried out in more polar solvents such as MeOH, i-PrOH and DMSO (entries 14–16). To our delight, varying the ratio of 1a/2a from 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.2 to 1.5:1 gave almost quantitative yield (99%, entry 17). Finally, the optimal conditions were achieved when the reaction was performed by using 2.0 equiv. of Na2CO3 and HFIP as a solvent.
:1.2 to 1.5:1 gave almost quantitative yield (99%, entry 17). Finally, the optimal conditions were achieved when the reaction was performed by using 2.0 equiv. of Na2CO3 and HFIP as a solvent.
|
|
|||
|---|---|---|---|
| Entry | Base | Solvent | Yieldb (%) |
|
a If not further mentioned, the reaction was carried out with 1a (0.1 mmol), 2a (0.12 mmol) and the base (2.0 equiv.) in the HFIP (1.0 mL) at room temperature for 3 h.
b Isolated yield.
c
1a:2a = 1.5:1. HFIP = hexafluoroisopropanol. TFE = 2,2,2-trifluoroethanol.
|
|||
| 1 | K2CO3 | HFIP | 86 |
| 2 | Cs2CO3 | HFIP | 80 |
| 3 | Na2CO3 | HFIP | 94 |
| 4 | NaOAc | HFIP | 53 |
| 5 | Et3N | HFIP | 27 |
| 6 | DIPEA | HFIP | 58 |
| 7 | DMAP | HFIP | 89 |
| 8 | NaOH | HFIP | 67 |
| 9 | Na2CO3 | TFE | 56 |
| 10 | Na2CO3 | Toluene | 26 |
| 11 | Na2CO3 | PhCF3 | 31 |
| 12 | Na2CO3 | C6F5CH3 | 19 |
| 13 | Na2CO3 | Et2O | 32 |
| 14 | Na2CO3 | MeOH | — |
| 15 | Na2CO3 | i-PrOH | — |
| 16 | Na2CO3 | DMSO | — |
| 17 | Na 2 CO 3 | HFIP | 99 |
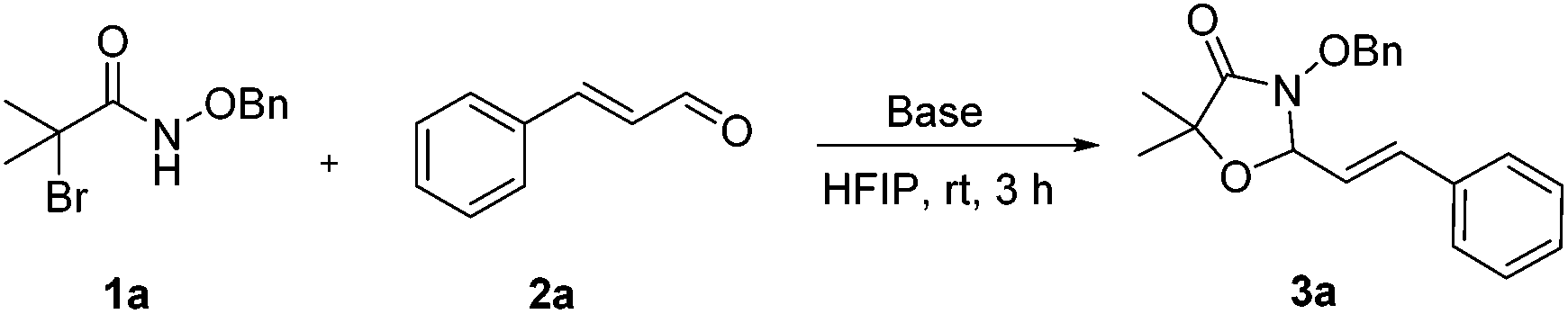
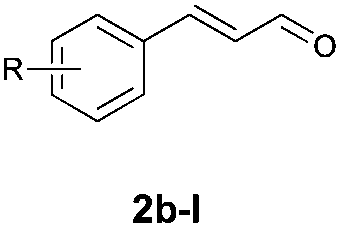
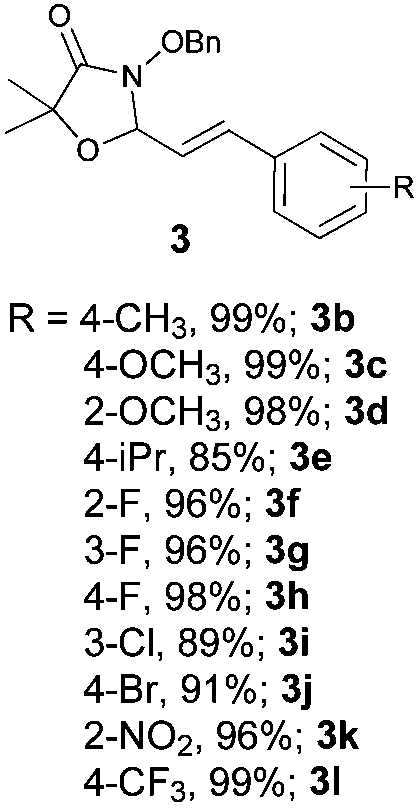
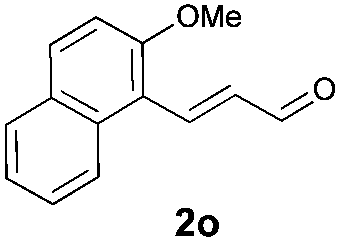
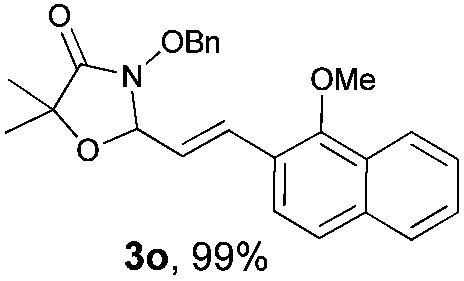
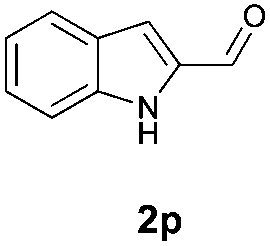
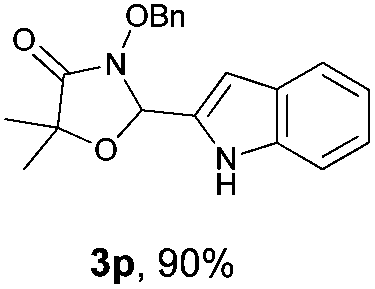
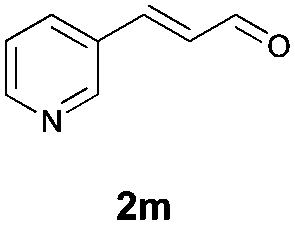
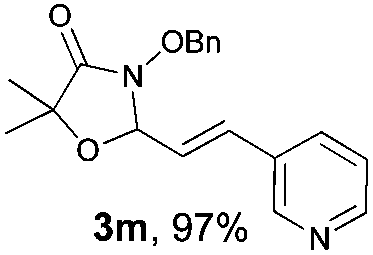
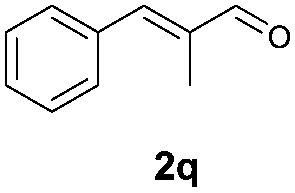
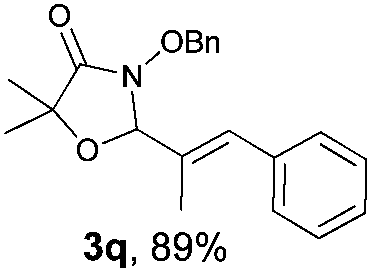
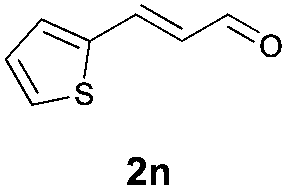
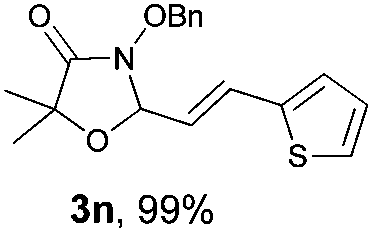
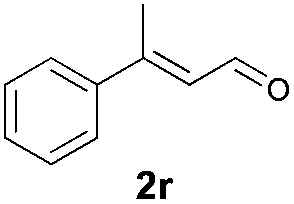
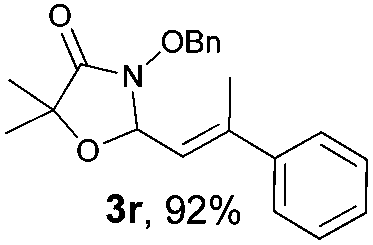
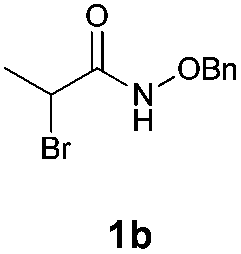
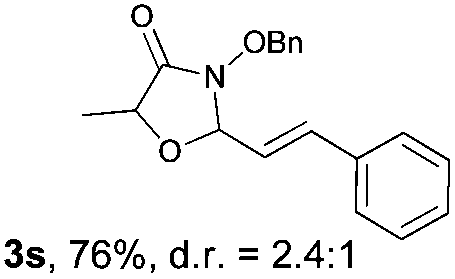
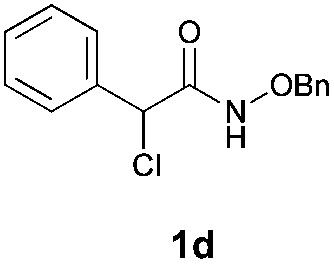
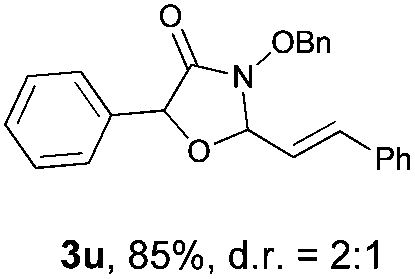
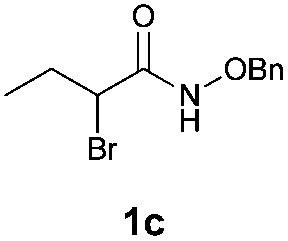
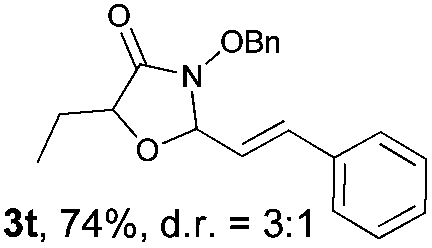
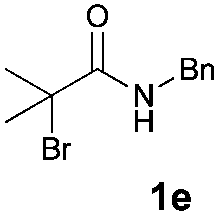
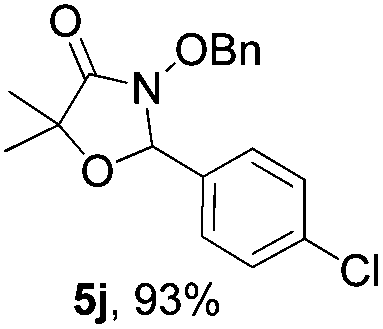
With the optimal reaction conditions in hand, we then evaluated the scope of aldehydes. A series of substituents on the phenyl ring of enals 2 with N-(benzyloxy)-2-bromo-2-methylpropanamide 1a were tested and are summarized in Table 2. Briefly, the substituent pattern and the electronic nature of enals 2 have limited effects on reaction conversion. The corresponding products were obtained in good to high yields (3b–l, 85–99% yields). On replacement of the β-phenyl substituent with heteroaryl, such as 3-pyridinyl, 2-thiophenyl, 2-methoxynaphthalenyl, and 1H-2-indole-yl, the reactions could still undergo smoothly to afford the desired products in satisfying yields (3m–3p, 90–99% yields). The methyl substituent located at the α- or β-positions of enals has little influence on reaction efficiency (3q and 3r, 89% and 92%, respectively). On the other hand, other types of α-halo O-alkylhydroxamates 1b–e with cinnamaldehyde 2a were also examined and displayed good performances. When R1 was alkyl or phenyl and R2 was hydrogen, the α-bromo/chloro O-alkylhydroxamates were found to be suitable partners to yield the desired products in high yields and with good diastereoselectivities (3s–u). Unfortunately, no reaction happened when the substrate N-benzyl-2-bromo-2-methylpropanamide 1e was used.14
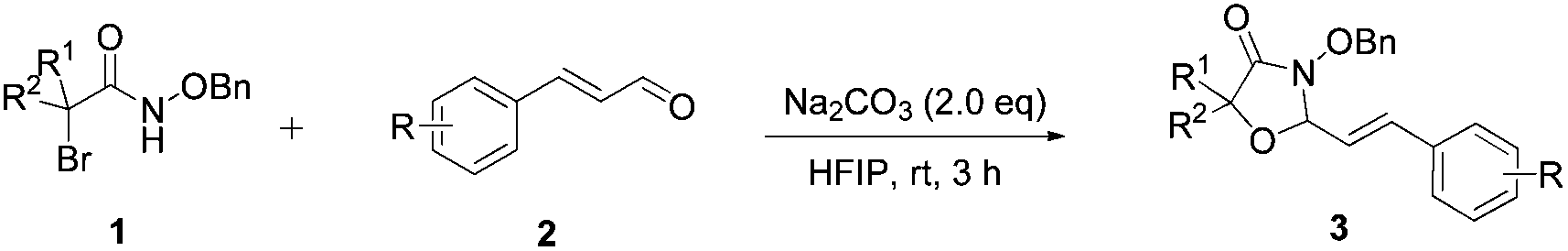
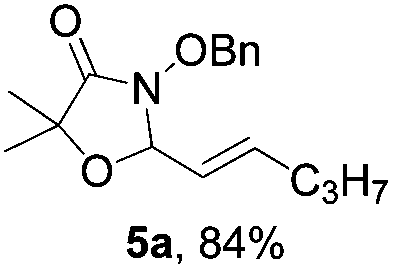
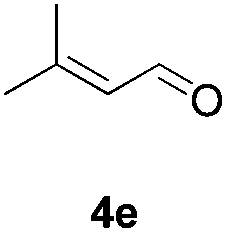
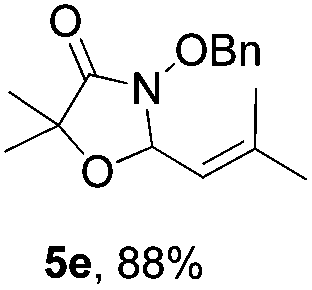
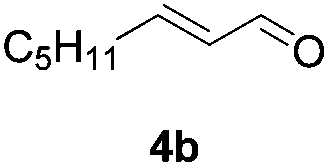
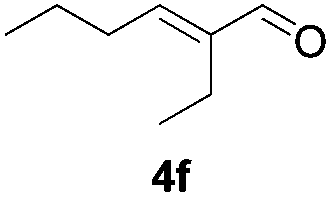
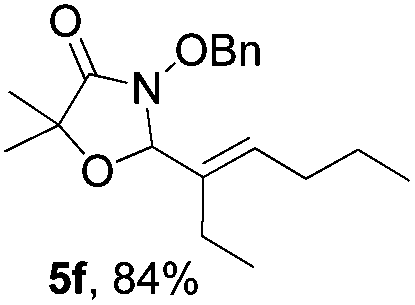
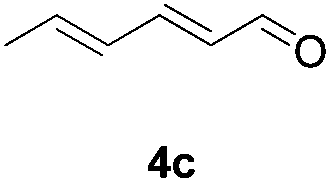
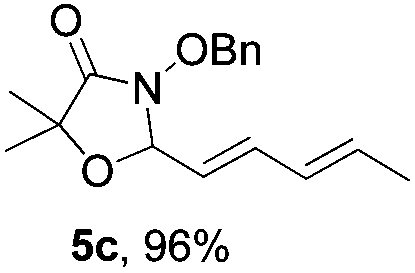
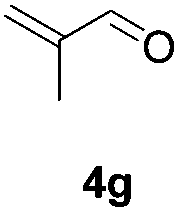
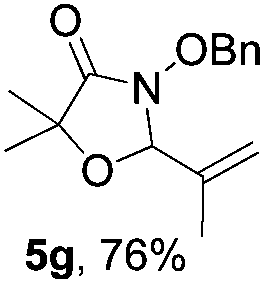
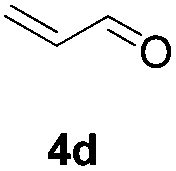
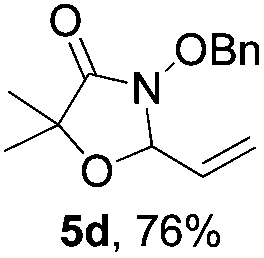
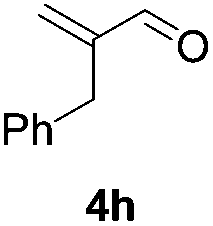
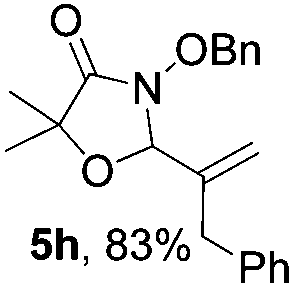
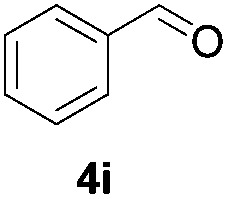
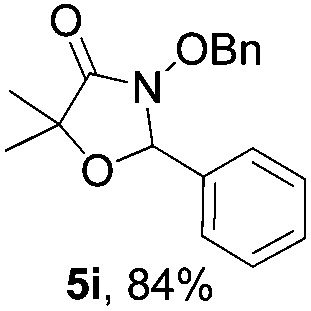
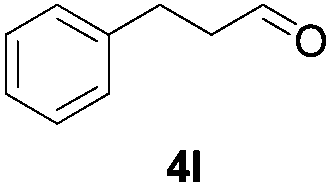
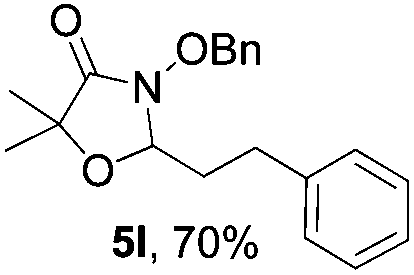
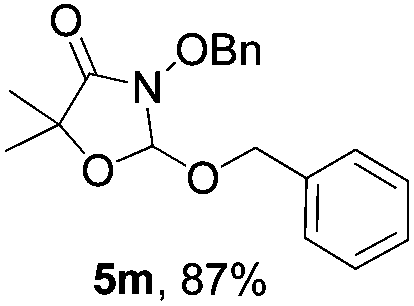
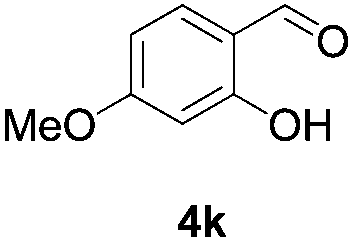
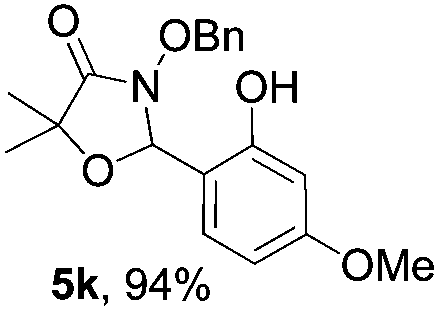
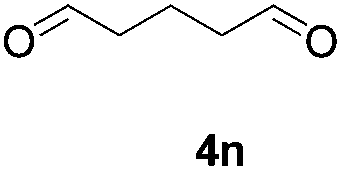
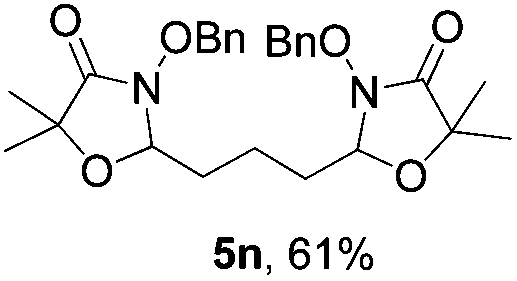
Further investigations on the feasibility of β-alkyl-type enals and simple aldehydes were also conducted. All the tested substrates worked well and provided [3 + 2] products in moderate to high yields (Table 3). α-Substituted or β-mono-/disubstituted enals were all tolerated (4a–h, 62–88% yields). Pleasingly, simple aldehydes directly bearing aryl or alkyl groups (4i–n) reacted well with 1a under standard conditions afforded the corresponding products in good to high yields (5i–n, 61–94%). To further demonstrate the utility of this methodology, we carried out a gram-scale synthesis (Scheme 2a). Further oxidation of 3a could selectively and efficiently afford functionalized diol 6 and epoxide 7 (Schemes 2b and c). Hydrogenation of 3a efficiently generated product 8 (Scheme 2d).
 | ||
| Scheme 2 (a) Standard conditions; (b) OsO4, NMO, MeCN/H2O/acetone = 1:1:2, r.t., 16 h; (c) m-CPBA, CH2Cl2, 72 h; (d) H2, Pd/C, EtOAc. | ||
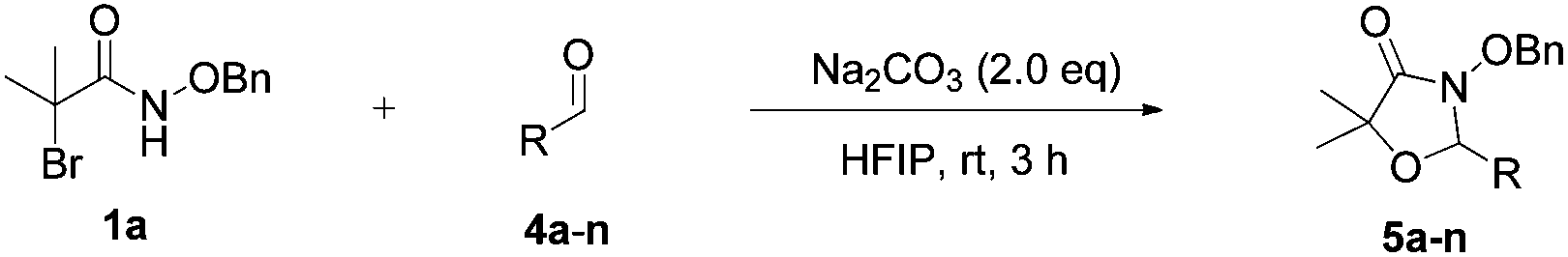

Conclusions
We have developed the example of catalyst-free [3 + 2] cycloaddition of an aza-oxyallylic cation with aldehydes. This protocol provides straightforward access to the corresponding biologically important and highly functionalized oxazolidin-4-ones in good to excellent yields under ambient conditions. The progress of an asymmetric version of this cycloaddition and the application of this methodology are ongoing in our laboratory.Acknowledgements
The authors acknowledge the financial support from the National Natural Science Foundation of China (21272043, 21672121), the Science and Technology Planning Project of Guangdong Province (2007A020300007-10, 2011B090400573) and the Guangdong Natural Science Foundation (S2011010004967).Notes and references
- For selected review and examples, see: (a) M. R. Barbachyn, D. K. Hutchinson, S. J. Brickner, M. H. Cynamon, S. P. Klemens, S. E. Glickman, K. C. Grega, S. K. Hendges, D. S. Toops, C. W. Ford and G. E. Zurenko, J. Med. Chem., 1996, 39, 680 CrossRef CAS PubMed; (b) S. Iwama, M. Segawa, S. Fujii, K. Ikeda and S. Katsumura, Bioorg. Med. Chem. Lett., 1998, 8, 3495 CrossRef CAS PubMed; (c) M. J. Genin, D. K. Hutchinson, D. A. Allwine, J. B. Hester, D. E. Emmert, S. A. Garmon, C. W. Ford, G. E. Zurenko, J. C. Hamel, R. D. Schaad, D. Stapert, B. H. Yagi, J. M. Friis, E. M. Shobe and W. J. Adams, J. Med. Chem., 1998, 41, 5144 CrossRef CAS PubMed; (d) K. S. Jandu, V. Barrett, M. Brockwell, D. Cambridge, D. R. Farrant, C. Foster, H. Giles, R. C. Glen, A. P. Hill, H. Hobbs, A. Honey, G. R. Martin, J. Salmon, D. Smith, P. Woollard and D. L. Selwood, J. Med. Chem., 2001, 44, 681 CrossRef CAS PubMed; (e) M. R. Barbachyn and C. W. Ford, Angew. Chem., Int. Ed., 2003, 42, 2010 CrossRef CAS PubMed; (f) V. R. Macherla, J. Liu, M. Sunga, D. J. White, J. Grodberg, S. Teisan, K. S. Lam and B. C. M. Potts, J. Nat. Prod., 2007, 70, 1454 CrossRef CAS PubMed; (g) N. V. Shymanska, I. H. An and J. G. Pierce, Angew. Chem., Int. Ed., 2014, 53, 5401 CrossRef CAS PubMed.
- B. M. Crowley, C. A. Stump, D. N. Nguyen, C. M. Potteiger, M. A. McWherter, D. V. Paone, A. G. Quigley, J. G. Bruno, D. Cui, J. C. Culberson, A. Danziger, C. Fandozzi, D. Gauvreau, A. L. Kemmerer, K. Menzel, E. L. Moore, S. D. Mosser, V. Reddy, R. B. White, C. A. Salvatore, S. A. Kane, I. M. Bell, H. G. Selnick, M. E. Fraley and C. S. Burgey, Bioorg. Med. Chem. Lett., 2015, 25, 4777 CrossRef CAS PubMed.
- M. Tadesse, J. Svenson, M. Jaspars, M. B. Strøm, M. H. Abdelrahman, J. H. Andersen, E. Hansen, P. E. Kristiansen, K. Stensvåg; and T. Haug, Tetrahedron Lett., 2011, 52, 1804 CrossRef CAS.
- P.-L. Shao, X.-Y. Chen and S. Ye, Angew. Chem., Int. Ed., 2010, 49, 8412 CrossRef CAS PubMed.
- S. Dong, X.-H. Liu, Y. Zhu, P. He, L. Lin and X.-M. Feng, J. Am. Chem. Soc., 2013, 135, 10026 CrossRef CAS PubMed.
- S. R. Smith, C. Fallan, J. E. Taylor, R. McLennan, D. S. B. Daniels, L. C. Morrill, A. M. Z. Slawin and A. D. Smith, Chem. – Eur. J., 2015, 21, 10530 CrossRef CAS PubMed.
- A. El Bouakher, R. Le Goff, J. Tasserie, J. Lhoste, A. Martel and S. Comesse, Org. Lett., 2016, 18, 2383 CrossRef CAS PubMed.
- (a) I. Lengyel and J. C. Sheehan, Angew. Chem., Int. Ed. Engl., 1968, 7, 25 CrossRef CAS; (b) I. Lengyel and R. V. Mark, Acta Chim. Acad. Sci. Hung., 1974, 81, 475 CAS; (c) G. L'Abbe, Angew. Chem., Int. Ed. Engl., 1980, 19, 276 CrossRef; (d) P. J. Stang and G. H. Anderson, Gazz. Chim. Ital., 1995, 125, 329 CAS; (e) A. D. Cohen, B. M. Showalter and J. P. Toscano, Org. Lett., 2004, 6, 401 CrossRef CAS PubMed.
- (a) C. S. Jeffrey;, K. L. Barnes;, J. A. Eickhoff; and C. R. Carson, J. Am. Chem. Soc., 2011, 133, 7688 CrossRef PubMed; (b) A. Acharya, J. A. Eickhoff and C. S. Jeffrey, Synthesis, 2013, 1825 CAS; (c) K. L. Barnes, A. K. Koster and C. S. Jeffrey, Tetrahedron Lett., 2014, 55, 4690 CrossRef CAS.
- (a) M. C. DiPoto, R. P. Hughes and J. Wu, J. Am. Chem. Soc., 2015, 137, 14861 CrossRef CAS PubMed; (b) A. Acharya, D. Anumandla and C. S. Jeffrey, J. Am. Chem. Soc., 2015, 137, 14858 CrossRef CAS PubMed; (c) W. Ji, L. Yao and X. Liao, Org. Lett., 2016, 18, 628 CrossRef CAS PubMed.
- (a) L. Chen, G.-M. Yang, J. Wang, Q.-F. Jia, J. Wei and Z.-Y. Du, RSC Adv., 2015, 5, 76696 RSC; (b) Q.-F. Jia, L. Chen, G.-M. Yang, J. Wang, J. Wei and Z.-Y. Du, Tetrahedron Lett., 2015, 56, 7150 CrossRef CAS.
- During the preparation of this manuscript, a [3 + 2] cycloaddition reaction of in situ formed azaoxyallyl cations with aldehydes was reported, see: K.-F. Zhang, C. Yang, H.-Q. Yao and A.-J. Lin, Org. Lett., 2016, 18, 4618 CrossRef CAS PubMed.
- (a) Hexafluoroisopropanol was proved to increase the yield of reactions proceeding through reactive oxyallyl cationic intermediates; see: A. G. Myers; and J. K. Barbay, Org. Lett., 2001, 3, 425 CrossRef; (b) M. Harmata;, C. Huang;, P. Rooshenas; and P. R. Schreiner, Angew. Chem., Int. Ed., 2008, 47, 8696 CrossRef PubMed.
- An alkoxy donor group on the nitrogen atom could potentially stabilize the aza-oxyallylic cation intermediate. See: (a) Y. Kikugawa, Heterocycles, 2009, 78, 571 CrossRef CAS; (b) D. J. Wardrop and E. G. Bowen, in Nitrenes and Nitrenium Ions, ed. D. E. Falvey and A. D. Gudmundsdottir, WILEY-VCH, Weinheim, 2013, p. 347 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6qo00526h |
| This journal is © the Partner Organisations 2017 |