
Reductive insertion of sulfur dioxide for the synthesis of trifluoromethyl thiolsulphonates through a one-pot reaction of aniline and trifluoromethanesulfanylamide†
Jie
Sheng
a,
Yuewen
Li
b and
Guanyinsheng
Qiu
*b
aCollege of Food Science and Technology, Shanghai Ocean University, 999 Huchenghuan Road, Shanghai, China
bCollege of Biological, Chemical Science and Engineering, Jiaxing University, 118 Jiahang Road, Jiaxing 314001, China. E-mail: 11110220028@fudan.edu.cn
First published on 20th October 2016
Abstract
A bismuth(III) chloride-mediated one-pot reaction of anilines, sulfur dioxide, and trifluoromethanesulfanylamide is reported herein, leading to antifungal trifluoromethyl thiolsulphonates. This transformation employs N-aminomorphine as an additive and provides the final products in good yields with a broad functional group tolerance. It is believed that bismuth(III) chloride facilitates the formation of “CF3S+”, and N-aminomorphine plays a critical role in providing electrons to catalyse the cross-coupling of in situ generated aryldiazonium, sulfur dioxide and trifluoromethanesulfanylamide.
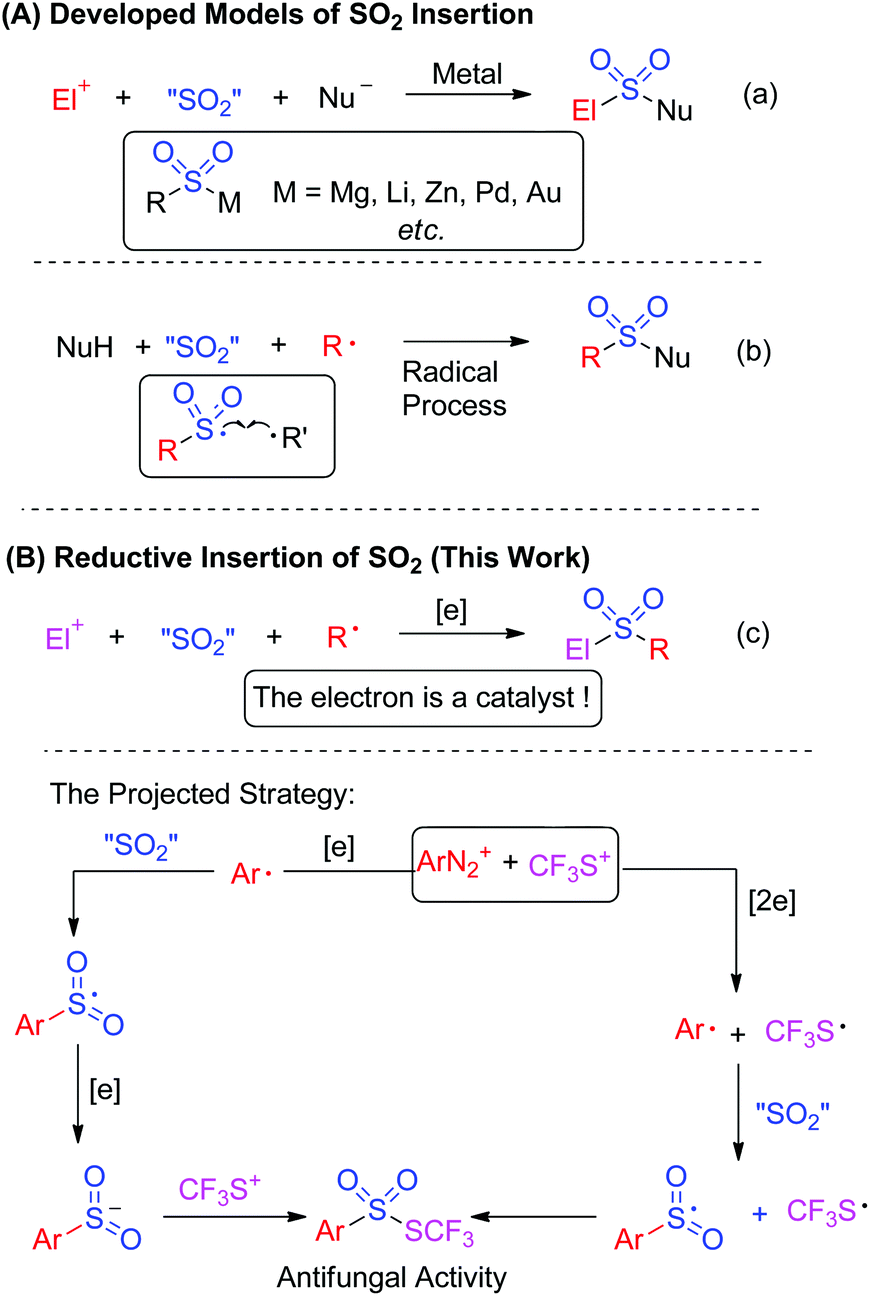
Currently, the insertion of sulfur dioxide into small molecules has been used as an efficient method for the introduction of sulfonyl groups in organic synthesis.1 As illustrated in Scheme 1A, two models were established for sulfur dioxide fixation. Approaches including metal-catalyzed or metal-free insertion of sulfur dioxide have appeared so far.
 | ||
| Scheme 1 Recent development of sulfur dioxide fixation. | ||
Basically, the first model of SO2 fixation was described as organometallic species-based nucleophilic insertion. The organometallic species mainly included Grignard reagents, organolithium, organozinc, in situ generated organopalladium and organogold species (Scheme 1a).2 The SO2-incorporated organosulfonylmetallic species was trapped by electrophiles or nucleophiles to offer the final products. An elegant example involving palladium-catalyzed SO2 fixation of aryl iodides, which was reported by Willis and co-workers for the preparation of aryl N-aminosulfonamides, represented an unprecedented advance in the chemistry of SO2 fixation.2a Distinctively, Mascitti and Toste succeeded in installing SO2 into an in situ generated arylgold species from aryl boronic acid.2b Here the formed arylsulfonylgold species was converted into sulfinate through ligand exchange, and was trapped by benzyl bromide for the formation of sulfones.
Another version of fixing sulfur dioxide is the radical pathway.3 In this process, sulfur dioxide was introduced by radical insertion. The generated sulfonyl radical was trapped by another radical to afford the final product (Scheme 1b). An initial example was highlighted by Wu's group, which was focused on insertion of sulfur dioxide into an aryl radical for the synthesis of N-aminosulfonamides.3a The aryl radical was derived from aryldiazonium via hydrazine-involved single electron transfer, which provided a hydrazine radical in the meantime. Cross-coupling between the sulfonyl radical and the hydrazine radical delivered the final N-aminosulfonamides. By altering the aryl radical precursor to aryl triazene, copper catalysis enabled the synthesis of sulfonamides, where the arylsulfonyl radical generated from sulfur dioxide insertion of aryl radical was postulated as an intermediate. More importantly, the nucleophiles trapping the sulfonyl radical were expanded to amines.3b
Encouraged by the information mentioned above, it can be concluded that the developed sulfur dioxide fixation formally occurred to a nucleophile and an electrophile or a nucleophile and a radical precursor. So, we would like to exploit reductive insertion of sulfur dioxide between an electrophile and a radical precursor for the synthesis of other useful architectures (Scheme 1c). In order to realize this projected strategy, injecting an electron into the reaction was required, and the electron was thus described as a catalyst to trigger the reaction.4
On the other hand, thiolsulphonates are very important organic sulphur compounds due to their excellent performance in antibacterial and antifungal properties.5 Among thiolsulphonates, trifluoromethyl thiolsulphonates have attracted much more interest of chemists, since high hydrophobicity of the trifluoromethyl thio (CF3S) moiety has been recognized.6 Traditionally, trifluoromethyl thiolsulphonates were prepared from the electrophilic trifluoromethylthiolation of sulphinates. Such approaches required the use of accessibility-limiting sulfinates and the toxic or synthesis-difficult “CF3S” reagent, therefore restricting its application and generality.5a To date, studies on synthetic methodology developments towards trifluoromethyl thiolsulphonates have rarely appeared although they have great utilization potential.5,6
As a consequence, we envisioned that insertion of sulfur dioxide and trifluoromethylthiolation7 could be combined in one pot for the preparation of trifluoromethyl thiolsulphonate from anilines (Scheme 1c). In this transformation, the in situ generated aryldiazonium salt from aniline was a radical precursor, and trifluoromethanesulfanylamide (PhNHSCF3) was an electrophile. Therefore, this paper is herein the first example on fixation of SO2 by an electrophile and a radical precursor. More importantly, we hypothesized that aryldiazonium was reduced by an electron to an aryl radical, followed by SO2 insertion to produce the arylsulfonyl radical. This sulfonyl radical was further reduced by a second electron, leading to sulfinate, and the subsequential electrophilic trifluoromethylthiolation gave the final thiolsulphonates. Alternatively, the electrons reduced the aryldiazonium and in situ generated “CF3S+” to the aryl radical and “CF3S˙”, respectively. SO2 insertion and cross-coupling between the two radicals also provided thiolsulphonate (Scheme 1c). To verify the hypothesis, we started to test the feasibility of this proposed transformation. Based on our design, finding a suitable electron donor was the most important step. According to Wu's results,3a hydrazines such as N-aminomorphine could serve as a reductant to reduce aryldiazonium to the aryl radical. Consequently, N-aminomorphine was employed as an electron donor in this paper.
During the past several decades, various reaction systems have witnessed diazotization of aniline to diazonium salt. Among them, a combination of BF3·OEt2 with tert-butyl nitrite (tBuONO) enabled the generation of diazonium salts with extremely high reaction efficiency.8 Diazotization of anilines was thus not optimized in this paper. Condition screenings towards SO2 insertion and trifluoromethylthiolation were evaluated accordingly.
In the previous papers, it was reported that Lewis acid bismuth(III) chloride (BiCl3) were favorable for the formation of CF3S+ from PhNHSCF3.9 As a result, the model reaction was initially carried out in acetonitrile in the presence of 1.3 equiv. BiCl3 and 1.0 equiv. N-aminomorphine at 80 °C. As expected, the desired trifluoromethyl thiolsulphonate 3a was achieved in 58% (entry 1, Table 1). Motivated by this positive result, other result-affecting factors were also evaluated. As illustrated in Table 1, the BiCl3 loading made a significant impact on the model reaction. By increasing the loading of BiCl3 from 1.3 equiv. to 1.5 equiv., the yield of the product 3a was improved to 64% (entry 3, Table 1). Further increase of the BiCl3 amount did not give a better outcome (entry 4, Table 1). The decrease of BiCl3 amount drastically retarded the reaction, leading to 3a in 38% (entry 2, Table 1). Subsequently, the temperature was examined. To our delight, the efficiency of the reaction was further improved when the reaction was carried out in 100 °C, providing 3a in 74% yield (entry 7, Table 1). Reducing the reaction temperature was not favorable for the yields of the model reaction (entries 5 and 6, Table 1). Other activators in the formation of CF3S+ including BF3, TsOH, and FeCl3etc. were also exploited (entries 8–10, Table 1). However, no better yields were afforded accordingly. Additionally, the solvent effect was also detected. Surprisingly, the solvent affected the model reaction significantly. For example, the reaction gave rise to the desired product 3a in 26% yield with recovery of aniline when changing the solvent from acetonitrile to tetrahydrofuran. It was believed that the use of a suitable solvent was critical for the in situ generation of diazonium salt from aniline. Similar results were observed when other solvents such as DMSO, EtOH, toluene and DCE were used (entries 12–15, Table 1). No improvements were gained when the N-aminomorphine loading was increased (data not shown in Table 1). To the best of our knowledge, our method described here represents an alternative procedure for the synthesis of biologically interesting trifluoromethyl thiolsulphonates. The comparatively simple and safe reaction operation and easy substrate accessibility of our method were impressive. Additionally, this transformation mentioned above initiated studies on trapping of the arylsulfonyl radical with an electrophile.
|
|
||||
|---|---|---|---|---|
| Entry | Solvent | Additive | T (°C) | Yield (%) |
| a Isolated yield based on PhNHSCF32a. b After the in situ generation of diazonium salt, DABCO·(SO2)2, morpholin-4-amine, PhNHSCF3 and the additive was added directly under N2 protection. | ||||
| 1 | CH3CN | BiCl3 (1.3 equiv.) | 80 | 58 |
| 2 | CH3CN | BiCl3 (1.0 equiv.) | 80 | 38 |
| 3 | CH3CN | BiCl3 (1.5 equiv.) | 80 | 64 |
| 4 | CH3CN | BiCl3 (3.0 equiv.) | 80 | 64 |
| 5 | CH3CN | BiCl3 (1.5 equiv.) | 25 | 27 |
| 6 | CH3CN | BiCl3 (1.5 equiv.) | 50 | 50 |
| 7 | CH 3 CN | BiCl 3 (1.5 equiv.) | 100 | 74 |
| 8 | CH3CN | BF3·OEt2 (1.5 equiv.) | 100 | 48 |
| 9 | CH3CN | TsOH (1.5 equiv.) | 100 | 60 |
| 10 | CH3CN | FeCl3 (1.5 equiv.) | 100 | 17 |
| 11 | THF | BiCl3 (1.5 equiv.) | 100 | 26 |
| 12 | DMSO | BiCl3 (1.5 equiv.) | 100 | n.d. |
| 13 | Toluene | BiCl3 (1.5 equiv.) | 100 | 36 |
| 14 | EtOH | BiCl3 (1.5 equiv.) | 100 | Trace |
| 15 | DCE | BiCl3 (1.5 equiv.) | 100 | 47 |
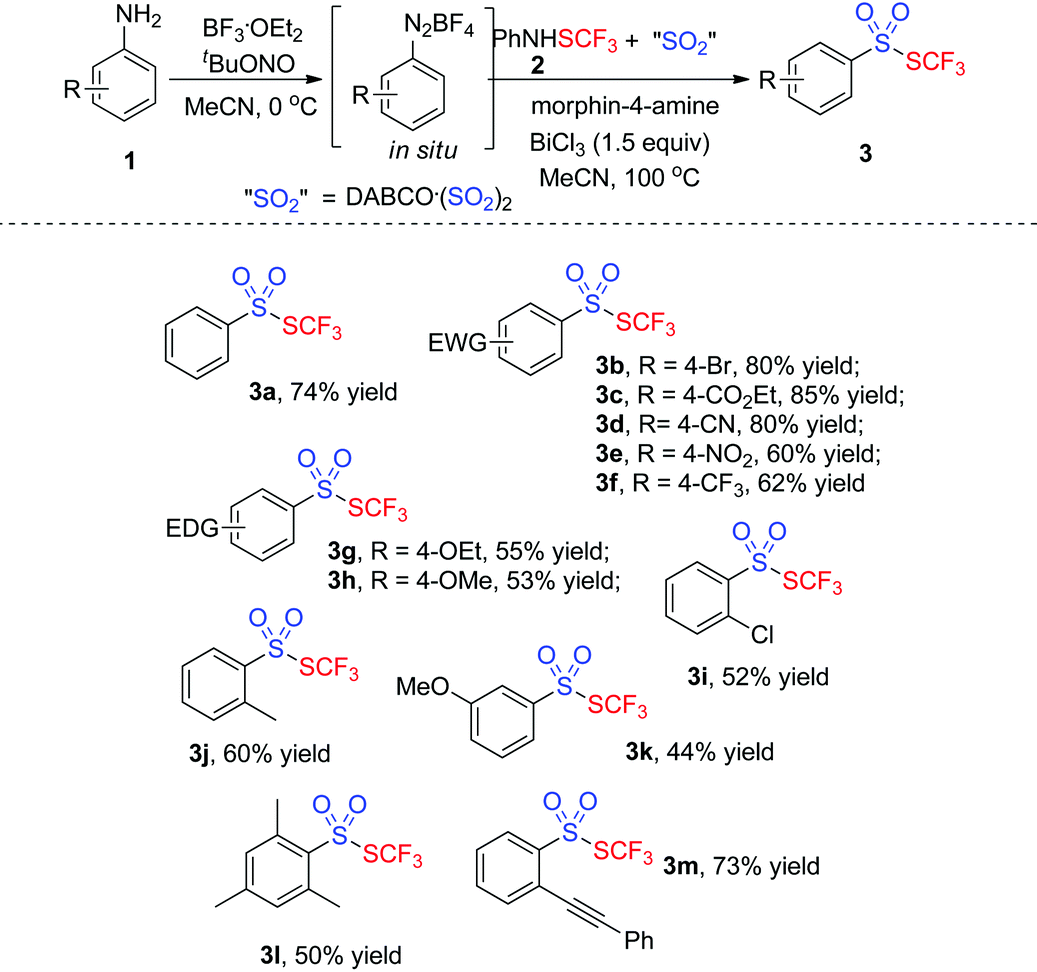
With the optimized conditions in hand (entry 7, Table 1), we then explored the scope of the projected transformation. As illustrated in Table 2, a series of trifluoromethyl thiolsulphonates 3 were achieved. Various anilines with a para substituent were compatible for the reactions, resulting in the corresponding thiolsulphonates 3 in 53–85% yields. Generally, anilines with an electron-deficient group were more favorable for the formation of thiolsulphonates. For instance, the reaction of 4-ester-connected aniline provided the product 3c in 85% yield while the reaction using 4-methoxylaniline gave 3h in 53% yield. The substrates with an ortho or a meta substituent were also suitable for the reactions. Interestingly, the tandem reaction from 2-alkynylaniline uniquely worked well under standard conditions, giving the corresponding thiolsulphonate 3m in good yield. Sterically hindered trimethylphenylamine was compatible for this reaction, producing thiolsulphonate 3l in 50% yield. However, heteroaromatic thiolsulphonates were not obtained by this procedure.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a
a
| a Isolated yield based on PhNHSCF3. |
|---|
|
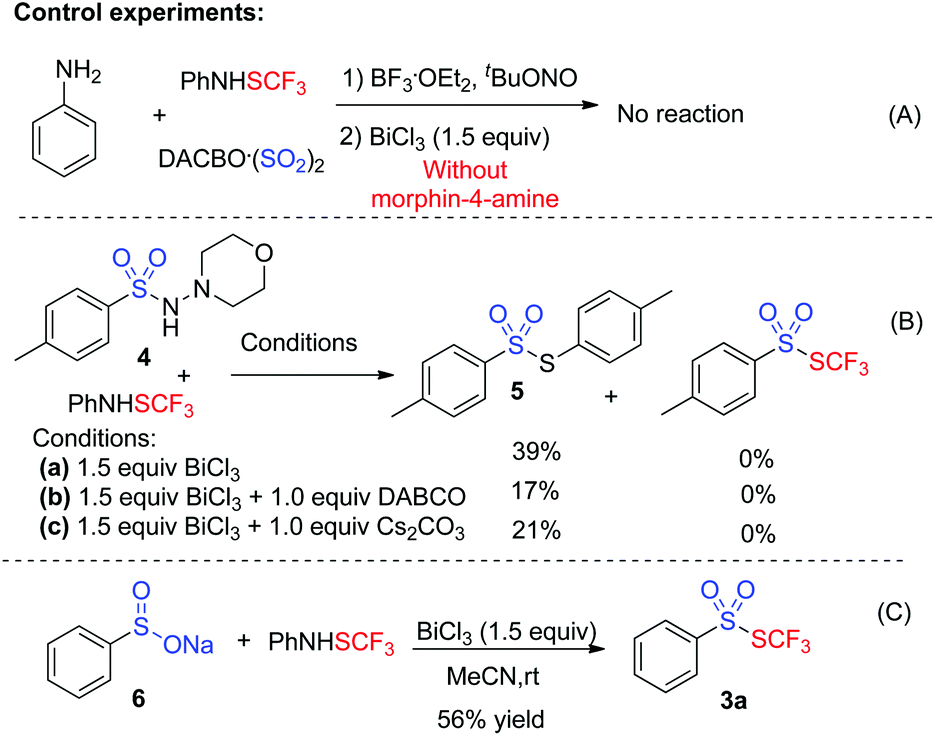
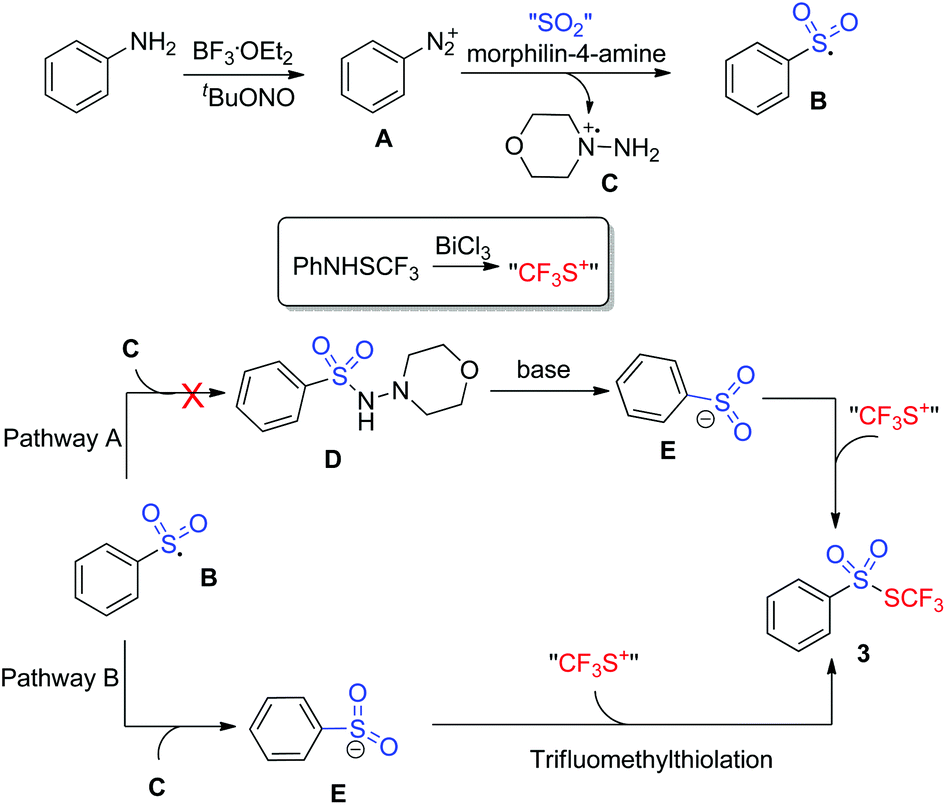
To gain insights into the mechanism, several control experiments were carried out. As presented in Scheme 2, the blank experiment without morphin-4-amine showed a negative result, thus indicating that morphin-4-amine served as a critical additive (Scheme 2A). Based on the above results and the previous findings from Wu's group,3a a stepwise pathway involving N-aminosulfonamide D as a key intermediate was proposed to understand the reaction (pathway A, Scheme 3) since N-aminosulfonamide D easily converted into sulfinate through base-assisted N–S bond cleavage.10 To support this hypothesis, the reactions of N-aminosulfonamide 4 and PhNHSCF3 were run using bismuth(III) chloride with or without the base. However, S-aryl benzenesulfonothioate 5 instead of the desired thiolsulphonate 3 was observed, which inferred that N-aminosulfonamide 4 was not the intermediate. Inspired by these results, pathway A was not plausible. Subsequently, pathway B was postulated. In pathway B, sulfinate was described as an intermediate since the BiCl3-promoted reaction of sodium sulfinate 6 and PhNHSCF3 gave the expected thiolsulphonate 3a in 56% yield (Scheme 2C). In this process, N-aminomorphine played a pivotal role. According to the results from Wu's group,3aN-aminomorphine facilitated the formation of the aryl radical from aryldiazonium. This aryl radical was trapped by sulfur dioxide to provide arylsulfonyl radical B. We reasoned that sulfinate E was generated from the reduction of the arylsulfonyl radical by the hydrazine radical cation C. Then electrophilic trifluoromethylthiolation of sulfinate E afforded the product 3a.11
 | ||
| Scheme 2 Control experiments. | ||
 | ||
| Scheme 3 Proposed pathway for the designed reaction. | ||
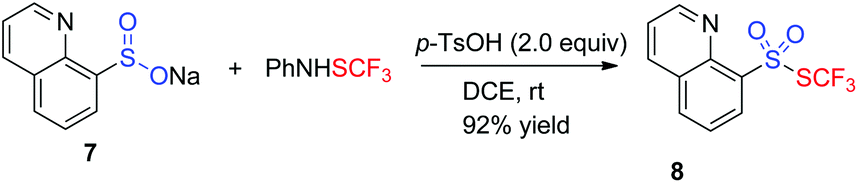
Given the fact that sulfinate was a plausible intermediate, the reaction of sulfinates and PhNHSCF3 was applied to synthesize heteroaromatic thiolsulphonates (Table 3). Based on the results from Shen's group,11b various result-affecting factors including additive, solvent, and temperature were evaluated accordingly. To our delight, the expected heteroaryl thiolsulphonate 8 was achieved by altering the additive to 2.0 equiv. 4-methylbenzenesulfonic acid in 1,2-dichloroethane at room temperature.
a
| a Isolated yield based on sulfinate 7. |
|---|
|
Conclusion
To sum up, we have developed a novel and efficient approach for the synthesis of trifluoromethyl thiolsulphonates via a sequential tandem reaction of diazotization, radical sulfur dioxide insertion, and electrophilic trifluoromethylthiolation. The tandem reactions start from aniline, sulfur dioxide, and trifluoromethanesulfanylamide. Control experiments implied that sulfinate was a key intermediate. N-Aminomorphine played a dual role in the reaction. One as the facilitator for the formation of an aryl radical and the other as a reductant to reduce the arylsulfonyl radical into sulfinate. It is believed that this transformation represents the first example of reductive insertion of sulfur dioxide between an electrophile and a radical precursor. Other transformations based on sulfur dioxide insertion for the synthesis of more useful architectures, particularly biologically interesting scaffolds, are ongoing in our lab. The results will be reported in due course.Experimental section
General procedure for the synthesis of trifluoromethyl thiolsulphonates 3 from aniline
Aniline 1 (0.45 mmol) and BF3·Et2O (0.54 mmol) were added into CH3CN (1.0 mL), followed by dropwise addition of tBuONO (0.54 mmol) at 0 °C. After 5 minutes, the above mixture was slowly added into a mixture of DABCO·(SO2)2 (0.21 mmol), morpholin-4-amine (0.40 mmol), PhNHSCF3 (0.39 mmol) and BiCl3 (0.6 mmol) in CH3CN (1 mL). Then the mixture was stirred at 100 °C for 6–12 hours. After completion of the reaction as indicated by TLC, the reaction mixture was filtered by using a sand core funnel with silica gel and washed by using CH2Cl2. The residue was concentrated in vacuo and purified by column chromatography on silica gel to provide the product 3.General procedure for the synthesis of trifluoromethyl thiolsulphonates 8 from sulfinates
PhNHSCF32 (0.3 mmol) was added to a mixture of sufinate 7 (0.2 mmol) and 4-methylbenzenesulfonic acid (0.4 mmol) in DCE (2 ml). The mixture was stirred at room temperature for 8 h. The volatiles were removed and the residue was purified by flash column chromatography (SiO2) to 8.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) 11b.
1H NMR (400 MHz, CDCl3) δ 9.14 (d, J = 4.2 Hz, 1H), 8.48 (d, J = 7.4 Hz, 1H), 8.33 (d, J = 8.4 Hz, 1H), 8.21 (d, J = 8.2 Hz, 1H), 7.71–7.73 (m, 1H), 7.64 (dd, J = 8.3, 4.3 Hz, 1H); 19F NMR (376 MHz, CDCl3) δ − 38.25; 13C NMR (101 MHz, CDCl3) δ 151.6, 143.0, 140.5, 136.7, 135.5, 130.7, 129.2, 127.8 (q, J = 312.0 Hz), 125.3, 122.9; HRMS (EI) calcd for C10H6F3NO2S2+: 292.9787 (M+), found: 292.9792.
11b.
1H NMR (400 MHz, CDCl3) δ 9.14 (d, J = 4.2 Hz, 1H), 8.48 (d, J = 7.4 Hz, 1H), 8.33 (d, J = 8.4 Hz, 1H), 8.21 (d, J = 8.2 Hz, 1H), 7.71–7.73 (m, 1H), 7.64 (dd, J = 8.3, 4.3 Hz, 1H); 19F NMR (376 MHz, CDCl3) δ − 38.25; 13C NMR (101 MHz, CDCl3) δ 151.6, 143.0, 140.5, 136.7, 135.5, 130.7, 129.2, 127.8 (q, J = 312.0 Hz), 125.3, 122.9; HRMS (EI) calcd for C10H6F3NO2S2+: 292.9787 (M+), found: 292.9792.
Acknowledgements
Financial supports from the Natural Science Foundation of China (no. 21502069), the Natural Science Foundation of Zhejiang Province (no. LQ15B020006) and the Ph.D. Scientific Research Starting Foundation of Jiaxing University (no. 70515015) and Shanghai Ocean University (no. A2-0203-00-100338) are gratefully acknowledged. Generous support of human resources from Prof. Jie Wu (Fudan University, China) is very gratefully acknowledged.Notes and references
- For recent reviews, see: (a) G. Liu, C. Fan and J. Wu, Org. Biomol. Chem., 2015, 13, 1592 RSC; (b) C. S. Richards-Taylor, D. C. Blakemore and M. C. Willis, Chem. Sci., 2014, 5, 222 RSC; (c) W. E. Truce and A. M. Murphy, Chem. Rev., 1951, 48, 69 CrossRef CAS PubMed.
- For selected examples on transitional metal-catalyzed SO2 fixation, see: (a) B. Nguyen, E. J. Emmet and M. C. Willis, J. Am. Chem. Soc., 2010, 132, 16372 CrossRef CAS PubMed; (b) M. Johnson, S. Bagley, N. Mankad, R. Bergman, V. Mascitti and F. D. Toste, Angew. Chem., Int. Ed., 2014, 53, 4404 CrossRef CAS PubMed. For selected examples on SO2 fixation of organometallicS, see: (c) A. S. Deeming, C. J. Rusell, A. J. Hennessy and M. C. Willis, Org. Lett., 2014, 16, 150 CrossRef CAS PubMed; (d) B. N. Rocke, K. B. Bahnck, M. Herr, S. Lavergne, V. Mascitti, C. Perreault, J. Polivkova and A. Shanya, Org. Lett., 2014, 16, 154 CrossRef CAS PubMed; (e) E. J. Emmett, B. R. Hayter and M. C. Willis, Angew. Chem., Int. Ed., 2013, 52, 12679 CrossRef CAS PubMed; (f) W. Li, H. Li, P. Langer, M. Beller and X.-F. Wu, Eur. J. Org. Chem., 2014, 3101 CrossRef.
- (a) D. Zheng, Y. An, Z. Li and J. Wu, Angew. Chem., Int. Ed., 2014, 53, 2451 CrossRef CAS PubMed; (b) W. Li, M. Beller and X.-F. Wu, Chem. Commun., 2014, 50, 9513 RSC; (c) D. Zheng, Y. Li, Y. An and J. Wu, Chem. Commun., 2014, 50, 8886 RSC; (d) Y. An, D. Zheng and J. Wu, Chem. Commun., 2014, 50, 11746 RSC. For photoinduced SO2 fixation, see: (e) Y. Li, D. Zheng, Z. Li and J. Wu, Org. Chem. Front., 2016, 3, 574 RSC.
- For recent reviews, see: (a) A. Studer and D. P. Curran, Nat. Chem., 2014, 6, 765 CrossRef CAS PubMed; (b) A. Studer and D. P. Curren, Angew. Chem., Int. Ed., 2016, 55, 58 CrossRef CAS PubMed.
- (a) L. D. Small, J. H. Bailey and C. J. Cavallito, J. Am. Chem. Soc., 1949, 71, 3565 CrossRef CAS; (b) J.-B. Feng, J.-L. Gong and X.-F. Wu, RSC Adv., 2014, 4, 29273 RSC; (c) J.-B. Feng and X.-F. Wu, ChemistryOpen, 2016, 5, 315 CrossRef CAS PubMed; (d) J.-B. Feng and X.-F. Wu, Org. Biomol. Chem., 2016, 14, 6951 RSC.
- (a) S. S. Block and J. P. Weidner, Nature, 1967, 214, 478 CrossRef CAS PubMed; (b) J. P. Weidner and S. S. Block, J. Med. Chem., 1967, 10, 1167 CrossRef CAS PubMed.
- For recent selected reviews, see: (a) S. Barata-Vallejo, S. Bonesi and A. Postigo, Org. Biomol. Chem., 2016, 14, 7150 RSC; (b) F. Toulgoat, S. Alazet and T. Billard, Eur. J. Org. Chem., 2014, 2415 CrossRef CAS; (c) C. Ni, M. Hu and J. Hu, Chem. Rev., 2015, 115, 765 CrossRef CAS PubMed; (d) X.-H. Xu, K. Matsuzaki and N. Shibata, Chem. Rev., 2015, 115, 731 CrossRef CAS PubMed; (e) X. Yang, T. Wu, R. J. Phipps and F. D. Toste, Chem. Rev., 2015, 115, 826 CrossRef CAS PubMed; (f) X. Shao, C. Xu, L. Lu and Q. Shen, Acc. Chem. Res., 2015, 48, 1227 CrossRef CAS PubMed. For recent selected examples, see: (g) T. Liu, Q. Ding, G. Qiu and J. Wu, Tetrahedron, 2016, 72, 1472 CrossRef CAS; (h) H. Chachignon, M. Maeno, H. Kondo, N. Shibata and D. Cahard, Org. Lett., 2016, 18, 2467 CrossRef CAS PubMed; (i) K. Jouvin, C. Matheis and L. J. Goossen, Chem. – Eur. J., 2015, 21, 14324 CrossRef CAS PubMed; (j) R. Honeker, J. B. Ernst and F. Glorius, Chem. – Eur. J., 2015, 21, 8047 CrossRef CAS PubMed; (k) Q. Wang, Z. Qi, F. Xie and X. Li, Adv. Synth. Catal., 2015, 357, 355 CrossRef CAS; (l) A. Hass and U. Niemann, Chem. Ber., 1977, 110, 67 CrossRef.
- For recent review, see: L. He, G. Qiu, Y. Gao and J. Wu, Org. Biomol. Chem., 2014, 12, 6965 CAS . For recent selected examples: (a) M. B. Andrus and C. Song, Org. Lett., 2001, 3, 3761 CrossRef CAS PubMed; (b) F. L. Callonec, E. Fouquet and F.-X. Felpin, Org. Lett., 2011, 13, 2646 CrossRef PubMed; (c) X.-F. Wu, H. Neumann and M. Beller, Chem. Commun., 2011, 47, 7959 RSC; (d) J.-J. Dai, C. Fang, B. Xiao, J. Yi, J. Xu, Z.-J. Liu, L. Liu and Y. Fu, J. Am. Chem. Soc., 2013, 135, 843 Search PubMed; (e) D. Qiu, H. Meng, L. Jin, S. Wang, S. Tang, X. Wang, F. Mo, Y. Zhang and J. Wang, Angew. Chem., Int. Ed., 2013, 52, 11581 CrossRef CAS PubMed; (f) Z. Xia, J. Huang, Y. He, J. Zhao, J. Lei and Q. Zhu, Org. Lett., 2014, 16, 2546 CrossRef CAS PubMed; (g) N. Chernyak and S. Buchwald, J. Am. Chem. Soc., 2012, 134, 12466 CrossRef CAS PubMed; (h) X.-F. Wu, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2011, 50, 11142 CrossRef CAS PubMed; (i) X.-F. Wu, J. Schranck, H. Neumann and M. Beller, Tetrahedron Lett., 2011, 52, 3702 CrossRef CAS.
- For selected examples, see: (a) J. Sheng, S. Li and J. Wu, Chem. Commun., 2014, 50, 578 RSC; (b) D.-Q. Chen, P. Cao, P.-X. Zhou, X.-R. Song, Y.-F. Qiu, X.-Y. Liu and Y.-M. Liang, Chem. Commun., 2015, 51, 6637 RSC; (c) Q. Xiao, H. Zhu, G. Li and Z. Chen, Adv. Synth. Catal., 2014, 356, 3809 CrossRef CAS; (d) R. Mao, D. Zheng, H. Xia and J. Wu, Org. Chem. Front., 2016, 3, 693 RSC.
- (a) C. S. Richards-Taylor, D. C. Blakemore and M. C. Willis, Chem. Sci., 2014, 5, 222 RSC; (b) M. W. Johnson, S. W. Bagley, N. P. Mankad, R. G. Bergman, V. Mascitti and F. D. Toste, Angew. Chem., Int. Ed., 2014, 53, 4404 CrossRef CAS PubMed; (c) Y. Luo, X. Pan, C. Chen, L. Yao and J. Wu, Chem. Commun., 2015, 51, 180 RSC; (d) D. M. Lemal, T. W. Rave and S. D. McGregor, J. Am. Chem. Soc., 1963, 85, 1944 CrossRef CAS; (e) D. M. Lemal, F. Menger and E. Coats, J. Am. Chem. Soc., 1964, 86, 2395 CrossRef CAS; (f) L. A. Carpino, J. Am. Chem. Soc., 1957, 79, 4427 CrossRef CAS; (g) R. Ballini, E. Marcantoni and M. Petrini, Tetrahedron, 1989, 45, 6791 CrossRef CAS.
- (a) L. M. Yagupolskii, A. V. Matsnev, R. K. Orlova, B. G. Deryabkin and Y. L. Yagupolskii, J. Fluorine Chem., 2008, 19, 131 CrossRef; (b) X. Shao, C. Xu, L. Lu and Q. Shen, J. Org. Chem., 2015, 80, 3012 CrossRef CAS PubMed; (c) M. Jereb and D. Dolenc, RSC Adv., 2015, 5, 58292 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedure, characterization data, 1H and 13C NMR spectra of compounds 3. See DOI: 10.1039/c6qo00530f |
| This journal is © the Partner Organisations 2017 |