
Synthesis of carbamates from amines and N-tosylhydrazones under atmospheric pressure of carbon dioxide without an external base†
Ji Young
Hong
,
Ue Ryung
Seo
and
Young Keun
Chung
*
Intelligent Textile System Research Center, and Department of Chemistry, College of Natural Sciences, Seoul National University, Seoul 08826, Korea. E-mail: ykchung@snu.ac.kr; Fax: (+82)-2-889-0310; Tel: (+82)-2-880-6662
First published on 19th April 2016
Abstract
The methodology for the synthesis of carbamates via the base-promoted coupling of carbon dioxide, amines, and N-tosylhydrazones using potassium carbonate as the base and the pressure of 4 MPa of carbon dioxide has been published by Jiang et al. (2015). We demonstrated that this method provides the desired carbamates under just 1 atm of carbon dioxide without the addition of a base promoter. Thus, carbamates were successfully synthesized under mild reaction conditions in moderate to high yields.
Organic carbamates have shown versatile potential in medicinal, agricultural chemicals, and coating applications.1 Conventional methods for the synthesis of carbamates are mainly based on the use of toxic phosgene and isocyanates.2 Thus, considerable interest has been generated in the development of safe and efficient approaches to the preparation of carbamates. Recently, a variety of phosgene- and isocyanate-free methods have been developed.3
Among them, the novel base-promoted three-component coupling reaction of CO2, amines, and N-tosylhydrazones to form carbamates under transition-metal-free conditions, described by Jiang et al.4 is notable. The reaction provides a wide range of organic carbamates from readily available substrates with excellent functional group tolerance, a wide substrate scope, and a facile work-up procedure. However, the reaction is not practical due to the high pressure of carbon dioxide (4 MPa) and the rather harsh conditions (0.5 mmol N-tosylhydrazone, 5 mmol amine, 1.5 mmol K2CO3, 4 MPa CO2, 3 mL MeCN/H2O (15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v), 120 °C, 24 h). Therefore, the development of a milder method for the synthesis of organic carbamates remains a challenging goal.
:1 v/v), 120 °C, 24 h). Therefore, the development of a milder method for the synthesis of organic carbamates remains a challenging goal.
While studying the use of a polymeric catalyst for organic reactions,5 the three-component coupling reaction reported by Jiang et al.4 attracted our attention. Thus, we explored the reaction in the presence of our polymeric catalyst to identify mild reaction conditions. We speculated that the use of the polymeric catalyst, poly(NHC)s, could lead to the formation of carbamates under an atmospheric pressure of CO2. We were confident that a judicious choice of reaction conditions would lead to a favorable outcome. Thus, we studied the reaction in the presence of poly(NHC)s to find the mild reaction conditions. During this endeavor, it was discovered that the reaction proceeds without any extra base or catalyst under an atmospheric pressure of carbon dioxide at 80 °C in reasonably high yields comparable to those obtained using the original conditions reported by Jiang et al.4
The reaction of N-tosylhydrazone 1a with pyrrolidine (2a) in the presence of poly(NHC)/ZnBr2/DBU under 1 atm of carbon dioxide in DMF at 80 °C for 18 h was investigated as a model reaction for the synthesis of carbamates (eqn (1)).
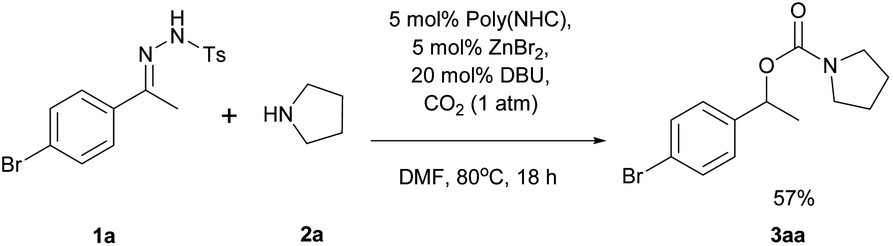 | (1) |
To our delight, carbamate (3aa) was isolated in 57% yield under 1 atm of CO2 supplied by a balloon, without any specialized gas manipulation. Encouraged by this observation, the reaction parameters were screened to maximize the yield of 3aa. Selected data related to the screening of reaction solvents are shown in Table 1.
a
|
|
|||||
|---|---|---|---|---|---|
| Entry | Additive | Base | Solvent | Time (h) | Yieldb (%) |
|
a Reaction conditions: 1 mmol 1a, 10 mmol 2a, 5 mol% additive, 20 mol% base, 1.5 mL solvent, 80 °C, 1 atm of CO2.
b Isolated yield.
c Reaction temperature: 100 °C.
d MeCN/H2O (v/v, 3.0 mL:0.2 mL).
|
|||||
| 1 | Poly(NHC), ZnBr2 | DBU | DMF | 18 | 57 |
| 2 | ZnBr2 | — | DMF | 18 | 59 |
| 3 | — | — | DMF | 18 | 57 |
| 4 | — | — | 1,4-Dioxane | 18 | 60 |
| 5 | — | — | Toluene | 18 | 61 |
| 6 | — | — | THF | 18 | 60 |
| 7 | — | — | DCE | 18 | 26 |
| 8 | — | — | — | 18 | 62 |
| 9 | — | — | CH3NO2 | 18 | 75 |
| 10 | — | — | CH3NO2 | 6 | 71 |
| 11 | — | — | CH3NO2 | 3 | 70 |
| 12 | — | — | CH3NO2 | 2 | 67 |
| 13c | — | — | CH3NO2 | 2 | 64 |
| 14d | — | — | MeCN/H2O | 18 | 63 |
Interestingly, the reaction proceeded in the presence of ZnBr2 only and without any additives. However, the yield was highly sensitive to the reaction solvent. In 1,4-dioxane, toluene, or THF, the yield was ca. 60% but just 26% in dichloroethane. In addition, when the reaction was performed neat without any solvent, the yield remained high (62%). When the reaction was conducted in a mixture solvent of acetonitrile and water (3 mL:0.2 mL) as Jiang et al. used,4 the yield was 63%. The best result was observed when the reaction was performed in nitromethane (75%). Shortening the reaction time to 6 h, 3 h, and 2 h provided yields of 71%, 70%, and 67%, respectively. In contrast, the yield was dependent upon the order of addition of the reactants, with a higher yield obtained when the reactants were added in the order: amine, CO2, and N-tosylhydrazone.7 Finally, when the reaction temperature was raised or lowered, no improvement in the yield was observed (data not shown; see the ESI†). The optimum reaction conditions were established as follows: 1 mmol 1a, 10 mmol 2a, 1.5 mL CH3NO2, 1 atm CO2, 80 °C, and a reaction time of 18 h.
Using the optimized reaction conditions, the substrate scope of the N-tosylhydrazones was then examined by varying the hydrazone moiety (Table 2). For most of the substrates, the reaction time was 18 h; for the substrates derived from 1-(4-nitrophenyl)ethan-1-one (1g), 4-acetylbenzonitrile (1h), and 1-(pyridin-4-yl)ethan-1-one (1r), the reaction time was shortened to 3 h because these compounds decomposed under the reaction conditions. As expected, various functional groups on the aryl ring of the N-tosylhydrazone were tolerated. When the functional group at the 4-position on the aryl group of N-tosylhydrazones derived from acetophenone was screened, all the substrates except a nitro- or nitrile substituent were found to be good substrates, giving high yields (3aa, 3da–3la: 58–75%). N-Tosylhydrazones derived from benzaldehyde were also found to be good substrates (3ma–3pa: 66–71%) and the best yield was observed with a methyl substituent (3pa, 71%). A steric effect was observed for substrates derived from acetophenone bearing 2-bromo and 3-bromoaryl substituents (3cavs.3ba: 60% vs. 70%). However no steric effect was observed for similar substrates bearing 1-naphthyl and 2-naphthyl groups (3tavs.3sa: 76% vs. 75%).
a
| a Reaction conditions: 1 mmol N-tosylhydrazone, 10 mmol 2a, 1.5 mL CH3NO2, 18 h, 80 °C, 1 atm of CO2. Yields of isolated products are given. Data from Angew. Chem., Int. Ed. 2015, 54, 3084 are given in parentheses. b Reaction time: 3 h. c 1-(1-(4-Nitrophenyl)ethyl)pyrrolidine (23%) and 1-ethyl-4-nitrobenzene (10%) were found to be a major byproduct and a minor by-product, respectively. |
|---|
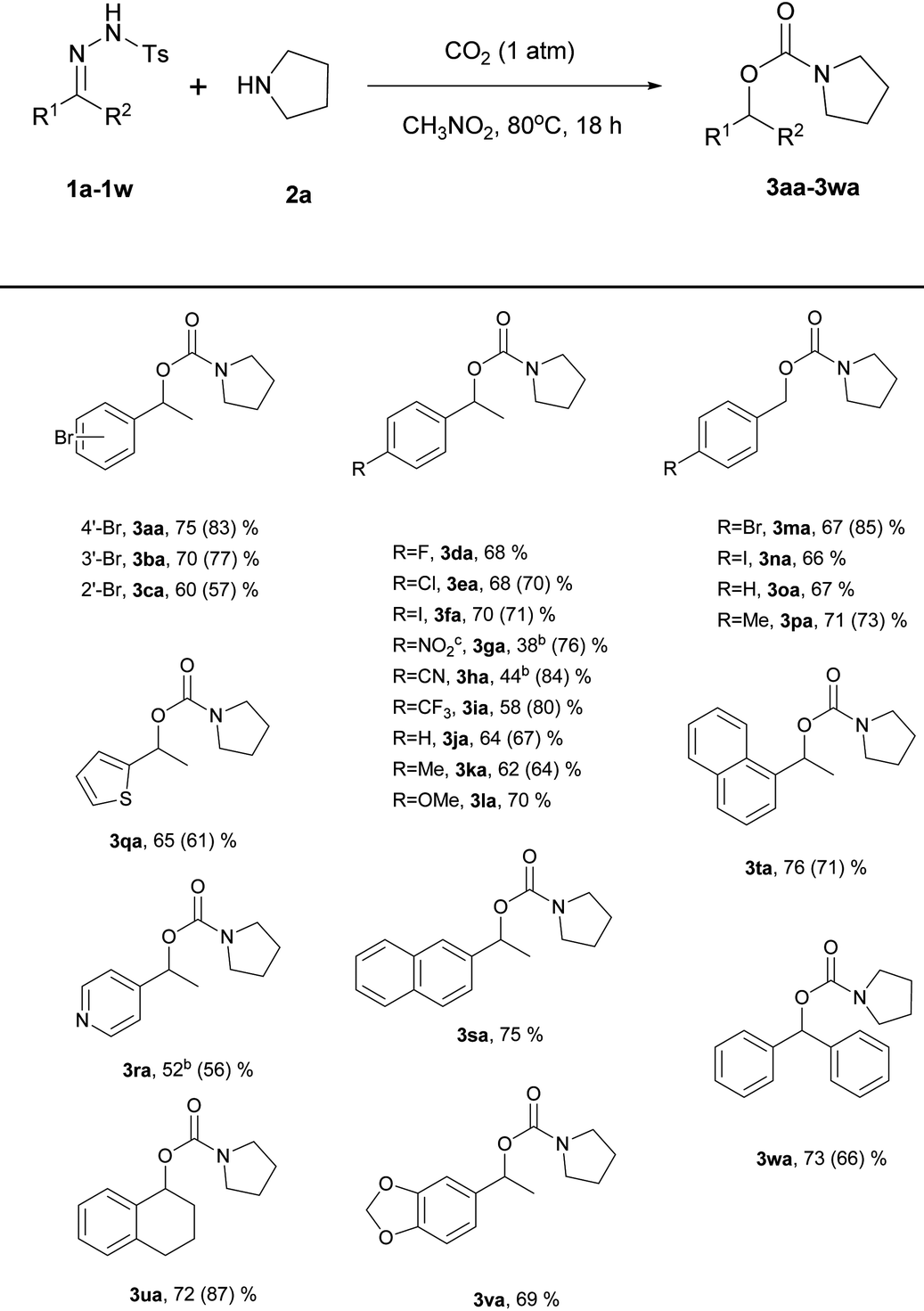
|
N-Tosylhydrazones derived from 3,4-dihydronaphthalen-1(2H)-one (1u), 1-(benzo[1,3]dioxol-5-yl)ethan-1-one (1v), and benzophenone (1w) were good substrates, giving an approximate 69–73% yield of the desired products (3ua–3wa). Substrates containing heteroarene groups, such as thiophene (1q) and pyridine (1r), gave carbamates in reasonable yields (3qa, 65%; 3ra, 52%). Importantly, the results obtained under the new reaction conditions were comparable to those obtained using Jiang's reaction conditions in most cases. However, substrates derived from 1-(4-nitrophenyl)ethan-1-one (1g), 4-acetylbenzonitrile (1h), and 1-(4-(trifluoromethyl)phenyl)ethan-1-one (1i) were poor substrates and afforded lower yields (3ga, 38% vs. 76%; 3ha, 44% vs. 84%; 3ia, 58% vs. 80%). In the cases of 3ga and 3ha, poor yields were observed compared to the original paper. We envisioned that the activity of the carbon center of the nitrile and nitro hydrazone derivatives may be quite high. Due to their high reactivity, byproducts were formed. However, when the reaction was carried out under a high pressure of CO2, there might be some restrictions in the generation of diverse active nucleophilic species that could attack the carbon center of the hydrazones. Thus, the yields are not highly sensitive to the N-tosylhydrazone derivatives under high pressures of CO2. According to Jiang's report,4N-tosylhydrazones derived from aliphatic ketones or aldehydes failed to yield even a trace of the desired products. No reaction was observed with an aliphatic N-tosylhydrazone derived from 2-butanone under our reaction conditions.
The substrate scope of amines was also investigated (Table 3).
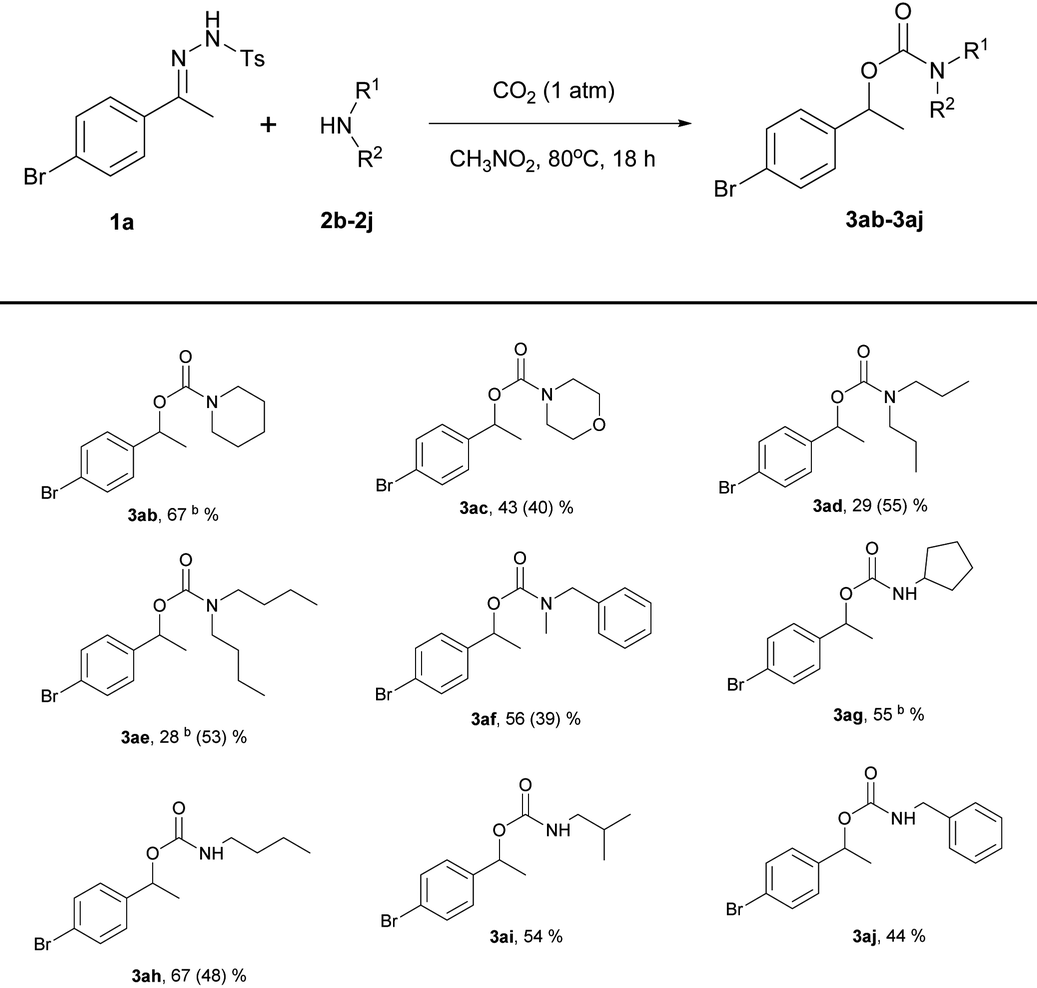
The reactions proceeded smoothly with secondary and primary amines. For example, reactions using piperidine (2b) and cyclopentylamine (2g) were complete within 3 h, affording the desired products in moderate to good yields under the present reaction conditions; the longer reaction times had no influence on the yields. In contrast, dipropylamine (2d) and dibutylamine (2e) afforded rather poor yields of the desired carbamates (3ad, 29%; 3ae, 28%); these isolated yields were lower than those obtained under Jiang's reaction conditions (3ad, 29% vs. 55%; 3ae, 28% vs. 53%). However, N-benzylmethylamine (2f) and n-butylamine (2h) gave higher yields (3af, 56% vs. 39%; 3ah, 67% vs. 48%) than those obtained using Jiang's reaction conditions. Piperidine (2b), which has a structure similar to that of pyrrolidine (2a), was a good substrate, yielding 67% of the expected product (3ab). When the yield was not high, a considerable amount of adduct from the reaction of hydrazone with amine was obtained as a byproduct.
Although little mechanistic information has been obtained right now, the above results suggest that the formation of a carbocation via the aid of carbonic acid from the reaction of water and carbon dioxide may not occur under our reaction conditions.4,6 The mechanism may follow the Wolff–Kishner-type reduction.8 A secondary amine, pyrrolidine, reacts with carbon dioxide to form a carbamate. The carbamate may act as a nucleophilic reagent, generating an intermediate azo compound. The intermediate picks up a proton to produce the final product (Scheme 1).
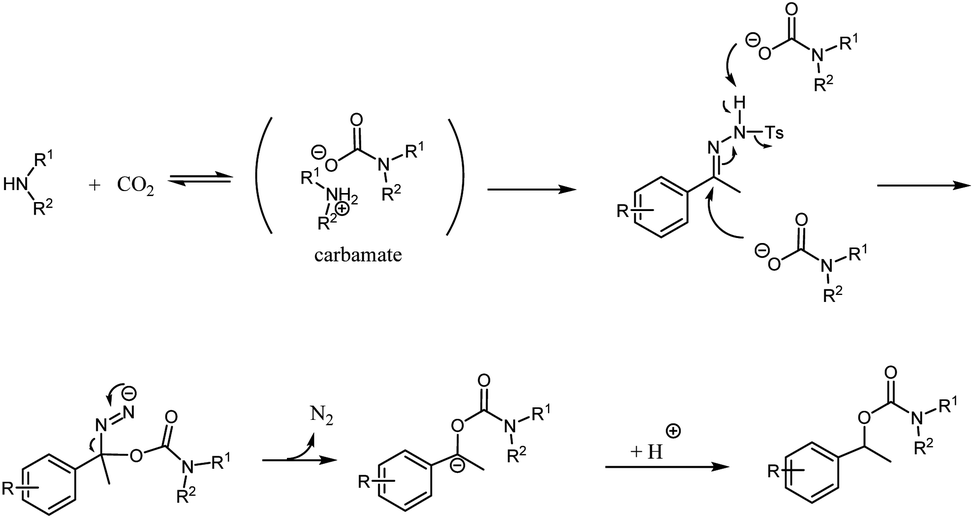 | ||
| Scheme 1 Proposed mechanism. | ||
Finally, it is worth noting that the reaction may be scaled up (eqn (2)).
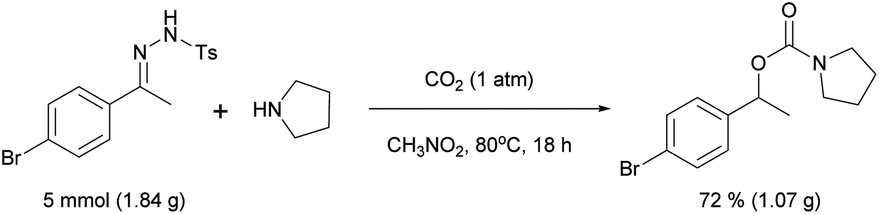 | (2) |
When 1.84 g of 1a (5 mmol) was reacted with 2a (50 mmol), 1.07 g (72% yield) of 3aa was isolated. Therefore, the scale-up of the procedure did not result in a reduction in the yield.
Conclusions
In conclusion, we have demonstrated a method for the synthesis of carbamates from readily available N-tosylhydrazones, amines, and carbon dioxide in moderate to high yields under mild reaction conditions. Compared to the original reaction conditions reported by Jiang et al.4 (3 equiv. K2CO3, 4 MPa CO2, 120 °C, and 24 h), the much milder reaction conditions of the present modified method (1 atm CO2, 80 °C, and 18 h) provide the desired carbamates without any considerable loss in yield for most substrates. This reaction is applicable to a gram-scale reaction.This work was supported by a National Research Foundation of Korea (NRF) grant funded by the Korean government (2014R1A5A1011165 and 2007-0093864). JYH and URS are recipients of the BK21 plus fellowship.
Notes and references
- For reviews, see: (a) A. K. Ghosh and M. Brindisi, J. Med. Chem., 2015, 58, 2895 CrossRef CAS PubMed; (b) D. Chaturvedi, Tetrahedron, 2012, 68, 15 CrossRef CAS.
- (a) S. Ozaki, Chem. Rev., 1972, 72, 457 CrossRef; (b) H. Babad and A. G. Zeiler, Chem. Rev., 1973, 73, 75 CrossRef CAS; (c) D. Chaturvedi, N. Mishra and V. Mishra, Curr. Org. Synth., 2007, 4, 308 CrossRef CAS.
- (a) K. Kurita, T. Matsumara and Y. Iwakura, J. Org. Chem., 1976, 41, 2076 CrossRef; (b) K. Kurita, T. Matsumara and Y. Iwakura, Org. Synth., 1980, 59, 195 CAS; (c) K. Kurita, T. Matsumara and Y. Iwakura, J. Org. Chem., 1985, 50, 715 CrossRef; (d) L. Cotarka, P. Delgu, A. Mardelli and V. Sunjuc, Synthesis, 1996, 553 CrossRef; (e) S.-I. Kim, F. Chu, E. E. Dueno and K. W. Jung, J. Org. Chem., 1999, 64, 4578 CrossRef CAS PubMed; (f) R. N. Salvatore, V. L. Flanders, D. Ha and K. W. Jung, Org. Lett., 2000, 2, 2797 CrossRef CAS PubMed; (g) R. Srivastava, M. D. Manju and P. Ratnasamy, Catal. Lett., 2004, 97, 41 CrossRef CAS; (h) R. Juárez, P. Concepción, A. Corma, V. Fornés and H. García, Angew. Chem., Int. Ed., 2012, 122, 1308 CrossRef; (i) S. Grego, F. Aricò and P. Tundo, Org. Process Res. Dev., 2013, 17, 679 CrossRef CAS; (j) S. Kumar and S. L. Jain, New J. Chem., 2013, 37, 2935 RSC; (k) O. Kreye, H. Mutlu and M. A. R. Meier, Green Chem., 2013, 15, 1431 RSC; (l) E. Delebecq, J.-P. Pascault, B. Boutevin and F. Ganachaud, Chem. Rev., 2013, 113, 80 CrossRef CAS PubMed; (m) L. Maisonneuve, O. Lamarzelle, E. Rix, E. Grau and H. Cramail, Chem. Rev., 2015, 115, 12407 CrossRef CAS PubMed.
- W. Xiong, C. Qi, H. He, L. Ouyang, M. Zhang and H. Jiang, Angew. Chem., Int. Ed., 2015, 54, 3084 CrossRef CAS PubMed.
- (a) U. R. Seo and Y. K. Chung, Adv. Synth. Catal., 2014, 356, 1955 CrossRef CAS; (b) U. R. Seo and Y. K. Chung, RSC Adv., 2014, 4, 32371 RSC; (c) J. Y. Chung, U. R. Seo, S. Chun and Y. K. Chung, ChemCatChem, 2016, 8, 318 CrossRef CAS.
- (a) J. Barluenga, P. Moriel, C. Valdes and F. Aznar, Angew. Chem., Int. Ed., 2007, 46, 5587 CrossRef CAS PubMed; (b) Z. Shao and H. Zhang, Chem. Soc. Rev., 2012, 41, 560 RSC.
- When we followed the following order, pyrrolidine, hydrazone and then carbon dioxide, we observed the formation of 1-(1-(4-bromophenyl)ethyl)pyrrolidine as a by-product and a lower reaction yield.
- (a) M. Renoud-Grappin, C. Vanucci and G. Lhomet, J. Org. Chem., 1994, 59, 3902 CrossRef CAS; (b) O. Huttenloch, E. Laxman and H. Waldmann, Chem. – Eur. J., 2002, 8, 3767 CrossRef; (c) D. Stead, P. O'Brien and A. J. Sanderson, Org. Lett., 2005, 7, 4459 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6qo00111d |
| This journal is © the Partner Organisations 2016 |