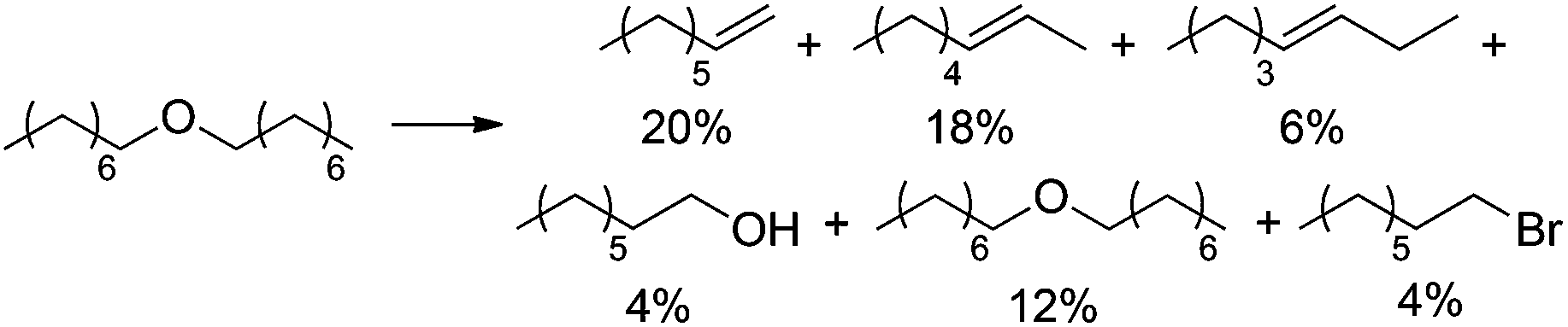
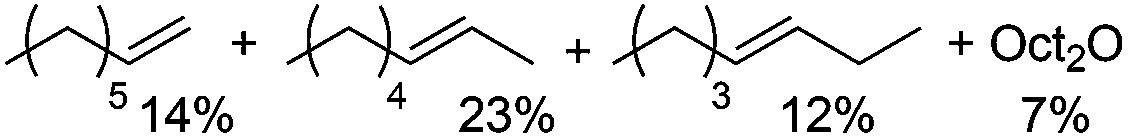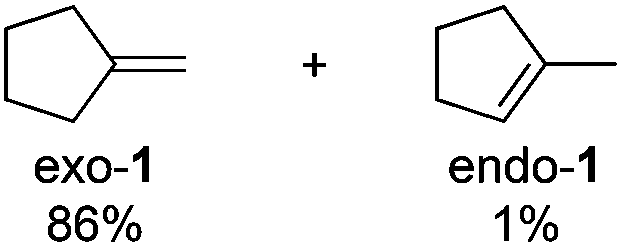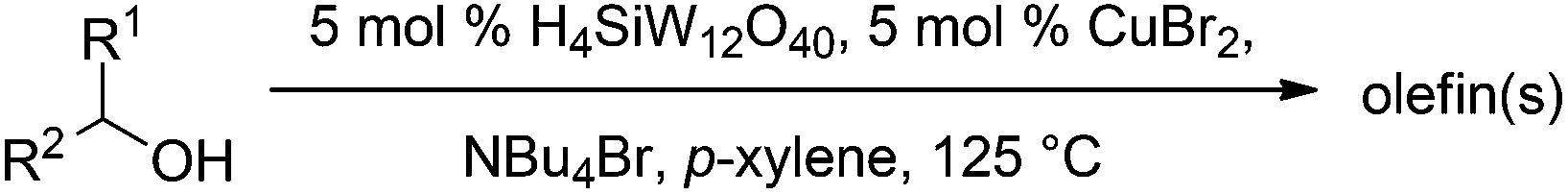DOI:
10.1039/C6QO00069J
(Research Article)
Org. Chem. Front., 2016,
3, 701-708
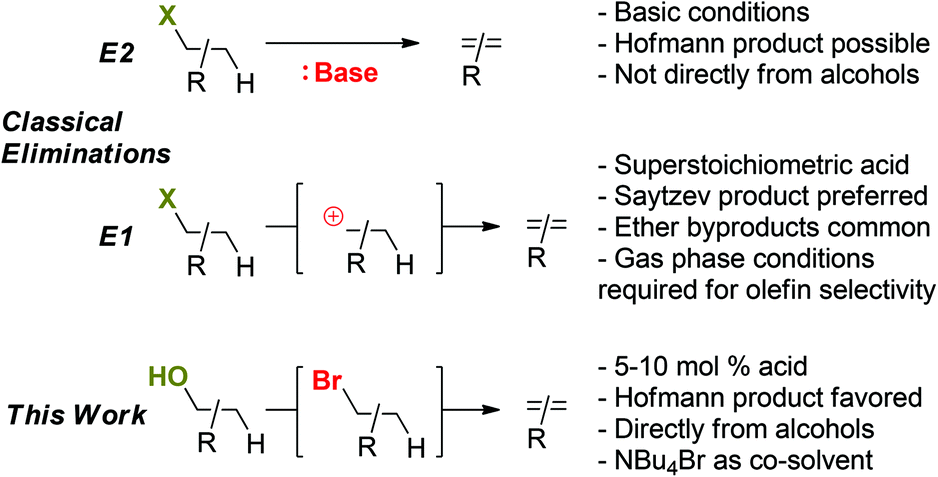
Low temperature dehydrations of non-activated alcohols via halide catalysis†
Received
16th February 2016
, Accepted 6th April 2016
First published on 7th April 2016
Abstract
Activating kinetically inert C–O bonds such as in primary alcohols is an important challenge for the transformation of biomass-derived feedstocks. The herein described methodology addresses this issue through a combination of halide and acid catalysis. The novel mechanistic pathway proposed based on detailed experimental studies enables selective olefin formation from alcohols – as opposed to ether formation – at relatively low temperatures. Suitable substrates are tertiary, secondary, and even primary alcohols. Furthermore, the observed selectivity for the Hoffman elimination product and the realization of a non-rearranging Friedel–Crafts alkylation suggest that the reaction medium with high concentrations of halide (NBu4Br) enables reaction outcomes that cannot be obtained through carbocation intermediates.
Introduction
Transitioning the energy and organic materials economy from a fossil-fuel based enterprise towards sustainable production is one of the greatest challenges of our time.1–3 With regard to the sustainable production of widely used chemicals, major issues are the availability and transport of carbon-containing raw materials4,5 other than crude oil together with a fundamental chemical challenge: the ubiquity of oxygen atoms in most biologically derived feedstocks.6–11 Therefore, deoxygenation reactions have received much attention during the last decade using various approaches such as pre-treatment of biomass12,13 and hydrodeoxygenations.14–16 Most of these processes are only operational at temperatures higher than 150 °C.17–20
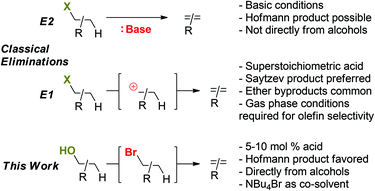
Herein, we describe an approach to deoxygenation chemistry that employs a combination of Lewis acid, Brønsted acid, and nucleophilic halide catalysis. The established protocol activates challenging primary C–O bonds in alcohols and ethers, providing access to deoxygenated products (olefins); secondary and tertiary alcohol functionalities are also rapidly converted to the corresponding olefins under analogous conditions. In contrast to classical alcohol dehydrations with mineral acids, relatively low temperatures (125 °C) and only catalytic amounts (5 mol%) of Brønsted and Lewis acid catalysts are required; furthermore, the major product obtained are olefins (as opposed to ethers) even with primary alcohol substrates. Differing from classical acid-promoted substitution and elimination pathways for alcohols proceeding through carbocation intermediates, mechanistic studies suggest a halide-catalyzed pathway with alkyl halides as intermediates (Scheme 1). Importantly, these observations imply a new mechanistic role of halide additives as nucleophilic catalysts for converting molecules with C–O bonds such as biomass-derived materials.
 |
| | Scheme 1 Mechanisms of classical elimination reactions and the herein proposed, halide-catalyzed elimination mechanism. | |
Results and discussion
Dehydration in organic solvents
We initially hypothesized that the activation of primary alcohols could be achieved using Brønsted and Lewis acid catalysis, in analogy to previous reports by others.21–26 However, classical Brønsted (TfOH, HCl, HBr, HI, H2SO4, H3PO4, HNO3) or Lewis acids (Cu(OTf)2, CuBr2, FeCl3, Fe(OTf)3, BiBr3, BiI3) or a combination of these potential catalysts in organic solvents (PhCl, PhBr) provided less than 1% yield of olefinic or ether product even at 125 °C, using the primary alcohol 1-octanol as substrate.
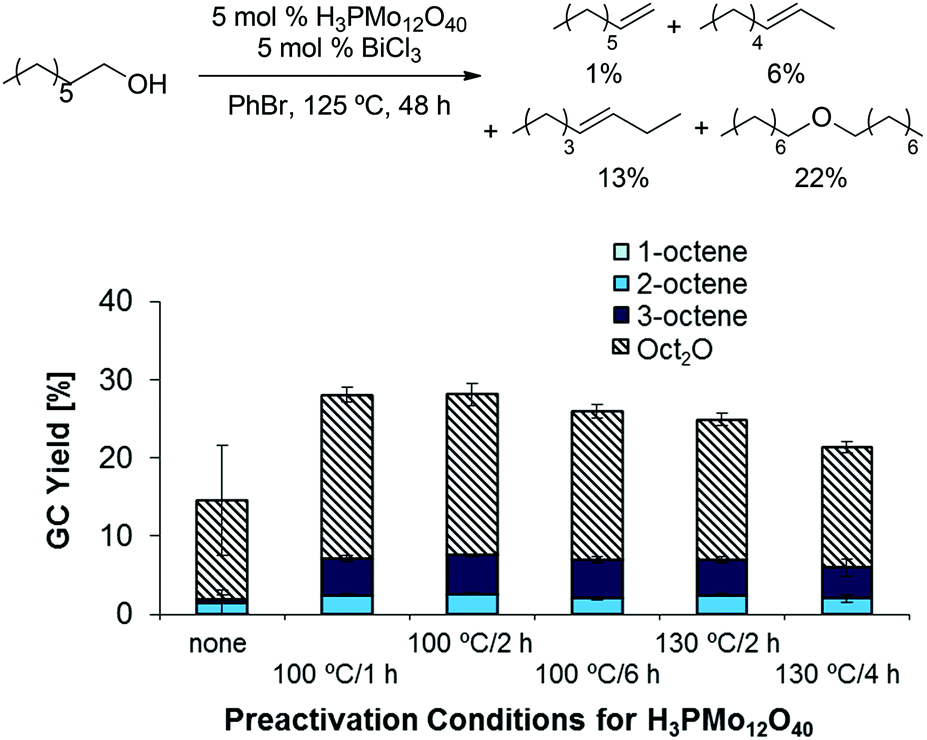
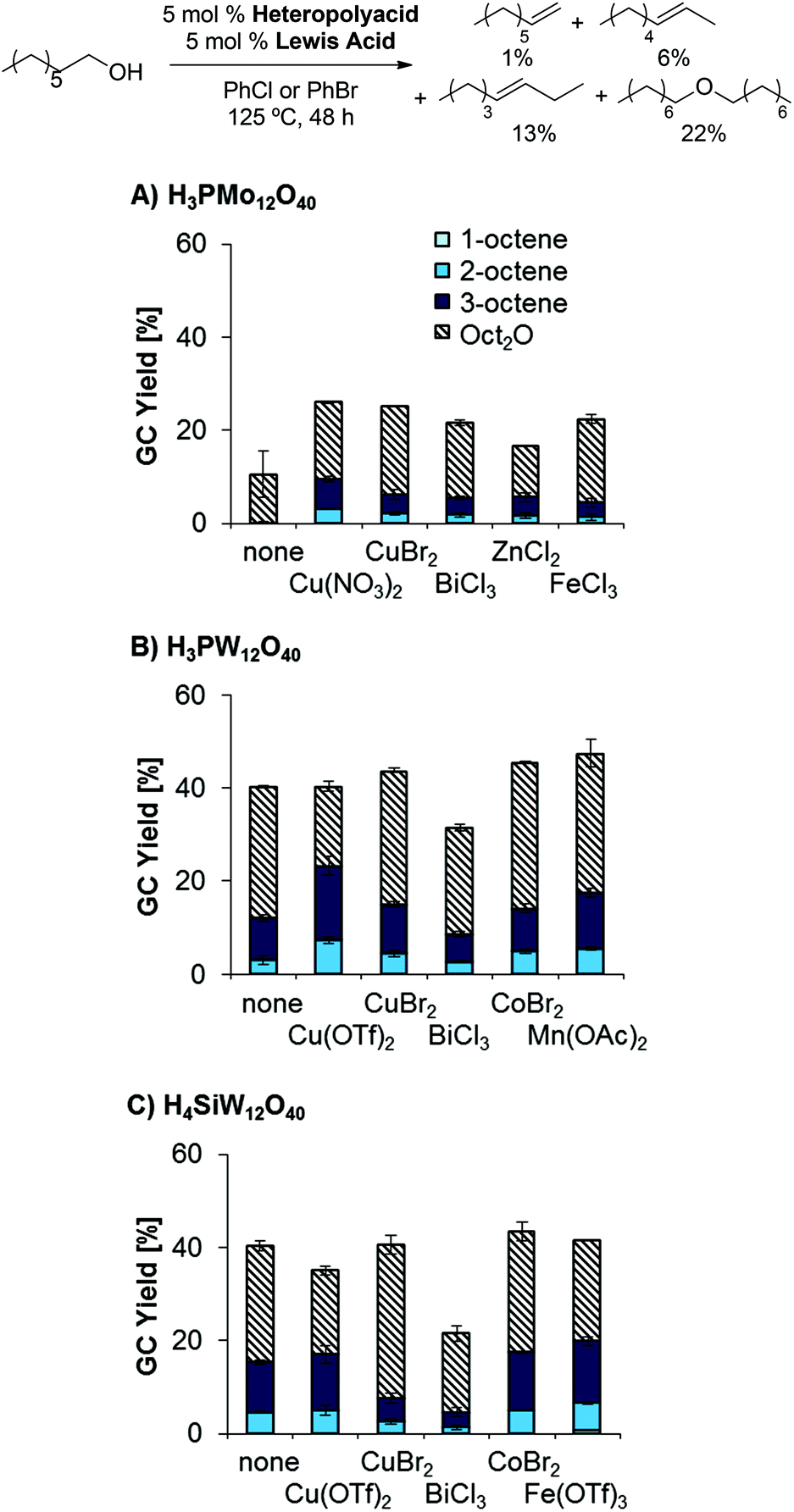
As classical Brønsted acids did not result in the desired reactivity, we explored the use of heteropolyacids, which are known to catalyze dehydrations at relatively high temperatures and in the gas phase.26,27 Interestingly, combinations of H3PMo12O40 as Brønsted acid catalyst and BiCl3 as Lewis acid catalyst afforded slightly higher yields than the previously tested classical mineral acid catalysts; however, a preference for ether formation and low reproducibility of data was problematic in these reactions (see left-most column in Scheme 2). This challenge was addressed by heat pretreatment of H3PMo12O40, affording higher overall product yields and higher reproducibility (Scheme 2). Pretreating the corresponding W-based heteropolyacids H3PW12O40 and H4SiW12O40 resulted in similarly high product yields (see ESI, Table 1†). Moreover, the use of other Lewis acidic co-catalysts provided significant yields (up to 16%) of the desired olefins as a mixture of isomers and 28% ether (Scheme 3; see also ESI, Tables 2–7†).
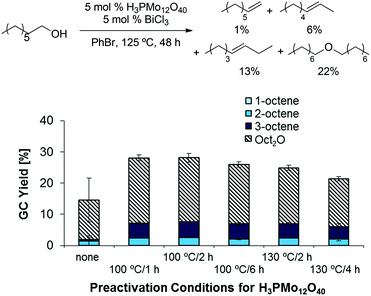 |
| | Scheme 2 Influence of H3PMo12O40 preactivation on dehydration yields and reproducibility. Conditions: H3PMo12O40 (5.0 mol%), BiCl3 (10.0 mg, 0.0318 mmol, 5.0 mol%), 1-octanol (0.100 ml, 82.7 mg, 0.635 mmol, 1.00 equiv.), PhBr (1.0 mL), 125 °C, 48 h. | |
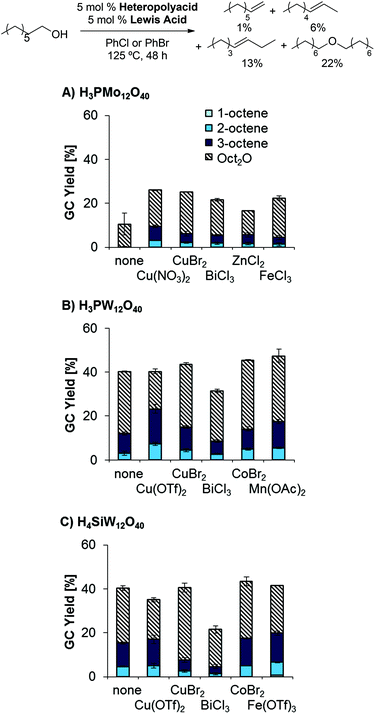 |
| | Scheme 3 Influence of Lewis acid on dehydration yields and product distribution. Conditions: (A) H3PMo12O40 (5.0 mol%), Lewis acid (5.0 mol%), 1-octanol (1.0 equiv.), PhCl (1.0 mL), 125 °C, 48 h; (B) like A, but H3PW12O40 (5.0 mol%), PhBr (1.0 mL); (C) like A, but H4SiW12O40 (5.0 mol%), PhBr (1.0 mL). | |
Unfortunately, none of these strategies provided high yields of olefins with 1-octanol as substrate. Even under optimized conditions, large amounts of the ether (17–38%) were observed after 48 h at 125 °C with typically only low olefin yields. Other organic solvents did not improve the reactivity profile further (no products in DMA, PhCN, DMSO, or AcOH; mostly ether in p-xylene, decane, and decalin). Overall, we considered ether formation a non-productive outcome, as this reaction represents oligomerization instead of depolymerization, which would be unfavorable for biomass deoxygenations.
Dehydrations in ionic liquids
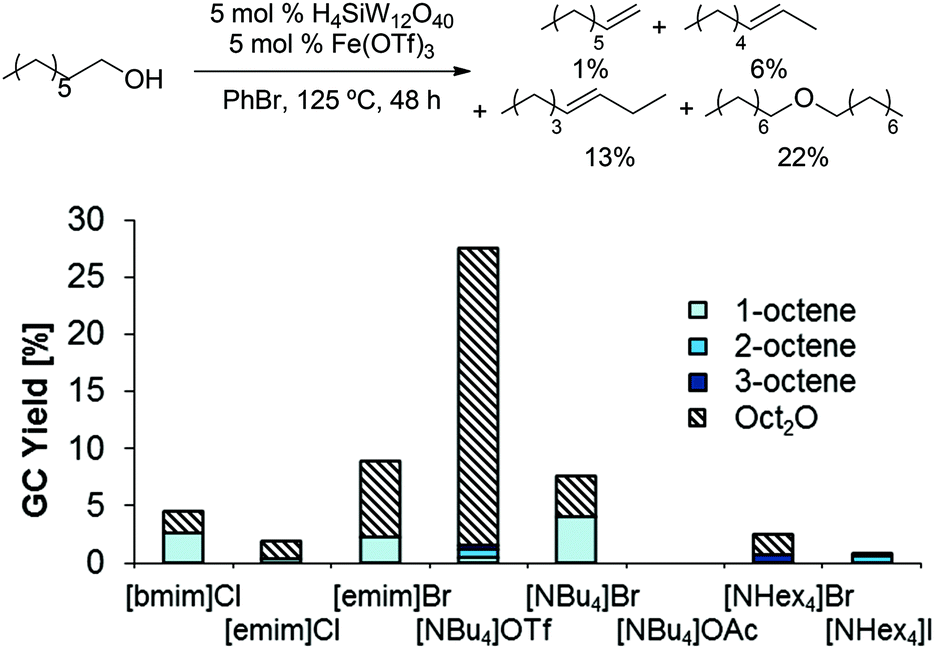
In order to address the relatively low olefin yields obtained above, we reasoned that ionic liquid solvents might promote deoxygenations by furthering the formation of charged species which are common intermediates in dehydrations.28,29 Thus, we evaluated a combination of a Brønsted acid (H3PW12O40) and Lewis acid (BiCl3) in several ionic liquid solvents (Scheme 4); indeed, several of these reactions (in [bmim]Cl, [emim]Br, [NBu4]Br) produced 1-octene in up to 4% yield. Among all tested ionic liquids, [NBu4]Br showed the highest selectivity for 1-octene (1-octene/Oct2O = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1; 4% each).
:1; 4% each).
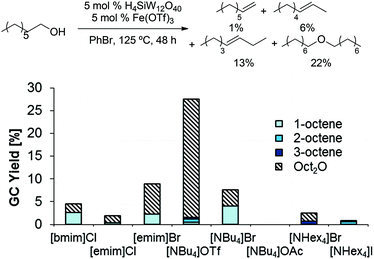 |
| | Scheme 4 BiCl3/H3PW12O40 catalyzed dehydration of 1-octanol in ionic liquids. Conditions: H3PW12O40 (5.0 mol%), BiCl3 (5.0 mol%), 1-octanol (1.0 equiv.), ionic liquid (1.0 g), para-xylene (0.10 mL), 48 h. All product yields were determined by calibrated GC analysis. | |
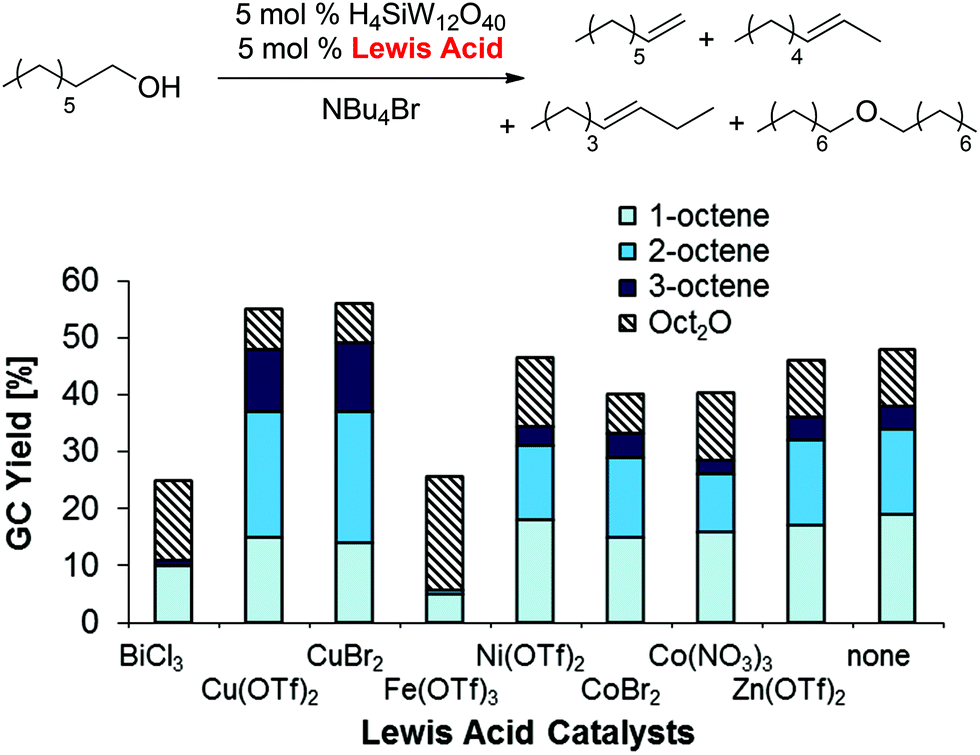
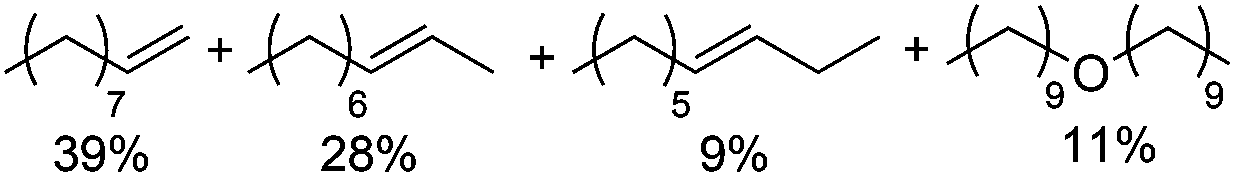
Optimization of the Lewis acid (Scheme 5), organic co-solvent, and heteropolyacid (see ESI†) resulted in significantly higher yields and selective formation of olefin products (50%; 1-/2-/3-octene 7:5:1) with only 7% dioctyl ether in the reaction mixture after 48 h. These results correspond to an olefin:ether selectivity of up to 7:1, which is contrary to the typical selectivities obtained in non-ionic organic solvents that favor ether formation (see for example Scheme 3 above).30
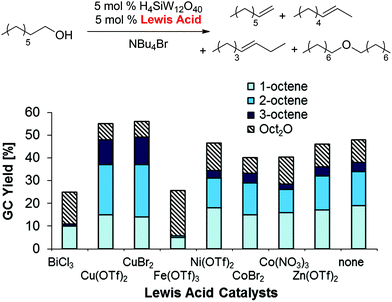 |
| | Scheme 5 Lewis acid optimization for 1-octanol dehydration in NBu4Br. Conditions: H4SiW12O40 (5.0 mol%), co-catalyst (5.0 mol%), 1-octanol (1.0 equiv.), NBu4Br (1.0 g), para-xylene (0.10 mL), 48 h. All product yields were determined by calibrated GC analysis. | |
Influence of Brønsted and Lewis acid catalyst loading
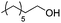
After establishing the acid-catalyzed dehydration in ionic liquids as a potential pathway for deoxygenation, we further investigated the influence of the Brønsted and Lewis acid catalysts on dehydration reactivity and selectivity (Table 1). To this effect, product yields with CuBr2 and H4SiW12O40 as catalysts under the optimized conditions (5 mol% each; entry 1) were compared to the yields obtained with lower Lewis or Brønsted acid loadings (entries 2 and 4) as well as in the absence of CuBr2 (entry 3) or H4SiW12O40 (entry 5). Lowering the H4SiW12O40 loading (entries 4/5) led to an almost inactive catalytic system, while lowering the CuBr2 loading (entries 2/3) resulted in only slightly lower olefin yields, but in higher ether yields and reduced olefin isomerization. These data suggest that the presence of Brønsted acid is crucially important for the formation of 1-octene, while CuBr2 potentially plays a role in olefin isomerization and ether cleavage.
Table 1 Effect of Lewis- and Brønsted-acid catalyst loading on dehydration activitya
|

|
| Entry |
mol% H4SiW12O40/CuBr2 |
% Yield 1-/2-/3-octene |
% Yield Oct2O |
|
Conditions: 0 to 5.0 mol% H4SiW12O40, 0 to 5.0 mol% CuBr2, 1.00 equiv. 1-octanol, 1.00 g NBu4Br, 0.10 mL p-xylene, 125 °C, 48 h. All product yields were determined by calibrated GC analysis.
|
| 1 |
5/5 |
14/23/12 |
7 |
| 2 |
5/1 |
15/16/6 |
8 |
| 3 |
5/0 |
19/15/4 |
10 |
| 4 |
1/5 |
3/0/0 |
7 |
| 5 |
0/5 |
0/0/0 |
2 |
Influence of solvent and NBu4Br volumes
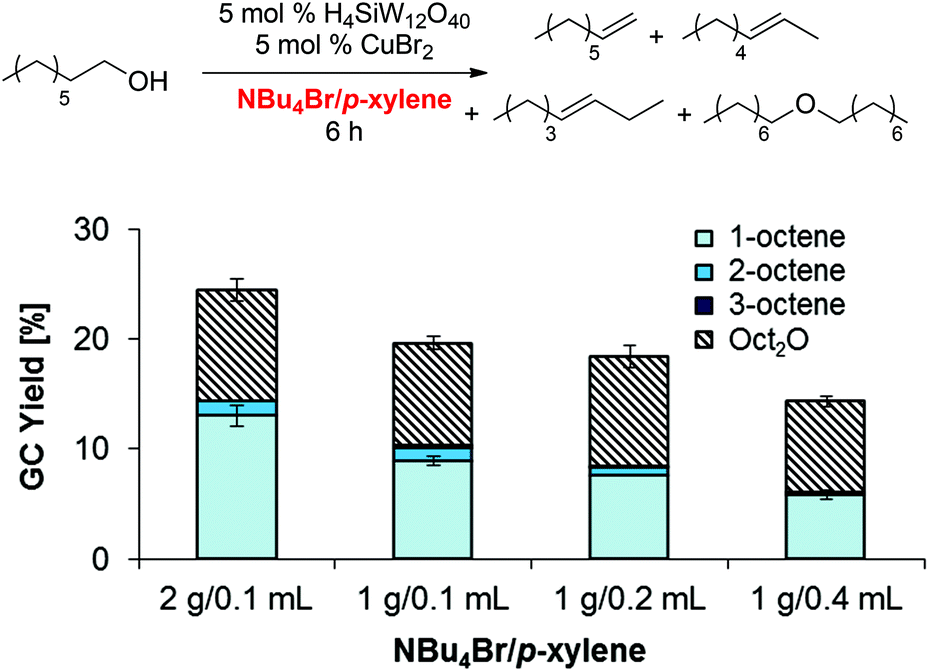
Other factors that can affect dehydration reactivity and selectivity are the relative concentrations of NBu4Br and p-xylene, which form two separate phases under the reaction conditions. In order to provide insight into the role of each solvent, product yields of olefins and dioctyl ether were determined after 6 h. These conditions were chosen to minimize potential effects of olefin isomerization on the reaction rates and product distributions (Scheme 6).
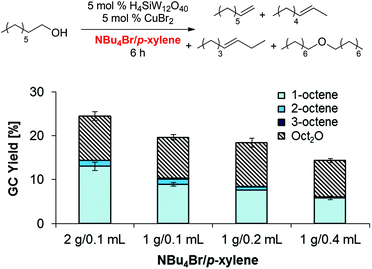 |
| | Scheme 6 Influence of NBu4Br and p-xylene loading on product yield and selectivity after 6 h. Conditions: H4SiW12O40 (91.5 mg, 31.8 μmol, 5.0 mol%), CuBr2 (7.1 mg, 32 μmol, 5.0 mol%), 1-octanol (0.100 ml, 82.7 mg, 635 μmol, 1.00 equiv.), NBu4Br (1.00 g or 2.00 g), para-xylene (0.10, 0.20, or 0.40 mL), 125 °C. | |
Overall, the obtained data suggest that (i) an increase in NBu4Br loading from 1 g to 2 g enhances both the product yield (up to 24%) and the selectivity for olefin (up to 1.4:1 olefin/Oct2O as compared to 1.1:1 with 1 g NBu4Br) and (ii) that a higher p-xylene loading (0.1 → 0.2 → 0.4 mL) results in lower overall activity as well as higher selectivity for ether formation (down to 0.7:1 olefin/Oct2O). Based on these observations, we conclude that NBu4Br is beneficial for olefin formation over ether formation.
Substrate scope of dehydration
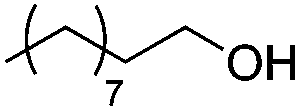
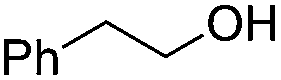
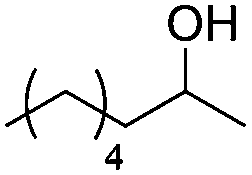
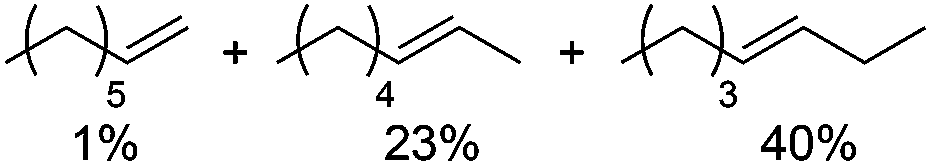
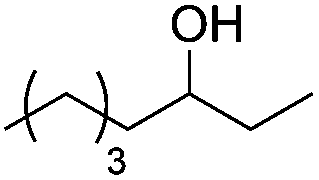
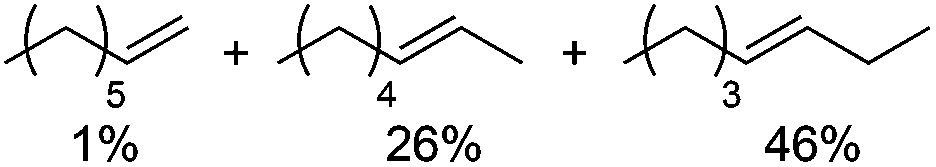
Next, we explored the generality of the established conditions in a substrate scope study (Table 2). Primary alcohols (entries 1 to 3) undergo dehydration readily (49% to 91% olefin yields), while secondary alcohols (entries 4 to 6) are converted to a mixture of olefins in similar yields (64% to 89%) but without any ether side products.
|
|
| Entry |
Substrate |
Product(s)a |
|
General conditions: 5.0 mol% H4SiW12O40, 5.0 mol% CuBr2, alcohol (635 μmol, 1.00 equiv.), 1.00 g NBu4Br, 0.10 mL para-xylene, 125 °C, 48 h. All product yields were determined by calibrated GC analysis, following product identification by GCMS and verification of product identity with original samples.
Two additional octene isomers were detected by GCMS (see ESI); however, since their identity was not established unambiguously, their yields are not represented in this table.
2.00 g NBu4Br were used.
24 h.
6 h.
1 h.
1 h, no CuBr2, 100 °C.
|
| 1a,b |
|
|
| 2c |
|
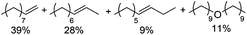
|
| 3 |

|
|
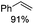
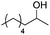
| 4b,d |
|
|
| 5b,e |
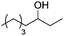
|
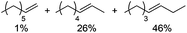
|
| 6f |
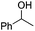
|
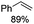
|
| 7g |
|
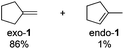
|
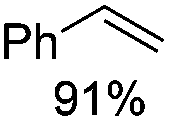
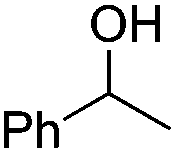
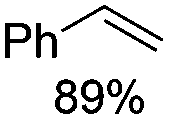
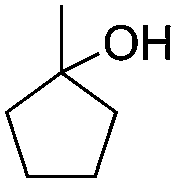
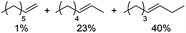
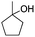
Notably, ideal reaction times for secondary alcohols can be significantly shorter (e.g. 1 h for 1-phenyl-ethanol; entry 6) than for primary alcohols. In these experiments, longer reaction time decrease the yields obtained for the resulting olefins, which is likely due to acid-catalyzed oligomerization. Remarkably, very high yields of styrene can be obtained from either the primary or secondary alcohol substrate (entries 3/6). This indicates that the olefin moiety in styrene is more stable against decomposition under the reactions conditions, possibly through π-conjugation with the phenyl substituent. The tertiary alcohol 1-methylcyclopentan-1-ol (entry 7) smoothly undergoes conversion to a mixture of olefins after a very short reaction time (1 h). This reaction proceeds at only 100 °C and shows selectivity for the less substituted olefin product exo-1 with an exocyclic double bond. These types of olefins are typically not obtained under acidic conditions and have not been accessible directly from alcohols prior to this work.31
Detection of halogenated intermediates
As both the selectivity for olefins from primary alcohol substrates at relatively low temperatures (125 °C) and the selectivity for exo-1 from 1-methylcyclopenan-1-ol are unusual features for transformations of alcohols under acidic conditions, we investigated the reaction mixtures for potential intermediates to obtain clues about the operating mechanism. Notably, all reaction mixtures employing 1-, 2-, or 3-octanol as substrates contained the corresponding brominated compounds (1-, 2-, or 3-bromooctane); furthermore, we identified PhCHBrCH3 in a reaction with PhCH(OH)CH3 as substrate (Scheme 7). These data indicate that alkyl bromides are potential intermediates in the herein developed protocol. Interestingly, 3-bromooctane was also detected in a reaction mixture employing 2-octanol as a substrate, while 2-bromooctane was identified in a reaction that employed 3-octanol as substrate (see ESI† for GCMS data). This suggests that the observed isomerization of olefins proceeds through the formation of isomeric bromooctanes, implying that the formation of olefins by elimination seems to be reversible under the reaction conditions.
 |
| | Scheme 7 Detected alkyl bromide intermediates. | |
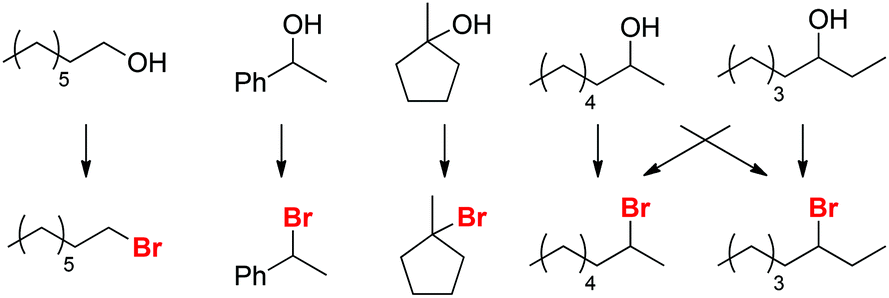
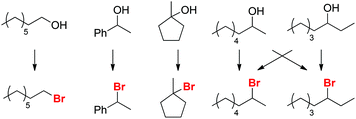
Selectivity for Saytzev product (exo-1)
Based on these findings, we postulate that alkyl bromide intermediates can be expected to be involved in the selective formation of the exocyclic olefin product exo-1; indeed, traces of the corresponding tertiary alkyl bromide intermediate are detected in reaction mixtures after 15 min. The time profile of the reaction forming exo-1 and endo-1 (Scheme 8) further suggests that exo-1 is the kinetic product, as the selectivity for exo-1 is high during the first 2 h, but errodes at longer reaction times. These data imply that the more thermodynamically favored isomer endo-1 is formed by isomerization of exo-1. Similar selectivities for exo-olefins are usually only obtained in E2 eliminations in the presence of sterically bulky, strong bases (e.g. KOtBu).31
 |
| | Scheme 8 Time dependence of formation for exo-1 and endo-1. Conditions: H4SiW12O40 (91.5 mg, 31.8 μmol, 5.0 mol%), alcohol (635 μmol, 1.00 equiv.), NBu4Br (1.00 g, 3.10 mmol, 4.88 equiv.), para-xylene (0.10 mL), 125 °C. | |
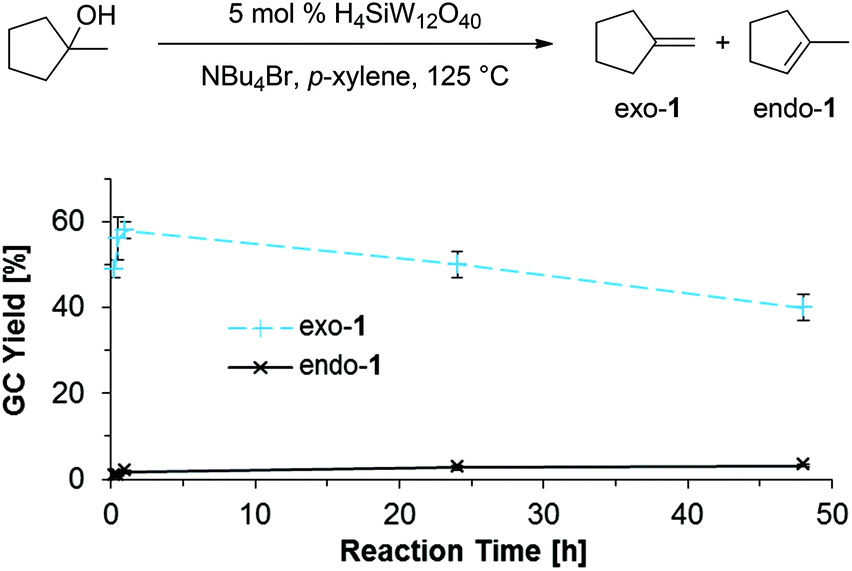
In contrast to E2 eliminations under basic conditions, the classical mechanism for the acid-catalyzed dehydration of tertiary alcohols suggests a carbocation intermediate 3, as indicated in Scheme 9 (left side).
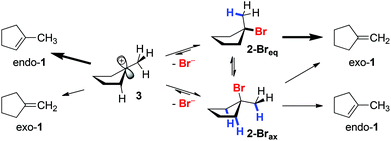 |
| | Scheme 9 Conformational analysis of eliminations from brominated intermediates 2-Breq and 2-Brax. | |
However, in the presence of high concentrations of Br− (such as in NBu4Br as reaction medium), 3 is likely trapped rapidly through nucleophilic attack to form 2-Br. Once formed, loss of Br− from 2-Br can be expected to be strongly kinetically disfavored in the presence of high concentrations of Br−. Even if E1 elimination through intermediate 3 would proceed, thermodynamically more favorable endo-1 would be expected as the major product, which is not consistent with the observed selectivity for exo-3. Thus, we consider 3 an unlikely intermediate for olefin formation.
The alternative mechanism for olefin formation, E2 elimination directly from the alkyl bromide intermediates (Scheme 9, right side), can proceed through at least two possible conformations: 2-Breq, in which the bromine substituent takes on a pseudo-equatorial position, and 2-Brax, in which the C–Br bond is oriented in a pseudo-axial position. A concerted E2 elimination pathway can favor the olefin isomer exo-1 for two reasons: (i) 2-Breq could be the favored conformer; as only one C–H bond (shown in blue) in 2-Breq is anti-periplanar to the leaving group, E2 elimination from 2-Breq would exclusively form exo-1. (ii) Alternatively, Curtin–Hammett control of selectivity32 for exo-1 might occur if the transition states leading from 2-Breq or 2-Brax to exo-1 are significantly lower in energy than the transition state leading to endo-1.
Mechanistic investigations
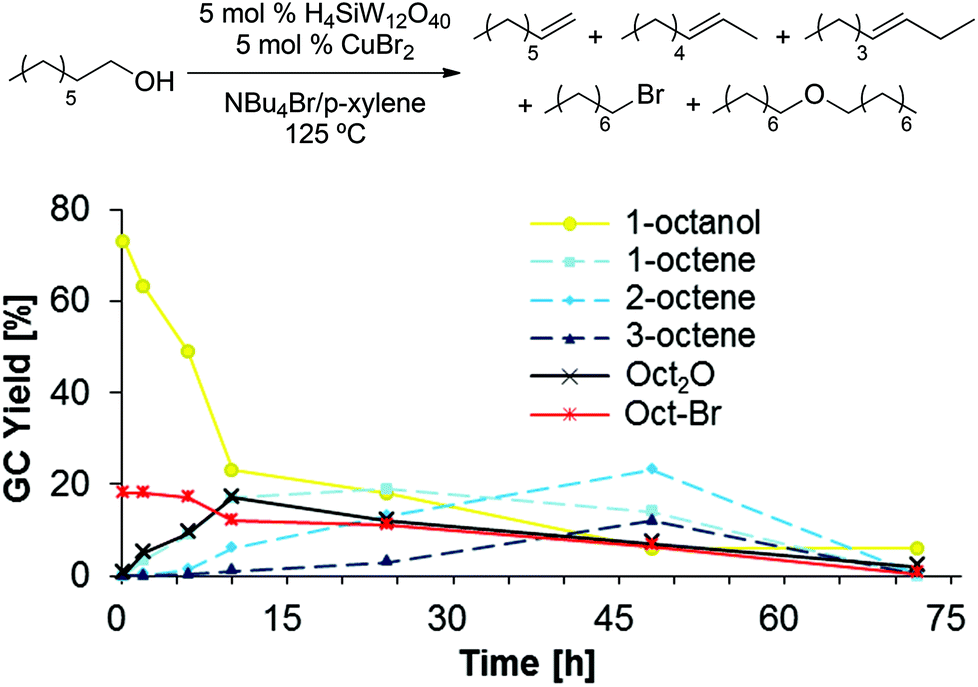
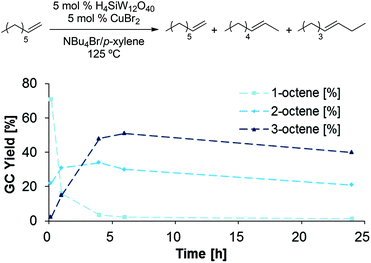
In order to gain further insight into the mechanism of dehydration, we followed product formation and substrate consumption over a period of 72 h (Scheme 10), using 1-octanol as substrate. Interestingly, the first product detected in the reaction mixture after only 10 min is 1-bromooctane. 1-Octene and dioctyl ether are only present in significant quantities after 2 h, while the onset of 2- and 3-octene formation is further delayed; thus, we speculate that the internal olefins are formed through acid-catalyzed olefin isomerization from 1-octene.33 Notably, the formation of dioctyl ether is reversible, as the amount of detected ether product decreases after a reaction time of 10 h. Finally, very little product is detected after 72 h, suggesting product decomposition, which likely proceeds through olefin oligomerization.34‡
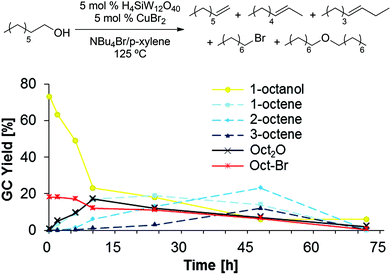 |
| | Scheme 10 Time study of 1-octanol dehydration in NBu4Br/p-xylene. Conditions: H4SiW12O40 (5.0 mol%), CuBr2 (5.0 mol%), 1-octanol (1.0 equiv.), NBu4Br (1.0 g), para-xylene (0.10 mL). Standard deviations and mass balance of products are omitted for clarity (see ESI†). All product yields were determined by calibrated GC analysis. | |
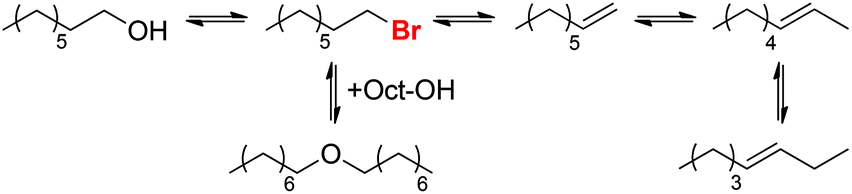
This study suggests that the reaction proceeds through initial formation of 1-bromooctane from 1-octanol (Scheme 11), followed by elimination or nucleophilic substitution to form 1-octene or dioctyl ether. The similar rates of 1-octene and Oct2O formation during the first 10 h of the reaction further support the hypothesis of a common alkyl bromide intermediate for both processes.
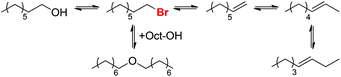 |
| | Scheme 11 Proposed halide catalysis mechanism for 1-octanol dehydration. | |
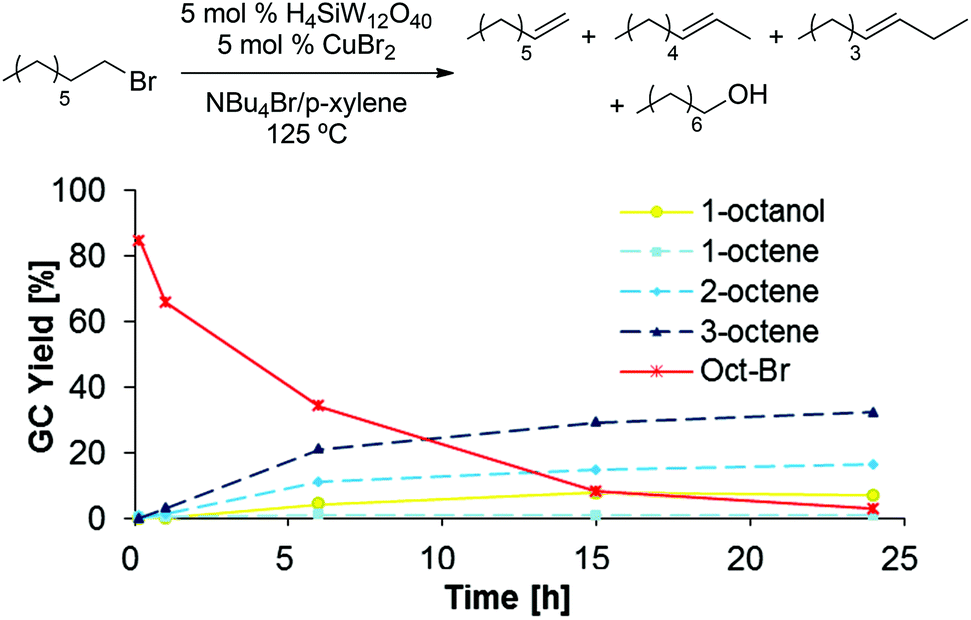
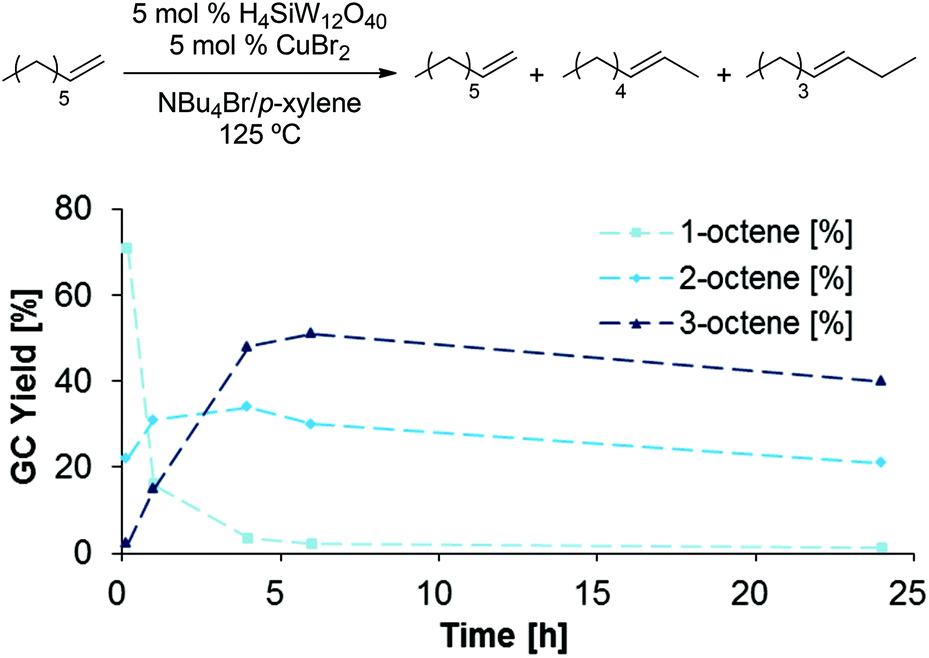
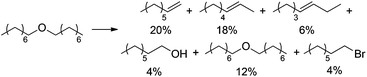
To gain further insight in the elementary steps proposed in Scheme 11, several potential intermediates were subjected to the established dehydration conditions (5 mol% H4SiW12O40, 5 mol% CuBr2, 1 g NBu4Br, 0.1 mL p-xylene, 125 °C, 48 h). 1-Bromooctane underwent rapid elimination in these experiments, producing 2- and 3-octene as the major products, while only small amounts of 1-octene, 1-octanol, or remaining 1-bromooctane were detected after 24 h (Scheme 12). When 1-octene was reacted in analogous fashion (Scheme 13), 2- and 3-octene were observed after only 10 min, suggesting that isomerization is fast. Furthermore, the negative slopes observed for all products in Scheme 13 after 6 h suggest that 1-, 2-, and 3-octene are not stable under the reaction conditions and decompose, likely by oligomerization.34–37 When subjecting dioctyl ether to the reaction conditions (Scheme 14), only 12% of remaining ether substrate were detected after 48 h; however, despite the sluggish transformation, significant amounts of olefins (44% overall) were obtained.
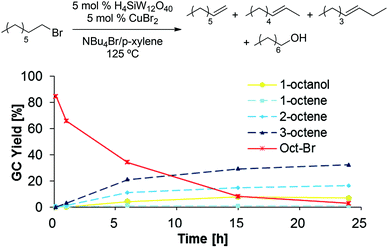 |
| | Scheme 12 1-Bromooctane conversion to olefins under dehydration conditions. | |
 |
| | Scheme 13 Isomerization of 1-octene under dehydration conditions. | |
 |
| | Scheme 14 C–O activation of dioctylether. Conditions: 0.5 equiv. Oct2O (1.0 equiv. 1-octanol building blocks), H4SiW12O40 (5.0 mol%), CuBr2 (5.0 mol%), NBu4Br (1.00 g), para-xylene (0.10 mL), 48 h. Yields of monomeric products are calculated based on 1 equiv. of 1-octanol building blocks present in 0.5 equiv. Oct2O. All product yields were determined by calibrated GC analysis. | |
In combination, these data suggest that: (i) HBr elimination from alkyl bromides to form olefins is fast under the reaction conditions;38§ (ii) that the nucleophilic substitutions forming 1-bromooctane or dioctyl ether from 1-octanol are in principle reversible reactions; and (iii) that 2-octene and 3-octene are formed by isomerization of 1-octene, with the increased thermodynamic stability of internal olefins likely serving as the driving force.39,40
All these conclusions support the halide-catalyzed mechanism for alcohol dehydration outlined in Scheme 11 above. Moreover, these studies in combination with the finding that bromide is crucial to the herein described reactivity suggests bromide involvement as nucleophile, leaving group, and/or base in the proposed substitution/elimination sequence, providing an alternative, halide-catalyzed pathway to classical, acid catalyzed alcohol dehydrations through carbocation intermediates. As such, the lower reactivity of chloride-based ionic liquids (see Scheme 4 above) can be rationalized by the lower nucleophilicity of chloride vs. bromide.41,42
Probing for carbocations: Friedel–Crafts alkylation
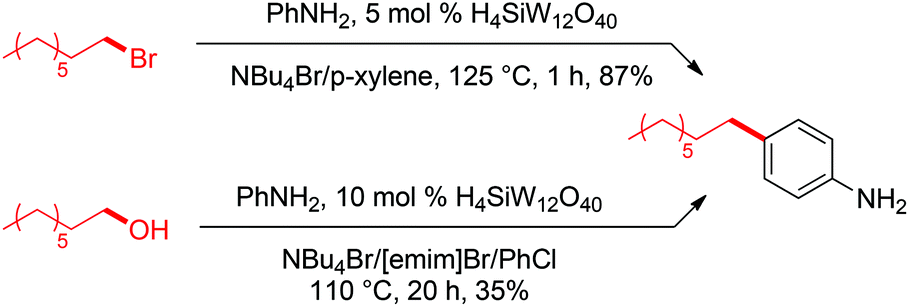
Based on the above postulated involvement of bromide as nucleophile, we reasoned that high concentrations of NBu4Br might aid in suppressing rearrangements that are a consequence of the formation of carbocations as intermediates. Thus, we explored the synthetic utility of the developed reaction conditions for Friedel–Crafts alkylations whose selectivity for linear alkyl arene typically suffers from such rearrangements.43 First, we reacted 1-bromooctane with aniline under conditions equivalent to our best C–O activation conditions. To our delight, high-yielding formation of para-n-octyl aniline (87%) was observed (Scheme 15). In a second experiment, we subjected 1-octanol to modified C–O activation conditions and added aniline after 4 h, which resulted in 35% of para-n-octyl aniline after an additional reaction time of 16 h.44 Remarkably, no secondary alkyl arenes (formed by isomerization of potential cationic intermediates) were detected in these reactions, indicating that carbocations are likely very short-lived species in the employed halide-based ionic liquid. Furthermore, no traces of ortho-n-octyl aniline were detected, providing high selectivity for the para-product. This is remarkable, since previously established reactions between 1-octanol and aniline typically suffer from the formation of such isomers.44
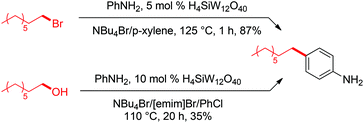 |
| | Scheme 15 Non-isomerizing Friedel–Crafts alkylation (top) and C–O activation/C–C bond formation (bottom). Product yields were determined by calibrated GC analysis. | |
Conclusions
In conclusion, we have established a new mode of reactivity for catalytic C–O activation, which likely proceeds through concurrent acid and nucleophilic bromide catalysis. As similar catalytic pathways have only been reported for reactions of CO245 or highly activated C–OH functionalities,46,47 the herein described methodology is – to the best of our knowledge – the first example of catalytic C–O activation (i.e. without the use of additional stoichiometric additive such as phosphines) through covalent halide catalysis applicable to simple, unactivated alcohols. Importantly, our findings are expected to contribute to a better understanding of beneficial halide effects observed widely in the conversion of biomass-derived substrates.6,7,48
Acknowledgements
We thank Worcester Polytechnic Institute and NSF for funding (Start-up funds to M. H. E.; REU fellowship EEC-1359064 to N. G.). We also acknowledge Juan M. Venegas for inital studies with ionic liquids and heteropolyacids and Dr. Remya P. Narayanan for editorial assistance.
Notes and references
- P. S. Nigam and A. Singh, Prog. Energy Combust. Sci., 2011, 37, 52 CrossRef CAS.
- N. S. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15729 CrossRef CAS PubMed.
- M. Stöcker, Angew. Chem., Int. Ed., 2008, 47, 9200 CrossRef PubMed.
- C. Panoutsou, J. Eleftheriadis and A. Nikolaou, Energy Policy, 2009, 37, 5675 CrossRef.
- A. Messineo, R. Volpe and A. Marvuglia, Energy, 2012, 45, 613 CrossRef CAS.
- R. Carrasquillo-Flores, M. Käldström, F. Schüth, J. A. Dumesic and R. Rinaldi, ACS Catal., 2013, 3, 993 CrossRef CAS.
- M. Mascal, S. Dutta and I. Gandarias, Angew. Chem., Int. Ed., 2014, 53, 1854 CrossRef CAS PubMed.
- R. Ma, W. Hao, X. Ma, Y. Tian and Y. Li, Angew. Chem., Int. Ed., 2014, 53, 7310 CrossRef CAS PubMed.
- S. K. Hanson, R. Wu and L. A. Silks, Angew. Chem., Int. Ed., 2012, 51, 3410 CrossRef CAS PubMed.
- B. Katryniok, S. Paul and F. Dumeignil, ACS Catal., 2013, 3, 1819 CrossRef CAS.
- C. H. Zhou, H. Zhao, D. S. Tong, L. M. Wu and W. H. Yu, Catal. Rev., 2013, 55, 369 CAS.
- M. E. Himmel, S.-Y. Ding, D. K. Johnson, W. S. Adney, M. R. Nimlos, J. W. Brady and T. D. Foust, Science, 2007, 315, 804 CrossRef CAS PubMed.
- S. Stephanidis, C. Nitsos, K. Kalogiannis, E. F. Iliopoulou, A. A. Lappas and K. S. Triantafyllidis, Catal. Today, 2011, 167, 37 CrossRef CAS.
- S. Mukkamala, M. C. Wheeler, A. R. P. van Heiningen and W. J. DeSisto, Energy Fuels, 2012, 26, 1380 CrossRef CAS.
- J. N. Chheda and J. A. Dumesic, Catal. Today, 2007, 123, 59 CrossRef CAS.
- Z.-P. Yan, L. Lin and S. Liu, Energy Fuels, 2009, 23, 3853 CrossRef CAS.
- Y. K. Lugo-José, J. R. Monnier and C. T. Williams, Appl. Catal., A, 2014, 469, 410 CrossRef.
- S. G. Wettstein, D. M. Alonso, E. I. Gürbüz and J. A. Dumesic, Curr. Opin. Chem. Eng., 2012, 1, 218 CrossRef CAS.
- Y. Liu, C. Luo and H. Liu, Angew. Chem., Int. Ed., 2012, 51, 3249 CrossRef CAS PubMed.
- A. M. Ruppert, K. Weinberg and R. Palkovits, Angew. Chem., Int. Ed., 2012, 51, 2564 CrossRef CAS PubMed.
- T. Nishiguchi, N. Machida and E. Yamamoto, Tetrahedron Lett., 1987, 28, 4565 CrossRef CAS.
- T. Nishiguchi and C. Kamio, J. Chem. Soc., Perkin Trans. 1, 1989, 707 RSC.
- K. Laali, R. J. Gerzina, C. M. Flajnik, C. M. Geric and A. M. Dombroski, Helv. Chim. Acta, 1987, 70, 607 CrossRef CAS.
- J. Ahmad and K. B. Astin, Langmuir, 1990, 6, 1098 CrossRef CAS.
- J. A. Dias, S. C. L. Dias and N. E. Kob, J. Chem. Soc., Dalton Trans., 2001, 228 RSC.
- D. Varisli, T. Dogu and G. Dogu, Chem. Eng. Sci., 2007, 62, 5349 CrossRef CAS.
- V. Kozhevnikov, Chem. Rev., 1998, 98, 171–198 CrossRef CAS PubMed; P. Vázquez, L. Pizzio, C. Cáceres, M. Blanco, H. Thomas, E. Alesso, L. Finkielsztein, B. Lantaño, G. Moltrasio and J. Aguirre, J. Mol. Catal. A: Chem., 2000, 161, 223–232 CrossRef; A. Martin, U. Armbruster and H. Atia, Eur. J. Lipid Sci. Technol., 2012, 114, 10–23 CrossRef.
- M. J. Janik, J. Macht, E. Iglesia and M. Neurock, J. Phys. Chem. C, 2009, 113, 1872 CAS.
- W. Yan, X. Ye, N. G. Akhmedov, J. L. Petersen and X. Shi, Org. Lett., 2012, 14, 2358 CrossRef CAS PubMed.
- Two additional octene isomers were identified in these reaction mixtures by GCMS analysis (see ESI, page S32†). However, since their identity is unclear (possibly 4-E-octene or Z-octene isomers), their contribution to the overall olefin yields are not shown in the schemes in this manuscript).
- R. A. Bartsch and J. Zavada, Chem. Rev., 1980, 80, 453–494 CrossRef CAS; L. M. T. Frija and C. A. M. Afonso, Tetrahedron, 2012, 68, 7414–7421 CrossRef.
- J. I. Seeman, J. Chem. Educ., 1986, 63, 42 CrossRef CAS.
- A. Corma, Chem. Rev., 1995, 95, 559 CrossRef CAS.
-
J. R. Johnson and J. D. Burrington, US5710225A, The Lubrizol Corporation, United States, 1998 Search PubMed.
- A. Onopchenko, B. L. Cupples and A. N. Kresge, Ind. Eng. Chem. Prod. Res. Dev., 1983, 22, 182 CrossRef CAS.
- Y. Gu, F. Shi and Y. Deng, Catal. Commun., 2003, 4, 597 CrossRef CAS.
- A. de Klerk, Ind. Eng. Chem. Res., 2005, 44, 3887 CrossRef CAS.
- T. Boudewijns, M. Piccinini, P. Degraeve, A. Liebens and D. De Vos, ACS Catal., 2015, 5, 4043 CrossRef CAS.
- A. Seayad, M. Ahmed, H. Klein, R. Jackstell, T. Gross and M. Beller, Science, 2002, 297, 1676 CrossRef CAS PubMed.
- D. R. Anderson, T. Ung, G. Mkrtumyan, G. Bertrand, R. H. Grubbs and Y. Schrodi, Organometallics, 2008, 27, 563 CrossRef CAS PubMed.
- C. G. Swain and C. B. Scott, J. Am. Chem. Soc., 1953, 75, 141 CrossRef CAS.
- N. L. Lancaster, P. A. Salter, T. Welton and G. B. Young, J. Org. Chem., 2002, 67, 8855 CrossRef CAS PubMed.
- Y. Xiao and S. V. Malhotra, J. Mol. Catal. A: Chem., 2005, 230, 129–133 CrossRef CAS.
- R. N. Gedye, J. Bozic, P. M. Durbano and B. Williamson, Talanta, 1989, 36, 1055–1058 CrossRef CAS PubMed.
- C. Lescot, D. U. Nielsen, I. S. Makarov, A. T. Lindhardt, K. Daasbjerg and T. Skrydstrup, J. Am. Chem. Soc., 2014, 136, 6142 CrossRef CAS PubMed.
- I. T. Raheem, P. S. Thiara, E. A. Peterson and E. N. Jacobsen, J. Am. Chem. Soc., 2007, 129, 13404 CrossRef CAS PubMed.
- G. Bergonzini, C. S. Schindler, C.-J. Wallentin, E. N. Jacobsen and C. R. J. Stephenson, Chem. Sci., 2014, 5, 112 RSC.
- H. Zhao, J. E. Holladay, H. Brown and Z. C. Zhang, Science, 2007, 316, 1597 CrossRef CAS PubMed;
M. Mascal and S. Dutta, in Selective Catalysis for Renewable Feedstocks and Chemicals, ed. K. M. Nicholas, Springer International Publishing, 2014, vol. 353, pp. 41–83 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6qo00069j |
| ‡ Product decomposition likely occurs through olefin polymerization or oligomerization. For an industrially relevant example of heteropolyacid catalyzed olefin polymerization, see ref. 34. However, preliminary GCMS and LCMS studies of the described reaction mixtures have so far not been able to definitively account for all decomposition products. |
| § Analogous acceleration of eliminations by halide-based ionic liquids has been documented recently in the literature; see ref. 48. |
|
| This journal is © the Partner Organisations 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.