
A novel zinc based binary catalytic system for CO2 utilization under mild conditions†
Willi
Desens
,
Christina
Kohrt
,
Anke
Spannenberg
and
Thomas
Werner
*
Leibniz-Institut für Katalyse e. V. (LIKAT Rostock), Albert-Einstein-Str. 29a, 18059 Rostock, Germany. E-mail: Thomas.Werner@catalysis.de
First published on 7th January 2016
Abstract
Herein we report the development of a novel binary catalytic system for the addition of CO2 to epoxides based on zinc iodide and an imidazolium salt. The influence of the nature of the imidazolium ion and the anion has been studied in a test reaction. The optimized system exhibits excellent activity and selectivity. Furthermore, the reaction can be performed under very mild (30 °C) and solvent free conditions yielding the desired cyclic carbonates in up to 99%.
 Thomas Werner | Thomas Werner studied chemistry at the Technische Universität Berlin, Germany, and at the Northumbria University in Newcastle upon Tyne, UK. After receiving his Diploma from the Technische Universität Berlin in 2001 he moved together with Prof. J. Christoffers to the Universität Stuttgart, Germany, where he worked on cerium catalyzed transformations of β-dicarbonyl compounds in the presence of molecular oxygen. He completed his Ph.D. in 2004 and joined shortly after, the group of Prof. A. G. M. Barrett at the Imperial College London, UK, for postdoctoral work on biomimetic approaches to the antifungal agent, 15G256β. In 2006 he joined the Coating and Colorants business unit of Evonik (former Degussa) as the head of R&D laboratory. In 2008 he relocated to Leibniz-Institut für Katalyse e. V. in Rostock, Germany, where he is currently working towards his habilitation in the group of Prof. M. Beller. Since October 2015 he is a visiting Professor at the Ernst Moritz Arndt University of Greifswald. His research interests include phosphorus based organocatalysis, especially catalytic reactions based on PIII/PV redox cycling and the utilization of carbon dioxide as a C1 building block. |
Introduction
Establishing a chemical industry based on carbon dioxide as a C1 building block is one scenario for the future to close the gap between decreasing fossil feedstocks and the increasing demand on chemical products.1 Recently efforts have been greatly intensified to develop efficient methods for the synthesis of useful products from this nonhazardous, economically feasible and environmentally benign building block.2 Particularly the atom economic conversion of epoxides and carbon dioxide into cyclic carbonates is one of the most promising routes for carbon dioxide fixation into value added products (Scheme 1).3 Cyclic carbonates are used in numerous applications ranging from polar aprotic solvents,4 electrolytes in lithium-ion batteries,5 synthetic building blocks,6 intermediates in fine chemical industry, industrial lubricants,7 components in oils and paints,3e raw materials for polycarbonates and isocyanate free polyurethanes.8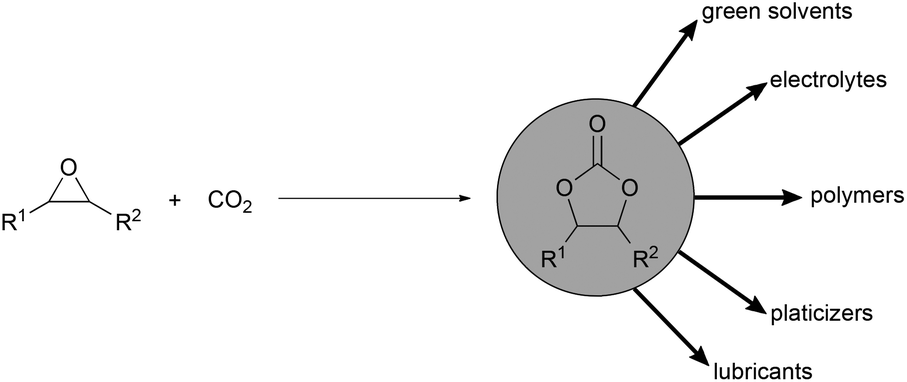 | ||
| Scheme 1 Synthesis and application of cyclic carbonates. | ||
Numerous catalysts9 have been developed for the coupling of carbon dioxide to epoxides. Most frequently employed systems are based on transition metal catalysts,10 alkali metal salts,11 carbenes,12 quaternary ammonium13 or phosphonium salts,14 functional polymers,15 and ionic liquids.16 A variety of metal salts and complexes have been reported in combination with ionic liquids as binary systems.9a,d,f,17 The Lewis acidic activation of the epoxide by the metal species and subsequent ring opening by the counter anion of the ionic liquid is well accepted in the literature. In those systems simple zinc salts are often used due to their low cost and high efficiency. Kim and co-workers reported imidazolium zinc tetrahalides as highly active catalysts.18 They also investigated the effect of the role of the Lewis acidic species in the presence of imidazolium salts. Subsequently the groups of Xia19 and Arai20 studied similar systems and proposed a mechanism involving the activation of the epoxide by zinc followed by nucleophilic ring opening by the counter anion of the imidazolium salt. Thereafter a series of onium salts have been utilized in combination with zinc salts as catalysts.9d,21 Most recently Sun et al. developed zinc based task specific ionic liquids for the coupling of carbon dioxide with epoxides to form cyclic carbonates.22 Even though great advances have been made in the development of ionic liquid zinc-based systems, in most cases, high reaction temperatures ≥100 °C and CO2 pressures >1.0 MPa are needed. We developed one and two component catalyst systems for the synthesis of cyclic carbonates under mild conditions based on alkali metal and bifunctional onium salts.11a,c,13a,14 In general, we observed the superior activity of iodides compared to other counter anions.
Thus we envisioned that a zinc iodide based binary catalytic system should be especially efficient even at temperatures well below 100 °C.
Results and discussion
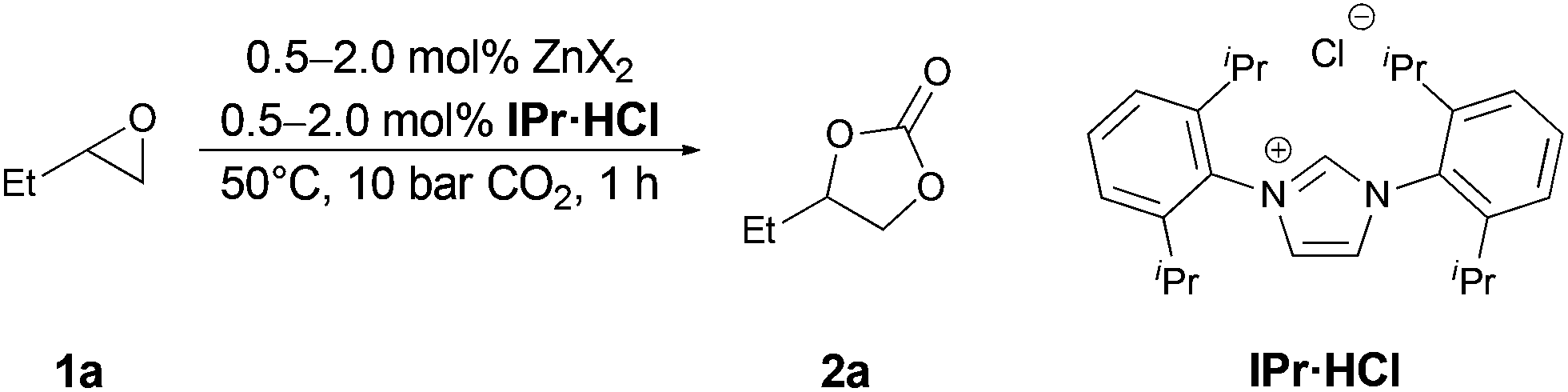
Initially we tested various simple zinc salts as potential catalysts to set a benchmark. Since our aim was to develop a system, which efficiently catalyzes the addition of CO2 under mild conditions, we set the initial screening parameters to 50 °C, 1.0 MPa CO2 pressure and 1 h reaction time under solvent free conditions (Table 1). We choose the conversion of 1,2-epoxybutane (1a) with CO2 to 1,2-butylene carbonate (2a) as test reaction. As expected without any catalyst the reaction does not proceed and no product formation was observed (entry 1). In the presence of simple zinc salts and absence of a co-catalyst no conversion was observed (except for ZnI2 see below).23 The co-catalyst 1,3-bis(2,6-diisopropylphenyl)imidazolium chloride (IPr·HCl) was also not active under these reaction conditions (entry 2). Subsequently, a combination of zinc salts and co-catalyst IPr·HCl were applied (entries 3–8). Thereby, zinc salts bearing non-nucleophilic anions showed only very low activity (entries 3–5). However, using zinc halides and IPr·HCl moderate to excellent yields were obtained (entries 6–8). The activity of the salts increased in the order Cl < Br < I, which has previously been observed for binary catalytic systems11d,18 and bifunctional catalysts.13a,14,24 Quantitative conversion of model substrate 1a was achieved with the combination of ZnI2 and IPr·HCl, which was chosen for further investigations (entry 8). Notably this was the only salt in which 2a could be detected in traces even in the absence of IPr·HCl. Employing 1.0 mol% catalyst and co-catalyst, 99% of 2a was obtained (entry 9). Further reduction of the catalyst amount to 0.5 mol% led to reduction in yield to 82% of 2a (entry 10).|
|
|||
|---|---|---|---|
| Entry | ZnX2 | ZnX2/IPr·HCl | Yield 2aa/% |
a Reaction conditions: 25 mmol epoxide 1a, 0.5–2.0 mol% catalyst and co-catalyst (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), T = 50 °C, p(CO2) = 1.0 MPa, t = 1 h. Yield determined by GC-FID with n-hexadecane as internal standard.
b Only 2.0 mol% IPr·HCl was employed.
c Yield in bracket was obtained in the absence of IPr·HCl. :1), T = 50 °C, p(CO2) = 1.0 MPa, t = 1 h. Yield determined by GC-FID with n-hexadecane as internal standard.
b Only 2.0 mol% IPr·HCl was employed.
c Yield in bracket was obtained in the absence of IPr·HCl.
|
|||
| 1 | — | — | 0 |
| 2 | —b | 2.0 mol% | 0 |
| 3 | Zn(OAc)2 | 2.0 mol% | 3 |
| 4 | Zn(BF4)2 | 2.0 mol% | 0 |
| 5 | Zn(acac)2 | 2.0 mol% | 2 |
| 6 | ZnCl2 | 2.0 mol% | 13 |
| 7 | ZnBr2 | 2.0 mol% | 70 |
| 8 | ZnI2 | 2.0 mol% | >99 (1)c |
| 9 | ZnI2 | 1.0 mol% | >99 |
| 10 | ZnI2 | 0.5 mol% | 82 |
Having identified zinc iodide as the most active zinc salt we turned our attention to investigate the influence of both the structure and counter anion of the imidazolium salt (Table 2). First we studied the impact of the anion by employing various 1-ethyl-3-methylimidazolium (EMIm) salts as co-catalysts in the test reaction (entries 1–6). As expected EMIm salts with non-nucleophilic counter anions showed only low conversions <10% (entries 1–3). However in the presence of [EMIm]Cl the desired product 2a was obtained in 75% yield (entry 4). Interestingly the respective bromide gave lower yields of only 62% of 2a (entry 5). In this series the best yield was obtained when [EMIm]I was utilized (entry 6). Subsequently we turned our attention to the impact of the imidazolium species (entries 7–8). Dimethylimidazolium chloride [DMIm]Cl gave 2a in 57% yield while 1,3-bis(2,4,6-trimethylphenyl)imidazolium chloride (IMes·HCl) gave the product in very good yield of 85% (entry 8). We also employed a thiazolium chloride, which showed only low conversion and yield, thus underlining the impact of the cationic structure (entry 9). Since iodide proved to be the most promising anion and IMes·HCl and IPr·HCl bearing the most active imidazolium structures, we decided to prepare the corresponding iodide salts IMes·HI and IPr·HI.
| Entry | Co-catalyst | Yield 2aa/% |
|---|---|---|
| a Reaction conditions: 25 mmol epoxide 1a, 0.5 mol% ZnI2, 0.5 mol% co-catalyst, T = 50 °C, p(CO2) = 1.0 MPa, t = 1 h. Yield determined by GC-FID with n-hexadecane as internal standard. | ||
| 1 | [EMIm][BF4] | 1 |
| 2 | [EMIm][PF6] | 0 |
| 3 | [EMIm][FeCl4] | 9 |
| 4 | [EMIm]Cl | 75 |
| 5 | [EMIm]Br | 62 |
| 6 | [EMIm]I | 81 |
| 7 | [DMIm]Cl | 57 |
| 8 | IMes·HCl | 85 |
| 9 |
|
4 |
Both salts can be prepared by anion exchange starting from the commercially available chlorides (Scheme 2).25 In the presence of a large excess of NaI in acetone/water the desired products IMes·HI and IPr·HI could be isolated in 51% and 61% yield, respectively. Notably, after recrystallization IMes·HI could be obtained as yellow crystals suitable for X-ray crystallographic analysis (Fig. 1).
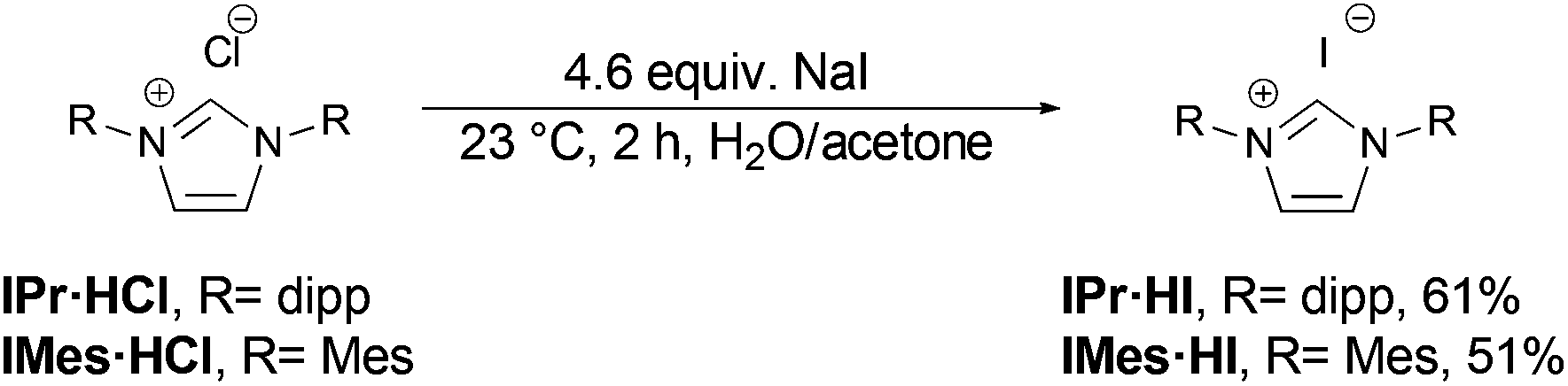 | ||
| Scheme 2 Synthesis of imidazolium iodides IPr·HI and IMes·HI from chlorides by anion exchange. | ||
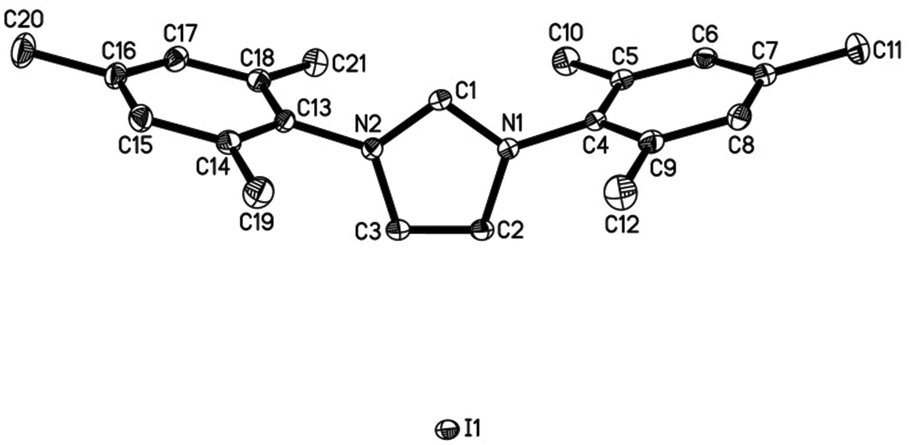 | ||
| Fig. 1 ORTEP representation of the molecular structure of IMes·HI in the crystal. Displacement ellipsoids correspond to 30% probability. Hydrogen atoms are omitted for clarity (see ESI† for crystallographic data). | ||
We employed the prepared imidazolium salts IMes·HI and IPr·HI under the standard conditions of our test reaction (Table 3, entries 1 and 2). In both cases the yields were slightly improved compared to the respective chlorides. We further evaluated the influence of catalyst concentration, reaction temperature, CO2 pressure and reaction time. Notably, the reaction can be performed even at 30 °C. Even though the amount of catalyst had to be increased to 1 mol% after 1 h, 1.0 MPa CO2 pressure, the obtained yield for 2a was 80% (entries 3 and 4). A further experiment was performed with IPr·HI at room temperature, the yield dropped significantly to 33% (entry 5). At 30 °C the extension of the reaction time to 3 h and 5 h led to product yields of 96% and >99% respectively (entries 6 and 7). The reduction of the pressure to 0.5 MPa and 0.1 MPa led to slightly reduced product yields of 97% and 83% respectively (entries 8 and 9).
| Entry | ZnI2/co-catalyst (1:1) |
Co-catalyst | T/°C | p(CO2)/MPa | t/h | Yield 2aa/% |
|---|---|---|---|---|---|---|
| a Reaction conditions: 25 mmol epoxide 1a, T = 30–50 °C, p(CO2) = 0.1–1.0 MPa, t = 1–5 h. Yield determined by GC-FID with n-hexadecane as internal standard. | ||||||
| 1 | 0.5 mol% | IMes·HI | 50 | 1.0 | 1 | 85 |
| 2 | 0.5 mol% | IPr·HI | 50 | 1.0 | 1 | 87 |
| 3 | 1.0 mol% | IMes·HI | 30 | 1.0 | 1 | 80 |
| 4 | 1.0 mol% | IPr·HI | 30 | 1.0 | 1 | 80 |
| 5 | 1.0 mol% | IPr·HI | 25 | 1.0 | 1 | 33 |
| 6 | 1.0 mol% | IPr·HI | 30 | 1.0 | 3 | 96 |
| 7 | 1.0 mol% | IPr·HI | 30 | 1.0 | 5 | >99 |
| 8 | 1.0 mol% | IPr·HI | 30 | 0.5 | 5 | 97 |
| 9 | 1.0 mol% | IPr·HI | 30 | 0.1 | 5 | 83 |
Based on the results of parameter screening we determined the standard reaction conditions for the evaluation of the substrate scope (Table 4). Initially we employed 1 mol% zinc iodide in a 1:1 ratio with IPr·HI as a co-catalyst at 30 °C, 1.0 MPa CO2 pressure for 5 h. Since some substrates gave only low to moderate yields, the catalyst amount was increased to 5 mol% which led to significantly improved yields in those cases. Alkyl functionalized cyclic carbonates 2a–2d could be obtained in yields of 83–99% (entries 1–8). Also aryl derivatives 2e and 2f could be isolated in excellent yields up to 93% (entries 9–12). Several glycidyl ethers 1g–1j could be smoothly converted leading to the respective products in yields up to 99% (entries 13–20). Interestingly the potential monomer 2k was obtained in 94% isolated yield as well as unsaturated substrate 1l was converted to 2l and isolated in 95% (entries 21–24). Other functionalized substrates bearing chloride 1m, bromide 1n and hydroxy 1o were also converted (entries 25–30). Moreover, internal epoxides 1p–1r were reacted with CO2 under more drastic reaction conditions (entries 31–34). However, due to more steric hindrance13a,14,26 significant lower yields up to 54% of their corresponding cyclic carbonates 2p–2r were determined.
|
|
||||
|---|---|---|---|---|
| Entry | ZnI2/IPr·HI (1:1) |
Product | Yield 2a/% |
|
| a Reaction conditions: epoxide 1, 1 or 5 mol% ZnI2, 1 or 5 mol% IPr·HI, T = 30 °C, p(CO2) = 1.0 MPa, t = 5 h. Isolated yield. b Yield determined by 1H NMR. c T = 75 °C. d T = 75 °C, p(CO2) = 5.0 MPa. | ||||
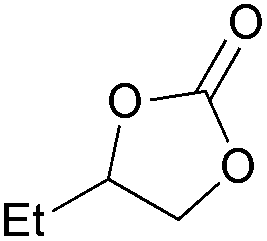
| 1 | 1 mol% |
|
2a | 99 |
| 2 | 5 mol% | — | ||
| 3 | 1 mol% |

|
2b | 94 |
| 4 | 5 mol% | — | ||
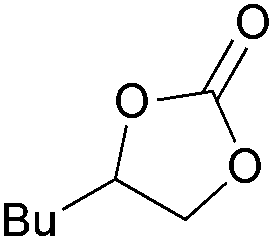
| 5 | 1 mol% |
|
2c | 99 |
| 6 | 5 mol% | — | ||
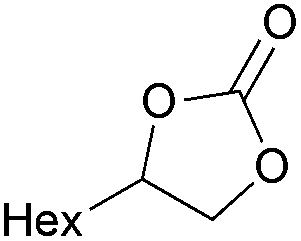
| 7 | 1 mol% |
|
2d | 6 |
| 8 | 5 mol% | 83 | ||
| 9 | 1 mol% |
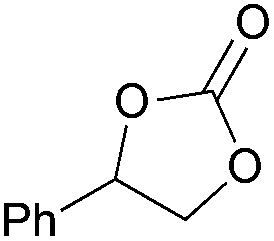
|
2e | 76b |
| 10 | 5 mol% | 93 | ||
| 11 | 1 mol% |
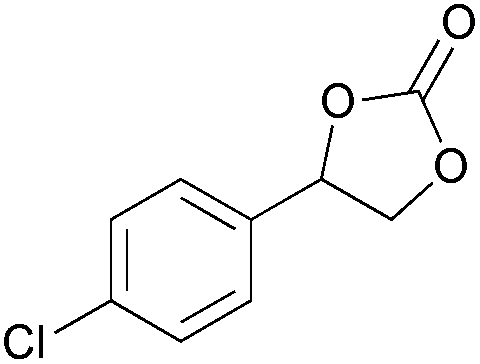
|
2f | 30b |
| 12 | 5 mol% | 87 | ||
| 13 | 1 mol% |

|
2g | 6 |
| 14 | 5 mol% | 99 | ||
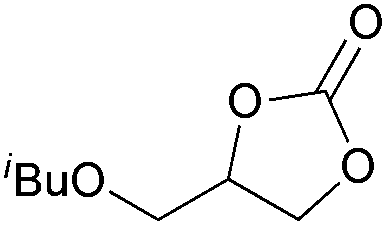
| 15 | 1 mol% |
|
2h | 9b |
| 16 | 5 mol% | 72 | ||
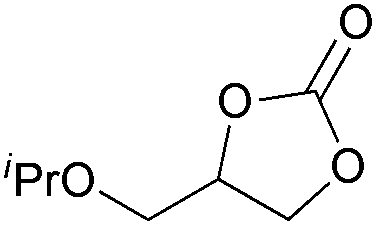
| 17 | 1 mol% |
|
2i | 14b |
| 18 | 5 mol% | 99 | ||
| 19 | 1 mol% |
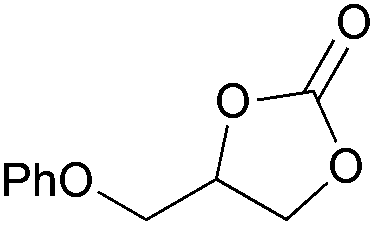
|
2j | 64b |
| 20 | 5 mol% | 85 | ||
| 21 | 1 mol% |
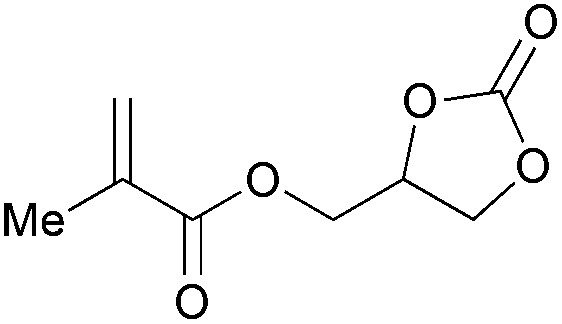
|
2k | 71b |
| 22 | 5 mol% | 94 | ||
| 23 | 1 mol% |
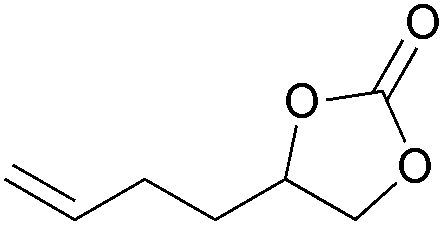
|
2l | 5b |
| 24 | 5 mol% | 95 | ||
| 25 | 1 mol% |
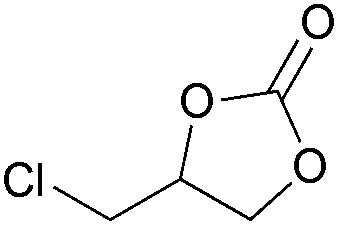
|
2m | 90 |
| 26 | 5 mol% | — | ||
| 27 | 1 mol% |
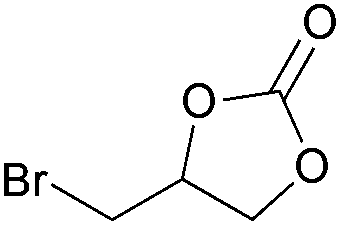
|
2n | 38 |
| 28 | 5 mol% | — | ||
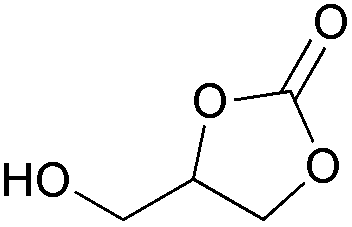
| 29 | 1 mol% |
|
2o | 43b |
| 30 | 5 mol% | 67 | ||
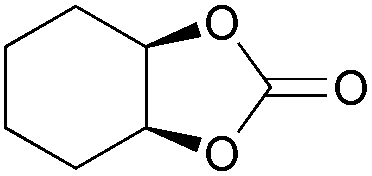
| 31 | 1 mol% |
|
2p | 18b,c |
| 32 | 1 mol% | 36d | ||
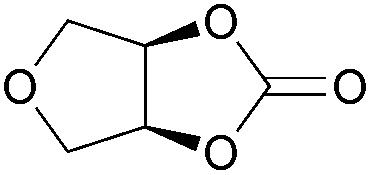
| 33 | 1 mol% |
|
2q | 8c |
| 34 | 1 mol% |
|
2r | 54d |
Proposed mechanism
We assume that Lewis acidic zinc halide coordinates to the oxygen atom to activate the epoxide. Similar activations of epoxides by Lewis acidic metal species have been reported previously.10f,11b,21b,27 The activation of the epoxide by the imidazolium cation via hydrogen bonding is also possible.9a,16a We observed a significant downfield shift of the C2 proton in the 1H NMR spectra when the epoxide was added to a mixture of ZnI2 and IPr·HI.23 Since no conversion is observed when one of the components is used alone under the reaction conditions, we assume a cooperative activation of the epoxide by the Lewis acidic zinc species and the imidazolium cation as a hydrogen donor. Subsequent ring opening by the iodide of the imidazolium salt through a nucleophilic attack on the less sterically hindered carbon atom forms an alkoxide. Insertion of carbon dioxide into the zinc–oxygen bond and subsequent ring closure yields the desired cyclic carbonate. This proposal is in accordance with the mechanisms of other zinc based binary systems (Scheme 3).9a,18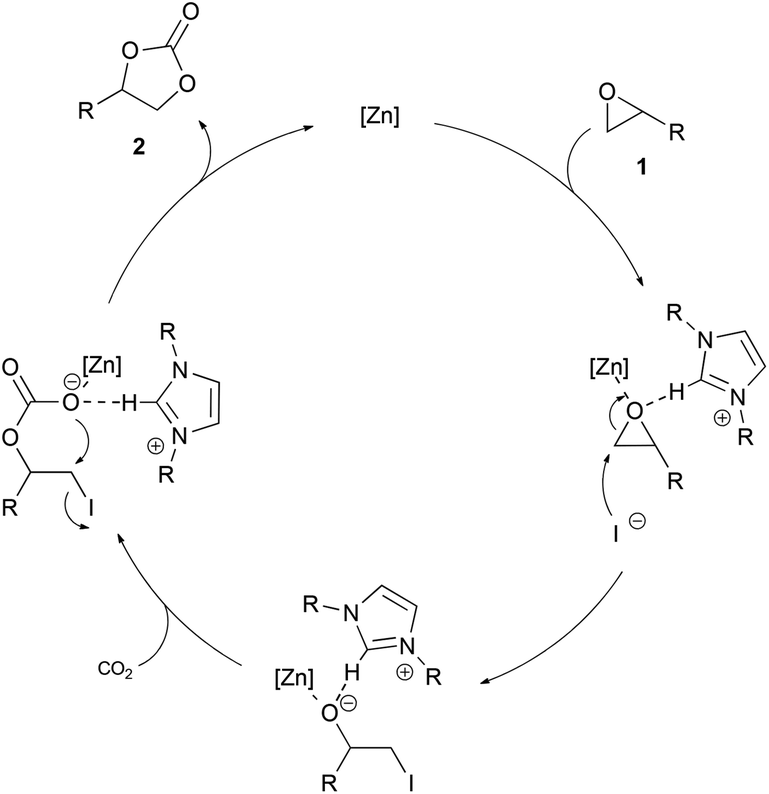 | ||
| Scheme 3 Possible reaction mechanism for the synthesis of cyclic carbonates catalyzed by ZnI2 and IPr·HI. | ||
Conclusion
We developed an efficient binary catalytic system based on zinc iodide and an imidazolium salt for the addition of CO2 to epoxides. This system allows the conversion of various epoxides into the corresponding cyclic carbonates in yields up to 99% under very mild and solvent free conditions. We showed that in this system the iodide anion is a superior nucleophile compared to the usually employed anions, bromide and chloride, respectively. Besides the anion, the structure of the cation also has a large influence on the reaction outcome. Based on those results catalytic systems based on zinc iodide which efficiently catalyze the addition of CO2 to epoxide under ambient conditions might emerge in the future.Experimental
Coupling reaction of CO2 with epoxides 1 (GP1)
A 45 mL stainless steel autoclave equipped with a magnetic stirrer was charged with zinc iodide (0.01 equiv.), IPr·HI (0.01 equiv.) and epoxide 1 (1.00 equiv.). The reactor was sealed and pressurized with 1.0 MPa of carbon dioxide until the equilibrium was reached. Subsequently the reactor was warmed to 30 °C for 5 hours. The autoclave was cooled with an ice bath below 20 °C and the CO2 was slowly liberated from the vessel. The crude reaction mixture was extracted with dichloromethane and filtered over a silica gel plug. After the removal of all volatiles under reduced pressure, the cyclic carbonates 2 were obtained.Coupling reaction of CO2 with epoxides 1 (GP2)
A 45 mL stainless steel autoclave equipped with a magnetic stirrer was charged with zinc iodide (0.05 equiv.), IPr·HI (0.05 equiv.) and epoxide 1 (1.00 equiv.). The reactor was sealed and pressurized with 1.0 MPa of carbon dioxide until the equilibrium was reached. Subsequently the reactor was warmed to 30 °C for 5 hours. The autoclave was cooled with an ice bath below 20 °C and the CO2 was slowly liberated from the vessel. The crude reaction mixture was extracted with dichloromethane and filtered over a silica gel plug. After the removal of all volatiles under reduced pressure, the cyclic carbonates 2 were obtained.IPr·HI and IMes·HI were synthesized according to known procedures.25
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) ppm.
O) ppm.
O) ppm.
O) ppm.
O) ppm.
O) ppm.
O) ppm.
O) ppm.
O) ppm.
O), 157.7 (C) ppm.
O), 166.56 (CO) ppm.
O) ppm.
O) ppm.
O) ppm.
O) ppm.
O).
O).
O).
:1, 0 °C) and dried under vacuum to yield the title compound (3.23 g, 6.24 mmol, 61%) as a colorless solid. 1H NMR (300 MHz, DMSO-d6): δ = 1.17 (d, J = 6.84 Hz, 12H), 1.27 (d, J = 6.78 Hz, 12H), 2.36 (pent, J = 6.79 Hz, 4H), 7.53–7.55 (m, 4H), 7.67–7.72 (m, 2H), 8.58 (d, J = 1.47 Hz, 2H), 10.18 (t, J = 1.47 Hz, 1H) ppm. 13C{1H} NMR (75 MHz, DMSO-d6): δ = 23.1 (4 × CH3), 24.1 (4 × CH3), 28.6 (4 × CH), 124.6 (4 × CH), 126.2 (2 × CH), 130.0 (2 × C), 131.8 (2 × CH), 139.2 (CH), 144.8 (4 × C) ppm; elemental analysis calcd (%) for C27H37IN2 (516.51) C 62.79, H 7.22, I 24.57, N 5.42; found C 62.70, H 7.30, I 24.63, N 5.56.
O) ppm.
O) ppm.
O) ppm.
O) ppm.
O) ppm.
O) ppm.
O) ppm.
O) ppm.
O) ppm.
O), 157.7 (C) ppm.
O), 166.56 (CO) ppm.
O) ppm.
O) ppm.
O) ppm.
O) ppm.
O).
O).
O).
:1, 0 °C) and dried under vacuum to yield the title compound (3.23 g, 6.24 mmol, 61%) as a colorless solid. 1H NMR (300 MHz, DMSO-d6): δ = 1.17 (d, J = 6.84 Hz, 12H), 1.27 (d, J = 6.78 Hz, 12H), 2.36 (pent, J = 6.79 Hz, 4H), 7.53–7.55 (m, 4H), 7.67–7.72 (m, 2H), 8.58 (d, J = 1.47 Hz, 2H), 10.18 (t, J = 1.47 Hz, 1H) ppm. 13C{1H} NMR (75 MHz, DMSO-d6): δ = 23.1 (4 × CH3), 24.1 (4 × CH3), 28.6 (4 × CH), 124.6 (4 × CH), 126.2 (2 × CH), 130.0 (2 × C), 131.8 (2 × CH), 139.2 (CH), 144.8 (4 × C) ppm; elemental analysis calcd (%) for C27H37IN2 (516.51) C 62.79, H 7.22, I 24.57, N 5.42; found C 62.70, H 7.30, I 24.63, N 5.56.
Acknowledgements
We wish to thank the Federal Ministry of Research and Education (BMBF) for financial support (Chemische Prozesse und stoffliche Nutzung von CO2: Technologien für Nachhaltigkeit und Klimaschutz, grant 01 RC 1004A).References
- (a) N. Yang and R. Wang, J. Cleaner Prod., 2015, 103, 784–792 CrossRef CAS; (b) R. Wennersten, Q. Sun and H. Li, J. Cleaner Prod., 2015, 103, 724–736 CrossRef CAS; (c) C. Maeda, Y. Miyazaki and T. Ema, Catal. Sci. Technol., 2014, 4, 1482–1497 RSC; (d) B. M. Bhanage and M. Arai, Transformation and Utilization of Carbon Dioxide, Springer, Berlin-Heidelberg, 2014 Search PubMed; (e) M. Aresta, A. Dibenedetto and A. Angelini, in CO2 Chemistry, ed. M. Aresta and R. V. Eldik, 2014, vol. 66, pp. 259–288 Search PubMed; (f) E. A. Quadrelli, G. Centi, J.-L. Duplan and S. Perathoner, ChemSusChem, 2011, 4, 1194–1215 CrossRef CAS PubMed; (g) G. A. Olah, G. K. S. Prakash and A. Goeppert, J. Am. Chem. Soc., 2011, 133, 12881–12898 CrossRef CAS PubMed; (h) G. Centi, G. Iaquaniello and S. Perathoner, ChemSusChem, 2011, 4, 1265–1273 CrossRef CAS PubMed.
- (a) G. Fiorani, W. S. Guo and A. W. Kleij, Green Chem., 2015, 17, 1375–1389 RSC; (b) I. Omae, Coord. Chem. Rev., 2012, 256, 1384–1405 CrossRef CAS; (c) M. Hölscher, C. Gürtler, W. Keim, T. E. Müller, M. Peters and W. Leitner, Z. Naturforsch: Chem. Sci., 2012, 67b, 961–975 Search PubMed; (d) M. Peters, B. Köhler, W. Kuckshinrichs, W. Leitner, P. Markewitz and T. E. Müller, ChemSusChem, 2011, 4, 1216–1240 CrossRef CAS PubMed; (e) M. Aresta, Carbon Dioxide as Chemical Feedstock, Wiley-VCH, Weinheim, 2010 Search PubMed; (f) T. Sakakura, J.-C. Choi and H. Yasuda, Chem. Rev., 2007, 107, 2365–2387 CrossRef CAS PubMed.
- (a) C. Martín, G. Fiorani and A. W. Kleij, ACS Catal., 2015, 5, 1353–1370 CrossRef; (b) A. Dibenedetto and A. Angelini, in Co2 Chemistry, ed. M. Aresta and R. V. Eldik, 2014, vol. 66, pp. 25–81 Search PubMed; (c) D. J. Darensbourg, in Co2 Chemistry, ed. M. Aresta and R. V. Eldik, 2014, vol. 66, pp. 1–23 Search PubMed; (d) W. Dai, S. Luo, S. Yin and C. Au, Front. Chem. Eng. China, 2010, 4, 163–171 CrossRef CAS; (e) T. Sakakura and K. Kohno, Chem. Commun., 2009, 1312–1330 RSC.
- (a) P. Neubert, S. Fuchs and A. Behr, Green Chem., 2015, 17, 4045–4052 RSC; (b) B. Schäffner, F. Schäffner, S. P. Verevkin and A. Börner, Chem. Rev., 2010, 110, 4554–4581 CrossRef PubMed; (c) J. Bayardon, J. Holz, B. Schäffner, V. Andrushko, S. Verevkin, A. Preetz and A. Börner, Angew. Chem., Int. Ed., 2007, 46, 5971–5974 CrossRef CAS PubMed.
- (a) M. Philipp, R. Bernhard, H. A. Gasteiger and B. Rieger, J. Electrochem. Soc., 2015, 162, A1319–A1326 CrossRef CAS; (b) O. Crowther, D. Keeny, D. M. Moureau, B. Meyer, M. Salomon and M. Hendrickson, J. Power Sources, 2012, 202, 347–351 CrossRef CAS; (c) R. F. Nelson and R. N. Adams, J. Electroanal. Chem., 1967, 13, 184–187 CrossRef CAS; (d) R. Jasinski, J. Electroanal. Chem., 1967, 15, 89–91 CrossRef CAS.
- M. Aresta and A. Dibenedetto, Dalton Trans., 2007, 2975–2992 RSC.
- L. Zhang, Y. Luo, Z. Hou, Z. He and W. Eli, J. Am. Oil Chem. Soc., 2014, 91, 143–150 CrossRef CAS.
- (a) G. Rokicki, P. G. Parzuchowski and M. Mazurek, Polym. Adv. Technol., 2015, 26, 707–761 CrossRef CAS; (b) S. Jalilian and H. Yeganeh, Polym. Bull., 2015, 72, 1379–1392 CrossRef CAS; (c) D. Miloslavskiy, E. Gotlib, O. Figovsky and D. Pashin, Int. Lett. Chem., Phys. Astron., 2014, 20–29 CrossRef CAS; (d) M. M. Mazurek, P. G. Parzuchowski and G. Rokicki, J. Appl. Polym. Sci., 2014, 131, 39764 CrossRef.
- (a) B.-H. Xu, J.-Q. Wang, J. Sun, Y. Huang, J.-P. Zhang, X.-P. Zhang and S.-J. Zhang, Green Chem., 2015, 17, 108–122 RSC; (b) J. W. Comerford, I. D. V. Ingram, M. North and X. Wu, Green Chem., 2015, 17, 1966–1987 RSC; (c) M. Cokoja, M. E. Wilhelm, M. H. Anthofer, W. A. Herrmann and F. E. Kühn, ChemSusChem, 2015, 8, 2436–2454 CrossRef CAS PubMed; (d) Q. He, J. W. O'Brien, K. A. Kitselman, L. E. Tompkins, G. C. T. Curtis and F. M. Kerton, Catal. Sci. Technol., 2014, 4, 1513–1528 RSC; (e) M. North, R. Pasquale and C. Young, Green Chem., 2010, 12, 1514–1539 RSC; (f) J. Sun, S.-i. Fujita and M. Arai, J. Organomet. Chem., 2005, 690, 3490–3497 CrossRef CAS.
- (a) M. Taherimehr, J. P. C. C. Sertã, A. W. Kleij, C. J. Whiteoak and P. P. Pescarmona, ChemSusChem, 2015, 8, 1034–1042 CrossRef CAS PubMed; (b) A. Chen, C. Chen, Y. Xiu, X. Liu, J. Chen, L. Guo, R. Zhang and Z. Hou, Green Chem., 2015, 17, 1842–1852 RSC; (c) M. Adolph, T. A. Zevaco, C. Altesleben, S. Staudt and E. Dinjus, J. Mol. Catal. A: Chem., 2015, 400, 104–110 CrossRef CAS; (d) R. Luo, X. Zhou, W. Zhang, Z. Liang, J. Jiang and H. Ji, Green Chem., 2014, 16, 4179–4189 RSC; (e) M. A. Fuchs, S. Staudt, C. Altesleben, O. Walter, T. A. Zevaco and E. Dinjus, Dalton Trans., 2014, 43, 2344–2347 RSC; (f) A. Monassier, V. D'Elia, M. Cokoja, H. Dong, J. D. A. Pelletier, J.-M. Basset and F. E. Kühn, ChemCatChem, 2013, 5, 1321–1324 CrossRef CAS; (g) R. Ma, L.-N. He and Y.-B. Zhou, Green Chem., 2016 10.1039/C5GC01826A.
- (a) W. Desens, C. Kohrt, M. Frank and T. Werner, ChemSusChem, 2015, 8, 3815–3822 CrossRef CAS PubMed; (b) J. Martínez, J. A. Castro-Osma, A. Earlam, C. Alonso-Moreno, A. Otero, A. Lara-Sánchez, M. North and A. Rodríguez-Diéguez, Chem. – Eur. J., 2015, 21, 9850–9862 CrossRef PubMed; (c) T. Werner and N. Tenhumberg, J. CO2 Util., 2014, 7, 39–45 CrossRef CAS; (d) K. R. Roshan, A. C. Kathalikkattil, J. Tharun, D. W. Kim, Y. S. Won and D. W. Park, Dalton Trans., 2014, 43, 2023–2031 RSC; (e) J. Song, B. Zhang, P. Zhang, J. Ma, J. Liu, H. Fan, T. Jiang and B. Han, Catal. Today, 2012, 183, 130–135 CrossRef CAS; (f) P. Ramidi, P. Munshi, Y. Gartia, S. Pulla, A. S. Biris, A. Paul and A. Ghosh, Chem. Phys. Lett., 2011, 512, 273–277 CrossRef CAS.
- (a) R. Lindner, M. L. Lejkowski, S. Lavy, P. Deglmann, K. T. Wiss, S. Zarbakhsh, L. Meyer and M. Limbach, ChemCatChem, 2014, 6, 618–625 CrossRef CAS; (b) X. Liu, C. Cao, Y. Li, P. Guan, L. Yang and Y. Shi, Synlett, 2012, 1343–1348 CAS.
- (a) H. Büttner, K. Lau, A. Spannenberg and T. Werner, ChemCatChem, 2015, 7, 459–467 CrossRef; (b) M. E. Wilhelm, M. H. Anthofer, M. Cokoja, I. I. E. Markovits, W. A. Herrmann and F. E. Kühn, ChemSusChem, 2014, 7, 1357–1360 CrossRef CAS PubMed.
- (a) H. Büttner, J. Steinbauer and T. Werner, ChemSusChem, 2015, 8, 2655–2669 CrossRef PubMed; (b) T. Werner and H. Büttner, ChemSusChem, 2014, 7, 3268–3271 CrossRef CAS PubMed.
- (a) C. Kohrt and T. Werner, ChemSusChem, 2015, 8, 2031–2034 CrossRef CAS PubMed; (b) J. Wang, J. G. Wei Yang, G. Yi and Y. Zhang, Chem. Commun., 2015, 51, 15708–15711 RSC; (c) K. Cui, Z. Liang, J. Zhang and Y. Zhang, Synth. Commun., 2014, 45, 712–723 Search PubMed; (d) W. Zhang, Q. Wang, H. Wu, P. Wu and M. He, Green Chem., 2014, 16, 4767–4774 RSC; (e) B. Song, L. Guo, R. Zhang, X. Zhao, H. Gan, C. Chen, J. Chen, W. Zhu and Z. Hou, J. CO2 Util., 2014, 6, 62–68 CrossRef CAS; (f) T.-Y. Shi, J.-Q. Wang, J. Sun, M.-H. Wang, W.-G. Cheng and S.-J. Zhang, RSC Adv., 2013, 3, 3726–3732 RSC; (g) A. J. R. Amaral, J. F. J. Coelho and A. C. Serra, Tetrahedron Lett., 2013, 54, 5518–5522 CrossRef CAS.
- (a) M. H. Anthofer, M. E. Wilhelm, M. Cokoja, I. I. E. Markovits, A. Pothig, J. Mink, W. A. Herrmann and F. E. Kuhn, Catal. Sci. Technol., 2014, 4, 1749–1758 RSC; (b) A.-L. Girard, N. Simon, M. Zanatta, S. Marmitt, P. Goncalves and J. Dupont, Green Chem., 2014, 16, 2815–2825 RSC; (c) J. Tharun, G. Mathai, R. Roshan, A. C. Kathalikkattil, K. Bomi and D.-W. Park, Phys. Chem. Chem. Phys., 2013, 15, 9029–9033 RSC.
- (a) M. S. Liu, F. X. Wang, L. Shi, L. Liang and J. M. Sun, RSC Adv., 2015, 5, 14277–14284 RSC; (b) M. Liu, B. Liu, L. Shi, F. Wang, L. Liang and J. Sun, RSC Adv., 2015, 5, 960–966 RSC; (c) M. Liu, L. Liang, T. Liang, X. Lin, L. Shi, F. Wang and J. Sun, J. Mol. Catal. A: Chem., 2015, 408, 242–249 CrossRef CAS; (d) Y.-L. Hu, M. Lu and X.-L. Yang, RSC Adv., 2015, 5, 67886–67891 RSC.
- (a) J. Palgunadi, O. S. Kwon, H. Lee, J. Y. Bae, B. S. Ahn, N.-Y. Min and H. S. Kim, Catal. Today, 2004, 98, 511–514 CrossRef CAS; (b) H. S. Kim, J. J. Kim, H. Kim and H. G. Jang, J. Catal., 2003, 220, 44–46 CrossRef CAS.
- (a) L.-F. Xiao, F.-W. Li, J.-J. Peng and C.-G. Xia, J. Mol. Catal. A: Chem., 2006, 253, 265–269 CrossRef CAS; (b) F. Li, L. Xiao, C. Xia and B. Hu, Tetrahedron Lett., 2004, 45, 8307–8310 CrossRef CAS.
- J. Sun, S.-i. Fujita, F. Zhao and M. Arai, Green Chem., 2004, 6, 613–616 RSC.
- (a) S.-i. Fujita, M. Nishiura and M. Arai, Catal. Lett., 2010, 135, 263–268 CrossRef CAS; (b) S.-S. Wu, X.-W. Zhang, W.-L. Dai, S.-F. Yin, W.-S. Li, Y.-Q. Ren and C.-T. Au, Appl. Catal., A., 2008, 341, 106–111 CrossRef CAS; (c) H. Xie, S. Li and S. Zhang, J. Mol. Catal. A: Chem., 2006, 250, 30–34 CrossRef CAS; (d) J. Sun, S.-I. Fujita, F. Zhao and M. Arai, Appl. Catal., A., 2005, 287, 221–226 CrossRef CAS.
- M. Liu, F. Wang, L. Shi, L. Liang and J. Sun, RSC Adv., 2015, 5, 14277–14284 RSC.
- See supportings.
- (a) J.-Q. Wang, W.-G. Cheng, J. Sun, T.-Y. Shi, X.-P. Zhang and S.-J. Zhang, RSC Adv., 2014, 4, 2360–2367 RSC; (b) W. Cheng, Q. Su, J. Wang, J. Sun and F. Ng, Catalysts, 2013, 3, 878–901 CrossRef; (c) J. Sun, S. Zhang, W. Cheng and J. Ren, Tetrahedron Lett., 2008, 49, 3588–3591 CrossRef CAS.
- C. Gibard, H. Ibrahim, A. Gautier and F. Cisnetti, Organometallics, 2013, 32, 4279–4283 CrossRef CAS.
- A. Decortes, M. Martinez Belmonte, J. Benet-Buchholz and A. W. Kleij, Chem. Commun., 2010, 46, 4580–4582 RSC.
- (a) C. J. Whiteoak, N. Kielland, V. Laserna, F. Castro-Gómez, E. Martin, E. C. Escudero-Adán, C. Bo and A. W. Kleij, Chem. – Eur. J., 2014, 20, 2264–2275 CrossRef CAS PubMed; (b) W. Clegg, R. W. Harrington, M. North and R. Pasquale, Chem. – Eur. J., 2010, 16, 6828–6843 CrossRef CAS PubMed; (c) F. Wang, C. Xu, Z. Li, C. Xia and J. Chen, J. Mol. Catal. A: Chem., 2014, 385, 133–140 CrossRef CAS; (d) J. K. Lee, Y. J. Kim, Y.-S. Choi, H. Lee, J. S. Lee, J. Hong, E.-K. Jeong, H. S. Kim and M. Cheong, Appl. Catal., B, 2012, 111–112, 621–627 CrossRef CAS.
- J. A. Castro-Osma, C. Alonso-Moreno, A. Lara-Sanchez, J. Martinez, M. North and A. Otero, Catal. Sci. Technol., 2014, 4, 1674–1684 CAS.
- K. M. Tomczyk, P. A. Gunka, P. G. Parzuchowski, J. Zachara and G. Rokicki, Green Chem., 2012, 14, 1749–1758 RSC.
- C. J. Whiteoak, A. H. Henseler, C. Ayats, A. W. Kleij and M. A. Pericas, Green Chem., 2014, 16, 1552–1559 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1435552. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5qo00356c |
| This journal is © the Partner Organisations 2016 |