
Cobalt(II)-catalyzed oxidative esterification of aldehydes: a cooperative effect between cobalt and iodide ions†
Ya-Fei
Guo
,
Bao-Hua
Xu
*,
Ting
Li
,
Lei
Wang
and
Suo-Jiang
Zhang
*
Beijing Key Laboratory of Ionic Liquids Clean Process, Key Laboratory of Green Process and Engineering, Institute of Process Engineering, Chinese Academy of Sciences, Beijing 100190, PR China. E-mail: bhxu@ipe.ac.cn; sjzhang@ipe.ac.cn
First published on 16th November 2015
Abstract
The efficient cobalt(II) catalyzed oxidative alkoxylation of aldehydes, a method directly leading to the corresponding esters, is presented. Mechanism studies provide extensive insights into the cobalt mediated decomposition of TBHP in the presence of the iodide ion. The in situ generated hypoiodites (IO−/IO2−) mainly from conversion of I3− to IO3− account for a cooperative effect with the oxygen centered radical species to make the important hydrogen abstraction of hemiacetal intermediates more selective, thereby offering high efficiency.
Introduction
Developing cobalt catalysts for the selective functionalization of C–H bonds is a conceptually and economically attractive strategy that overcomes the need for precious metals and enables new transformations.1 Cross-coupling reactions by low-valent cobalt have emerged as a versatile approach.2–6 However, the requisite reductive conditions limit the functional group tolerance.4–6 As an alternative strategy, the high-valent-cobalt-catalyzed coupling reaction is attracting increasing attention and was developed very recently.7–9 For instance, Co(II)-catalyzed C–H bond activation in the presence of an oxidant has been applied in the construction of various C–O, C–N and C–C bonds.8,9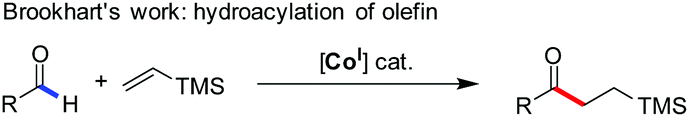 | (1) |
 | (2) |
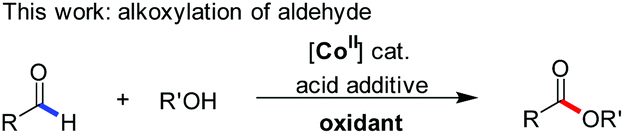 | (3) |
 | (4) |
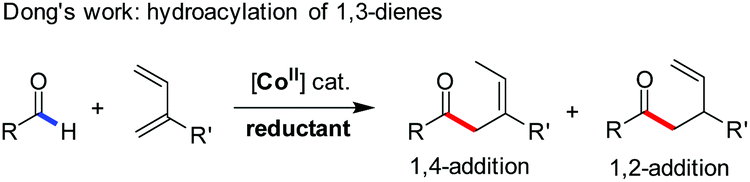
Despite this significant progress, only two examples of Co(I)-catalyzed functionalization of the formyl C–H bond in aldehydes have been reported (eqn (1) and (2)). These include Brookhart's oxidative C–H activation3 and Dong's oxidative cyclization,6 respectively. Interestingly, the unusual β-hydride elimination1 occurred on the cyclic [Co(III)–O–CH] intermediate in the latter case, probably due to the ring tension thereof. Independent of this, the oxidative strategy by using an external oxidant, albeit representing an efficient mode of reactivity towards aldehydes,10 appears reluctant in the desired cross-coupling with other organic substrates. Therein, the dehydrogenative cross-coupling of aldehydes and alkanols (eqn (3)) is more challenging because of the fact that alkanols are easily transformed to the corresponding aldehydes (further to acids) or ketones through radical mechanisms.11 Even the intermediate hemiacetal–metal complexes were formed before the undesired decomposition of each substrate, the subsequent β-hydrogen elimination leading to esters might be a limitation in the case of cobalt1 compared with palladium or nickel catalysis.12 Therefore, cobalt-catalyzed direct oxidative esterification of aldehydes with alcohols warrants further study.
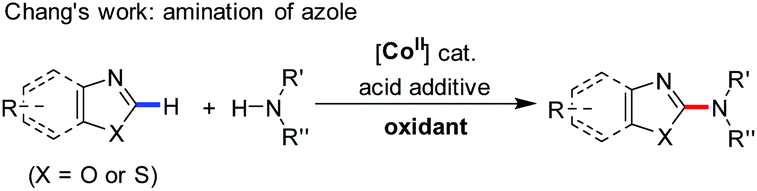
Previous studies demonstrated that organic radical species, generated from the reactions of some transition-metal complexes or iodides with tert-butyl hydroperoxide (TBHP), exhibit high reactivity as oxidizing agents in the oxidative coupling of aldehydes/alcohols.13 Chang and coworkers once reported Co(II)/T-HYDRO catalyzed dehydrogenative C–N cross-coupling in the presence of AcOH (eqn (4), T-HYDRO is the trademark name for 70 wt% tert-butyl hydroperoxide solution in water).9b Nucleophilic attack of amines was found to be more facile by using an acid additive. Noteworthily, subtle differences in the ligands of cobalt complexes were known to result in extreme differences in the catalytic cycles of peroxide decomposition,14 thereby tuning the efficiency of hydrogen abstraction. Based on these results, we speculated that certain Co(II) species would catalyze the oxidative esterification of aldehydes when they are used in combination with TBHP and proper acids. Herein, we disclose the first CoI2-catalyzed direct alkoxylation of aldehydes (eqn (3)). Interestingly, investigations attribute the high efficiency to a cooperative effect between the iodide chelating ligand and the cobalt ion in the catalytic cycle.
Results and discussion
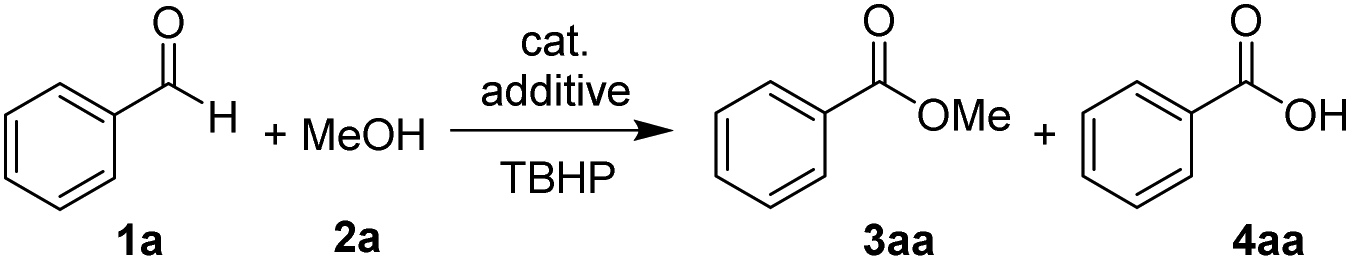
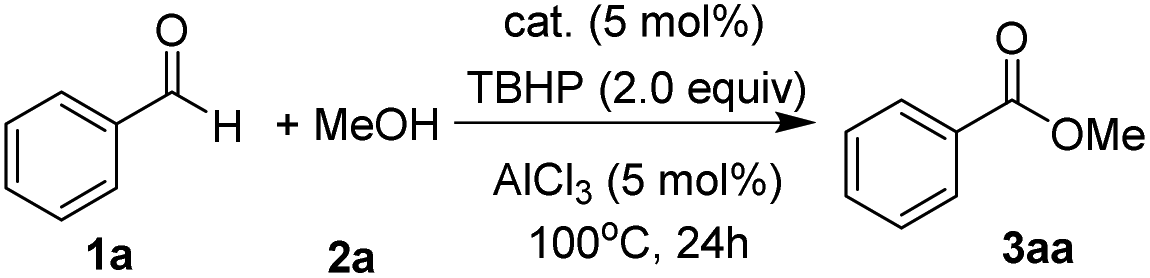
Our study started with developing a Co(II)/TBHP system in the oxidative coupling of benzaldehyde (1a) and MeOH (2a). As expected, the choice of acid additives and their amount employed turned out to be crucial for the selectivity (Table 1, entries 1–9). The Brønsted acid AcOH tends to form an undesired carboxylic acid (entry 1), the origin of which will be discussed later in the paper. In contrast, the Lewis acid AlCl3 was the most effective in the desired product ester formation among the various acid co-catalysts screened (entries 2–5), although the mass balance still consisted of a part of carboxylic acid (4aa). Moreover, cobalt species other than CoI2 exhibited reduced catalytic activity (entries 9–13). And the ester yields were not improved by either decreasing or increasing the equivalent of MeOH (entries 8 and 9) and TBHP (entries 14 and 15). Under the optimized conditions, unsubstituted benzaldehyde (1a) smoothly reacted with MeOH (2a) to provide methyl carboxylate 3aa in a high yield of 94% (entry 13).|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Cat. | 2a (equiv.) | Additive | Conv. (%) | Yieldb (%) | |
| 3aa | 4aa | |||||
| a General conditions: 1a (1 mmol), cat. (5 mol%), additive (5 mol%), TBHP (2 equiv.), 100 °C, 24 h. b Yields determined by GC with biphenyl as the internal standard. c AlCl3 (10 mol%). d TBHP: 1.5 equiv. e TBHP: 4.0 equiv. | ||||||
| 1 | Co(OAc)2·4H2O | 8.0 | HOAc | 100 | 15 | 84 |
| 2 | Co(OAc)2·4H2O | 8.0 | InBr3 | 95 | 57 | 35 |
| 3 | Co(OAc)2·4H2O | 8.0 | BF3·Et2O | 78 | 41 | 31 |
| 4 | Co(OAc)2·4H2O | 8.0 | In(OTf)3 | 62 | 39 | 20 |
| 5 | Co(OAc)2·4H2O | 8.0 | AlCl3 | 98 | 59 | 38 |
| 6 | Co(OAc)2·4H2O | 8.0 | — | 97 | 37 | 58 |
| 7c | Co(OAc)2·4H2O | 8.0 | AlCl3 | 75 | 29 | 40 |
| 8 | Co(OAc)2·4H2O | 4.0 | AlCl3 | 100 | 42 | 53 |
| 9 | Co(OAc)2·4H2O | 16.0 | AlCl3 | 87 | 55 | 30 |
| 10 | CoCl2 | 8.0 | AlCl3 | 88 | 64 | 23 |
| 11 | Co(acac)2 | 8.0 | AlCl3 | 81 | 60 | 15 |
| 12 | Co(OAc)2 | 8.0 | AlCl3 | 95 | 62 | 31 |
| 13 | CoI2 | 8.0 | AlCl3 | 98 | 94 | 3 |
| 14d | CoI2 | 8.0 | AlCl3 | 89 | 83 | 1 |
| 15e | CoI2 | 8.0 | AlCl3 | 100 | 52 | 44 |
To explore the substrate scope, we examined a range of aldehydes (1) in the coupling with MeOH (2a) under the optimized reaction conditions (Table 2, entries 1–16). It was observed that electronic variation of the substituents at the para- and meta-positions of benzaldehyde did not significantly affect the reaction efficiency. The corresponding methyl carboxylate products of benzaldehyde substituted with a methoxy, nitro, and methyl group were obtained in satisfactory yields (entries 2–8). Benzaldehyde bearing electron-donating groups such as methoxy at the ortho-position could also be employed as facile substrates that provided the corresponding ester in a high yield (entry 10). However, the one with electron-withdrawing groups at the same position such as chloride performed poorly in conversion (entry 11). Not only methyl aromatic carboxylates but also methyl aliphatic carboxylates were readily obtained in acceptable yields (entries 12 and 13). In addition, various heteroaromatic aldehydes were tolerated in such a catalytic system and afforded their methyl carboxylates in moderate yields (entries 14–16). We next examined the scope of alcohol reactant in the cobalt-catalyzed oxidative esterification of aldehydes (Table 2, entries 17–20). In the case of benzaldehyde (1a), the desired alkoxylate products were obtained in moderate yields when branched alcohols were employed instead of linear ones.
|
|
||||
|---|---|---|---|---|
| Entry | Substrate 1 (R1) | Substrate 2 (R2) | Conv. (%) | Product 3 (yieldb (%)) |
| a General conditions: 1 (1 mmol), 2 (8 mmol), cat (5 mol%), AlCl3 (5 mol%), TBHP (2.0 mmol), 100 °C, 24 h. b Isolated yields. c Yields determined by GC-MS. | ||||
| 1 | 1a (Ph) | 2a (Me) | 97 | 3aa (94) |
| 2 | 1b (4-NO2C6H4) | 2a (Me) | 100 | 3ba (80) |
| 3 | 1c (4-OMeC6H4) | 2a (Me) | 100 | 3ca (94) |
| 4 | 1d (4-MeC6H4) | 2a (Me) | 100 | 3da (89) |
| 5 | 1e (4-ClC6H4) | 2a (Me) | 96 | 3ea (88) |
| 6 | 1f (3-NO2C6H4) | 2a (Me) | 95 | 3fa (92) |
| 7c | 1g (3-OMeC6H4) | 2a (Me) | 100 | 3ga (92) |
| 8 | 1h (3-MeC6H4) | 2a (Me) | 100 | 3ha (93) |
| 9 | 1i (3-ClC6H4) | 2a (Me) | 100 | 3ia (94) |
| 10 | 1j (2-OMeC6H4) | 2a (Me) | 89 | 3ja (85) |
| 11 | 1k (2-ClC6H4) | 2a (Me) | 54 | 3ka (49) |
| 12 | 1l (n-C6H13) | 2a (Me) | 97 | 3la (65)c |
| 13 | 1m (Ph(CH2)2) | 2a (Me) | 95 | 3ma (72) |
| 14 | 1n (4-pyridine) | 2a (Me) | 93 | 3na (75) |
| 15 | 1o (2-furyl) | 2a (Me) | 91 | 3oa (66) |
| 16 | 1p (2-thiophene) | 2a (Me) | 97 | 3pa (69) |
| 17 | 1a (Ph) | 2b (n-Et) | 98 | 3ab (96) |
| 18 | 1a (Ph) | 2c (n-Bu) | 94 | 3ac (94) |
| 19 | 1a (Ph) | 2d (i-Pr) | 90 | 3ad (73) |
| 20 | 1a (Ph) | 2e (t-Bu) | 100 | 3ae (58) |
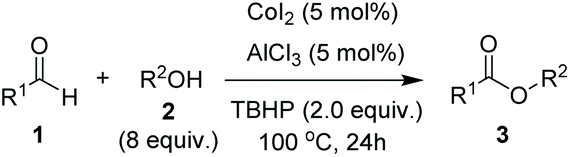
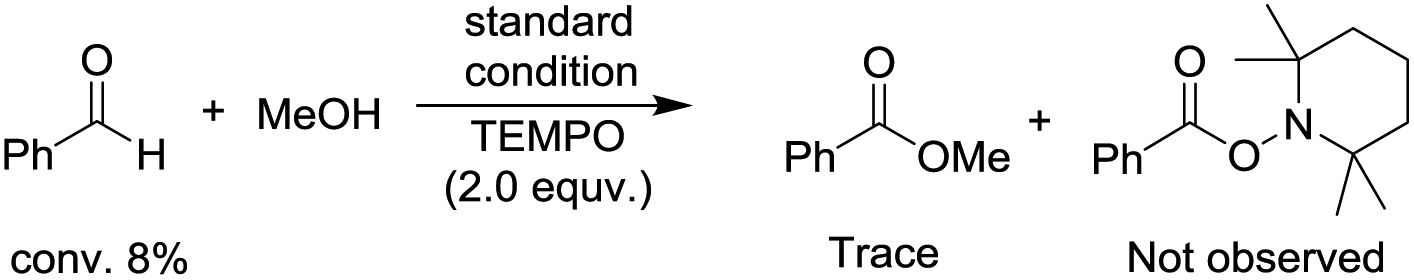
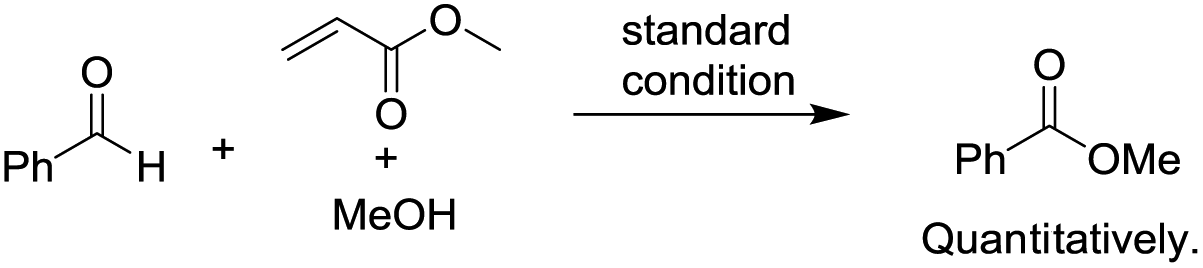
To begin investigating the mechanism of this Co-catalyzed oxidative coupling of alcohols with aldehydes, the single electron transfer (SET) process was considered. The formation of ester was completely suppressed when two equivalents of TEMPO were introduced into the reaction under standard conditions, but no TEMPO adduct of acyl radicals was obtained (eqn (5)). Importantly, cross-coupling did not occur between the aldehyde and the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond in the presence of 16 equiv. of methyl acrylate (eqn (6)), thus disfavoring an acyl radical pathway.15 Moreover, it was also demonstrated that the carboxylic acid (4aa) was formed as a side product rather than an intermediate in the oxidative esterification since only traces of the corresponding methyl carboxylate (3aa, conversion: 10%, yield: 8%) were obtained under standard conditions by reacting 4aa with MeOH (8 equiv.). Combined with the fact that the reaction is accelerated by a strong-Lewis acid, these results indicate that the reaction may proceed via a hemiacetal13a pathway.
C bond in the presence of 16 equiv. of methyl acrylate (eqn (6)), thus disfavoring an acyl radical pathway.15 Moreover, it was also demonstrated that the carboxylic acid (4aa) was formed as a side product rather than an intermediate in the oxidative esterification since only traces of the corresponding methyl carboxylate (3aa, conversion: 10%, yield: 8%) were obtained under standard conditions by reacting 4aa with MeOH (8 equiv.). Combined with the fact that the reaction is accelerated by a strong-Lewis acid, these results indicate that the reaction may proceed via a hemiacetal13a pathway.
 | (5) |
 | (6) |
Notably, some alkali16 and organic iodides17 readily react with TBHP. As an alternative to the conventional understanding, pioneering work reported by Ishihara disclosed that the in situ generated hypoiodites (IO−/IO2−) are the catalytically active species for the oxidative carbon–oxygen cross-coupling.17a Encouraged by this discovery, many achievements on the new bond formation have been obtained recently by employing the iodide/TBHP oxidation system.17b Interestingly, transition-metal catalysis in the presence of iodide either as a chelating ligand or additive often shows distinguished reactivity from the others.18 Although it was ever interpreted as a result of the iodine-assisted effect,18h,i the reaction details were unclear. Indeed, in our study, both n-Bu4NI and KI facilitated this transformation well, albeit providing different yields with respect to specific substituents (Table S1†). Thus, the uncertainty of whether Co(II) was involved in the catalytic cycle arose.
To address this issue, we performed various control experiments and spectroscopic analysis. A cyclic voltammogram (CV) obtained from an acetonitrile (CH3CN) solution of CoI2 (1 mM) revealed the presence of two reversible signals at E1/2 −0.07 V and E1/2 0.32 V (vs. Ag+/Ag) corresponding to 3I−/I3− and I3−/3/2I2, respectively, and a broad irreversible Co2+/Co3+ wave at a relatively higher positive Eox ∼ 1.80 V (vs. Ag+/Ag) (Fig. 1). These iodide oxidative features are nearly the same as those observed with a solution containing non-transition-metal iodides (NaI and n-Bu4NI, Fig. S2†), however, the oxidative potential of the Co2+/Co3+ couple is much higher compared with that detected for the chlorine-ligated analogue (CoCl2). These CV data indicated that the iodide chelating ligand is relatively more sensitive to oxidants other than the Co(II) ion, on the other hand, Co(III)I2 is capable of oxidizing I− and I3− ions if formed.
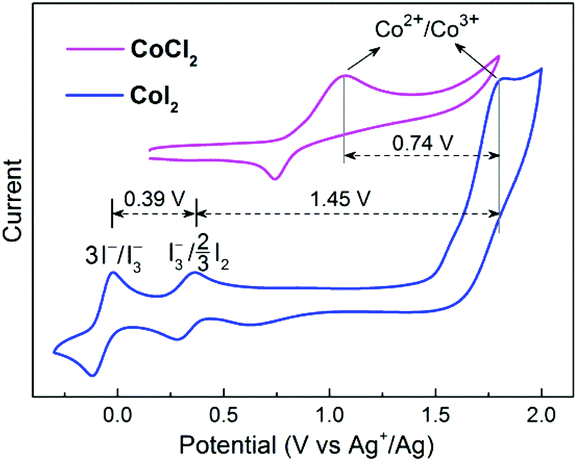 | ||
| Fig. 1 Cyclic voltammograms of 1 mM CoI2 and CoCl2. Conditions: 0.1 M LiClO4 in CH3CN, under Ar, 50 mV s−1 scan rate. | ||
The lack of reactivity between halogen-ligated Co(II) and TBHP was subsequently demonstrated by UV-visible spectroscopic study, indicating oxidation did not occur in the reaction with CoCl2 at 100 °C for 30 min (Fig. 2A). But it readily proceeded in the case of CoI2 even at lower temperature. Representative spectra were obtained during oxidation of CoI2 in CH3CN at 60 °C (Fig. 2B). The absorption band of the I− ion at 247 nm decreased with time. Instead, the transient iodide species referenced at 289 nm and 362 nm19 emerged and reached their potential concentration limit within 5 min. Importantly, the absorption band at a range from 600 nm to 800 nm typically assigned to tetrahedral complexes of halogenated Co(II)20 deceased sharply, coinciding with a broad signal appeared at around 650 nm. Moreover, the obtained spectrum was also different from Co(II)I2 plus I2 (Fig. S5†). The charge variation on the cobalt ion was, therefore, most likely involved.
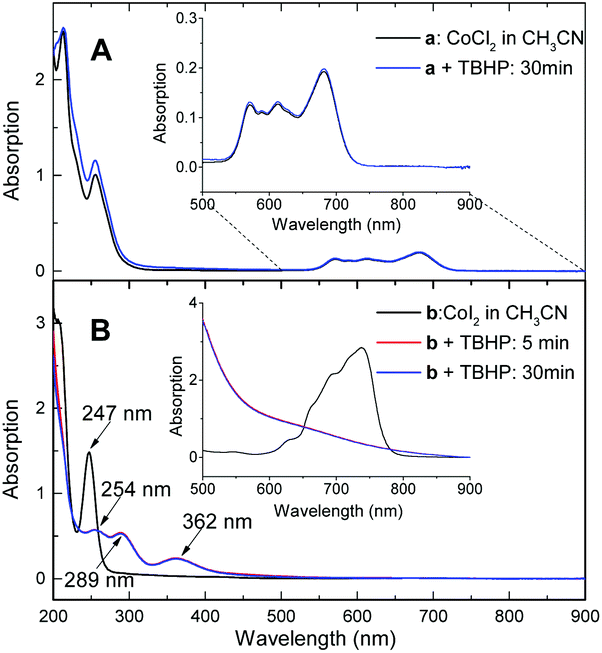 | ||
Fig. 2 UV-visible spectra acquired from monitoring the reaction time courses of the oxidation of CoI2 and CoCl2 by TBHP ((A) 0.05 mM CoCl2, CoCl2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :TBHP = 1:26, reaction at 100 °C; (B) 0.05 mM CoI2, CoI2:TBHP = 1:26, reaction at 60 °C; insert: CoI2: 4.0 mM, CoI2:TBHP = 1:7, reaction at 25 °C.). :TBHP = 1:26, reaction at 100 °C; (B) 0.05 mM CoI2, CoI2:TBHP = 1:26, reaction at 60 °C; insert: CoI2: 4.0 mM, CoI2:TBHP = 1:7, reaction at 25 °C.). | ||
Co2p XPS (X-ray photoelectron spectroscopy) spectra were then acquired for the reaction residue of CoI2 and TBHP (20 equiv.) to elucidate the electronic state variation of the cobalt ion during the catalytic cycle. Upon reaction at 60 °C for 6 h, the sample showed a Co2p spectrum of the main peak at 781.0 eV, integrating about 49% of the overall signal area, and minor satellites at 783.5, 787.0 and 790.5 eV, with ΔESO (spin-orbital splitting) contributions of 15.4 eV (Fig. 3A). The XPS Co2p spectral features of CoI2 (Fig. 3B), on the other hand, showed a higher ΔESO (16.0 eV), with high intensity satellites, as expected for 3d ions with unquenched orbital momentum.21 This difference in ΔESO of the Co2p spectrum suggested the presence of ls-Co(III) (ls = low spin) in the oxidized sample.22
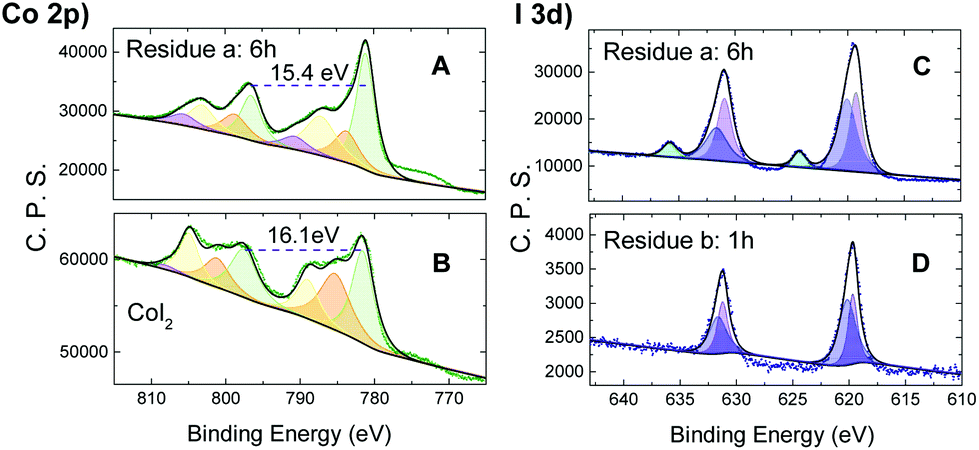 | ||
| Fig. 3 The Co2p (left) and the I3d (right) XPS spectrum of the catalyst residue and raw CoI2 along with deconvolution of the photopeaks and the corresponding best fitting lines (reaction conditions for residue a: 0.1 M CoI2, 2.0 M TBHP, CH3CN as solvent, 6 h at 60 °C; reaction conditions for residue a: 2.6 × 10−3 M; CoI2:TBHP = 1:20, CH3CN as solvent, 1 h at 60 °C). | ||
In addition, the corresponding I3d XPS spectral study of the residue (Fig. 3C and D) showed two separated bands at both I 3d5/2 and 3d3/2 levels. The narrow and asymmetric band at the relatively lower level was deconvoluted into two peaks of intensity ratio ca. 1.4:1 with different binding energies, containing values assigned to I2 at 620.0 eV (ref. 23) and I− at 619.2 eV. Note that the I3− ion is a resonating system consisting of the I− ion and I2 molecule and the 3d core-level spectra for some I3− show a band composed of 2:1 double-component peaks. This detected iodine species was, therefore, attributed to a mixture of I3− and unconverted I− ions. Independent of this, the higher energy iodate, IO3−, was directly formed (Fig. 3C) as illustrated by the I 3d5/2 band typically located at 624.3 eV.23 In comparison, this band for IO3− was not detected in the residue of n-Bu4NI with TBHP under identical conditions (Fig. S6†). These observations can be interpreted as a result of an oxidative conversion from I− to IO3−via I3−, leading to a mixture with different ratios depending on the conditions. It's understandable that no iodate emerged in the residue (Fig. 3D) after the reaction of the substrates of reduced concentration (CoI2: 2.6 × 10−3 M; CoI2:TBHP = 1:20) for a shorter time (1 h). Noteworthily, the rapid generation and subsequent disproportionation of the transient hypoiodites (IO−/IO2−) account for the transformation either from I− to I3− or the subsequent from I3− to IO3−.17a,19 Thus, albeit undetected, hypoiodites were poised to be in the catalytic cycle.
Kinetic evidence that CoI2 is quite different from KI and n-Bu4NI as the catalyst in the decomposition of TBHP was obtained by comparing their specific rate on iodide oxidation. Indicated by the literature and our cross experiments (Fig. 3, S3 and S4†), the oxidative conversion from I− to I3− was supposed to occur in each case at the initial stage, especially with employing dilute substrates. These oxidations were monitored at 362 nm by the UV-visible spectrum which was assigned to the in situ generated transient hypoiodites (IO− and IO2−) and relatively stable I3−.19
It was found that the cobalt mediated iodide oxidation proceeds much faster than the other two non-transitional metal cases from the initial slope of the plots (Fig. 4A). Additionally, the saturated capacity of transient iodides at equilibrium in the case of either KI or n-Bu4NI was not significantly affected (Fig. 4B). In contrast, the oxidized species of iodide was found to be quickly predominant in the solution of CoI2 at a lower equivalent of TBHP and reached the potential limit with around only two equivalents to [I−], a 20-fold lower present in the catalytic reaction. Specifically, with increased concentration of TBHP to eight equivalents, the absorption at 362 nm decreased sharply (Fig. 4B). It's attributable to the second oxidative conversion from I3− to IO3−. Thus, the iodide chelating ligand in CoI2 was prone to be quickly converted to an inert I3− ion and the hypoiodites thereof formed were not kinetically competent to serve as the active site unless the subsequent generation occurred during the deep oxidation to IO3−.
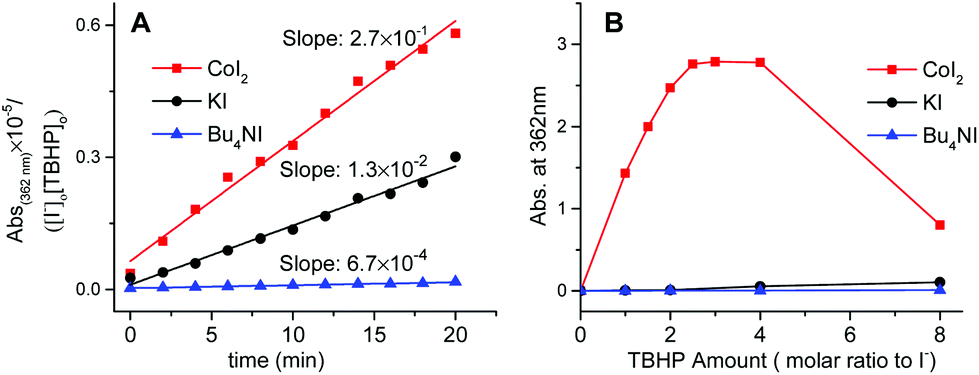 | ||
| Fig. 4 (A) Plot of the initial slope acquired from monitoring the reaction time course of the oxidation of iodides by TBHP at 25 °C. (B) Plot of the potential amount limit of IO−/I3− produced by the reaction of iodides ([I−]: 0.4 mM) with various concentrations of TBHP at 60 °C. | ||
Collectively, the reaction of CoI2 with TBHP was poised to start with oxidative transformation of the iodide chelating ligand to the active intermediate, while the charge variation on cobalt remained at this stage (Scheme 1). It seems likely that the active Co(II)OH species were thereof formed and initiated another pathway to decompose TBHP. In this emerged hydroxyl system, the cobalt is cycling between oxidation states +II and +III. And as soon as the Co(III) species were formed, they reacted as one-electron oxidant to accelerate the transformation speed of I− to I3− and thus making the second oxidative conversion to IO3− possible.
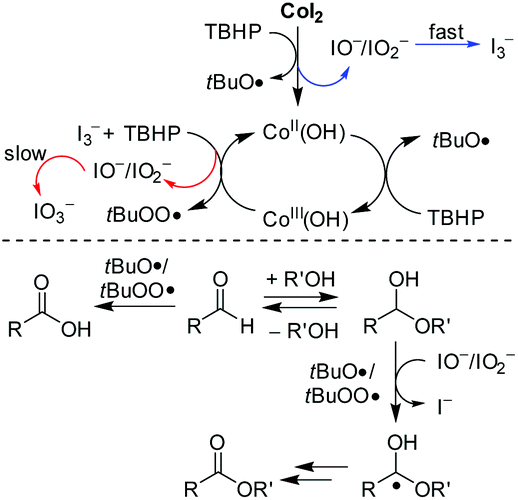 | ||
| Scheme 1 Proposed mechanism for CoI2 catalyzed oxidative esterification of aldehydes. | ||
The subsequent control experiment by using Co(OH)2 instead of CoI2 as the catalyst proceeded in a reduced efficiency leading to 3aa in a yield of 70% (Table 3, entry 2). We, therefore, could not completely rule out a catalytic role of the iodide. And the in situ generated hypoiodites account for the catalytically active species making the important hydrogen abstraction from hemiacetals more selective (Scheme 1), however, they are probably mainly from the second oxidative conversion from I3− to IO3− under Co(II) mediation. In consistence with this assumption, the reaction by using a combination of Co(OH)2 with either n-Bu4NI or n-Bu4NI3 as the catalyst provided a comparable yield as CoI2 (entries 5 and 6). But the performance was not improved upon direct addition of either molecular iodine or iodate additive (entries 3 and 4). In these cases without efficient iodide additives, carboxylic acid 4aa was formed as the main side product (entries 2, 3 and 4).
|
|
|||
|---|---|---|---|
| Entry | Cat. | Additive (mol%) | Yieldb (%) |
| a General conditions: 1a (1 mmol). b Yields determined by GC with biphenyl as the internal standard. c Yield of 4aa: 2: 29%; 3: 31%; 4: 30%. | |||
| 1 | CoI2 | — | 94 |
| 2c | Co(OH)2 | — | 70 |
| 3c | Co(OH)2 | I2 (5) | 68 |
| 4c | Co(OH)2 | AgIO3 (10) | 66 |
| 5 | Co(OH)2 | n-Bu4NI3 (10) | 90 |
| 6 | Co(OH)2 | n-Bu4NI (10) | 89 |
Conclusions
The present study reports a practical CoI2 catalyzed system for the oxidative C–O cross-coupling of various aldehydes and alcohols. It is the first homogeneous inexpensive cobalt catalyst system for oxidative esterification of aldehydes. The methods are compatible with substrates bearing a variety of functional groups, including electron-poor or rich aromatic aldehydes, as well as aliphatic and heterocyclic aldehydes. And, the methods exhibit highly favorable practical characteristics: a solvent free system, a rather low alcohol/aldehyde ratio and the catalyst components are inexpensive and commercially available reagents. More importantly, this study has provided extensive insights into the cobalt mediated decomposition of TBHP in the presence of the iodide ion. The in situ generated hypoiodites mainly from I3− to IO3− account for a cooperative effect with oxygen centered radical species to offer the highly efficient conversion.Acknowledgements
The authors are grateful to the National Key Basic Research Program of China (or 973 Program, No. 2015CB251401), the National Natural Science Foundation of China (No. 21476240), the Special Funds of the National Natural Science Foundation of China (No. 21127011) and the CAS 100-Talent Program (2014), and the Strategic Priority Research Program of the Chinese Academy of Sciences (XDA07070600) for financial support.References
- For recent reviews on Co-catalyzed C–H bond functionalization, see: (a) B. Su, Z.-C. Cao and Z.-J. Shi, Acc. Chem. Res., 2015, 48, 886 CrossRef CAS PubMed; (b) O. Daugulis, J. Roane and L. D. Tran, Acc. Chem. Res., 2015, 48, 1053 CrossRef CAS PubMed; (c) K. Gao and N. Yoshikai, Acc. Chem. Res., 2014, 47, 1208 CrossRef CAS PubMed; (d) L. Ackermann, J. Org. Chem., 2014, 79, 8948 CrossRef CAS PubMed; (e) A. A. Kulkarni and O. Daugulis, Synthesis, 2009, 4087 CAS. For other selected reviews on Co-catalyzed cross-coupling reactions, see: (f) G. Cahiez and A. Moyeux, Chem. Rev., 2010, 110, 1435 CrossRef CAS PubMed; (g) C. Gosmini, J. M. Begouin and A. Moncomble, Chem. Commun., 2008, 3221 RSC.
- J. V. Obligacion, S. P. Semproni and P. J. Chirik, J. Am. Chem. Soc., 2014, 136, 4133 CrossRef CAS PubMed.
- (a) C. P. Lenges, M. Brookhart and P. S. White, Angew. Chem., Int. Ed., 1999, 38, 552 CrossRef CAS; (b) M. Brookhart, B. E. Grant, C. P. Lenges, M. H. Prosenc and P. S. White, Angew. Chem., Int. Ed., 2000, 39, 1676 CrossRef CAS; (c) C. P. Lenges and M. Brookhart, J. Am. Chem. Soc., 1997, 119, 3165 CrossRef CAS; (d) C. P. Lenges, P. S. White and M. Brookhart, J. Am. Chem. Soc., 1998, 120, 6965 CrossRef CAS.
- (a) Z. H. Ding and N. Yoshikai, Angew. Chem., Int. Ed., 2013, 52, 8574 CrossRef CAS PubMed; (b) K. Gao and N. Yoshikai, J. Am. Chem. Soc., 2013, 135, 9279 CrossRef CAS PubMed; (c) P.-S. Lee, T. Fujita and N. Yoshikai, J. Am. Chem. Soc., 2011, 133, 17283 CrossRef CAS PubMed; (d) Q. Chen, L. Ilies and E. Nakamura, J. Am. Chem. Soc., 2011, 133, 428 CrossRef CAS PubMed; (e) L. Ilies, Q. Chen, X. Zeng and E. Nakamura, J. Am. Chem. Soc., 2011, 133, 5221 CrossRef CAS PubMed; (f) B. Li, Z.-H. Wu, Y.-F. Gu, C.-L. Sun, B.-Q. Wang and Z.-J. Shi, Angew. Chem., Int. Ed., 2011, 50, 1109 CrossRef CAS PubMed; (g) H. Li, C.-L. Sun, M. Yu, D.-G. Yu, B.-J. Li and Z.-J. Shi, Chem. – Eur. J., 2011, 17, 3593 CrossRef CAS PubMed; (h) W. Liu, H. Cao, J. Xin, L. Jin and A. Lei, Chem. – Eur. J., 2011, 17, 3588 CrossRef CAS PubMed; (i) W. Song and L. Ackermann, Angew. Chem., Int. Ed., 2012, 51, 8251 CrossRef CAS PubMed; (j) B. Punji, W. Song, G. A. Shevchenko and L. Ackermann, Chem. – Eur. J., 2013, 19, 10605 CrossRef CAS PubMed.
- T. Andou, Y. Saga, H. Komai, S. Matsunaga and M. Kanai, Angew. Chem., Int. Ed., 2013, 52, 3213 CrossRef CAS PubMed.
- Catalysis in the presence of a reductant: Q.-A. Chen, D. K. Kim and V. M. Dong, J. Am. Chem. Soc., 2014, 136, 3772 CrossRef CAS PubMed.
- (a) T. M. Figg, S. Park, J. Park, S. Chang and D. G. Musaev, Organometallics, 2014, 33, 4076 CrossRef CAS; (b) D. Zhao, J. H. Kim, L. Stegemann, C. A. Strassert and F. Glorius, Angew. Chem., Int. Ed., 2015, 54, 4508 CrossRef CAS PubMed; (c) H. Lu, C. Li, H. Jiang, C. L. Lizardi and X. P. Zhang, Angew. Chem., Int. Ed., 2014, 53, 7028 CrossRef CAS.
- (a) L.-B. Zhang, X.-Q. Hao, S.-K. Zhang, Z.-J. Liu, X.-X. Zheng, J.-F. Gong, J.-L. Niu and M.-P. Song, Angew. Chem., Int. Ed., 2015, 54, 272 CrossRef CAS PubMed; (b) X. Wu, K. Yang, Y. Zhao, H. Sun, G. Li and H. Ge, Nat. Commun., 2015, 51, 6462 CrossRef PubMed; (c) L. Grigorjeva and O. Daugulis, Angew. Chem., Int. Ed., 2014, 53, 10209 CrossRef CAS PubMed.
- (a) K. Hirano, T. Iwahama, S. Sakaguchi and Y. Ishii, Chem. Commun., 2000, 2457–2458 RSC; (b) J. Y. Kim, S. H. Cho, J. Joseph and S. Chang, Angew. Chem., Int. Ed., 2010, 49, 9899 CrossRef CAS PubMed.
- (a) L. I. Simándi, Advances in Catalytic activation of Dioxygen by Metal Complexes, Kluwer academic publishers, 2002, eBook ISBN: 0-306-47816-1; Print ISBN: 1-4020-1074-5 Search PubMed; (b) C. Chatgilialoglu, D. Crich, M. Komatsu and I. Ryu, Chem. Rev., 1999, 99, 1991 CrossRef CAS PubMed; (c) E. Watanabe, A. Kaiho, H. Kusama and N. Iwasawa, J. Am. Chem. Soc., 2013, 135, 11744 CrossRef CAS PubMed.
- (a) V. B. Sharma, S. L. Jain and B. Sain, Tetrahedron Lett., 2003, 44, 383 CrossRef CAS; (b) S. I. Murahashi, T. Naota and N. Hirai, J. Org. Chem., 1993, 58, 7318 CrossRef CAS; (c) H. C. Tung and D. T. Sawyer, J. Am. Chem. Soc., 1990, 112, 8214 CrossRef CAS; (d) G. Zhang, K. V. Vasudevan, B. L. Scott and S. K. Hanson, J. Am. Chem. Soc., 2013, 135, 8668 CrossRef CAS PubMed; (e) A. Sobkowiak and D. T. Sawyer, J. Am. Chem. Soc., 1991, 113, 9520 CrossRef CAS.
- For Pd, see: (a) S. Gowrisankar, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2011, 50, 5139 CrossRef CAS PubMed; (b) C. Liu, S. Tang, L. Zheng, D. Liu, H. Zhang and A. Lei, Angew. Chem., Int. Ed., 2012, 51, 5662 CrossRef CAS PubMed; (c) A. M. Whittaker and V. M. Dong, Angew. Chem., Int. Ed., 2015, 54, 1312 CrossRef CAS PubMed.
- (a) W.-J. Yoo and C.-J. Li, Tetrahedron Lett., 2007, 48, 1033 CrossRef CAS; (b) S. K. Rout, S. Guin, K. K. Ghara, A. Banerjee and B. K. Patel, Org. Lett., 2012, 14, 3982 CrossRef CAS PubMed; (c) J. Wang, C. Liu, J. Yuan and A. Lei, Chem. Commun., 2014, 50, 4736 RSC; (d) A. S. K. Hashmi, C. Lothschutz, M. Ackermann, R. Doepp, S. Anantharaman, B. Marchetti, H. Bertagnolli and F. Rominger, Chem. – Eur. J., 2010, 16, 8012 CrossRef CAS PubMed; (e) G. S. Kumar, C. U. Maheswari, R. A. Kumar, M. L. Kantam and K. R. Reddy, Angew. Chem., Int. Ed., 2011, 50, 11748 CrossRef CAS PubMed.
- (a) F. A. Chavez, J. M. Rowland, M. M. Olmstead and P. K. Mascharak, J. Am. Chem. Soc., 1998, 120, 9015 CrossRef CAS; (b) E. Spier, U. Neuenschwander and I. Hermans, Angew. Chem., Int. Ed., 2013, 52, 1581 CrossRef CAS PubMed.
- (a) C. Chatgilialoglu, D. Crich, M. Komatsu and I. Ryu, Chem. Rev., 1999, 99, 1991 CrossRef CAS PubMed; (b) Z. Liu, J. Zhang, S. Chen, E. Shi, Y. Xu and X. Wan, Angew. Chem., Int. Ed., 2012, 51, 3231 CrossRef CAS PubMed; (c) W. Wei, C. Zhang, Y. Xu and X. Wan, Chem. Commun., 2011, 47, 10827 RSC.
- (a) M. Karimi, D. Saberi, K. Azizi, M. Arefi and A. Heydari, Tetrahedron Lett., 2014, 55, 5351 CrossRef CAS; (b) K. R. Reddy, M. Venkateshwar, C. U. Maheswari and S. Prashanthi, Synth. Commun., 2010, 46, 186 Search PubMed; (c) K. R. Reddy, C. U. Maheswari, M. Venkateshwar and M. L. Kantam, Eur. J. Org. Chem., 2008, 3619 CrossRef CAS; (d) J. Zhao, P. Li, C. Xia and F. Li, Chem. Commun., 2014, 50, 4751 RSC.
- (a) M. Uyanik, H. Hayashi and K. Ishihara, Science, 2014, 345, 291 CrossRef CAS PubMed; (b) G. Wang, Q.-Y. Yu, S.-Y. Chen and X.-Q. Yu, Org. Biomol. Chem., 2014, 12, 414 RSC; (c) M. Uyanik, H. Okamoto, T. Yasui and K. Ishihara, Science, 2010, 328, 1376 CrossRef CAS PubMed.
- (a) Y. Ding, X. Zhang, D. Zhang, Y. Chen, Z. Wu, P. Wang, W. Xue, B. Song and S. Yang, Tetrahedron Lett., 2015, 56, 831 CrossRef CAS; (b) X.-F. Wu, M. Sharif, A. Pews-Davtyan, P. Langer, K. Ayub and M. Beller, Eur. J. Org. Chem., 2013, 2783 CrossRef CAS; (c) S. Yang, H. Yan, X. Ren, X. Shi, J. Li, Y. Wang and G. Huang, Tetrahedron, 2013, 69, 6431 CrossRef CAS; (d) H. Xie, Y. Liao, S. Chen, Y. Chen and G.-J. Deng, Org. Biomol. Chem., 2015, 13, 6944 RSC; (e) A. B. Khemnar and B. M. Bhanage, Org. Biomol. Chem., 2014, 12, 9631 RSC; (f) Y. F. Zhu and Y. Y. Wei, Eur. J. Org. Chem., 2013, 4503 CrossRef CAS; (g) W.-J. Yoo and C.-J. Li, J. Am. Chem. Soc., 2006, 128, 13064 CrossRef CAS PubMed. Iodide as an additive: (h) J.-B. Feng, D. Wei, J.-L. Gong, X. Qi and X.-F. Wu, Tetrahedron Lett., 2014, 55, 5082 CrossRef CAS; (i) T. Wang, L. Yuan, Z. Zhao, A. Shao, M. Gao, Y. Huang, F. Xiong, H. Zhang and J. Zhao, Green Chem., 2015, 17, 2741 RSC.
- J. C. Wren, J. Paquette, S. Sunder and B. L. Ford, Can. J. Chem., 1986, 64, 2284 CrossRef CAS.
- G. J. Janz, A. E. Marcinkowsky and H. V. Venkatasetty, Electrochim. Acta, 1963, 8, 867 CrossRef CAS.
- B. J. Tan, K. J. Klabunde and P. M. A. Sherwood, J. Am. Chem. Soc., 1991, 113, 855 CrossRef CAS.
- (a) T. Ivanova, A. Naumkin, A. Sidorov, I. Eremenko and M. Kiskin, J. Electron Spectrosc. Relat. Phenom., 2007, 156–158, 200 CrossRef CAS; (b) G. Poneti, L. Poggini, M. Mannini, B. Cortigiani, L. Sorace, E. Otero, P. Sainctavit, A. Magnani, R. Sessoli and A. Dei, Chem. Sci., 2015, 6, 2268 RSC.
- P. M. A. Sherwood, J. Chem. Soc., Faraday Trans. 2, 1976, 72, 1805 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5qo00293a |
| This journal is © the Partner Organisations 2016 |