
Chemoselective catalytic reduction of conjugated α,β-unsaturated ketones to saturated ketones via a hydroboration/protodeboronation strategy†
Wen
Ding
a and
Qiuling
Song
*ab
aInstitute of Next Generation Matter Transformation, College of Chemical Engineering at Huaqiao University, Xiamen, China
bNational Laboratory for Molecular Sciences, Institute of Chemistry, Beijing, 100190, P. R. China. E-mail: qsong@hqu.edu.cn; Fax: +86-592-6162990
First published on 26th October 2015
Abstract
A novel copper-catalyzed chemoselective reduction of a carbon–carbon double or triple bond to a carbon–carbon single bond in α,β-unsaturated ketones is developed. This reaction proceeds under hydrogen gas or stoichiometric metal hydride free conditions. Saturated ketones were obtained in good to excellent yields with a broad substrate scope. Mechanistic studies reveal that the two hydrogen atoms come from H2O in the system. Thus this reaction represents a highly efficient, and remarkably chemoselective strategy.
Arylboronic acids or arylboronic esters are widely employed as coupling partners in cross-coupling reactions for the formation of a C–C bond due to their ready accessibility.1 In contrast, the cross-coupling reaction of alkylboronic esters with alkyl electrophiles, and the protodeboronation of organoboronic esters have rarely been investigated. Among them, the cross-coupling reaction between alkylboronic esters and alkyl electrophiles has attracted more attention compared to protodeboronation. For instance, Fu et al. reported pioneering work on the Pd-catalyzed2 coupling reaction of alkyl 9-BBN reagents with inert alkyl electrophiles and the Ni-catalyzed3 coupling reaction of alkyl 9-BBN reagents with secondary alkyl halides. Very recently, Shibata4 and Morken5 successfully developed novel alkylboron compounds, respectively, to employ them in the cross-coupling reaction. Moreover, the Cu-catalyzed cross coupling reaction of alkylboronic esters with primary alkyl electrophiles was also achieved.6
However, progress on protodeboronation of alkylboronic ester moves slowly. There are only few known reports of protodeboronation of alkylboronic ester, yet all have made excellent contributions to modern organic chemistry. For example, Aggarwal7 has well described his “F–B bond strategy” for protodeboronation of tertiary alkylboronic esters, which was successfully applied to the synthesis of 1,1-diarylalkanes7a and hydroxyphthioceranic acid;7b Renaud8 reported his classical TBC (4-tert-butylcatechol) system for protodeboronation of alkylboronic esters, which has been applied to reduce olefins. Apart from the above-mentioned seminal studies, there are no more precedents on this topic, although protodeboronation of alkylboronic esters has become a fascinating area for the synthetic community.
Chemoselective reduction of α,β-unsaturated ketones leading to saturated ones is one of the most prevalent reactions in organic synthesis yet crucial due to its wide applications in the synthesis of industrial chemicals and pharmaceuticals.9–11 1,2-Reduction and full reduction usually occur as the side reactions in the course of conjugated reduction of α,β-unsaturated carbonyl compounds, so it is always highly desirable to increase the chemoselectivity on such reductions. Great advances have been made in this field. Several expensive transition-metal catalyst systems, such as rhodium,12 ruthenium,13 palladium,14 iridium15 and other metal complexes,16 have been developed and applied to the reduction of α,β-unsaturated carbonyl compounds. In 1988, Stryker et al.17 developed an inexpensive copper catalyst (CuH), known as Stryker's Reagent, which has been widely used as an effective reagent in the highly regioselective conjugated reductions of various carbonyl derivatives. Usually a hydride transfer mechanism is involved in the majority of the reported reduction methods, leading to a formidable requirement of sheltering from air (O2).18 Meanwhile, utilizing H2O as the hydrogen source is scarcely reported owing to the difficulty in the formation of a negative hydrogen from H2O in a reductive reaction. Very recently, we discovered that the C–B bond of alkylboronic esters can be easily transformed into a C–H bond in the presence of K2CO3 or Cs2CO3 at 90 °C (see the ESI†), which stands in contrast to previous work (Scheme 1).
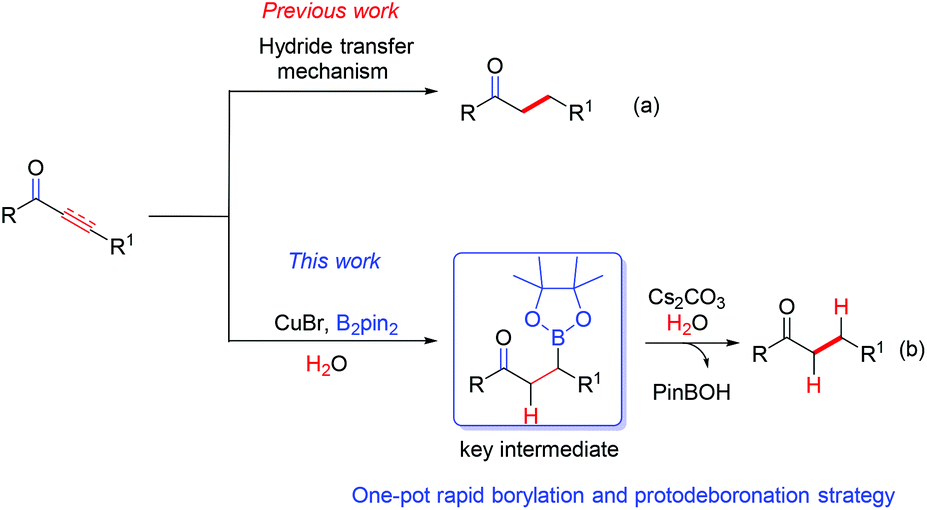 | ||
| Scheme 1 Strategies on conjugated reduction of α,β-unsaturated ketones. | ||
Based on the understanding of our novel protodeboronation pathway, we would like herein to report the first domino reaction of a highly chemoselective conjugated reduction of α,β-unsaturated ketones by utilizing costless but quite efficient H2O as the hydrogen source via a protodeboronation strategy without a special requirement of reaction atmosphere. Thus, a novel, simple and efficient borylation/protodeboronation reaction was developed to lead to the highly chemoselective reduction of the C–C unsaturated bond. More importantly, this reaction shows great chemoselectivity over other unconjugated C–C multiple bonds and can be readily scaled up without loss of its efficiency.
Our initial investigation commenced with chalcone (1) as the model substrate to investigate the copper salt-catalyzed chemoselective reduction (Table 1 and Table S1 in the ESI†). Gratifyingly, both CuI and CuII salts demonstrated good activities on this novel transformation (Table 1, entries 1–7, 9 and 10) except CuO (Table 1, entry 8) and 86% or 88% of the desired product was obtained respectively by using 10 mol% of CuBr with B2pin2 (2 equiv.) and Cs2CO3 (1.5 equiv.) at 90 °C in toluene![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :THF = 4:1 (5 mL) under air or N2 in a sealed tube (Table 1, entries 2 and 3). Further screening of solvents and bases revealed that Cs2CO3 (1.5 equiv.) and toluene:THF = 4:1 (5 mL) are the optimal choices. When the reaction was conducted in one single solvent, inferior results were obtained. Temperature affected this reaction dramatically and when the temperature was decreased to 70 °C, the yield of the desired product 3 was dropped to 70% (Table 1, entry 20). Without CuBr, no reaction occurred; in the absence of the base, B2pin2 or Cs2CO3, no reaction happened either. Interestingly, solvents greatly affect the reaction. Reduction can even be directly performed in H2O although it is slower than organic solvents. Moreover, to our surprise, we later found that this reduction reaction gave as good a result in only 1 hour as in 22 hours (Table 1, entry 16).
:THF = 4:1 (5 mL) under air or N2 in a sealed tube (Table 1, entries 2 and 3). Further screening of solvents and bases revealed that Cs2CO3 (1.5 equiv.) and toluene:THF = 4:1 (5 mL) are the optimal choices. When the reaction was conducted in one single solvent, inferior results were obtained. Temperature affected this reaction dramatically and when the temperature was decreased to 70 °C, the yield of the desired product 3 was dropped to 70% (Table 1, entry 20). Without CuBr, no reaction occurred; in the absence of the base, B2pin2 or Cs2CO3, no reaction happened either. Interestingly, solvents greatly affect the reaction. Reduction can even be directly performed in H2O although it is slower than organic solvents. Moreover, to our surprise, we later found that this reduction reaction gave as good a result in only 1 hour as in 22 hours (Table 1, entry 16).
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Catalyst (10 mol%) | Base (1.5 equiv.) | Atmosphere | Temp. (°C) | Solvent (mL) | Time (22 h) | Yieldb (%) |
| a Reaction conditions: chalcone (1a, 0.25 mmol), B2pin2 (2 equiv.), Cu salt (10 mol%), base (1.5 equiv.), solvent (5 ml), 22 h, temp., corresponding atmosphere. b GC yield. c Isolated yield. d Without B2pin2 (2 equiv.). | |||||||
| 1 | CuBr | Cs2CO3 | O2 | 90 | Toluene (4)/THF (1) | 65 | |
| 2 | CuBr | Cs2CO3 | Air | 90 | Toluene (4)/THF (1) | 86c | |
| 3 | CuBr | Cs2CO3 | N2 | 90 | Toluene (4)/THF (1) | 88 | |
| 4 | CuBr2 | Cs2CO3 | Air | 90 | Toluene (4)/THF (1) | 71 | |
| 5 | CuI | Cs2CO3 | Air | 90 | Toluene (4)/THF (1) | 85 | |
| 6 | CuCl | Cs2CO3 | Air | 90 | Toluene (4)/THF (1) | 75 | |
| 7 | Cu(OAc)2 | Cs2CO3 | Air | 90 | Toluene (4)/THF (1) | 79 | |
| 8 | CuO | Cs2CO3 | Air | 90 | Toluene (4)/THF (1) | Trace | |
| 9 | Cu(OTf)2 | Cs2CO3 | Air | 90 | Toluene (4)/THF (1) | 83 | |
| 10 | CuCl2 | Cs2CO3 | Air | 90 | Toluene (4)/THF (1) | 68 | |
| 11 | CuBr | Cs2CO3 | Air | 90 | Toluene (5) | 38 | |
| 12 | CuBr | Cs2CO3 | Air | 90 | THF (5) | Trace | |
| 13 | CuBr | Cs2CO3 | Air | 90 | DMF (5) | 0 | |
| 14 | CuBr | Cs2CO3 | Air | 90 | Dioxane (5) | 41 | |
| 15 | CuBr | Cs2CO3 | Air | 90 | H2O (5) | 24 | |
| 16 | CuBr | Cs 2 CO 3 | Air | 90 | Toluene (4)/THF (1) | 1 h | 88 |
| 17 | — | Cs2CO3 | Air | 90 | Toluene (4)/THF (1) | 1 h | 0 |
| 18 | CuBr | — | Air | 90 | Toluene (4)/THF (1) | 1 h | 0 |
| 19d | CuBr | Cs2CO3 | Air | 90 | Toluene (4)/THF (1) | 1 h | 0 |
| 20 | CuBr | Cs2CO3 | Air | 70 | Toluene (4)/THF (1) | 1 h | 70 |
With the optimized conditions in hand, we explored the scope and limitations of the chemoselective reaction. Firstly, we surveyed different substituted chalcones. To our delight, all substituted chalcones were competent candidates in this transformation (Scheme 2) and the corresponding desired products were obtained in good to excellent yields. Special attention should be paid to halogen substituted chalcones (Scheme 2, 1b, 1d and 1f), since in each case, the corresponding products were afforded in >90% yield. Remarkably, α,β-unsaturated ketones with an additional substituent at the α-position (1m) performed well. α,β-Unsaturated ketones which contain the heterocycle (1l) and ester group (1n) are also tolerable with good yields in this strategy. The compound (1E,4E)-1,5-diphenylpenta-1,4-dien-3-one (dba) (1p), which is usually used as an excellent ligand in palladium-catalyzed reactions, is compatible with this system as well and can be reduced to the saturated product in a good yield. Moreover, yneone (1r) was reduced to a saturated ketone when the loading of the base and B2pin2 was doubled.
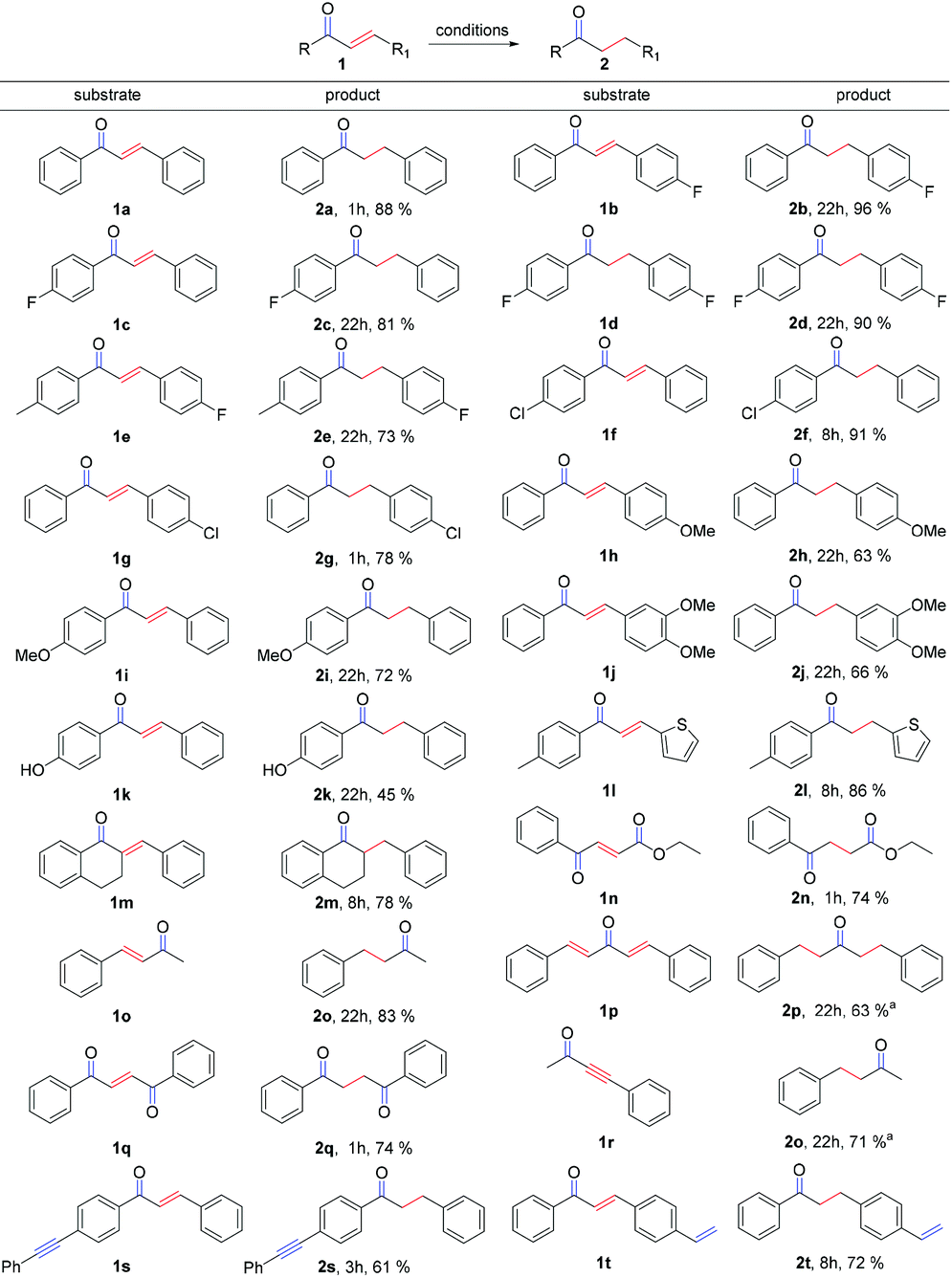 | ||
| Scheme 2 Substrate scope of the chemoselective reduction. Conditions: chalcone (1, 0.25 mmol), B2pin2 (2 equiv.), Cu salt (10 mol%), base (1.5 equiv.), solvent = toluene:THF = 4:1 (5 mL), 90 °C, air. aThe loadings of base and B2pin2 were doubled. | ||
Notably, this reaction showed very good chemoselectivity when various unsaturated C–C bonds are on the molecules: for example, the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C bond of 1s and the C
C bond of 1s and the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond of 1t were both untouched under the standard conditions, no corresponding LCu–Bpin adducts over the unconjugated C–C multiple bonds were ever detected, suggesting that the α,β-unsaturated carbonyl system is essential for the success of the reduction.
C bond of 1t were both untouched under the standard conditions, no corresponding LCu–Bpin adducts over the unconjugated C–C multiple bonds were ever detected, suggesting that the α,β-unsaturated carbonyl system is essential for the success of the reduction.
The chemoselective reduction of α,β-unsaturated ketones could be easily scaled up without decreasing their efficiency (Scheme 3), which clearly indicated the potential practical applications of this strategy.
 | ||
| Scheme 3 Scale-up reaction. | ||
To gain insight into the originality of the hydrogen source of the reduced product in this reaction system, chalcone (1a) in anhydrous solvent was subjected to D2O (4 equiv.) under the standard conditions (Scheme 4a). The result revealed that the deuterated ratio of the β-position is 82% and the deuterated ratio of the α-position is 155% (because of keto–enol tautomerism) (determined by 1H NMR and GC-MS, see details in the ESI†), while almost no reaction occurred when the experiment was performed in anhydrous solvent under the standard conditions (Scheme 4b), which clearly indicated that water should be the hydrogen source for this reduction reaction. Interestingly, when the radical scavenger (BHT or TEMPO) was added into the reaction, the reaction gave >99% yield of 3a (Scheme 4c and d), which reveal that a radical pathway may be involved in the protodeboronation process, although no free radical intermediate 7 (Scheme 6) was captured thus far.
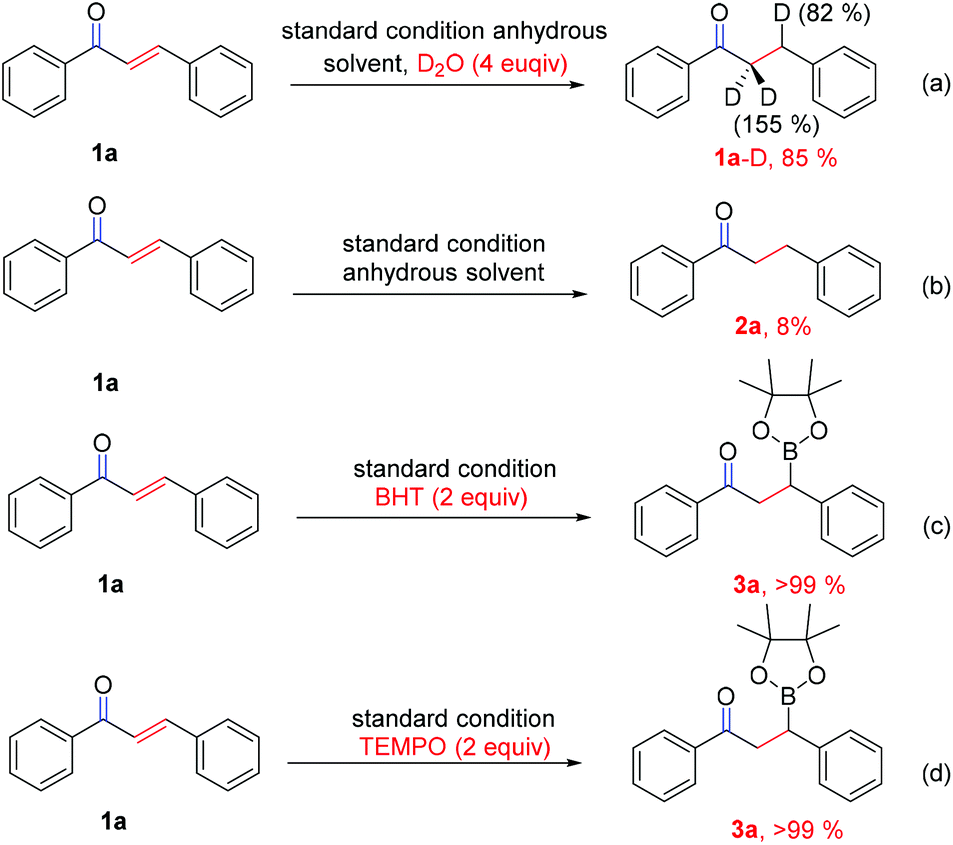 | ||
| Scheme 4 Hydrogen isotope labeling and radical trapping experiments. | ||
To determine the possible intermediate of this process, two control experiments were conducted (Scheme 5). 1,3-Diphenyl-3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)propan-1-one (3a) was subjected to various conditions: the standard conditions or in the absence of copper salt can both lead to the desired product 2a in ca. 75% yield (Scheme 5a and c), yet no reaction happened in the absence of the base (Scheme 5b), these results obviously demonstrate that 1,3-diphenyl-3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)propan-1-one (3) is the key intermediate in this chemoselective conjugated reduction of α,β-unsaturated carbonyl compounds.
 | ||
| Scheme 5 Control experiments. | ||
Based on the above control and isotope labeled experiments and the related literature report,19 we propose the mechanism (Scheme 6): in the presence of copper salt, base, and B2pin2, the α,β-unsaturated carbonyl compound (1) was firstly transformed into a borylated copper enolate (4), which was rapidly converted into boronic ester substituted ketones (3) by H2O. Then saturated compound 2 was obtained after protodeboronation of the key intermediate (3) in the presence of the base and H2O. The protodeboronation process was supposed to be relevant to an alkylboronic ester/water complex (5), which might furnish a species activated for O–H bond homolysis.20 This hypothesis was also supported by the work of Wood et al.,20 in which a related trimethyl-borane/water complex was formed.20
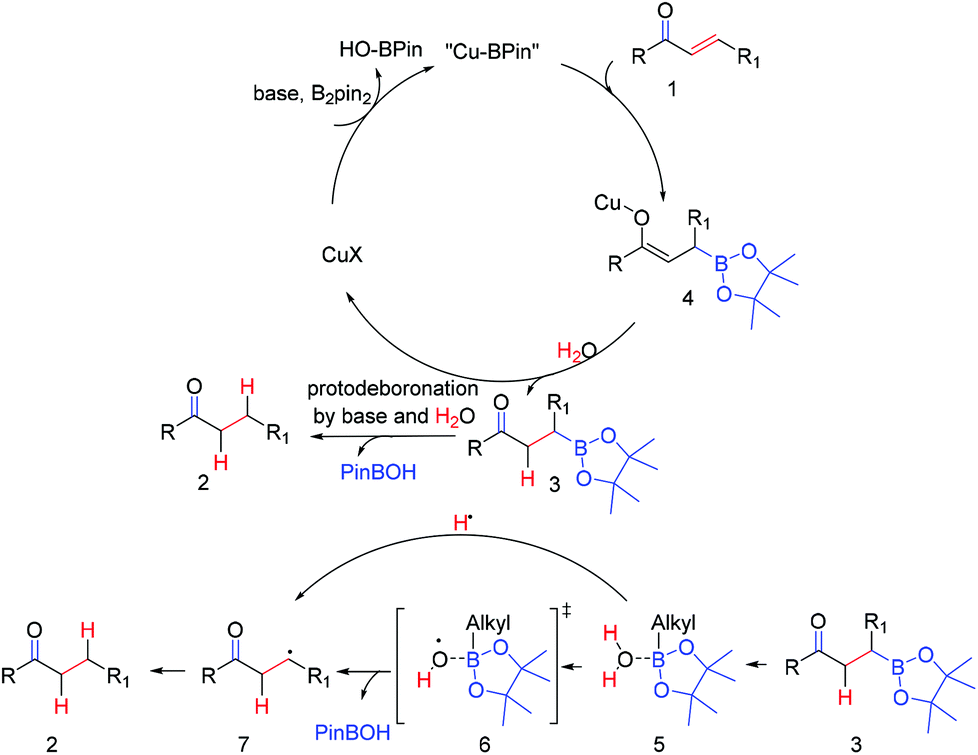 | ||
| Scheme 6 Proposed mechanism. | ||
Conclusions
In summary, we have developed the first copper-catalyzed conjugated reduction of α,β-unsaturated ketones making use of bis(pinacolato)diboron and H2O as a hydrogen source via a protodeboronation strategy under simple and mild conditions. This strategy is efficient, and highly chemoselective. It requires low catalyst loadings and tolerates various functional groups and can be easily scaled up without loss of its efficiency. Ongoing studies are directed at further reactivity, synthetic utility and mechanism of this catalytic system in our laboratory.Acknowledgements
Financial support from the National Natural Science Foundation of China (21202049), the Recruitment Program of Global Experts (1000 Talents Plan), the Fujian Hundred Talents Program and the Program of Innovative Research Team of Huaqiao University (Z14X0047) are gratefully acknowledged.Notes and references
- (a) N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457 CrossRef CAS; (b) P. Lloyd-Williams and E. Giralt, Chem. Soc. Rev., 2001, 30, 145 RSC; (c) S. Darses and J.-P. Genet, Chem. Rev., 2007, 108, 288 CrossRef PubMed; (d) R. Jana, T. P. Pathak and M. S. Sigman, Chem. Rev., 2011, 111, 1417 CrossRef CAS PubMed.
- (a) M. R. Netherton, C. Dai, K. Neuschüütz and G. C. Fu, J. Am. Chem. Soc., 2001, 123, 10099 CrossRef CAS PubMed; (b) J. H. Kirchhoff, C. Dai and G. C. Fu, Angew. Chem., 2002, 114, 2025 ( Angew. Chem., Int. Ed. , 2002 , 41 , 1945 ) CrossRef.
- (a) B. Saito and G. C. Fu, J. Am. Chem. Soc., 2007, 129, 9602 CrossRef CAS PubMed; (b) B. Saito and G. C. Fu, J. Am. Chem. Soc., 2008, 130, 6694 CrossRef CAS PubMed; (c) N. A. Owston and G. C. Fu, J. Am. Chem. Soc., 2010, 132, 11908 CrossRef CAS PubMed; (d) Z. Lu, A. Wilsily and G. C. Fu, J. Am. Chem. Soc., 2011, 133, 8154 CrossRef CAS PubMed; (e) S. L. Zultanski and G. C. Fu, J. Am. Chem. Soc., 2011, 133, 15362 CrossRef CAS PubMed; (f) A. Wilsily, F. Tramutola, N. A. Owston and G. C. Fu, J. Am. Chem. Soc., 2012, 134, 5794 CrossRef CAS PubMed.
- (a) K. Endo, T. Ohkubo, M. Hirokami and T. Shibata, J. Am. Chem. Soc., 2010, 132, 11033 CrossRef CAS PubMed; (b) K. Endo, T. Ohkubo and T. Shibata, Org. Lett., 2011, 13, 3368 CrossRef CAS PubMed; (c) K. Endo, T. Ohkubo, T. Ishioka and T. Shibata, J. Org. Chem., 2012, 77, 4826 CrossRef CAS PubMed.
- (a) C. Sun, B. Potter and J. P. Morken, J. Am. Chem. Soc., 2014, 136, 6534 CrossRef CAS PubMed; (b) K. Hong, X. Liu and J. P. Morken, J. Am. Chem. Soc., 2014, 136, 10581 CrossRef CAS PubMed.
- C.-T. Yang, Y. Fu, Z.-Q. Zhang, Y.-C. Liu and L. Liu, Angew. Chem., 2011, 123, 3990 ( Angew. Chem., Int. Ed. , 2011 , 50 , 3904 ) CrossRef.
- (a) S. Nave, R. P. Sonawane, T. G. Elford and V. K. Aggarwal, J. Am. Chem. Soc., 2010, 132, 17096 CrossRef CAS PubMed; (b) R. Rasappan and V. K. Aggarwal, Nat. Chem., 2014, 6, 810 CrossRef CAS PubMed; (c) S. Roesner, J. M. Casatejada, T. G. Elford, R. P. Sonawane and V. K. Aggarwal, Org. Lett., 2011, 13, 5740–5743 CrossRef CAS PubMed.
- (a) D. Pozzi, E. M. Scanlan and P. Renaud, J. Am. Chem. Soc., 2005, 127, 14204 CrossRef CAS PubMed; (b) G. Villa, G. Povie and P. Renaud, J. Am. Chem. Soc., 2011, 133, 5913 CrossRef CAS PubMed.
- (a) U. Leutenegger, A. Madin and A. Pfaltz, Angew. Chem., 1989, 101, 61 ( Angew. Chem. Int. Ed. Engl. , 1989 , 28 , 60 ) CrossRef CAS; (b) M. Misun and A. Pfaltz, Helv. Chim. Acta, 1996, 79, 961 CrossRef CAS; (c) D. S. Hays, M. Scholl and G. C. Fu, J. Org. Chem., 1996, 61, 6751 CrossRef CAS PubMed; (d) D. H. Appella, Y. Moritani, R. Shintani, E. M. Ferreira and S. L. Buchwald, J. Am. Chem. Soc., 1999, 121, 9473 CrossRef CAS; (e) Y. Moritani, D. H. Appella, V. Jurkauskas and S. L. Buchwald, J. Am. Chem. Soc., 2000, 122, 6797 CrossRef CAS.
- (a) E. Keinan and N. Greenspoon, J. Am. Chem. Soc., 1986, 108, 7314 CrossRef CAS; (b) H. Lee and M. An, Tetrahedron Lett., 2003, 44, 2775 CrossRef CAS; (c) G. Neri, L. Mercadante, A. Visco and S. Galvagno, Catal. Lett., 1994, 29, 379 CrossRef CAS.
- (a) G. Hughes, M. Kimura and S. L. Buchwald, J. Am. Chem. Soc., 2003, 125, 11253 CrossRef CAS PubMed; (b) B. H. Lipshutz and J. M. Servesko, Angew. Chem., 2003, 115, 4937 ( Angew. Chem., Int. Ed. , 2003 , 42 , 4789 ) CrossRef; (c) C. Czekelius and E. M. Carreira, Angew. Chem., 2003, 115, 4941 ( Angew. Chem., Int. Ed. , 2003 , 42 , 4793 ) CrossRef.
- (a) X. Li, L. Li, Y. Tang, L. Zhong, L. Cun, J. Zhu, J. Liao and J. Deng, J. Org. Chem., 2010, 75, 2981 CrossRef CAS PubMed; (b) Z. Baan, Z. Finta, G. Keglevich and I. Hermecz, Green Chem., 2009, 11, 1937 RSC; (c) X.-J. Du, Y.-H. Tang, X. Zhang and M. Lei, Chin. Chem. Lett., 2013, 24, 1083 CrossRef CAS.
- (a) Y. Sasson and J. Blum, Tetrahedron Lett., 1971, 12, 2167 CrossRef; (b) Y. Sasson and J. Blum, J. Org. Chem., 1975, 40, 1887 CrossRef CAS; (c) G. Descotes and D. Sinou, Tetrahedron Lett., 1976, 17, 4083 CrossRef; (d) T. Doi, T. Fukuyama, J. Horiguchi, T. Okamura and I. Ryu, Synlett, 2006, 721 CAS; (e) C. Mebi, R. Nair and B. Frost, Organometallics, 2007, 26, 429 CrossRef CAS.
- (a) Z. Baan, Z. Finta, G. Keglevich and I. Hermecz, Tetrahedron Lett., 2005, 46, 6203 CrossRef CAS; (b) A. Mori, Y. Miyakawa, E. Ohashi and T. Haga, Org. Lett., 2006, 8, 3279 CrossRef CAS PubMed; (c) P. Four and F. Guibe, Tetrahedron Lett., 1982, 23, 1825 CrossRef CAS; (d) E. Keinan and N. Greenspoon, Tetrahedron Lett., 1985, 26, 1353 CrossRef CAS; (e) E. Keinan and N. Greenspoon, J. Am. Chem. Soc., 1986, 108, 7314 CrossRef CAS; (f) H. Otsuka, E. Shirakawa and T. Hayashi, J. Chem. Soc., Chem. Commun., 2007, 1819 RSC; (g) A. Sharma, V. Kumar and A. K. Sinha, Adv. Synth. Catal., 2006, 348, 354 CrossRef CAS.
- (a) S. Sakaguchi, T. Yamaga and Y. Ishii, J. Org. Chem., 2001, 66, 4710 CrossRef CAS PubMed; (b) J. Blum, Y. Sasson and S. Iflah, Tetrahedron Lett., 1972, 13, 1015 CrossRef; (c) Y. Himeda, N. Onozawa-Komatsuzaki, S. Miyazawa, H. Sugihara, T. Hirose and K. Kasuga, Chem. – Eur. J., 2008, 14, 11076 CrossRef CAS PubMed.
- (a) A. Kosal and B. Ashfeld, Org. Lett., 2010, 12, 44 CrossRef CAS PubMed; (b) E. Keinan and D. Perez, J. Org. Chem., 1987, 52, 2576 CrossRef CAS; (c) D. Hays, M. Scholl and G. C. Fu, J. Org. Chem., 1996, 61, 6751 CrossRef CAS PubMed.
- (a) D. M. Brestensky, D. E. Huseland, C. McGettigan and J. M. Stryker, Tetrahedron Lett., 1988, 29, 3749 CrossRef CAS; (b) W. S. Mahoney, D. M. Brestensky and J. M. Stryker, J. Am. Chem. Soc., 1988, 110, 291 CrossRef CAS; (c) D. M. Brestensky and J. M. Stryker, Tetrahedron Lett., 1989, 30, 5677 CrossRef CAS; (d) T. M. Koenig, J. F. Daeuble, D. M. Brestensky and J. M. Stryker, Tetrahedron Lett., 1990, 31, 3237 CrossRef CAS; (e) For the first application of CuH in synthesis, see: H. Brunner and W. Miehling, J. Organomet. Chem., 1984, 275, C17–C21 CrossRef CAS.
- C. Deutsch, N. Krause and B. H. Lipshutz, Chem. Rev., 2008, 108, 2916 CrossRef CAS PubMed.
- (a) S. Kobayashi, P. Xu, T. Endo, M. Ueno and T. Kitanosono, Angew. Chem., 2012, 124, 12935 ( Angew. Chem., Int. Ed. , 2012 , 51 , 12763 ) CrossRef; (b) S. B. Thorpe, J. A. Calderone and W. L. Santos, Org. Lett., 2012, 14, 1918 CrossRef CAS PubMed; (c) Z. Li, Z. Wang, L. Zhu, X. Tan and C. Li, J. Am. Chem. Soc., 2014, 136, 16439 CrossRef CAS PubMed.
- D. A. Spiegel, K. B. Wiberg, L. N. Schacherer, M. R. Medeiros and J. L. Wood, J. Am. Chem. Soc., 2005, 127, 12513 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5qo00289c |
| This journal is © the Partner Organisations 2016 |