
How easy is CO2 fixation by M–C bond containing complexes (M = Cu, Ni, Co, Rh, Ir)?†
Sai V. C.
Vummaleti
a,
Giovanni
Talarico
b,
Steven P.
Nolan
*c,
Luigi
Cavallo
*a and
Albert
Poater
*d
aKAUST Catalysis Center, Physical Sciences and Engineering Division, King Abdullah University of Science and Technology, Thuwal 23955-6900, Saudi Arabia. E-mail: luigi.cavallo@kaust.edu.sa
bDipartimento di Scienze Chimiche, Università di Napoli Federico II, Via Cintia, 80126 Napoli, Italy
cChemistry Department, College of Science, King Saud University, Riyadh 11451, Saudi Arabia. E-mail: stevenpnolan@gmail.com
dInstitut de Química Computacional i Catàlisi and Departament de Química, Universitat de Girona, Campus Montilivi, 17071 Girona, Catalonia, Spain. E-mail: albert.poater@udg.edu
First published on 27th November 2015
Abstract
A comparison between different M–C bonds (M = Cu(I), Ni(II), Co(I), Rh(I) and Ir(I)) has been reported by using density functional theory (DFT) calculations to explore the role of the metal in the fixation or incorporation of CO2 into such complexes. The systems investigated are various metal based congeners of the Ir-complex 8 [(cod)(IiPr)Ir-CCPh], with a ligand scaffold based on cod and IiPr ligands (cod = 1,5-cyclooctadiene; IiPr = 1,3-bis(isopropyl)imidazol-2-ylidene). The results of this study show that the calculated CO2 insertion barriers follow the trend: Cu(I) (20.8 kcal mol−1) < Rh(I) (30.0 kcal mol−1) < Co(I) (31.3 kcal mol−1) < Ir(I) (37.5 kcal mol−1) < Ni(II) (45.4 kcal mol−1), indicating that the Cu(I) based analogue is the best CO2 fixer, while Ni(II) is the worst in the studied series.
The development of catalysts capable of utilising CO2 as feedstock,1–4 possibly in large volume transformations,5–7 is considered to be one of the key challenges in modern chemistry. However, the wide spread use of CO2 in chemical transformations is limited due to its chemical inertness.8 In the recent past, despite some attempts with metal-free processes,9,10 even organocatalyzed trapping of CO2 through C–C and C–O bond formation,11 progress has been made in exploring the CO2 reactivity with a large number of its reactions being insertion into a transition metal–X bond (where X = C, O, N) supported with appropriate ligands.12–17 These reactions are believed to be one of the most challenging organometallic reactions as CO2 is thermodynamically stable and kinetically inert.18–24 To this end, a novel organometallic complex as well as a chemical mechanism able to perform or explain the capture of CO2 would be extremely interesting and valuable in directing catalyst design efforts.25–46 Therefore, a thorough understanding of the mechanism of CO2 insertion into the M–X bonds is of fundamental importance and could guide advances in the development of this important reaction.
Despite the very recent large number of experimental47–52 and theoretical53–59 studies focusing on CO2 insertion, reports on direct comparison between theoretical and experimental results remain very limited.53,60,61 To partially fill this gap, we have recently reported density functional theory based investigations of the CO2 fixation mechanism as proposed by Nolan et al. for a new Ir(I) complex,61–63 [Ir(cod)IiPr)(OH)] (1) (cod = 1,5-cyclooctadiene; IiPr = 1,3-bis(isopropyl)imidazol-2-ylidene) and its derivatives containing Ir–O (complexes 3 and 4), Ir–N (complex 5), and Ir–C bonds (complexes 6–9; see Scheme 1).64 According to the experiments, complexes containing Ir–heteroatom bonds (1, 3–5) display high reactivity towards CO2, while complexes with Ir–C bonds (6–9) are completely unreactive towards the greenhouse gas.
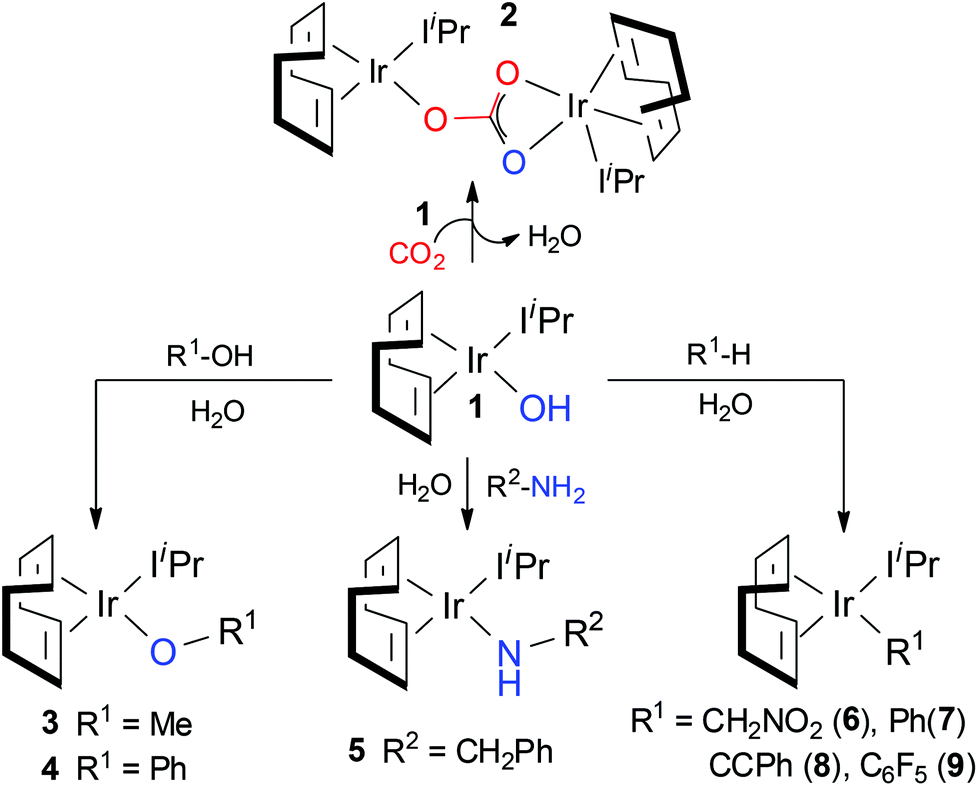 | ||
| Scheme 1 | ||
Based on the experimental findings, a plausible mechanism for CO2 insertion into the M–X bonds (X = O or N) proposed by Nolan et al. is outlined in Scheme 2. Considering complex A, the nucleophilic attack of the heteroatom X onto CO2 does not proceed via the oxygen atoms but through the carbon atom, involving a four-membered cyclic center, hypothetically structure B, i.e. most probably B is the transition state itself. The particular structural nature of 1 allows for a second molecule of 1 to react yielding a dimeric structural product 2; this occurs once the water molecule has been released from intermediate C.
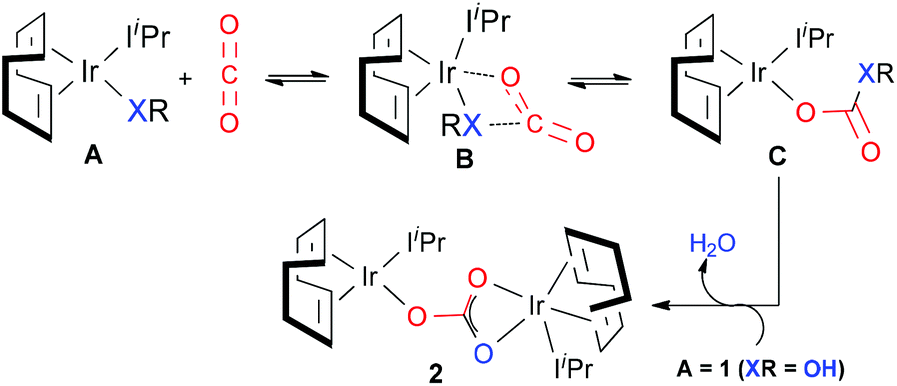 | ||
| Scheme 2 | ||
Our detailed investigation of the proposed mechanism in Scheme 2 for complex 1 revealed that the CO2 insertion step is the rate determining step rather than the subsequent dimerization step, C → 2 (15.8 kcal mol−1vs. 9.4 kcal mol−1, respectively).62 This finding contradicts the recent study by Truscott et al.61 as they argued that dimerization is probably the rate-limiting step for complex 1. In fact, we pointed out that the facile dimerization step is due to the cooperative nature of the iridium centers. Additionally, in agreement with experiments, the calculated low CO2 insertion barriers for complex 1 and its derivatives 3–5 (3 (11.8 kcal mol−1) < 4 (22.5 kcal mol−1) < 5 (17.7 kcal mol−1)) indicated that the CO2 insertion into the Ir–O and Ir–N bonds is kinetically facile. On the other hand, significantly higher barrier values were estimated for complexes 6–9 (ranges from 35.0 to 50.0 kcal mol−1), suggesting that CO2 insertion into the Ir–C bonds is kinetically more challenging and supports the experimental observation that the Ir–C bonds are unreactive towards CO2.
Having rationalized the behaviour of the systems proposed in the literature (1, 3–9), in the present study we turn our attention to the effect of changing the metal (but keeping the same ligand scaffold; cod and IiPr ligands) with the aim of finding one metal that can induce CO2 insertion into the M–C bonds. It is well documented in the literature that CO2 can be inserted into the M–C bonds of Rh and other transition metals such as Cu and Ni.65–67 To this end, we studied the CO2 insertion step for different metal (Cu(I), Ni(II)), Co(I) and Rh(I)68 based congeners of 8 [(cod)(IiPr)Ir-CCPh], which presented the lowest CO2 insertion barrier (37.5 kcal mol−1) among the investigated Ir–C bond containing complex in the series 6–9.
To clarify the role of the metal in the CO2 insertion DFT calculations were carried out. All DFT static calculations have been performed at the GGA level with the Gaussian09 set of programs,69 using the BP86 functional of Becke and Perdew.70,71 The electronic configuration of the molecular systems was described with the standard split-valence basis set with a polarization function of Ahlrichs and co-workers for H, C, N, and O (TZVP keyword in Gaussian).72 For Co, Cu, Ni, Rh and Ir, the quasi-relativistic Stuttgart/Dresden effective core potential was used, with the associated valence basis set (standard SDD keywords in Gaussian09).73–75 The geometry optimizations were carried out without symmetry constraints, and the characterization of the stationary points was performed by analytical frequency calculations. Single point calculations on the BP86 optimized geometries were performed using the M06 functional76 with the triple-ζ basis set of Ahlrichs for main-group atoms (TZVP keyword in Gaussian),77 while for Ir the SDD basis set has been employed. These single point calculations also used the polarizable continuous model PCM to address solvent effects, using benzene as a solvent.78,79 The reported free energies in this work include energies obtained at the M06/TZVP level corrected with zero-point energies, thermal corrections and entropy effects evaluated at 298 K by the BP86/SVP method in the gas phase.80,81
Fig. 1 presents the computed energy profile for the CO2 insertion into the M–C bond of different metal analogues of 8. The reference point is the energy of complex 8 analogues plus free CO2. For the selected metals, the calculated CO2 insertion barriers (for simplicity named as TS8, in analogy with our Ir(I)-complexes of Scheme 1) follows the trend: Cu(I) (20.8 kcal mol−1) < Rh(I) (30.0 kcal mol−1) < Co(I) (31.3 kcal mol−1) < Ir(I) (37.5 kcal mol−1) < Ni(II) (45.4 kcal mol−1), indicating that the Cu(I) based analogue is the best CO2 fixer, while Ni(II) is the worst among the studied metal series. However, from a thermodynamic stand point, the overall CO2 insertion reaction is endergonic in nature for all the examined metals (see the energy values for II8 in Fig. 1), being less endergonic in the case of Cu(I) (5.0 kcal mol−1) than Ni(II) (22.3 kcal mol−1). It is worth mentioning here that we calculated the CO2 insertion barrier for high spin Co(II) complex and is predicted to be 32.9 kcal mol−1, which is slightly higher in energy (by 1.6 kcal mol−1) than the calculated barrier for low spin Co complex (31.3 kcal mol−1).
 | ||
| Fig. 1 Computed free energy profile for the CO2 insertion into the M–C bond of different metal analogues of Ir(I)-complex 8 (where R1 = CCPh). Free energies are given in kcal mol−1. Ni(II) is a cationic complex (overall charge +1). | ||
A plausible explanation for the remarkable metal effect on the calculated CO2 insertion barriers can be drawn from the geometries of the TS structures (see Fig. 2) and by considering the Hammond postulate. In detail, the calculated high energy TS for Ni(II) can be considered as a textbook example of a “late TS” closer to the product (r(Ni–O) = 2.269 Å; see Fig. 2e). By changing the metal from Ni(II) to Cu(I), the transition state moves towards the reactants as the reaction becomes less endothermic. Thus, the calculated low energy TS for Cu(I) can be considered as an “early TS” closer to the reactants (r(Cu–O) = 3.053 Å; see Fig. 2a). Intermediate situations are reported for Rh and Ir (r(Rh–O) = 2.891 Å and r(Ir–O) = 2.716 Å; Fig. 2b and c, respectively).
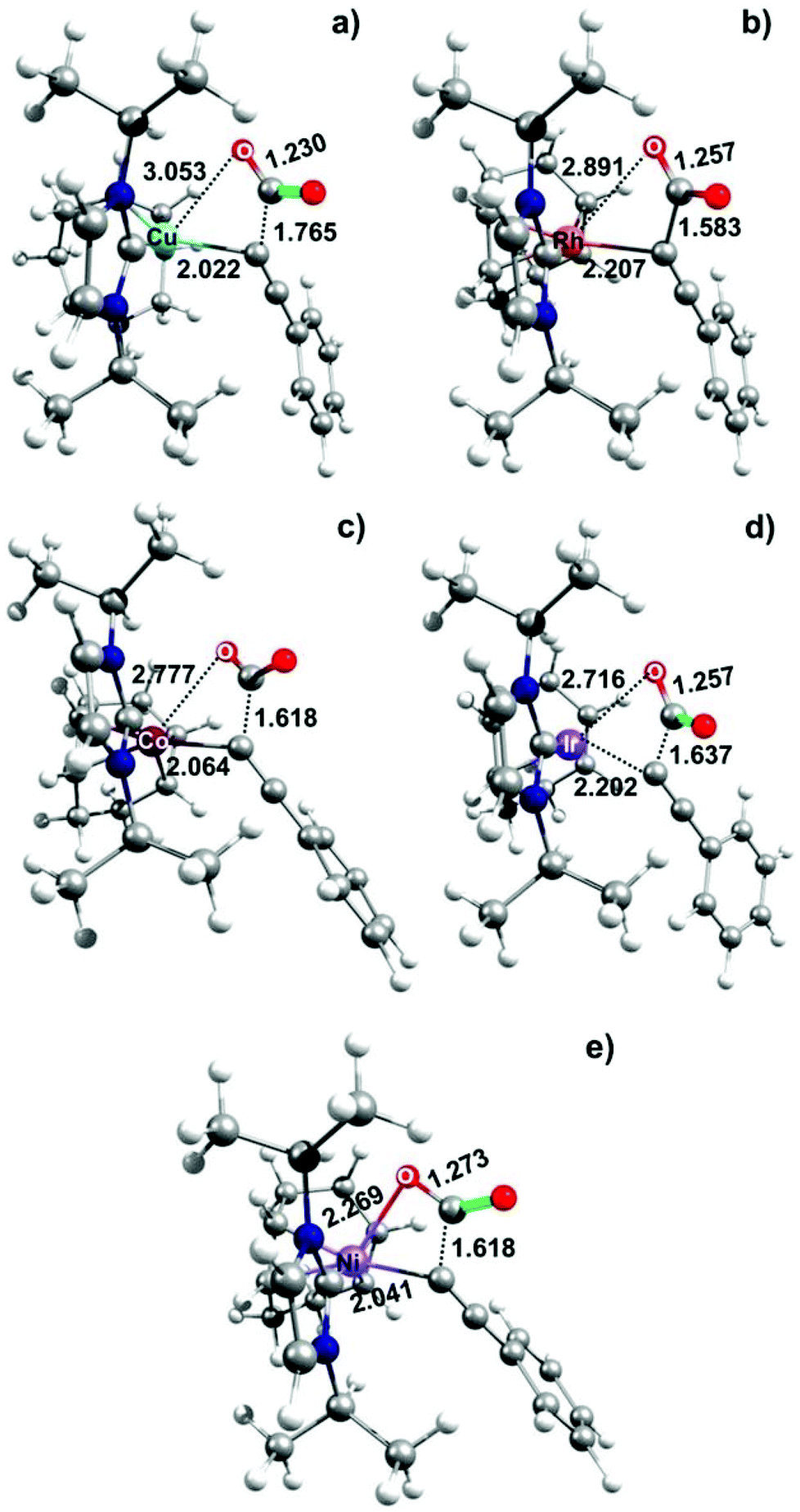 | ||
| Fig. 2 Calculated transition states of the CO2 fixation mechanism promoted by different metals analogues of Ir(I)-complex 8: (a) Cu(I); (b) Rh(I); (c) Co(I); (d) Ir(I); and (e) Ni(II). The most notable distances are given in Å. | ||
Having these results in hand, we then calculated the CO2 insertion barrier for the experimentally known Cu(I) complex, [Cu(IPr)(CCPh)],82 and further, to be compared with other group 11 metals Ag(I) and Au(I). The calculated CO2 insertion barrier follows the trend: Ag(I) (30.9 kcal mol−1) < Cu(I) (34.6 kcal mol−1) < Au(I) (37.0 kcal mol−1), indicating that the Ag(I) based analogue is the best CO2 fixer, followed by Cu(I) and Au(I) based analogues. However, the calculated CO2 insertion barriers are still above 30.0 kcal mol−1, suggesting that the CO2 fixation is kinetically challenging in the considered group 11 metal series. Calculations attempting to screen a longer series of ligands to characterize in detail how the CO2 insertion barrier is affected by the nature of the ligand are ongoing.82,83
Last but not least, even though the energy data include the correction for dispersion implicitly, the geometry optimization is not included, as previously reported,62,79,80 however new tests performed on the transition states for Ni and Cu displayed in Fig. 2 reveal an increase in the energy barrier of +0.4 kcal mol−1 for Ni and a decrease of 0.8 kcal mol−1 for Cu, which suggest that in absolute value the difference is small to negligible and that there is no obvious trend concerning the effect of dispersion on the CO2 insertion barriers.
Conclusions
We have explored the role of metals in the CO2 insertion into a M–C bond by considering various metal (M = Cu(I), Co(I), Rh(I) and Ni(II)) analogues of Ir(I)-complex 8. The results show that among the considered metals, Cu(I) was the best CO2 fixer with an estimated barrier of 20.8 kcal mol−1, while Ni(II) was found to be the least reactive with a barrier of 45.4 kcal mol−1. As a final note, the large spread in the activation energies estimated by changing the metal but with the same ligand scaffold (cod and IiPr ligands) strongly suggests that there is a large space to explore for a suitable combination of ligands and metals to add important components to improve/affect the thermodynamics of this fascinating reaction.Acknowledgements
A.P. thanks the Spanish MINECO for a project CTQ2014-59832-JIN, and European Commission for a Career Integration Grant (CIG09-GA-2011-293900). S.V.C.V., S.P.N. and L.C. thank King Abdullah University of Science and Technology (CCF-1974-3 project) for support. G.T. thanks the KAUST for the visiting contract KAUST-2015-C0546.Notes and references
- M. Aresta, Carbon Dioxide as Chemical Feedstock, Wiley-VCH, Weinheim, 2010 Search PubMed.
- B. Dutcher, M. Fan and A. G. Russell, ACS Appl. Mater. Interfaces, 2015, 7, 2137–2148 CAS.
- C. C. Wang, Y. Q. Zhang, J. Li and P. Wang, J. Mol. Struct., 2015, 1083, 127–136 CrossRef CAS.
- N. von der Assen, P. Voll, M. Peters and A. Bardow, Chem. Soc. Rev., 2014, 43, 7982–7994 RSC.
- V. D'Elia, A. A. Ghani, A. Monassier, J. Sofack-Kreutzer, J. D. A. Pelletier, M. Drees, S. V. C. Vummaleti, A. Poater, L. Cavallo, M. Cokoja, J. M. Basset and F. E. Kuhn, Chem. – Eur. J., 2014, 20, 11870–11882 CrossRef PubMed.
- V. D'Elia, H. Dong, A. J. Rossini, C. M. Widdifield, S. V. C. Vummaleti, Y. Minenkov, A. Poater, E. Abou-Hamad, J. D. A. Pelletier, L. Cavallo, L. Emsley and J. M. Basset, J. Am. Chem. Soc., 2015, 137, 7728–7739 CrossRef PubMed.
- M. Cokoja, C. Bruckmeier, B. Rieger, W. A. Herrmann and F. E. Kuhn, Angew. Chem., Int. Ed., 2011, 50, 8510–8537 CrossRef CAS PubMed.
- C. Song, Catal. Today, 2006, 115, 2–32 CrossRef CAS.
- J. Y. Hu, J. Ma, Q. G. Zhu, Z. F. Zhang, C. Y. Wu and B. X. Han, Angew. Chem., Int. Ed., 2015, 54, 5399–5403 CrossRef CAS PubMed.
- E. Theuergarten, T. Bannenberg, M. D. Walter, D. Holschumacher, M. Freytag, C. G. Daniliuc, P. G. Jones and M. Tamm, Dalton Trans., 2015, 43, 1651–1662 RSC.
- Z. Xin, C. Lescot, S. D. Friis, K. Daasbjerg and T. Skrydstrup, Angew. Chem., Int. Ed., 2015, 54, 6862–6866 CrossRef CAS PubMed.
- W. Leitner, Coord. Chem. Rev., 1996, 153, 257–284 CrossRef CAS.
- D. H. Gibson, Coord. Chem. Rev., 1999, 185–186, 335–355 CrossRef CAS.
- F. J. Fernández-Alvarez, M. Iglesias, L. A. Oro and V. Polo, ChemCatChem, 2013, 5, 3481–3494 CrossRef.
- G. E. Dobereiner, J. Wu, M. G. Manas, N. D. Schley, M. K. Takase, R. H. Crabtree, N. Hazari, F. Maseras and A. Nova, Inorg. Chem., 2012, 51, 9683–9693 CrossRef CAS PubMed.
- H.-W. Suh, T. J. Schmeier, N. Hazari, R. A. Kemp and M. K. Takase, Organometallics, 2012, 31, 8225–8236 CrossRef CAS.
- T. J. Schmeier, A. Nova, N. Hazari and F. Maseras, Chem. – Eur. J., 2012, 18, 6915–6927 CrossRef CAS PubMed.
- A. Behr, Angew. Chem., Int. Ed. Engl., 1988, 27, 661–678 CrossRef.
- T. Herskovitz and L. J. Guggenberger, J. Am. Chem. Soc., 1976, 98, 1615–1616 CrossRef CAS.
- D. W. Lee, C. M. Jensen and D. Morales-Morales, Organometallics, 2003, 22, 4744–4749 CrossRef CAS.
- G. Parkin, Chem. Rev., 2004, 104, 699–768 CrossRef CAS PubMed.
- X. Yin and J. R. Moss, Coord. Chem. Rev., 1999, 181, 27–59 CrossRef CAS.
- J. Langer, W. Imhof, M. J. Fabra, P. García-Orduña, H. Görls, F. J. Lahoz, L. A. Oro and M. Westerhausen, Organometallics, 2010, 29, 1642–1651 CrossRef CAS.
- M. Aresta and A. Dibenedetto, Dalton Trans., 2007, 2975–2992 RSC.
- R. A. Allred, L. H. McAlexander, A. M. Arif and L. M. Berreau, Inorg. Chem., 2002, 41, 6790–6801 CrossRef CAS PubMed.
- N. Kitajima, S. Hikichi, M. Tanaka and Y. Morooka, J. Am. Chem. Soc., 1993, 115, 5496–5508 CrossRef CAS.
- R. Johansson and O. F. Wendt, Organometallics, 2007, 26, 2426–2430 CrossRef CAS.
- Z.-W. Mao, F. W. Heinemann, G. Liehr and R. v. Eldik, Dalton Trans., 2001, 3652–3662 RSC.
- U. Singh, P. Babbar, P. Tyagi and T. Weyhermüller, Transition Met. Chem., 2008, 33, 931–940 CrossRef CAS.
- R. A. Jewsbury, Inorg. Chim. Acta, 1981, 49, 141–143 CrossRef CAS.
- V. D. Bianco, S. Doronzo and N. Gallo, Inorg. Nucl. Chem. Lett., 1979, 15, 187–189 CrossRef CAS.
- R. S. Haszeldine, Science, 2009, 325, 1647–1652 CrossRef CAS PubMed.
- A. Collado, A. Gómez-Suárez, P. B. Webb, H. Kruger, M. Buhl, D. B. Cordes, A. M. Z. Slawin and S. P. Nolan, Chem. Commun., 2014, 50, 11321–11324 RSC.
- B. P. Buffin, A. M. Arif and T. G. Richmond, J. Chem. Soc., Chem. Commun., 1993, 1432–1434 RSC.
- D. S. Glueck, L. J. N. Winslow and R. G. Bergman, Organometallics, 1991, 10, 1462–1479 CrossRef CAS.
- L. J. Newman and R. G. Bergman, J. Am. Chem. Soc., 1985, 107, 5314–5315 CrossRef CAS.
- S. Krogsrud, S. Komiya, T. Ito, J. A. Ibers and A. Yamamoto, Inorg. Chem., 1976, 15, 2798–2805 CrossRef CAS.
- M. H. Chisholm and M. Extine, J. Chem. Soc., Chem. Commun., 1975, 438–439 RSC.
- G. Gattow and W. Behrendt, Angew. Chem., Int. Ed. Engl., 1972, 11, 534–535 CrossRef CAS.
- M. Rahim, C. White, A. L. Rheingold and K. J. Ahmed, Organometallics, 1993, 12, 2401–2403 CrossRef.
- J. M. Seul and S. Park, Bull. Korean Chem. Soc., 2010, 31, 3745–3748 CrossRef CAS.
- S. Park, Bull. Korean Chem. Soc., 2001, 22, 1410–1412 CAS.
- D. J. Darensbourg, G. Groetsch, P. Wiegreffe and A. L. Rheingold, Inorg. Chem., 1987, 26, 3827–3830 CrossRef CAS.
- R. Johansson, M. Jarenmark and O. F. Wendt, Organometallics, 2005, 24, 4500–4502 CrossRef CAS.
- T. J. Schmeier, N. Hazari, C. D. Incarvito and J. A. Raskatov, Chem. Commun., 2011, 47, 1824–1826 RSC.
- M. T. Johnson, J. M. J. Van Rensburg, M. Axelsson, M. S. G. Ahlquist and O. F. Wendt, Chem. Sci., 2011, 2, 2373–2377 RSC.
- F.-Y. Yi, H.-L. Jiang and X.-M. Sun, Chem. Commun., 2015, 51, 8446–8449 RSC.
- G. G. Zhang, Z.-A. Lan and X. C. Wang, ChemCatChem, 2015, 7, 1422–1423 CrossRef CAS.
- T. Kurahashi and S. Matsubara, Acc. Chem. Res., 2015, 48, 1703–1716 CrossRef CAS PubMed.
- I. P. Koronaki, L. Prentza and V. Papaefthimiou, Renewable Sustainable Energy Rev., 2015, 50, 547–566 CrossRef CAS.
- D. Yu, S. P. Teong and Y. Zhang, Coord. Chem. Rev., 2015, 293, 279–291 CrossRef.
- J. Jeromenok, W. Böhlmann, C. Jäger and J. Weber, ChemistryOpen, 2013, 2, 17–20 CrossRef CAS PubMed.
- A. Paparo, J. S. Silvia, C. E. Kefalidis, T. P. Spaniol, L. Maron, J. Okuda and C. C. Cummins, Angew. Chem., Int. Ed., 2015, 54, 9115–9119 CrossRef CAS PubMed.
- S. Marmitt and P. F. B. Goncalves, J. Comput. Chem., 2015, 36, 1322–1333 CrossRef CAS PubMed.
- S. Gennen, M. Alves, R. Mereau, T. Tassaing, B. Gilbert, C. Detrembleur, C. Jerome and B. Grignard, ChemSusChem, 2015, 8, 1845–1849 CrossRef CAS PubMed.
- K. Sokolowski, W. Bury, A. Tulewicz, A. M. Cieslak, I. Justyniak, D. Kubicki, E. Krajewska, A. Milet, R. Moszynski and J. Lewinski, Chem. – Eur. J., 2015, 21, 5496–5503 CrossRef CAS PubMed.
- S. Rasul, D. H. Anjum, A. Jedidi, Y. Minenkov, L. Cavallo and K. Takanabe, Angew. Chem., Int. Ed., 2015, 54, 2146–2150 CrossRef CAS PubMed.
- T. Ema, Y. Miyazaki, J. Shimonishi, C. Maeda and J.-Y. Hasegawa, J. Am. Chem. Soc., 2014, 136, 15270–15279 CrossRef CAS PubMed.
- W. K. Offermans, C. Bizzarri, W. Leitner and T. E. Müller, Beilstein J. Org. Chem., 2015, 11, 1340–1351 CrossRef CAS PubMed.
- T. Fan, X. Chen and Z. Lin, Chem. Commun., 2012, 48, 10808–10828 RSC.
- B. J. Truscott, H. Kruger, P. B. Webb, M. Bühl and S. P. Nolan, Chem. – Eur. J., 2015, 21, 6930–6935 CrossRef CAS PubMed.
- S. V. C. Vummaleti, S. P. Nolan, L. Cavallo, G. Talarico and A. Poater, Eur. J. Inorg. Chem., 2015, 4653–4657 CrossRef CAS.
- B. J. Truscott, D. J. Nelson, C. Lujan, A. M. Z. Slawin and S. P. Nolan, Chem. – Eur. J., 2013, 19, 7904–7916 CrossRef CAS PubMed.
- B. J. Truscott, D. J. Nelson, A. M. Z. Slawin and S. P. Nolan, Chem. Commun., 2014, 50, 286–288 RSC.
- T. Suga, H. Mizuno, J. Takaya and N. Iwasawa, Chem. Commun., 2014, 50, 14360–14363 RSC.
- I. S. Kolomnikov, A. O. Gusev, T. S. Belopotapova, M. Kh. Grigoryan, T. V. Lysyak, Y. T. Struchkov and M. E. Vol'pin, J. Organomet. Chem., 1974, 69, C10–C12 CrossRef CAS.
- K. Ukai, M. Aoki, J. Takaya and N. Iwasawa, J. Am. Chem. Soc., 2006, 128, 8706–8707 CrossRef CAS PubMed.
- B. J. Truscott, G. C. Fortman, A. M. Z. Slawin and S. P. Nolan, Org. Biomol. Chem., 2011, 9, 7038–7041 CAS.
- M. J. Frisch, et al., Gaussian 09, Revision C.01, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- A. Becke, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef CAS.
- J. P. Perdew, Phys. Rev. B: Condens. Matter, 1986, 33, 8822–8824 CrossRef.
- A. Schaefer, H. Horn and R. Ahlrichs, J. Chem. Phys., 1992, 97, 2571–2577 CrossRef CAS.
- U. Haeusermann, M. Dolg, H. Stoll and H. Preuss, Mol. Phys., 1993, 78, 1211–1224 CrossRef.
- W. Kuechle, M. Dolg, H. Stoll and H. Preuss, J. Chem. Phys., 1994, 100, 7535–7542 CrossRef CAS.
- T. Leininger, A. Nicklass, H. Stoll, M. Dolg and P. Schwerdtfeger, J. Chem. Phys., 1996, 105, 1052–1059 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- V. Barone and M. Cossi, J. Phys. Chem. A, 1998, 102, 1995–2001 CrossRef CAS.
- J. Tomasi and M. Persico, Chem. Rev., 1994, 94, 2027–2094 CrossRef CAS.
- A. Poater, E. Pump, S. V. C. Vummaleti and L. Cavallo, J. Chem. Theory Comput., 2014, 10, 4442–4448 CrossRef CAS PubMed.
- J. P. Martínez, M. Solà and A. Poater, ChemistryOpen, 2015 DOI:10.1002/open.201500093.
- G. C. Fortman, A. M. Z. Slawin and S. P. Nolan, Organometallics, 2010, 29, 3966–3972 CrossRef CAS.
- A. H. Christian, P. Müller and S. Monfette, Organometallics, 2014, 33, 2134–2137 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Energies, Cartesian coordinates, and 3D view for all DFT optimized species. See DOI: 10.1039/c5qo00281h |
| This journal is © the Partner Organisations 2016 |