
Bromide anion-triggered visible responsive metallogels based on squaramide complexes†
Di
Wu
*,
Ruyong
Jiang
,
Liang
Luo
,
Zhen
He
and
Jingsong
You
*
Key Laboratory of Green Chemistry and Technology of Ministry of Education, College of Chemistry, Sichuan University, 29 Wangjiang Road, Chengdu 610064, China. E-mail: wood@scu.edu.cn; jsyou@scu.edu.cn
First published on 13th October 2016
Abstract
Driven by the cooperation of metal coordination and multiple hydrogen bonds, a squaramide-based bisimidazole ligand forms structurally novel metallogels involving one-dimensional (1D) nanofibrils with linearly and infinitely extended Cl–Cu or Br–Cu bonds wrapped up with organic ligands. The resulting soft material enables us to discriminate bromides from other salts and, of special significance, from other halides both in aqueous solutions and in powder forms by the naked-eye. Raman spectroscopy clearly unravels the discrimination mechanism associated with a key step of anion ligand exchange, which has witnessed the exceptional ability of Raman spectroscopy in the investigation of metallogels.
Introduction
Coordination polymer gels, also known as metallogels, have drawn extensive attention as a class of functional soft materials owing to their fascinating properties including metal-related photophysical, magnetic, catalytic, and redox properties as well as self-healing ability and host–guest interactions.1 Many interesting metallogels have been reported,2–4 including the gels driven by the coordination between metals and bisimidazole ligands L1–3 developed by us (Scheme 1).3 As a ligand to construct metallogels, the squaramide building block, a structural analogue of urea,2,5 has the following advantages.6 Firstly, squaramide has equal numbers of H-bond donors (N–H) and acceptors (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) and thus excels in the formation of multiple and self-complementary H-bonds. Secondly, the squaramides can pack with each other through π–π interactions between cyclobutenedione rings due to the planarity and aromaticity. More attractively, the two types of non-covalent interactions are fixed perpendicularly to each other by the square geometry of the cyclobutenedione ring, which would be useful to build anisotropic 3D architectures. However, despite extensive applications in catalytic, biological and medicinal realms, the squaramide derivatives remain underrepresented in metallogels. In this work, by combining a squaramide motif with two imidazole units, we assemble the ligand L4 that could have three types of intermolecular interaction capabilities including H-bonding, π–π stacking and metal coordination (Scheme 1).
O) and thus excels in the formation of multiple and self-complementary H-bonds. Secondly, the squaramides can pack with each other through π–π interactions between cyclobutenedione rings due to the planarity and aromaticity. More attractively, the two types of non-covalent interactions are fixed perpendicularly to each other by the square geometry of the cyclobutenedione ring, which would be useful to build anisotropic 3D architectures. However, despite extensive applications in catalytic, biological and medicinal realms, the squaramide derivatives remain underrepresented in metallogels. In this work, by combining a squaramide motif with two imidazole units, we assemble the ligand L4 that could have three types of intermolecular interaction capabilities including H-bonding, π–π stacking and metal coordination (Scheme 1).
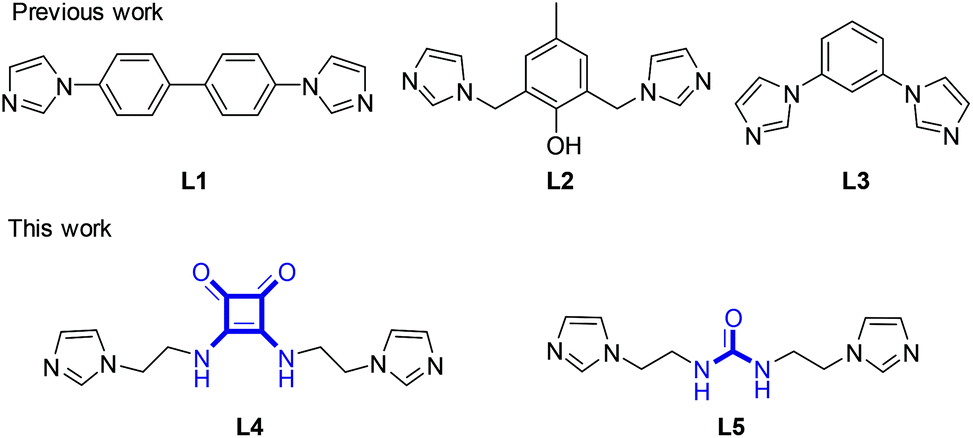 | ||
| Scheme 1 Molecular structures of ligands L1–5. | ||
Experimental
General remarks
1H and 13C NMR spectra were recorded by using a Bruker AV-400 or a Varian Inova spectrometer. The chemical shifts were calibrated using CDCl3, TMS or DMSO-d6 as the internal reference (for 1H, CDCl3: δ = 7.26 ppm; DMSO-d6: δ = 2.50 ppm; for 13C CDCl3: δ = 77.16 ppm; DMSO-d6: δ = 39.52 ppm). The powder X-ray diffraction (PXRD) was performed on a Shimadzu XRD-6100 diffractometer using Cu Kα radiation (where λ is 1.5406 Å) operated at 30 mA and 40 kV. The samples were mounted onto a zero background sample holder and the surface of the powders was leveled using a glass slide. A scan range of 2θ between 5° and 35°, a scan speed of 10° min−1 and a sampling width of 0.02° were employed. The single crystals were easy to lose the crystallized solvent molecules, and thus were covered by Paratone-N oil during data collection. A New Gemini (Dual, Cu at zero) EosS2 diffractometer was used to collect single crystal data at 293(2) K. The structure was solved by Olex2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 7 with the Superflip program8 and refined with the ShelXL9 package (least squares minimization). Due to the unresolvable disorder, residual electron densities possibly corresponding to the solvent molecules and the counterions of the framework were squeezed out.
7 with the Superflip program8 and refined with the ShelXL9 package (least squares minimization). Due to the unresolvable disorder, residual electron densities possibly corresponding to the solvent molecules and the counterions of the framework were squeezed out.
Transmission electron microscopy (TEM) was performed by using a Hitachi H-600, operating at 75 kV. The metallogel was dispersed in EtOH. A drop of this dispersion was placed on a carbon-coated copper grid, stained negatively with aqueous phosphotungstic acid, dried at room temperature and then subjected to TEM observation. A JSM-5900LV scanning electron microscope (SEM) was employed for imaging at 5.0 kV. The xerogel sample for SEM was prepared as follows. The gel was frozen in liquid nitrogen for ca. 10 min and then evaporated under vacuum at −55 °C for 20 h. The obtained xerogel was shielded with gold and then examined. Absorption spectra were not available using the gel sample either sandwiched between quartz plates or scattered in solvents. Diffuse reflectance UV-vis spectroscopy (DR-UV-vis) was performed by using a UV2100 using gel samples sandwiched between two quartz plates. Raman scattering spectra were recorded on a gel sample by using a Horiba HR800 spectrometer with a 532 nm argon ion laser as the exciting light. Elemental analyses were recorded on an EA Flash 1112 analyzer. The inductively coupled plasma-atomic emission spectroscopy (ICP-AES) measurements were performed on an IRIS advantage ER/S.
Chemicals
Unless otherwise noted, reagents were purchased and used without further purification. Diethyl squarate was prepared according to the literature.10L4 and L5 were synthesized via the route presented in Scheme 2.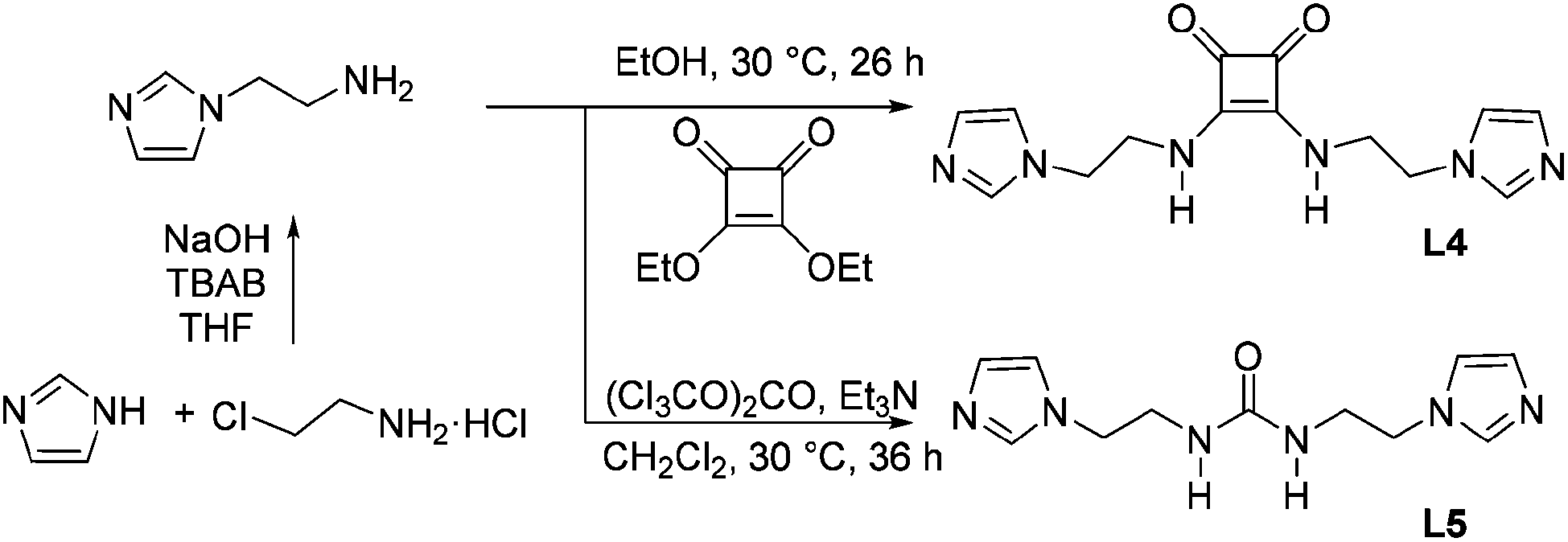 | ||
| Scheme 2 Synthetic routes for L4 and L5. | ||
:1, v/v) to provide 2-(1H-imidazol-1-yl)ethanamine as a pale yellow oil (2.0 g, 60%). 1H NMR (400 MHz, CDCl3): δ = 1.22 (s, 2H), 3.06 (t, J = 6.0 Hz, 2H), 4.01 (t, J = 6.0 Hz, 2H), 6.97 (s, 1H), 7.09 (s, 1H), 7.53 (s, 1H) ppm. 13C NMR (100 MHz, CDCl3): δ = 42.1, 49.5, 118.4, 128.7, 136.7 ppm.
:4, 5.0 mL) solution of L4 (0.40 mmol, 120.1 mg) was added into a MeOH/H2O (v/v = 10:4, 5.0 mL) solution of CuCl2·2H2O (0.2 mmol, 34.1 mg) at 50 °C to afford a turbid mixture. The mixture was kept at the same temperature and a MeOH/H2O (v/v = 10:4) solvent was added until the mixture became clear. After standing overnight at ambient temperature, the crystalline precipitation was filtered and dried under reduced pressure to obtain a green powder (101.6 mg, 69%). NMR spectra were not obtained due to the existence of the paramagnetic Cu(II) center. Elemental analysis and ICP analysis for C28H32Cl2CuN12O4: Calc. C 45.75, H 4.39, N 22.87, Cu 8.65%. Found C 45.10, H 4.83, N 23.35, Cu 8.79%.
The complex [Cu-L42]Br2 was obtained from CuBr2 (0.20 mmol, 44.7 mg) and L4 (0.40 mmol, 120.1 mg) similar to the complex [Cu-L42]Cl2 as a lavender crystalline powder (111.1 mg, 67%). NMR spectra were not obtained due to the existence of the paramagnetic Cu(II) center. Elemental analysis and ICP analysis for C28H32Br2CuN12O4: Calc. C 40.81, H 3.91, N 20.40, Cu 7.71%. Found C 40.81, H 3.98, N 21.28, Cu 7.61%.
Single crystal X-ray diffraction (SXRD)
Single crystals of [Cu-L42]Cl2 were isolated as the precipitate from a slowly cooled solution of L4 (0.2 mmol, 120.1 mg) and CuCl2·2H2O (0.2 mmol, 34.1 mg) in MeOH/H2O (v/v = 10:4). Crystal data: orthorhombic, Pnma, a = 27.9361(13), b = 25.4180(14), c = 6.0524(3) Å, V = 4297.7(4) Å3, Z = 4, T = 293.15 K, R(ref) = 0.0924(3221), wR2(ref) = 0.2698(3922), CCDC 1402219.
Single crystals of [Cu-L42]Br2 were isolated as the precipitate from a slowly cooled solution of L4 (0.40 mmol, 120.1 mg) and CuBr2 (0.20 mmol, 44.7 mg) in MeOH/H2O (v/v = 10:4). Crystal data: orthorhombic, Pcmn, a = 6.06932(11), b = 25.5373(6), c = 27.9983(6) Å, V = 4339.57(16) Å3, Z = 4, T = 293.15 K, R(ref) = 0.0980(3374), wR2(ref) = 0.3134(3967), CCDC 1402142.
Results and discussion
Gelation behavior
Gelation was initially observed after shaking the mixture of the methanol solutions (0.50 mL) of L4 (1.0 mL, 0.050 mmol) and CuCl2·2H2O with molar ratios of 1:0.6–1.4 at 50 °C. A green opaque gel (Gel-Cl) was yielded and no visible collapse was observed over several months at ambient temperature (Fig. S1 and S2†). In contrast, the urea derivative L5 gave rise to precipitation under the same conditions, possibly due to the absence of the π–π interactions between the planar and aromatic cyclobutenedione rings and/or the self-complementary H-bond donor and acceptor. L4 itself did not undergo gelation in various solvents including methanol, ethanol, water, dimethyl formamide (DMF), dimethyl sulphoxide (DMSO) and dimethylacetamide (DMAc), demonstrating that the coordination serves as one of the main driving forces of gelation. The testing of Cu(II) salts revealed that CuBr2 formed a dark purple gel (Gel-Br), whereas CuSO4·5H2O, Cu(OAc)2·H2O, Cu(NO3)2·3H2O, Cu(OOCCF3)2·H2O and Cu(OTf)2 could not provide any metallogels, suggesting that the gel formation is dependent on the nature of the anions. Gelation proceeded well in mixture solvents with methanol. A maximum ratio of 1:10 (v/v) of H2O:MeOH still led to gelation (Table S1†). Gel-Cl and Gel-Br used for the following measurements were prepared by mixing L4 and CuCl2·2H2O or CuBr2 (molar ratio 1:0.6) in MeOH, respectively. Gelators are 1.53 wt% for Gel-Cl and 1.68 wt% for Gel-Br, respectively.
Morphology of the metallogel
The morphology of Gel-Cl was next investigated by TEM and SEM (Fig. 1a and b). Tape-like fibers with widths of approximately 70–200 nm were observed in the TEM image. The SEM image of the xerogel depicts well-grown fibrillar networks with lengths of over ten micrometers. Interestingly, a majority of the fibers show parallel packing, illustrating an ordered assembly on a micrometer-scale.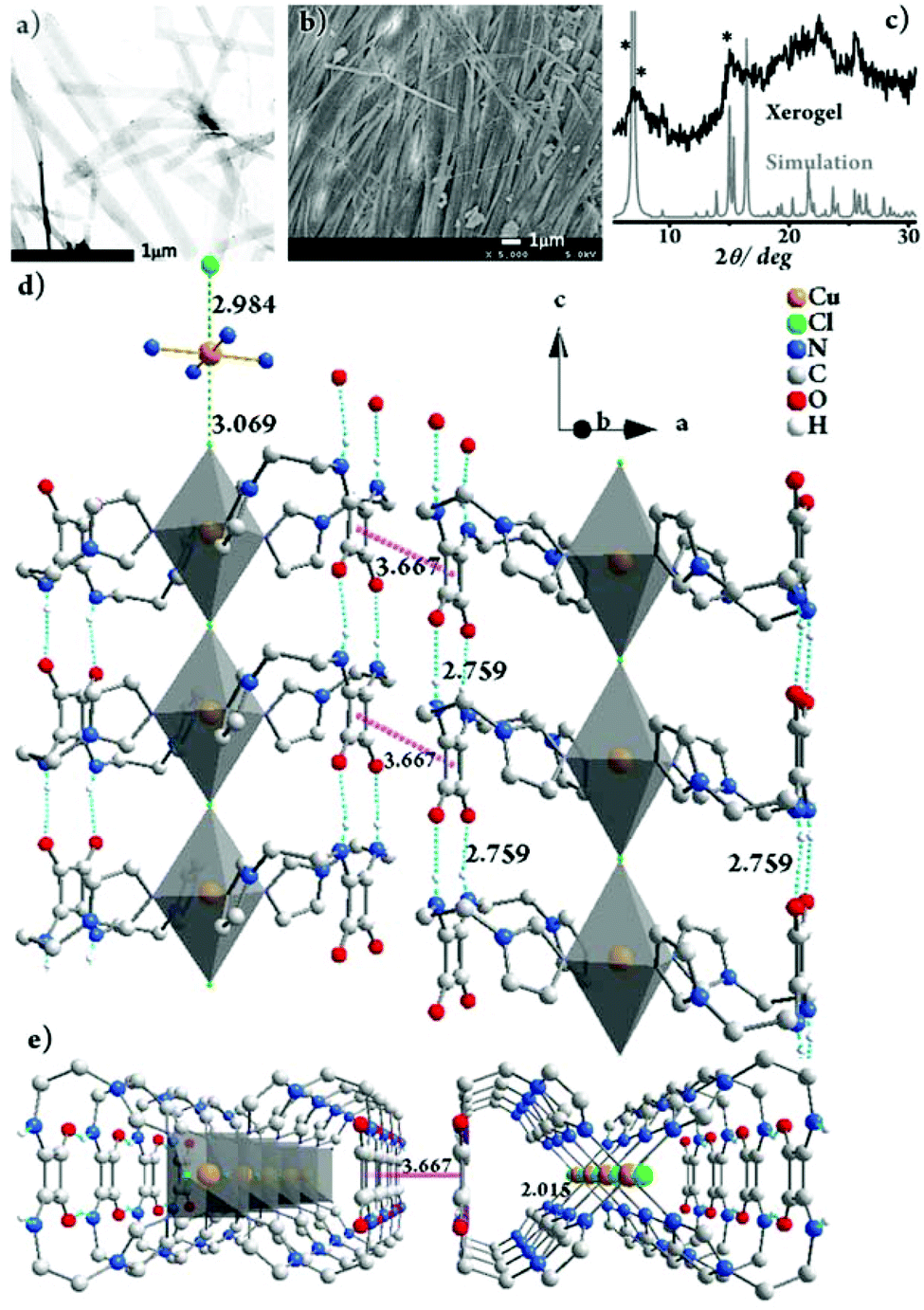 | ||
| Fig. 1 (a) TEM image of Gel-Cl, stained with phosphotungstic acid. (b) SEM image of the xerogel of Gel-Cl. (c) PXRD pattern of the xerogel of Gel-Cl and the simulated pattern from single crystal XRD data of [Cu-L42]Cl2. Star-marked peaks are at 6.33°, 6.96° and 14.64°, respectively. The packing structure of [Cu-L42]Cl2 in crystals viewed along (d) the b-axis, rotated by 20° along the c-axis and (e) the c-axis. The Cu:Cl molar ratio of 1:1 in the framework of single crystals suggests that there is another equivalent of Cl− dissociated from the framework. Residual electron densities possibly corresponding to the counterions were squeezed out due to the unresolvable disorder. H atoms are partially omitted for clarity. For details, see Fig. S3 and S4.† | ||
Gelation mechanism
To better understand the gelation mechanism, we tried to isolate the complex of L4 with CuCl2·2H2O under conditions similar to the gelating environment. Fortunately, the single crystals of [Cu-L42]Cl2 were obtained in H2O/MeOH (v/v = 4:10). The powder X-ray diffraction (PXRD) experiments were subsequently employed to investigate the freeze-dried Gel-Cl (xerogel) (Fig. 1c). Compared with the PXRD pattern simulated from single crystal XRD data, despite the broadening of the xerogel pattern due to less order and smaller particle size in the xerogel than in crystals,2,11 2θ peaks such as those at 6.33°, 6.96° and 14.64° similarly appear in both the patterns, implying the resemblance of molecular packing in the gel and in crystals.
The gelation mechanism of Gel-Cl is next proposed. Two L4 molecules bend over their flexible alkyl chains to bidentately coordinate the Cu center to four imidazolyl motifs, forming a central symmetric, figure eight-shaped mononuclear complex (Cu-L42)2+. Four chelating nitrogen atoms and a cupric center are situated in the crystallographic ab plane with a typical and identical N–Cu distance of 2.015 Å.12 Two Cl atoms axially coordinate to Cu with unusually long distances of 2.984 and 3.069 Å, forming an axially elongated octahedral configuration (Fig. 1d, e and S3, 4†).11 By sharing apical Cl atoms, CuN4Cl2 octahedra line up in a row along the crystallographic c-axis. The Cl–Cu bonds linearly and infinitely extend to form a one dimensional (1D) Cl–Cu line wrapped up with organic ligand L4, which is rarely reported.12 It could also be described as a Cl-bridged dicopper(II) complex with a short intermetallic distance of 6.053 Å, which may be appealing in the emerging fields of molecular spintronics and quantum computation.13 Vertically situated squaramides stretch their hydrogen donors (N–H) and acceptors (CO) along the c-axis to form four equivalent H-bonds with a N⋯O distance of 2.759 Å. The cooperation of metal coordination and multiple H-bonds is robust enough to give rise to 1D-fibril formation. A number of 1D-fibrils (approximately 35–140) parallelly aggregate to form 2D-sheets through the π–π interaction of squaramides with a distance of 3.667 Å between the centroids (Fig. 1d). 2D-sheets then pack into a 3D-framework through van der Waals interactions, solidifying solvents to form a gel (Fig. 2). Single crystals of [Cu-L42]Br2 were also successfully isolated for comparison. Resemblance is found in two single crystals with a similar intermetallic distance (6.069 Å in [Cu-L42]Br2), 1D-fibrils, 2D-sheets and 3D-frameworks (Fig. 3 and S5–7†). Different from the exceptional length of the Cl–Cu coordination bond,12 those of Br–Cu (3.010 and 3.059 Å) are consistent with the similar systems in the literature.14
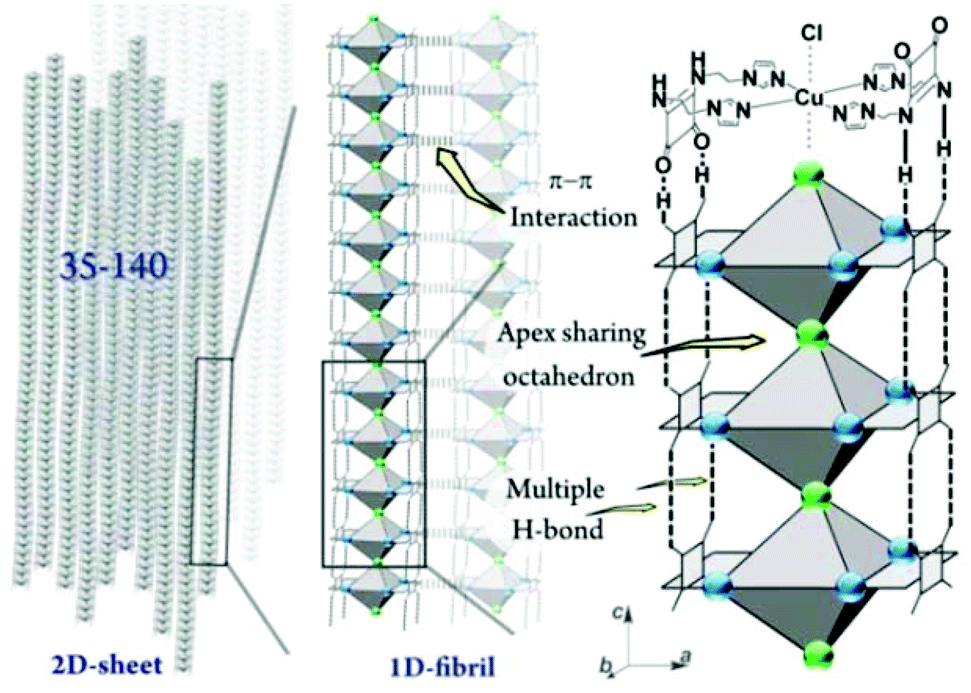 | ||
| Fig. 2 Schematic representation of the proposed gelation mechanism of Gel-Cl. | ||
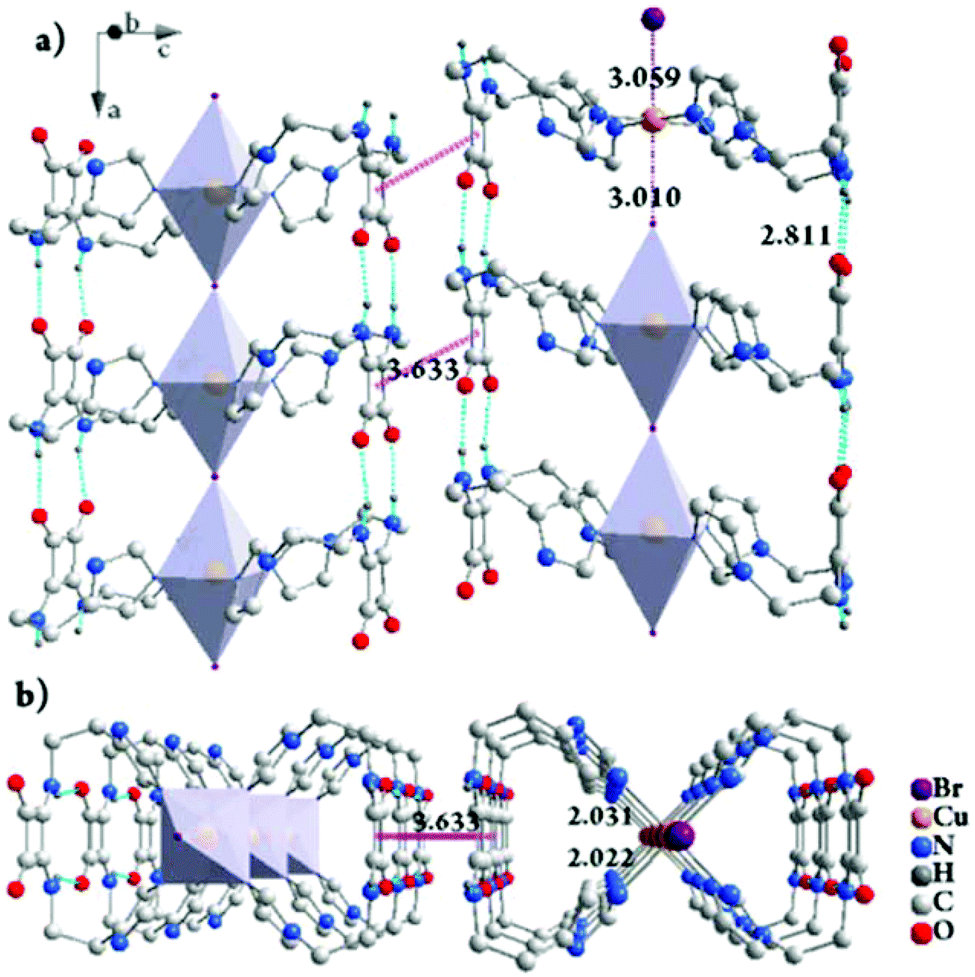 | ||
| Fig. 3 The packing structure of [Cu-L42]Br2 in crystals viewed along the b-axis, rotated by 20° along the a-axis (a) and the a-axis (b). The Cu:Br molar ratio of 1:1 in the framework of single crystals suggests that there is another equivalent of Br− dissociated from the framework. Residual electron densities possibly corresponding to the counterions were squeezed out due to the unresolvable disorder. H atoms are partially omitted for clarity. For details, see Fig. S6 and S7.† | ||
Bromide anion-triggered visible responses
Despite the environmental harmfulness, biological toxicity and wide distribution of bromide anions in aqueous solutions and in powder forms in the biosphere, a convenient method to discriminate bromide salts from other halides remains less developed.15 The challenge mainly lies in the chemical similarity of halides, which often show close chemical behaviors. Considering that two metallogels display structural resemblance and visually distinguishable color difference, we tested the feasibility of discriminating the Br− anion. First, aqueous solutions of various sodium salts were investigated. Gladly, a pale purple color immediately appeared (within ca. 4 seconds) upon the addition of a NaBr solution into Gel-Cl, whereas other anions led to blue color (Fig. 4a and S8–11†). The purple color could remain over three months, even after further adding chloride or drying in air (Fig. S12 and 13†). The generality of the Br− source is elucidated by adding a variety of bromide salts including inorganic, ammonium, phosphonium and imidazolium salts. To further expand the application scope, bromide powders were directly added into Gel-Cl. To our delight, the stimuli-responsive color change occurred and was even more easily observed than in solutions, which demonstrates an exceptional advantage of a wet and soft material. All these features make Gel-Cl a smart soft material for rapid and selective discrimination of bromides from other salts and, of special significance, from other halides.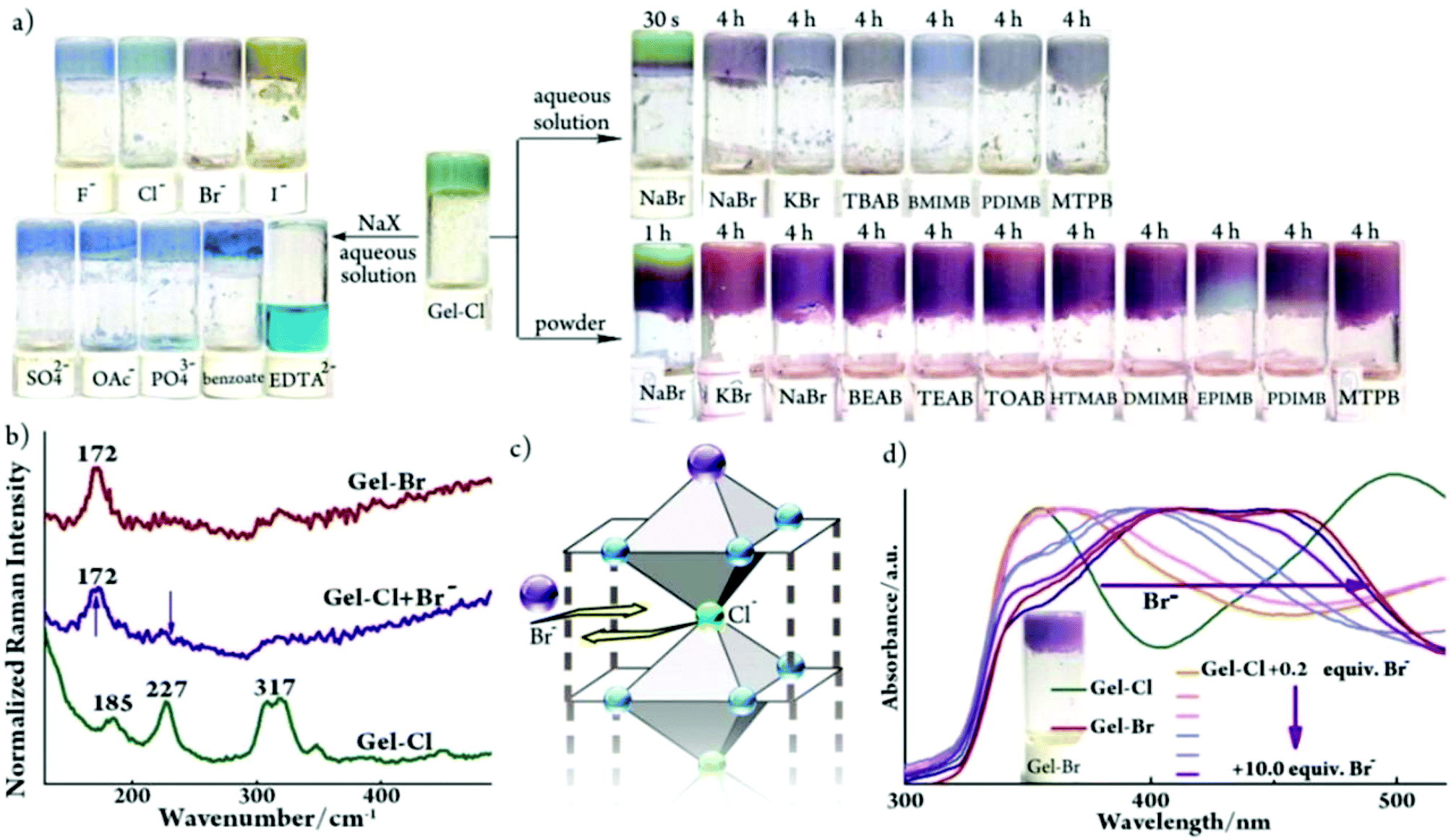 | ||
| Fig. 4 (a) Samples of Gel-Cl upon addition of aqueous sodium salts (2.4 mol L−1, 70 μL), aqueous bromide salts (2.4 mol L−1, 70 μL) and bromide powders (10.0 equiv.). TBAB: tetrabutylammonium bromide; BMIMB: 1-butyl-3-methylimidazolium bromide; PDIMB: 1-propyl-2,3-dimethylimidazolium bromide; MTPB: methyl triphenyl phosphonium bromide; BEAB: 2-bromoethyl-1-ammonium bromide; TEAB: tetra-ethylammonium bromide; TOAB: tetra-n-octylammonium bromide; HTMAB: hexadecyl trimethyl ammonium bromide; DMIMB: 1-dodecyl-3-methylimidazolium bromide; EPIMB: 1-ethyl-3-(4-N,N-dimethylphenyl)imidazolium bromide; and EDTA2−: ethylene diamine tetraacetate. (b) Raman and (d) DR-UV-vis spectra of Gel-Br and Gel-Cl before and after adding Br−. Inserted in (d) is a photo of Gel-Br. (c) Schematic representation of anion ligand exchange. For experimental details, see Fig. S8.† | ||
Mechanism for bromide anion-triggered responses
As illustrated in Fig. 1d, the crystal structure of [Cu-L42]Cl2 shows the exceptionally long Cu–Cl bonds, suggesting a relatively weak Cu–Cl coordination.12 Considering the structural similarity between [Cu-L42]Cl2 and [Cu-L42]Br2 and the rarely observed gel-to-gel transition of Gel-Cl after adding bromides, we envisage that the exogenous bromide anion could enter into the framework of a 1D-fibril and replace the Cl− anion that axially coordinates to the cupric center. It is then necessary to find evidence to confirm the coordination interaction between Br− and the Cu center in the gel. However, despite significant progress, it remains a huge challenge to understand the structure of the gel at the molecular level mainly because common techniques that are often used to investigate bond connections in the solid state and in solution are not applicable for metallogel samples.4 For example, the NMR technique could be invalidated by paramagnetic metal centers, including often used Cu(II), Fe(III), Mn(II) and Co(II). Raman spectroscopy is a useful tool to distinguish the Cu–Br coordination bond from the electrostatic interaction between Cu2+ and Br− as well as to detect the difference between Cu–Br and Cu–Cl coordination bonds.16 Thus, Raman spectroscopy was employed to gain insight into the discrimination mechanism. The sample of Gel-Cl + Br− for the Raman test was prepared by mixing solid NaBr (10 equiv. relative to Cl species in Gel-Cl) with Gel-Cl and aging overnight. As depicted in Fig. 4b, the addition of Br− into Gel-Cl leads to the disappearance of the peak at 227 cm−1 corresponding to the vibration of the Cu–Cl coordination bond and meanwhile causes a new peak at 172 cm−1 corresponding to the vibration of the Cu–Br coordination bond, which is similar to the pattern of Gel-Br.12 These observations suggest that the bromide-triggered color switching of Gel-Cl could mainly be attributed to the Cl− ligand exchange with Br− rather than the extra electrostatic interaction between dissociated bromide ions and frameworks (Fig. 4c).The diffuse reflectance UV-visible (DR-UV-vis) test was performed upon Gel-Cl samples overnight after adding increasing equivalents of solid NaBr. As shown in Fig. 4d, the addition of bromide into Gel-Cl gives rise to a substantial red-shift of absorption band edges from 435 to 560 nm, very close to that of Gel-Br. Based on the mechanism of anion ligand exchange, we propose that color switching could originate from either the changed d–d transition energies ascribed to the modification of the coordination environment of the cupric center or the ligand-to-metal charge transfer (LMCT) from different ligands (Cl− or Br−). Density functional theory (DFT) calculations were thus performed to show that the energy gaps between the HOMO and LUMO reduce dramatically when the ligand changes from Cl− to Br−, explaining the color change (Fig. S14 and S15†).
Conclusions
In conclusion, structurally novel metallogels are constructed by the assembly of squaramide-based bisimidazole ligand with copper(II) halides. The gelation mechanism involves the formation of one-dimensional (1D) nanofibrils with linearly and infinitely extended Cl–Cu or Br–Cu bonds wrapped up with organic ligands. Such 1D linear assemblies centered with a Cu(II)-containing line are rarely reported, especially in metallogels. Gel-Cl is selectively responsive to bromides to display purple color, which proceeds in a way of rarely reported gel-to-gel transition. The stability in the gel-to-gel transition is dependent on the structural similarity between the two frameworks. Based on this property, we develop a soft material for convenient and selective discrimination of bromide anions from other anions, especially for other halides. Raman spectroscopy offers insights into the recognition mechanism with a key step of anion ligand exchange. We believe that Raman spectroscopy would be an ideal tool to better understand the gelation mechanism.Acknowledgements
This work was supported by grants from the National NSF of China (no. 21432005 and 21321061). We acknowledge the Comprehensive Training Platform of Specialized Laboratory, College of Chemistry, Sichuan University for measurements.Notes and references
- For selected reviews, see: N. M. Sangeetha and U. Maitra, Chem. Soc. Rev., 2005, 34, 821 RSC; F. Fages, Angew. Chem., Int. Ed., 2006, 45, 1680 CrossRef CAS PubMed; M.-O. M. Piepenbrock, G. O. Lloyd, N. Clarke and J. W. Steed, Chem. Rev., 2010, 110, 1960 CrossRef PubMed; G. R. Whittell, M. D. Hager, U. S. Schubert and I. Manners, Nat. Mater., 2011, 10, 176 CrossRef PubMed; A. Y.-Y. Tam and V. W.-W. Yam, Chem. Soc. Rev., 2013, 42, 1540 RSC; J. Zhang and C.-Y. Su, Coord. Chem. Rev., 2013, 257, 1373 CrossRef; J. H. Jung, J. H. Lee, J. R. Silverman and G. John, Chem. Soc. Rev., 2013, 42, 924 RSC; T. Tu, W. Fang and Z. Sun, Adv. Mater., 2013, 25, 5304 CrossRef PubMed; S. Dong, B. Zheng, F. Wang and F. Huang, Acc. Chem. Res., 2014, 47, 1982 CrossRef PubMed.
- For selected samples of metallogels, see: J. B. Beck and S. J. Rowan, J. Am. Chem. Soc., 2003, 125, 13922 CrossRef CAS PubMed; M. Vondrova, T. M. McQueen, C. M. Burgess, D. M. Ho and A. B. Bocarsly, J. Am. Chem. Soc., 2008, 130, 5563 CrossRef PubMed; A. Y.-Y. Tam, K. M.-C. Wong and V. W.-W. Yam, J. Am. Chem. Soc., 2009, 131, 6253 CrossRef PubMed; X. Chen, Z. Huang, S.-Y. Chen, K. Li, X.-Q. Yu and L. Pu, J. Am. Chem. Soc., 2010, 132, 7297 CrossRef PubMed; H. Lee, S. H. Jung, W. S. Han, J. H. Moon, S. Kang, J. Y. Lee, J. H. Jung and S. Shinkai, Chem. – Eur. J., 2011, 17, 2823 CrossRef PubMed; T. Tu, W. Fang, X. Bao, X. Li and K. H. Dötz, Angew. Chem., Int. Ed., 2011, 50, 6601 CrossRef PubMed; L. Meazza, J. A. Foster, K. Fucke, P. Metrangolo, G. Resnati and J. W. Steed, Nat. Chem., 2013, 5, 42 CrossRef PubMed; L. Li, S. Xiang, S. Cao, J. Zhang, G. Ouyang, L. Chen and C. Su, Nat. Commun., 2013, 4, 1774 CrossRef PubMed; X. Yan, T. R. Cook, J. B. Pollock, P. Wei, Y. Zhang, Y. Yu, F. Huang and P. J. Stang, J. Am. Chem. Soc., 2014, 136, 4460 CrossRef PubMed; W. Fang, X. Liu, Z. Lu and T. Tu, Chem. Commun., 2014, 50, 3313 RSC; X.-Q. Wang, W. Wang, G.-Q. Yin, Y.-X. Wang, C.-W. Zhang, J.-M. Shi, Y. Yu and H.-B. Yang, Chem. Commun., 2015, 51, 16813 RSC; A. V. Zhukhovitskiy, M. Zhong, E. G. Keeler, V. K. Michaelis, J. E. P. Sun, M. J. A. Hore, D. J. Pochan, R. G. Griffin, A. P. Willard and J. A. Johnson, Nat. Chem., 2015, 8, 33 CrossRef PubMed; S. C. Grindy, R. Learsch, D. Mozhdehi, J. Cheng, D. G. Barrett, Z. Guan, P. B. Messersmith and N. Holten-Andersen, Nat. Mater., 2015, 14, 1210 CrossRef PubMed; M. Xue, Y. Lü, Q. Sun, K. Liu, Z. Liu and P. Sun, Cryst. Growth Des., 2015, 15, 5360 Search PubMed; M. Martínez-Calvo, O. Kotova, M. E. Möbius, A. P. Bell, T. McCabe, J. J. Boland and T. Gunnlaugsson, J. Am. Chem. Soc., 2015, 137, 1983 CrossRef PubMed; P. Chen, Q. Li, S. Grindy and N. Holten-Andersen, J. Am. Chem. Soc., 2015, 137, 11590 CrossRef PubMed; J. Sun, Y. Liu, L. Jin, T. Chen and B. Yin, Chem. Commun., 2016, 52, 768 RSC.
- S. Zhang, J. Lan, Z. Mao, R. Xie and J. You, Cryst. Growth Des., 2008, 8, 3134 Search PubMed; S. Zhang, S. Yang, J. Lan, S. Yang and J. You, Chem. Commun., 2008, 6170 RSC; S. Zhang, S. Yang, J. Lan, Y. Tang, Y. Xue and J. You, J. Am. Chem. Soc., 2009, 131, 1689 CrossRef CAS PubMed; L. Yang, L. Luo, S. Zhang, X. Su, J. Lan, C.-T. Chen and J. You, Chem. Commun., 2010, 46, 3938 RSC.
- J. Brandt, K. K. Oehlenschlaeger, F. G. Schmidt, C. Barner-Kowollik and A. Lederer, Adv. Mater., 2014, 26, 5758 CrossRef CAS PubMed.
- J. W. Steed, Chem. Soc. Rev., 2010, 39, 3686 RSC.
- R. I. Storer, C. Aciro and L. H. Jones, Chem. Soc. Rev., 2011, 40, 2330 RSC; J. Alemán, A. Parra, H. Jiang and K. A. Jørgensen, Chem.−Eur. J., 2011, 17, 6890 CrossRef PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339 CrossRef CAS.
- L. Palatinus and G. Chapuis, J. Appl. Crystallogr., 2007, 40, 786 CrossRef CAS; L. Palatinus and A. van der Lee, J. Appl. Crystallogr., 2008, 41, 975 CrossRef; L. Palatinus, S. J. Prathapa and S. van Smaalen, J. Appl. Crystallogr., 2012, 45, 575 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- H.-B. Zhou, J. Zhang, S.-M. Lü, R.-G. Xie, Z.-Y. Zhou, M. C. K. Choi, A. S. C. Chan and T.-K. Yang, Tetrahedron, 2001, 57, 9325 CrossRef CAS.
- M.-O. M. Piepenbrock, N. Clarke and J. W. Steed, Langmuir, 2009, 25, 8451 CrossRef CAS PubMed.
- Cu–Cl bond lengths are normally about 2.3–2.9 Å in CuN4Cl2 octahedra. See: M. Lagrenee, S. Sueur, J. P. Wignacourt, B. Mernari and A. Boukhari, J. Chim. Phys. Phys.-Chim. Biol., 1991, 88, 2075 Search PubMed; P. Comba, S. M. Luther, O. Maas, H. Pritzkow and A. Vielfort, Inorg. Chem., 2001, 40, 2335 CrossRef CAS PubMed; S. Banerjee and P. Dastidar, CrystEngComm, 2013, 15, 9415 RSC; R. Atencio, K. Ramirez, J. A. Reyes, T. Gonzalez and P. Silva, Inorg. Chim. Acta, 2005, 358, 520 CrossRef.
- J. Gómez-Herrero and F. Zamora, Adv. Mater., 2011, 23, 5311 CrossRef CAS; M. Castellano, R. Ruiz-García, J. Cano, J. Ferrando-Soria, E. Pardo, F. R. Fortea-Pérez, S.-E. Stiriba, M. Julve and F. Lloret, Acc. Chem. Res., 2015, 48, 510 CrossRef PubMed.
- Cu–Br bond lengths are normally about 2.8–3.2 Å in CuN4Br2 octahedra. See: C. Nather, M. Wriedt and I. Jess, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2002, 58, m107 Search PubMed; S. Bieller, A. Haghiri, M. Bolte, J. W. Bats, M. Wagner and H.-W. Lerner, Inorg. Chim. Acta, 2006, 359, 1559 CrossRef CAS; V. Kubicek, I. Rehor, J. Havlickova, J. Kotek, I. Cisarova, P. Hermann and I. Lukes, Eur. J. Inorg. Chem., 2007, 3881 CrossRef.
- F. Qu, N. B. Li and H. Q. Luo, Anal. Chem., 2012, 84, 10373 CrossRef CAS PubMed; L. Xu, Y. Xu, W. Zhu, Z. Xu, M. Chen and X. Qian, New J. Chem., 2012, 36, 1435 RSC.
- L. V. Stepakova, M. Y. Skripkin, L. V. Chernykh, G. L. Starova, L. Hajba, J. Mink and M. J. Sandström, Raman Spectrosc., 2008, 39, 16 CrossRef CAS; W. J. Gee and S. R. Batten, Chem. Commun., 2012, 48, 4830 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1402219 and 1402142. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6qi00386a |
| This journal is © the Partner Organisations 2016 |