
A sponge network-shaped Mn3O4/C anode derived from a simple, one-pot metal organic framework-combustion technique for improved lithium ion storage†
Balaji
Sambandam‡
,
Vaiyapuri
Soundharrajan‡
,
Jinju
Song
,
Sungjin
Kim
,
Jeonggeun
Jo
,
Duong Pham
Tung
,
Seokhun
Kim
,
Vinod
Mathew
and
Jaekook
Kim
*
Department of Materials Science and Engineering, Chonnam National University, 300 Yongbong-dong, Bukgu, Gwangju 500-757, South Korea. E-mail: jaekook@chonnam.ac.kr; Fax: +82-62-530-1699; Tel: +82-62-530-17
First published on 8th October 2016
Abstract
A sponge network-shaped Mn3O4 material is synthesized by a one-pot metal organic framework-combustion (MOF-C) technique for Li-ion battery anodes with improved performance. The as-synthesized ordered sponge network morphology is characterized by various techniques, such as powder X-ray diffraction, scanning electron microscopy, transmission electron microscopy, Raman spectroscopy, X-ray photoelectron spectroscopy, and N2 adsorption–desorption measurements. The one-pot synthesized Mn3O4 material shows a uniform amorphous graphitic carbon coating with few-nanometer thickness on the surface. This anode shows an initial discharge capacity of 1186 mA h g−1 and a reversible capacity of 768 mA h g−1 is maintained at an applied current density of 200 mA g−1 after 100 cycles. Sustained reversible capacities of 651 and 592 mA h g−1 are measured for the other two different current densities of 500 and 700 mA g−1, respectively, after 120 cycles, demonstrating the high stability of the anode. This unique morphology appears to contribute to the significantly high rate performance, as observed from the retained reversible capacity of 155 mA h g−1 at a very high current density of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 mA g−1, which is maintained for the next two subsequent sequences with a notable recovered capacity of 700 mA h g−1 for an intermediate current density of 400 mA g−1 after 175 cycles.
000 mA g−1, which is maintained for the next two subsequent sequences with a notable recovered capacity of 700 mA h g−1 for an intermediate current density of 400 mA g−1 after 175 cycles.
Introduction
At present, Li-ion batteries (LIBs) are one of the most promising and powerful sources for both electronics and electrical applications.1 Therefore research on their internal components including electrodes have taken center stage presently. In particular, suitable anode materials that can act as alternatives to graphite with high specific capacity and excellent electrical conductivity are highly in demand for carbon-free electric vehicles, hybrid electric vehicles, and plug-in hybrid electric vehicles.2,3 In this aspect, transition metal oxides (TMOs) are attractive due to their ease of preparation, relatively high theoretical specific capacitance, low cost, and stability through conversion reaction mechanisms.4,5 Among the metal oxides, Mn3O4 has been widely studied as an anode for LIB applications owing to its high specific capacity (its theoretical capacity is 937 mA h g−1, which is almost three times greater than that of graphite), low discharge potential, high abundance, low cost, and high stability. However, the intrinsic low electrical conductivity (10−7–10−8 S cm−1) and the drastic volume changes during electrochemical cycling tend to deteriorate the electrode performance of Mn3O4.6 Hence, different approaches including carbon-additive composites and metal ion doping have been employed by researchers in the last few years to overcome these barriers and improve the properties of Mn3O4.7–10Wang et al. reported a Mn3O4–graphene composite anode for lithium ion batteries; however, the synthetic process was complex and graphene is expensive.11 Hao et al. recently demonstrated that a Mn3O4 octahedral microparticle electrode, which was prepared by a de-alloying corrosion method, exhibited an ultralong cycle life with a capacity retention of 746 mA h g−1 and 620 mA h g−1 even after 500 cycles at current densities of 100 and 300 mA g−1, respectively.12 Moreover, various efforts to enhance the electrochemical properties of Mn3O4 by tuning particle morphologies are seen in the literature. Wang et al. found that an order-aligned Mn3O4 anode delivered an initial capacity of up to 637 mA h g−1 and maintained a capacity of 494 mA h g−1 after 100 cycles.13 In a step forward, Jian et al. developed hollow spheres of Mn3O4 in an anode that delivered a highly stable cycle performance with a capacity retention of ∼980 mA h g−1 over 140 cycles at a current density of 200 mA g−1, and showed an excellent rate capability of 300 mA h g−1 at a high current density of 10000 mA g−1.14 Gao et al. fabricated sponge-like Mn3O4 nanomaterials using a precipitation method, which exhibited a high initial reversible capacity of 869 mA h g−1 and good cyclability over 40 cycles.15 Even at 10C (1C = 937 mA h g−1), the material could deliver a capacity close to 500 mA h g−1. Recently, Alfaruqi et al. prepared a nanostructured Mn3O4/C anode material by a one-pot pyro-polyol technique that showed a stable capacity retention of 1141 mA h g−1 with 100% Coulombic efficiency (CE) at the 100th cycle at a low current density of 42 mA g−1.16 Park et al. reported Mn3O4 coaxial nanocables with carbon (hierarchically mesoporous carbon nanofibers) additives that delivered a high reversible capacity of 760 mA h g−1 at a current density of 100 mA g−1 for 50 cycles, whose specific capacity was greatly enhanced in the presence of the carbon additive.17 By a facile approach, Bai et al. prepared mesoporous Mn3O4 nanotubes which exhibited a reversible capacity as high as 641 mA h g−1 at a high current density of 500 mA g−1 after 100 cycles.18 In addition, a few more studies have been dedicated to the enhancement of the performance of Mn3O4 using carbon-based additives.19,20 Thus, most of the above mentioned Mn3O4 electrodes bear carbon additives in the form of graphite, mesoporous carbon, carbon quantum dots (QDs), carbon nanotubes (CNTs), and other available forms for improved performance. However, this desired morphological tuning is mostly achieved by tedious and/or time-consuming synthesis and/or post heat treatment processes. Also worth noting is that the realization of practical specific capacities as high as their theoretical values still remain challenging in the case of Mn3O4 electrodes. The above reasoning motivates the call for further strategical tuning of the lithium storage properties in this particular anode.
Therefore, with an aim to realize electrodes that can facilitate the promotion of better reactivity with lithium by utilizing simple strategies, herein we introduce a nanostructured Mn3O4 electrode with excellent cyclability and superior rate capability at high current densities. This sponge network-shaped Mn3O4 electrode is prepared by a simple, robust, and rapid technique called “metal organic framework-combustion” (MOF-C).21 The advantage of this technique is that this Mn3O4 electrode is directly synthesized under very short reaction times similar to that of microwave digestion. Also, the anodic performance of this sponge network-shaped electrode compares well with other reported studies with and without carbon additives as supporting materials. The presented technique may provide an opportunity for simple, scale-up, and cost-effective strategies for nanomaterial synthesis with very short reaction times.
Experimental section
Synthesis of Mn3O4/C with a sponge-network morphology
In a typical procedure, 0.5 mmol of manganese acetate was mixed with 0.5 mmol of terephthalic acid (benzene dicarboxylic acid, BDC) in 20 ml of DMF. After stirring for 20 min, 5 ml of paint thinner was added, and the mixture stirred for 10 more minutes, resulting in a homogeneous clear solution. This was then transferred into an aluminum boat made from commercially available aluminum foil, filled to 35–40% of its capacity, and then heated to 250–300 °C by simultaneous ignition with the help of an external igniter (caution: Take minimum volume to avoid bumping). The as-synthesized material was used directly for further characterization and electrochemical application.Structure and morphology characterization
Powder X-ray diffraction measurements (PXRD) were performed using a Shimadzu X-ray diffractometer (Cu Kα radiation, λ = 1.5406 Å). The surface morphology and the lattice fringe were analyzed by field emission scanning electron microscopy (FE-SEM, S-4700 Hitachi with an EDS detector) and field emission transmission electron microscopy (FE-TEM, Philips Tecnai F20 at 200 kV in KBSI Chonnam National University) equipped with selected area electron diffraction (SAED), respectively. The elemental oxidation states were examined by X-ray photoelectron spectroscopy (XPS, Thermo VG Scientific instrument, Multilab 2000 in the Chonnam Center for Research Facilities) using Al Kα as the X-ray source. The spectrometer was calibrated with respect to the C 1s peak binding energy of 284.6 eV. The surface area and pore size distribution of the samples were determined based on the nitrogen adsorption and desorption isotherms using Brunauer–Emmett–Teller method (BET, Micromeritics ASAP2010 Instrument Co., Norcross, GA, USA). Raman spectra were measured using a JASCO Raman spectrometer, NRS-5100 with a 532 nm laser line. Electrochemical impedance spectroscopy (EIS) measurements were carried out using an AUTOLAB potentiostat (PGSTAT302N) instrument. The carbon content was determined by Thermogravimetric analysis (TGA) using an SDT Q600 thermobalance (USA, Chonnam National University, South Korea) in air with a temperature change of 5 °C min−1.Electrochemical characterization
Electrochemical experiments were performed with 2032 coin-type cells using Li foil as the counter electrode. The working electrode was prepared by mixing the active material, Super P, and sodium alginate as a binder in deionized water to form a slurry at a weight ratio of 75:15:10. The obtained mixture was then uniformly pasted onto a pure Cu foil current collector and dried overnight under vacuum. The dried material was then pressed between stainless steel twin rollers at room temperature. The foil was then punched into circular discs (the active material mass loading was found to be 1.35–1.5 mg), and the coin cells were assembled with lithium metal as the counter electrode and a membrane (Celgard 2400), together with a glass fiber separator. The specific capacity was calculated based on the whole Mn3O4/C electrode inclusive of the carbon content. The electrolyte was 1 M LiPF6 in ethylene carbonate:dichloromethane (1:1 by volume). Charge/discharge cycles were performed on a BTS-2004H (Nagano, Japan) battery test instrument at 0.01–3.0 V. Cyclic voltammetry (CV) measurements with the electrode were performed using a Bio Logic Science Instrument (VSP 1075) at a scan rate of 0.2 mV s−1.
Results and discussion
The PXRD pattern of the sample synthesized by the MOF-C technique, in Fig. 1, corresponds to a tetragonal phase of Mn3O4 with high crystallinity. In other words, all of the diffraction peaks have been indexed exactly to the standard (bulk) hexagonal crystal system of Mn3O4 (JCPDS 80-0382). Specifically, the diffraction peaks at 2θ angles of 18.0, 28.9, 30.9, 32.2, 36.1, 38.1, 44.4, 50.9, 53.7, 56.0, 58.4, 60.0, and 64.4° correspond to the (101), (112), (200), (103), (211), (004), (220), (105), (312), (303), (321), (224), and (400) planes, respectively, which suggest the presence of highly pure Mn3O4 without any notable impurities. The morphology of the Mn3O4 anode prepared by a one-pot MOF-C technique was analyzed by FE-SEM. From Fig. 2a, one can see that the prepared material consists of a sponge-like network morphology in which the nanoparticles are extensively connected through the network. This morphology is most likely due to the initial evolution of the periphery of the extended network of the MOF. The magnified image in Fig. 2b shows a detailed view of the network in which nanoparticles of average diameters less than 50 nm are connected in addition to voids/pores. For further confirmation, FE-TEM studies were performed. The low-magnification image in Fig. 2c reveals that the Mn3O4 sponge network morphology is comprised of nanoparticles and connecting voids. Thus, the observed morphology is consistent with the observations in SEM studies. Detailed lattice information was obtained through high-resolution images. The high-magnification image in Fig. 2d, displays clear lattice fringes with a measured inter-planar distance value d of 0.25 nm, revealing that the (211) plane corresponds to the mother peak of the tetragonal phase of Mn3O4. Furthermore, the other high-magnification image in the inset of Fig. 2d clearly shows a few nanometers (2–3 nm) thick carbon layer being uniformly coated on the surface of a primary particle. It is highly feasible that this carbon coating results from the present synthetic procedure wherein the decomposition of the MOF network leads to carbon substitution in the lattice or in the interstitial positions and/or near the particle surface.22,23 The distinct lattice fringes from the surface to the core suggest that the carbon coating layer may display graphitic behavior, as is evident from additional TEM images (see ESI Fig. S1†). The unique morphology of the Mn3O4/C electrode appears to be related to the synthesis adopted in the present study. In fact, a detailed mechanism of the present MOF-C technique has already been proposed by us.21 Interestingly, we have observed that the obtained particle morphology is unique for each electrode material synthesized by this method. However, the identification of the origin of the peculiar electrode morphology requires further investigations.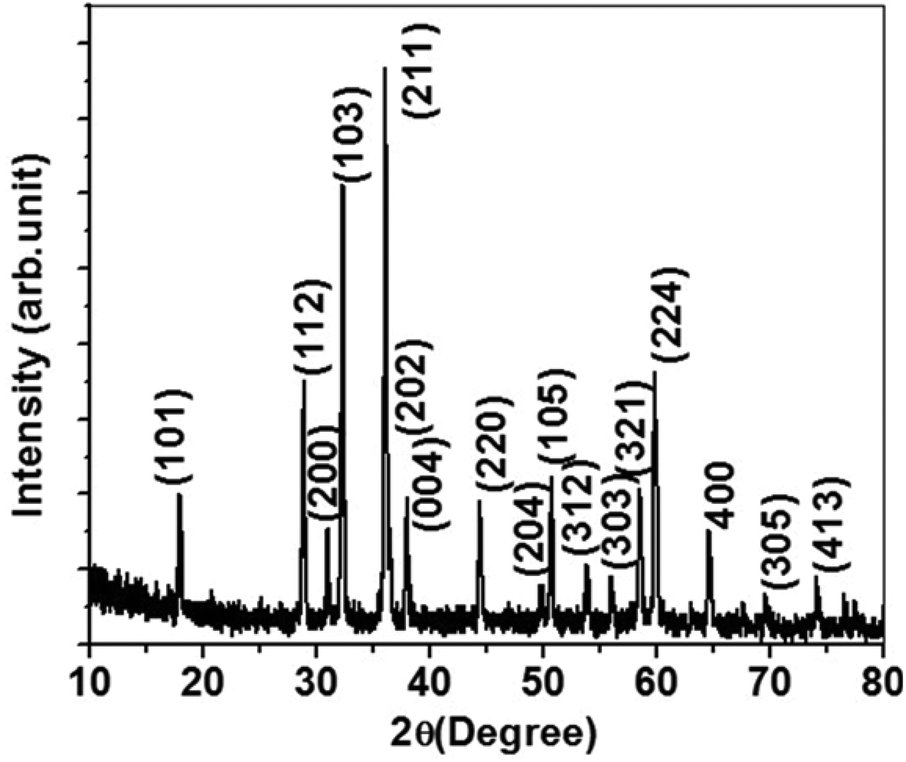 | ||
| Fig. 1 PXRD pattern of the as-synthesized Mn3O4 sponge network morphology derived from the MOF-C technique. | ||
 | ||
| Fig. 2 (a) Low and (b) high magnified SEM images of the as-synthesized Mn3O4 sponge network morphology, (c) low and (d) high magnification of TEM images. The inset in (d) reveals clear lattice fringes of a Mn3O4 anode with a uniform carbon coating on the surface as denoted by the arrow mark. | ||
For further confirmation on the coating behavior of carbon on the prepared Mn3O4/C electrode, the corresponding Raman spectrum was recorded and examined in detail. In general, the spectral features in the high frequency region consist of D and G bands which are normally observed at ∼1350 cm−1 and 1585 cm−1, respectively, where the former band represents the sp2 carbon atoms in a disordered (D) environment and the latter one is attributed to the carbon in the extended p conjugated graphite-like (G) arrangements. The intensity ratio of the D and G bands (ID/IG), i.e. the degree of graphitization, is an indication of the amount of graphitic carbon formed at the surface. The lower the ID/IG ratio, the higher the degree of graphitization.24 The enriched G band and the very low intense D band appear to suggest that carbon exists in a less disordered graphitic form (amorphous)8 on the surface (Fig. S2†). In addition, the peak broadening feature is due to the amorphous character with a reasonable sp3 content of carbon.25,26
The carbon content in the Mn3O4/C composite was estimated by TGA (Fig. S3†). Based on the mass loss, the carbon content was calculated to be 8.5%. It is appropriate to assume that this amount of carbon may be sufficient to account for the surface coating on the active material particles.
The surface chemistry, especially surface oxidation states of the sponge network shaped as-synthesized Mn3O4/C was analyzed by XPS measurement. The C 1s peak profile in Fig. 3a shows three distinct peaks at 284.6, 286.7, and 289.3 eV, which are due to adventitious carbon species. The peak at 284.6 eV is most likely due to the disordered graphitic amorphous carbon (C–C) and the remaining peaks at 286.8 and 289.3 eV can be due to the formation of C–OH (and C–O–C) and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (and COO) bonds, respectively.27 These carbon residue species are formed due to cleaving and subsequent condensation on the surface of the MOF network during the one-pot combustion synthesis at moderate temperatures. The deconvoluted oxygen profile (Fig. 3b) clearly describes the different environmental states of oxygen on the surface of the electrode material. The peak positions at 528.6, 530.5, 532.2, and 533.1 eV can be ascribed to the M–O (O2− lattice oxygen-unreconstructed), M–O (O2−reconstructed), CO (and COO, carbonate), and C–OH (and C–O–C, carbonate) species, respectively. The latter two deconvoluted peaks are carbonate-like species originating from the oxidized carbon atoms.28 The finest deconvolution of the Mn 2p spectrum is observed for the two pairs of spin–orbit doublets indicating the coexistence of Mn2+, Mn3+, and Mn4+ species, as shown in Fig. 3c. The spin–orbit splitting energy between the two major peaks was found to be 11.5 eV, which is in agreement with the energy levels of Mn 2p3/2 and Mn 2p1/2. The deconvoluted profile demonstrates the presence of various oxidation states for Mn on the surface of the Mn3O4 electrode. The peak positions in the 2p3/2 region at 640.1, 641.8, and 643.5 eV are indicative of the Mn oxidation states +2, +3, and +4, respectively. The small region left after deconvolution above ∼645 eV is due to the shakeup satellite region for the Mn2+ species, as denoted by an arrow in the same profile.29,30 As expected, the +3 oxidation state predominates over the other two oxidation states. The presence of variable oxidation states (normally +2 and +3) on the surface is quite common for Mn3O4 and, in this case, the available different oxidation states originate from the oxidation of the Mn3O4 surface at the high temperatures reached during the synthesis. The overall survey scan image given in Fig. 3d demonstrates the purity of this material.
O (and COO) bonds, respectively.27 These carbon residue species are formed due to cleaving and subsequent condensation on the surface of the MOF network during the one-pot combustion synthesis at moderate temperatures. The deconvoluted oxygen profile (Fig. 3b) clearly describes the different environmental states of oxygen on the surface of the electrode material. The peak positions at 528.6, 530.5, 532.2, and 533.1 eV can be ascribed to the M–O (O2− lattice oxygen-unreconstructed), M–O (O2−reconstructed), CO (and COO, carbonate), and C–OH (and C–O–C, carbonate) species, respectively. The latter two deconvoluted peaks are carbonate-like species originating from the oxidized carbon atoms.28 The finest deconvolution of the Mn 2p spectrum is observed for the two pairs of spin–orbit doublets indicating the coexistence of Mn2+, Mn3+, and Mn4+ species, as shown in Fig. 3c. The spin–orbit splitting energy between the two major peaks was found to be 11.5 eV, which is in agreement with the energy levels of Mn 2p3/2 and Mn 2p1/2. The deconvoluted profile demonstrates the presence of various oxidation states for Mn on the surface of the Mn3O4 electrode. The peak positions in the 2p3/2 region at 640.1, 641.8, and 643.5 eV are indicative of the Mn oxidation states +2, +3, and +4, respectively. The small region left after deconvolution above ∼645 eV is due to the shakeup satellite region for the Mn2+ species, as denoted by an arrow in the same profile.29,30 As expected, the +3 oxidation state predominates over the other two oxidation states. The presence of variable oxidation states (normally +2 and +3) on the surface is quite common for Mn3O4 and, in this case, the available different oxidation states originate from the oxidation of the Mn3O4 surface at the high temperatures reached during the synthesis. The overall survey scan image given in Fig. 3d demonstrates the purity of this material.
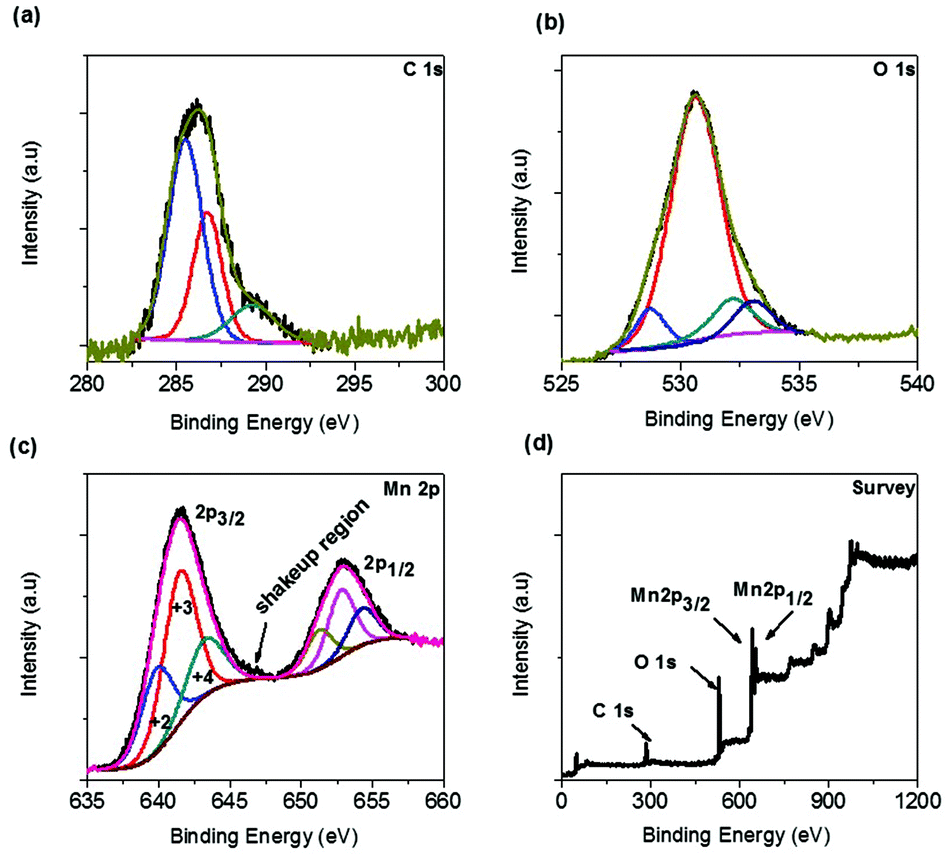 | ||
| Fig. 3 XPS profiles of the as-synthesized Mn3O4/C sponge network morphology. (a) C 1s, (b) O 1s, (c) Mn 2p and (d) survey scan. | ||
CV measurements for the sponge network-shaped Mn3O4/C anode were carried out over the voltage window of 0.01–3.0 V vs. Li/Li+ and the curves obtained for the first, second, and fifth cycles, in Fig. 4a, clearly depict the oxidation and reduction states of the electrode at a scan rate of 0.2 mV s−1. In the first cathodic scan (Fig. 4a), the broad peak that begins below 1.0 V and centered at 0.58 V is attributed to the reduction of Mn3+ into Mn2+ and the formation of a solid–electrolyte interface (SEI) layer (the decomposition of the electrolyte on the surface of the material). This broad peak subsequently disappears in the following cycles. The intensive sharp peak that appears in the low potential region is mainly attributed to the reduction of Mn2+ to Mn0 (metal) accompanied by the formation of Li2O.14,31 In the anodic scans, the consistent peak at around 1.32 V results from the simultaneous processes of metallic Mn to Mn2+/3+ (Mn3O4) oxidation and Li2O decomposition. The consistent cathodic and anodic peaks and overlapping patterns observed from the second cycle indicates the stability and reversibility of the electrochemical reaction with lithium in the present Mn3O4/C electrode.
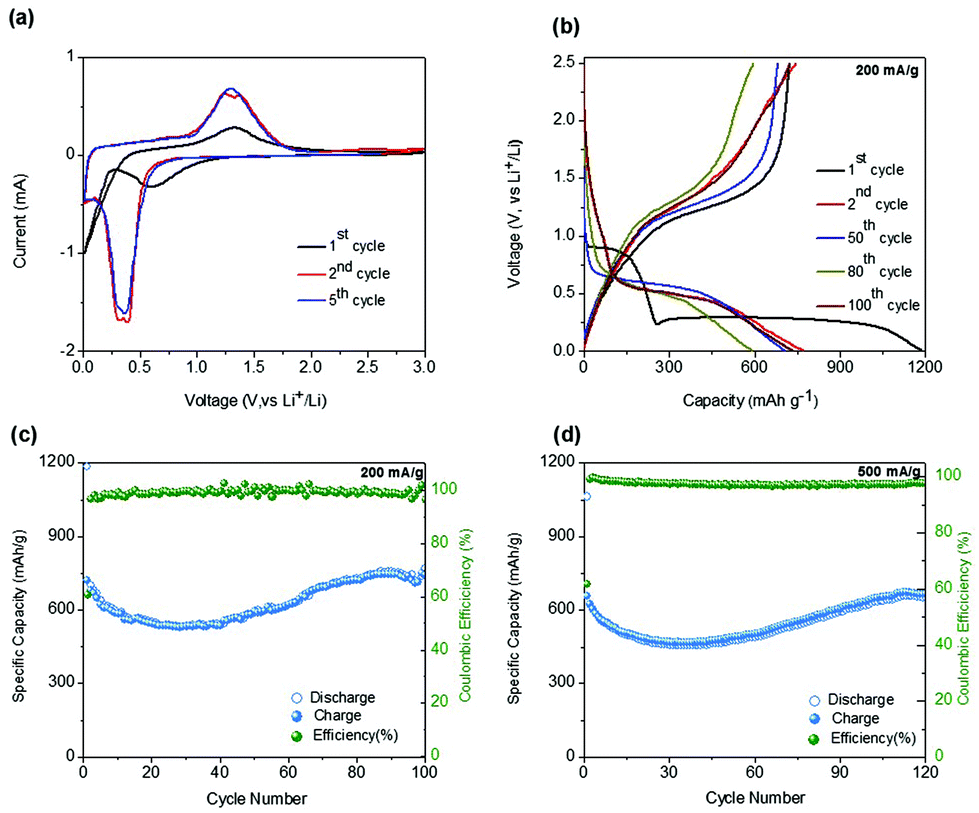 | ||
| Fig. 4 Electrochemical performances of the as-synthesized Mn3O4/C sponge network morphology anode. (a) CV pattern; (b) charge/discharge profiles at 200 mA g−1 current density; (c) and (d) cyclability curves at 200 and 500 mA g−1 current densities. | ||
Fig. 4b shows the galvanostatic charge and discharge performance of the Mn3O4/C electrode as examined at a current density of 200 mA g−1. The voltage profiles of the 1st, 2nd, 50th, 80th, and 100th cycles for the sponge-shaped Mn3O4/C electrode range from 0.005 V vs. Li/Li+ to 2.5 V vs. Li/Li+. In the first discharge curve, a notable sloping voltage is observed from 0.9 to 0.20 V. The early potential drop to 0.9 V observed only for the initial few cycles is due to the decomposition of the electrolyte and the formation of SEI layers, and the reduction of Mn3O4 to MnO (case 1: Mn3+ into Mn2+).10,32 A well-defined voltage plateau at 0.28 V, is mainly due to the lithiation reaction with Mn3O4. The second voltage drop near 0.22 V could have originated from the reduction to metallic Mn (case 2: Mn2+ into Mn0). In the anodic process, alternatively, the oxidation of Mn0 to Mn2+ (the reverse of case 2) and Mn2+ to Mn3+ (the reverse case of 1) occurs at around 1.1 and 1.5 V, respectively. Thus, at the end of the first cycle, the discharge and charge capacities are 1186 and 722 mA h g−1, respectively, and the corresponding CE is 60.8%. The low CE value is mainly attributed to the formation of the SEI layer in the initial cycle. The lithiation plateau moves to a higher voltage of 0.5 V in the second and subsequent cycles, which implies structural changes during the first cycle, as corroborated by the CV results. In the second cycle, the discharge and charge capacities were measured to be 702 and 685 mA h g−1 with a CE value greater than 97%. Nevertheless, the delivered discharge capacity is equivalent to 75% of the theoretical value (∼937 mA h g−1), as described by the reaction: Mn3O4 + 8Li+ + 8e− → 3Mn0 + 4Li2O. Interestingly, on prolonged cycling, the measured discharge capacities increased, as observed from the corresponding electrochemical profiles in Fig. 4b. In specific, the discharge capacities in the 50th, 80th, and 100th cycles were, respectively, 590, 731, and 768 mA h g−1 corresponding to 63%, 78% and 82% of theoretical capacity. The clear trend in variation of reversible capacities and the CE values during extended cycling can be observed in the cyclability plot shown in Fig. 4c. Specifically, the reversible capacity initially decreased to reach the lowest value of ∼550 mA h g−1 in the 30th cycle, beyond which the registered value increased gradually. In the 82nd cycle, a value of 733 mA h g−1 was registered and thereafter the capacity values tend to be saturated as stable capacities of 770 mA h g−1 with almost 100% CE (compared to just 61% in the initial cycle) are delivered in the 100th cycle. The electrochemical performance at a higher current density of 500 mA g−1 was also tested for 120 cycles with the present electrode and the cyclability results are plotted in Fig. 4d; the performance trend being similar to that observed for the current density profile at 200 mA g−1. The measured 1st cycle discharge and charge capacities were 1065 and 658 mA h g−1, respectively, with a considerable capacity loss (∼38%) associated with the formation of the SEI layer. On successive cycling, the capacity initially dropped to reach a value of 457 mA h g−1 in the 35th cycle; the value then increased to achieve ∼660 mA h g−1 at the 114th cycle, which was then sustained until the 120th cycle with a measured capacity of 651 mA h g−1. A notable CE% of ∼97% was maintained throughout the cycles, except for the first cycle, which showed a value of 61%. The corresponding selected charge/discharge profiles obtained for this current density are given in Fig. S4.†
The rate capability of this sponge network-shaped Mn3O4/C anode was studied at different current densities in the 0.01–2.5 V potential window, as shown in Fig. 5a. The observed average capacity was found to be 730 mA h g−1 at an initial applied current density of 100 mA g−1; however, the capacity value decreased with the increasing current density. Thus, the measured average capacities were 545, 485, 406, 350, 298, 239, and 155 mA h g−1 at current densities of 200, 400, 800, 1600, 3200, 6400, and 10000 mA g−1 respectively. When the applied current density was reduced to 400 mA g−1, the capacity was found to be 442 mA h g−1, which was lower than the average capacity (485 mA h g−1) at the same current density. However, the capacity value increased on successive cycling and reached 505 mA h g−1 in the 70th cycle. It is worth noting that the sustained performance at different current densities for the next two subsequent sequences demonstrates the stability of the sponge-shaped Mn3O4/C electrode. Meanwhile, the specific capacity for the intermediate current density of 400 mA g−1 reached a stable capacity of 704 mA h g−1 at the 175th cycle. This excellent performance is an attractive feature of this sponge-shaped Mn3O4/C electrode given that a robust and simple technique utilizing a short reaction time was adopted for the synthesis. The comparative electrochemical performances of Mn3O4 electrodes (with and without carbon additives) already reported in the literature are provided as a table in the ESI (Table S1†). This table illustrates the electrochemical performance of Mn3O4 anodes with different morphologies, with and without a carbon support, at different current densities at a given potential window for LIB application. These data further support that the performance of the present Mn3O4/C electrode competes well with that of other morphologies, especially from the point of view of rate capability. Fig. 5b shows the cycling performance of the Mn3O4 sponge network-shaped anode material at another high current density, 700 mA g−1. The performance at this current density is very similar to the cyclability performance at 200 and 500 mA g−1 current densities, as the capacity decreased initially and then increased with the increasing number of cycles. In the first cycle, the measured discharge and charge capacities were found to be 1155 and 639 mA h g−1, respectively, with a CE% value of 55.3%. The lowest registered discharge capacity was found to be 391 mA h g−1 at the end of the 53rd cycle with an improved CE% of 99.2%. The capacity gradually increased thereafter with the cycle number and a sustained capacity of 592 mA h g−1 was found after 120 cycles with a CE% close to 100% (Fig. S5†). On the whole, the performance of the anode at three different current densities (770, 651, and 592 mA h g−1 at 200, 500, and 700 mA g−1 for 100, 120, and 120 cycles, respectively) demonstrates the remarkable lithium storage properties of this electrode during the electrochemical reaction. The increased capacity behavior in all the cyclability curves could be attributed to the presence of a possible activation process of the electrode.21 In order to examine the structural stability of the present Mn3O4/C synthesized by the one-pot MOF-C technique, ex situ SEM measurements were performed on the electrode recovered after the completion of 120 cycles at 500 mA g−1. The image in Fig. 5c shows that the electrode has mostly retained the sponge network morphology even after prolonged cycling at high discharge/charge rates.
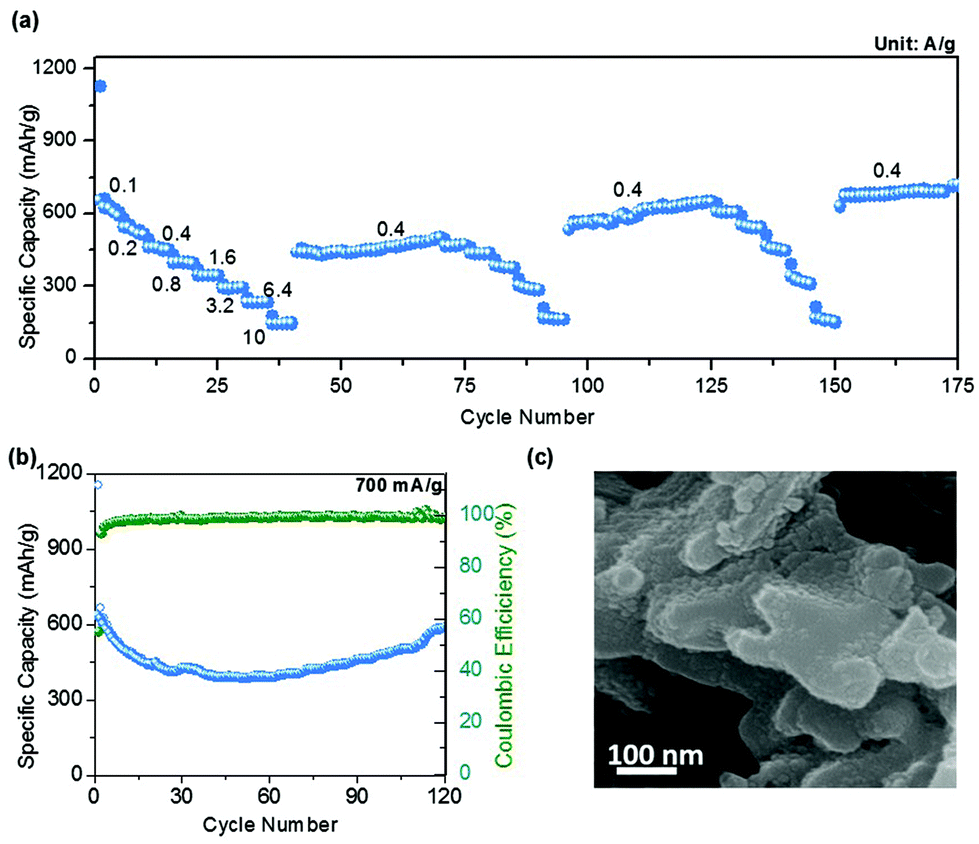 | ||
| Fig. 5 (a) Rate capability pattern at different current densities, (b) cyclability performance at 700 mA g−1 current density and (c) ex situ SEM image of the Mn3O4/C anode after 120 cycles at 500 mA g−1 current density. | ||
The surface area and pore size distribution were measured for this sponge network-shaped material using BET nitrogen adsorption–desorption analysis at liquid nitrogen temperature. The Mn3O4/C sample exhibited a surface area of 8.0 m2 g−1 and a pore volume of 0.06 cm3 g−1, as obtained from the Barrett–Joyner–Halenda (BJH) pore size distribution curve (Fig. S6†). The adsorption–desorption pattern follows a type III adsorption isotherm with a small hysteresis. The little or no knee of the isotherm close to P/Po ∼ 0.95 reveals the micro or macroporous nature of the material. A detailed observation of the isotherm suggests that, overall, the crystallization of the present material occurs in a specific pattern. Indeed, small pores tend to fuse together and form micropores or voids during crystallization, which also concurs well with the electron microscopy studies. To further understand the cell performance, Nyquist plots of the Mn3O4/C electrodes before and after 100 cycles at a current density of 200 mA g−1 were obtained by EIS measurements. The obtained plots (Fig. S7†) consist of two different parts: (a) the sloping line (inclined vertical line) in the low frequency region that is associated with the mass transfer of ions (diffusion of lithium ions into the active material), and (b) the semicircle in the high frequency region due to the charge-transfer resistance (charge transfer in the electron/ion conductive junction). The observation of similar patterns indicate that both electrodes (before and after 100 cycles) sustain a similar ionic conductivity, which is advantageous for long-term cycling. However, the sloping patterns are different, which indicates that solid-state diffusion of Li-ions in the latter case is not as significant as in the sample before cycling.
It is worth mentioning that the performance of the present Mn3O4/C anode prepared by the one-pot, rapid, simple and scalable technique displays intriguing electrochemical properties when compared with those observed for Mn3O4 electrodes already reported. For example, the reversible specific capacity of 770 mA h g−1 delivered by the present Mn3O4/C electrode at a current density of 200 mA g−1 is advantageous to those attained by flexible graphene/Mn3O4 nanocomposites (802 mA h g−1 at 100 mA g−1),33 rGO/Mn3O4 nanoparticles (810 mA h g−1 at 40 mA g−1),11 Mn3O4/C nanorods (723 mA h g−1 at 40 mA g−1),8 Mn3O4/ordered mesoporous carbon (773 mA h g−1 at 100 mA g−1),19 CNT/Mn3O4 nanoparticles (701.4 mA h g−1 at 93.6 mA g−1),9 CNT/Mn3O4 nanocrystals (709 mA h g−1 at 100 mA g−1),34 Mn3O4 nanoparticles on graphene (553 mA h g−1 at 120 mA g−1),35 and porous Mn3O4 nanorod/reduced graphene oxide hybrid paper (573 mA h g−1 at 100 mA g−1).36 However, although there are a few reports on Mn3O4 electrodes with/without carbon additives with enhanced storage properties,37–41 the significantly high rate performances of the present electrode (Table S1† and related references) reveal the remarkable advantage of the simple synthetic strategy adopted here to produce Mn3O4/C anodes. Very recently, a flexible full Li-ion battery fabricated with graphene/Mn3O4||LiMn2O4 has opened a practical application of this anode.33
Conclusions
A carbon-coated Mn3O4 (Mn3O4/C) electrode with a sponge network-shaped morphology was directly synthesized by a simple one-pot MOF-C technique in the present study. The synthetic strategy afforded the formation of a less disordered amorphous graphitic carbon coating with almost uniform thickness on the surface of the Mn3O4 particles. The prepared Mn3O4/C electrode with a unique morphology delivered stable capacities of 770, 651, and 592 mA h g−1 at current densities of 200, 500, and 700 mA g−1 after 100, 120, and 120 cycles, respectively. The excellent performance of the Mn3O4/C electrode was attributed to the unique particle morphology and the electronically conductive carbon combined with the facilitation of shorter Li-ion diffusion, a greater electrode/electrolyte contact area due to the notable surface area, and porous features of the nano-scale particles. The development of such nanostructured electrodes with unique morphologies and enhanced electrochemical performances motivates the re-visiting of already known/existing metal oxides by utilizing this simple, robust, and rapid synthetic technique. Such an effort will pave the way for not only identifying improved next generation electrodes for rechargeable battery applications but also for other applications including solar cells and photocatalysis.Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korean government (MSIP) (2014R1A2A1A10050821). This research was also supported by the Global Frontier Program through the Global Frontier Hybrid Interface Materials (GFHIM) of the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT & Future Planning (2013M3A6B1078875) or (2013-073298).Notes and references
- C. Liu, F. Li, L. P. Ma and H. M. Cheng, Adv. Mater., 2010, 22, E28–E62 CrossRef CAS PubMed.
- V. Etacheri, R. Marom, R. Elazari, G. Salitra and D. Aurbach, Energy Environ. Sci., 2011, 4, 3243–3262 CAS.
- S. Goriparti, M. Ermanno, F. D. Angelis, E. D. Fabrizio, R. P. Zaccaria and C. Capiglia, J. Power Sources, 2014, 257, 421–443 CrossRef CAS.
- S. Goriparti, M. N. Harish and S. Sampath, Chem. Commun., 2013, 49, 7234–7236 RSC.
- Y. Sun, J. Zhu, L. Bai, Q. Li, X. Zhang, W. Tong and Y. Xie, Inorg. Chem. Front., 2014, 1, 58–64 RSC.
- Y. Jiang, M. Hu, D. Zhang, T. Yuan, W. Sun, B. Xu and M. Yan, Nano Energy, 2014, 5, 60–66 CrossRef CAS.
- X. Ma, Y. Zhai, N. Wang, J. Yang and Y. Qian, RSC Adv., 2015, 5, 46829–46833 RSC.
- C. Wang, L. Yin, D. Xiang and Y. Qi, ACS Appl. Mater. Interfaces, 2012, 4, 1636–1642 CAS.
- S. Luo, H. Wu, Y. Wu, K. Jiang, J. Wang and S. Fan, J. Power Sources, 2014, 249, 463–469 CrossRef CAS.
- M. Jing, J. Wang, H. Hou, Y. Yang, Y. Zhang, C. Pan, J. Chen, Y. Zhu and X. Ji, J. Mater. Chem. A, 2015, 3, 16824–16830 CAS.
- H. Wang, L. Cui, Y. Yang, H. S. Casalongue, J. T. Robinson, Y. Liang, Y. Cui and H. Dai, J. Am. Chem. Soc., 2010, 132, 13978–13980 CrossRef CAS PubMed.
- Q. Hao, J. Wang and C. Xu, J. Mater. Chem. A, 2014, 2, 87–93 CAS.
- J. Wang, N. Du, H. Wu, H. Zhang, J. Yu and D. Yang, J. Power Sources, 2013, 222, 32–37 CrossRef CAS.
- G. Jian, Y. Xu, L. Lai, C. Wang and M. R. Zachariah, J. Mater. Chem. A, 2014, 2, 4627–4632 CAS.
- J. Gao, M. A. Lowe and H. D. Abruna, Chem. Mater., 2011, 23, 3223–3227 CrossRef CAS.
- M. H. Alfaruqi, J. Gim, S. Kim, J. Song, P. T. Duong, J. Jo, J. P. Baboo, Z. Xiu, V. Mathew and J. Kim, Chem. – Eur. J., 2016, 22, 2039–2045 CrossRef CAS PubMed.
- S. H. Park and W. J. Lee, J. Power Sources, 2015, 281, 301–309 CrossRef CAS.
- Z. Bai, N. Fan, Z. Ju, C. Guo, Y. Qian, B. Tang and S. Xiong, J. Mater. Chem. A, 2013, 1, 10985–10990 CAS.
- Z. Li, N. Liu, X. Wang, C. Wang, Y. Qi and L. Yin, J. Mater. Chem., 2012, 22, 16640–16648 RSC.
- Y. Luo, S. Fan, N. Hao, S. Zhong and W. Liu, Dalton Trans., 2014, 43, 15317–15320 RSC.
- B. Sambandam, V. Soundharrajan, V. Mathew, J. Song, S. Kim, J. Jo, D. P. Tung, S. Kim and J. Kim, J. Mater. Chem. A, 2016, 4, 14605–14613 CAS.
- H. Yue, Z. Shi, Q. Wang, Z. Cao, H. Dong, Y. Qiao, Y. Yin and S. Yang, ACS Appl. Mater. Interfaces, 2014, 6, 17067–17074 CAS.
- V. Soundharrajan, B. Sambandam, J. Song, S. Kim, J. Jo, S. Kim, S. Lee, V. Mathew and J. Kim, ACS Appl. Mater. Interfaces, 2016, 8, 8546–8553 CAS.
- Y. M. Foong, A. T. T. Koh, H. Y. Ng and D. H. C. Chua, J. Appl. Phys., 2011, 110, 054904 CrossRef.
- L. Wang, R. Zhang, U. Jansson and N. Nedfors, Sci. Rep., 2015, 5, 11119 CrossRef PubMed.
- A. C. Ferrari, Solid State Commun., 2007, 143, 47–57 CrossRef CAS.
- I. Nam, N. D. Kim, G. Kim, J. Park and J. Yi, J. Power Sources, 2013, 244, 56–62 CrossRef CAS.
- J. Liu, Q. Zhang, J. Yang, H. Ma, M. O. Tade, S. Wang and J. Liu, Chem. Commun., 2014, 50, 13971–13974 RSC.
- C. Drouet, C. Laberty, J. L. G. Fierro, P. Alphonse and A. Rousset, Int. J. Inorg. Mater., 2000, 2, 419–426 CrossRef CAS.
- Z. Bejia, M. Sun, L. S. Smiri, F. Herbst, C. Mangeney and S. Ammar, RSC Adv., 2015, 5, 65010–65022 RSC.
- F. Zheng, D. Zhu, X. Shi and Q. Chen, J. Mater. Chem. A, 2015, 3, 2815–2824 CAS.
- Y. Zhao, G. Chen, C. Yan, C. Lv, R. Wang and J. Sun, RSC Adv., 2015, 5, 106206–106212 RSC.
- J. G. Wang, D. Jin, R. Zhou, X. Li, X. Liu, C. Shen, K. Xie, B. Li, F. Kang and B. Wei, ACS Nano, 2016, 10, 6227–6234 CrossRef CAS PubMed.
- Z. H. Wang, L. X. Yuan, Q. G. Shao, F. Huang and Y. H. Huang, Mater. Lett., 2012, 80, 110–113 CrossRef CAS.
- I. A. Ayhan, Q. Li, P. Meduri, H. Oh, G. R. Bhimanapati, G. Yang, J. A. Robinson and Q. Wang, RSC Adv., 2016, 6, 33022–33030 RSC.
- S. K. Park, C. Y. Seong, S. Yoo and Y. Piao, Energy, 2016, 99, 266–273 CrossRef CAS.
- F. Ma, A. Yuan and J. Xu, ACS Appl. Mater. Interfaces, 2014, 6, 18129–18138 CAS.
- L. Su, X. Wu, J. Hei, L. Wang and Y. Wang, Part. Part. Syst. Charact., 2015, 32, 721–727 CrossRef CAS.
- N. Wang, J. Yue, L. Chen, Y. Qian and J. Yang, ACS Appl. Mater. Interfaces, 2015, 7, 10348–10355 CAS.
- X. Y. Fan, Y. Cui, P. Liu, L. Gou, L. Xu and D. L. Li, Phys. Chem. Chem. Phys., 2016, 18, 22224–22234 RSC.
- Z. Bai, X. Zhang, Y. Zhang, C. Guo and B. Tang, J. Mater. Chem. A, 2014, 2, 16755–16760 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional TEM images, Raman spectra, CHN analysis, charge/discharge profiles, surface area and EIS measurements. See DOI: 10.1039/c6qi00348f |
| ‡ These authors contributed equally. |
| This journal is © the Partner Organisations 2016 |