
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Insertion of organometallic moieties into peptides and peptide nucleic acids using alternative “click” strategies†
Cristina
Mari
,
Sandro
Mosberger
,
Nuria
Llorente
,
Sarah
Spreckelmeyer
and
Gilles
Gasser
 *
*
University of Zurich, Department of Chemistry, Winterthurerstrasse 190, 8057, Zurich, Switzerland. E-mail: gilles.gasser@chem.uzh.ch; Web: http://www.gassergroup.com Tel: +41 44 635 4630
First published on 8th January 2016
Abstract
The insertion of metal complexes in biologically active systems is of great interest in view of diagnostic and therapeutic applications as well as a precious tool to unveil biological mechanisms. Optimization of safe and biocompatible reactions is critical to achieve high functionalization efficiency. Herein we present the application of two modified versions of copper-catalyzed azide–alkyne cycloaddition (click) chemistry, namely a one-pot diazotransfer + azide–alkyne cycloaddition (one-pot click) and a copper-free photoactivated tetrazole–alkene cycloaddition (photoclick), for derivatization of peptides and peptide nucleic acids (PNAs) with ferrocene and cymantrene moieties. These metal fragments were chosen for their possible exploitation as redox and IR probes. We could demonstrate that one-pot click enables for efficient functionalization of propargyl-glycine and an alkyne-containing peptide with an amino-containing cymantrene precursor. In addition, we could show that photoclick allows for the insertion of maleimido-ferrocene into a peptide and a PNA sequence containing a tetrazole moiety.
 Gilles Gasser | Gilles Gasser completed a PhD thesis in supramolecular chemistry in the group of Prof. Helen Stoeckli-Evans at the University of Neuchâtel (2004). After post-doctoral stays at Monash University (Australia) with Prof. Leone Spiccia in bioinorganic chemistry and at the Ruhr-University Bochum (Germany) with Prof. Nils Metzler-Nolte in bioorganometallic chemistry, Gilles started his independent research career at the University of Zurich, first as Swiss National Science Foundation (SNSF) Ambizione fellow (2010) and then as a SNSF Assistant Professor (2011). Gilles is the recipient of several awards including the Werner Prize from the Swiss Chemical Society for outstanding independent chemical research (2015) and an ERC Consolidated Grant (2015). |
Introduction
The functionalization of biologically active molecules is of great importance nowadays in medicinal chemistry.1–3 In particular, their functionalization with metal-based compounds can confer novel properties to the formed bioconjugate compared to the biomolecule itself allowing for novel applications.4 Metal complexes can be used as both diagnostic and therapeutic tools in the clinical treatment of patients.5 The coupling of a targeting vector (i.e. peptide, antibody, etc.) to therapeutic/diagnostic metal-based drugs can, for example, increase their organ/cell selectivity.6–10 In a similar vein, the insertion of metal fragments into biologically active molecules can yield biocompatible and extremely useful tools for biological investigations, such as luminescent probes.11The application of copper-catalyzed azide–alkyne cycloaddition (CuAAC, commonly referred to as click chemistry) for bioconjugation has been extensively exploited due to the high versatility, selectivity and functional group tolerance of this synthetic technique.12 A great number of probes, including metal-containing probes, were “clicked” to an important variety of biologically active compounds such as peptides, antibodies, peptide nucleic acids (PNAs) or polymers.13–17 However, this reaction presents two important drawbacks from both a chemical as well as a biological point of view. Since an azide is one of the protagonists of this transformation, this functional group has to be installed on one of the two molecules to be coupled. Safety concerns about the isolation of azides (especially with low molecular weight compounds) are still actual nowadays due to the potential instability of this class of compounds.18 Therefore, from a chemical point of view, it would be more convenient to skip the azide isolation, for example, via an in situ insertion of this functionality. The second disadvantage of the CuAAC is its reliability on the presence of CuI as catalyst for its mechanism to take place. This situation represents a problem for in cellulo applications of the technique due to the toxicity of the metal.13,19 To address these limitations, a great effort has been made towards the optimization of new methodologies for click-mediated biocompatible conjugation.13,20,21 Since the absence of a Cu catalyst makes the click reaction very inefficient, an alternative activation mechanisms had to be developed. Among the most relevant examples, Strain-Promoted Inverse Electron Demand Diels–Alder Cycloaddition (SPIEDAC) and Strain-Promoted Alkyne–Azide Cycloaddition (SPAAC) exploit the formation of an unstrained product as reaction driving force, circumventing the need of Cu catalysis.19 Other approaches involve the use of light to form a reactive intermediate.21
Herein, we present, to the best of our knowledge, the first examples of the application of two modified versions of the classical CuAAC to introduce organometallic fragments into biologically active molecules, namely a peptide and a PNA oligomer (see Fig. 2). In particular, we focused our attention on two modified click reactions, namely the copper-catalyzed one-pot diazotransfer + azide–alkyne cycloaddition reaction (which will be called from now on in this article one-pot click)22 and the metal-free photoactivated tetrazole–alkene cycloaddition (referred to from now on as photoclick).23 The two organometallic components investigated in this study were a cymantrene derivative A and a ferrocene derivative B (Fig. 1),24,25 chosen for their potential use as IR (A) and redox (B) probes in a biological environment.26,27
 | ||
| Fig. 1 Structures of the organometallic complexes used in this paper and their simplified depictions. | ||
 | ||
| Fig. 2 Structures of the peptides and PNA to be “clicked”. All amine groups in peptide and PNA sequences are present as TFA salts. | ||
Results and discussion
One-pot click reaction
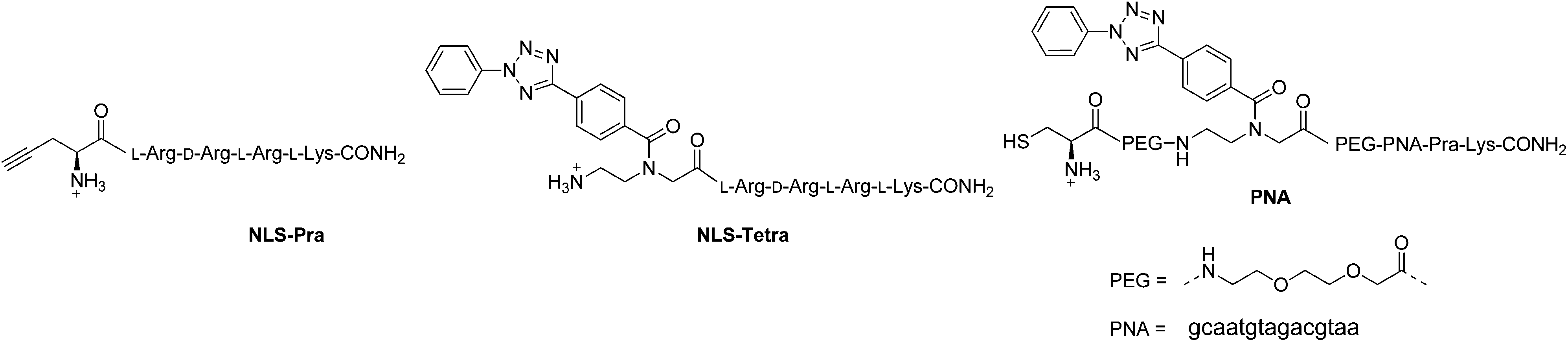
The one-pot click reaction developed by Wittmann et al. allows in a subtle manner to circumvent the isolation of the azide intermediate.22 More specifically, the one-pot click proceeds via two subsequent steps. First, an amine derivative is turned into an azide via a CuII catalyzed diazotransfer. This intermediate can then be clicked to an alkyne by the classical CuAAC reaction. The CuI catalyst for the click reaction is formed in situ by addition of a reducing agent to the mixture. In the present study, we applied this synthetic technique to the two amino-organometallic derivatives A and B. These molecules were clicked to an unnatural amino acid carrying a propargyl group (Fmoc-L-Pra-OH) and a short peptide containing this amino acid (NLS-Pra, Fig. 2). This small peptide is known for its nuclear targeting properties and has been used in the past by our group to deliver different metal complexes to the cell nucleus.6,28,29NLS-Pra was synthesized by standard solid phase synthesis and the alkynyl containing unnatural amino acid was inserted at the N-terminus.28 The original one-pot click procedure reported by Wittmann et al. was followed in this study with the exception of minor modifications: the amounts of copper salt, tris(benzyltriazolylmethyl)amine (TBTA) and sodium ascorbate were doubled and the reaction duration was extended to 1 h of microwave assisted refluxing, instead of 30 minutes. When A was used as the amine precursor, the reaction was found to be effective for labelling both the amino acid and the peptide (Fig. 3).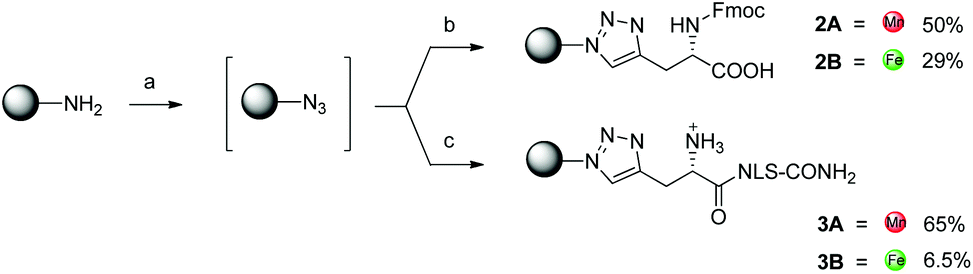 | ||
| Fig. 3 One-pot click reactions. (a) TfN3, CuSO4, NaHCO3, H2O/MeOH/DCM, 30 min, RT. (b) Fmoc-L-Pra-OH, TBTA, CuSO4, Na ascorbate, 1 h, MW 80 °C. (c) NLS-Pra, TBTA, CuSO4, Na ascorbate, 1 h, MW 80 °C. All amine groups in the peptide sequence are present as TFA salts. | ||
More specifically, product 2A was isolated in 50% yield and its structure confirmed by several analytical techniques. The UPLC-MS chromatogram showed one peak, whose integrated mass of m/z 694.2 is attributable to [M + H]+. The nature of 2A was further confirmed by HR ESI-MS, in which the peak of [M − H]− is visible. In addition, the IR spectrum clearly showed the characteristic vibrations of the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) O groups at 1921 and 2015 cm−1. However, due to the presence of traces amounts of a paramagnetic impurity, we were unable to obtain a good 1H-NMR spectrum. In the 13C-NMR, however, resonances at 55.73 and 29.22 ppm can be attributed to the CH2 and CH carbons between the newly formed triazole and the COOH moiety. The peptide derivative 3A was isolated via preparative RP-HPLC purification. UPLC-MS analysis showed one peak in the chromatogram, with the m/z values assigned to the product with multiple charges. MALDI-TOF analysis also showed the expected peak at m/z 1289.6 corresponding to [M + H]+, but also the typical fragmentation of [M + H − Mn(CO)3]+. Such a fragmentation was already reported for similar biomolecules.30 As further confirmation of the identity of the compound, the IR spectrum of 3A showed the vibrations of the CO at 1927 and 2017 cm−1. When the Fc precursor B was used as a reaction substrate, lower yields were observed for both the amino acid and the peptide labelling reactions. This is probably due to the instability of Fc-methylene compounds.31,32 The harsh conditions of the click reaction (80 °C for 1 h) most probably led to partial decomposition of the ferrocene derivative. Nevertheless, products 2B and 3B could be isolated in 29% and 6.5% yields, respectively. For 2B, the HR ESI-MS analysis showed the [M − H]− peak at m/z 575.1391 (calculated value: 575.1387). In addition, the 13C-NMR revealed the carbon signals of the newly formed triazole moiety at 144.70 and 124.25 ppm. Furthermore, 2D experiments correlated the resonance at 124.25 ppm to the broad peak at 7.77 ppm in the 1H-NMR, which could be then assigned to the triazole CH. For 3B, MALDI-TOF analysis showed the most intense peak at m/z 1172.6, which can be attributed to [M + H]+.
O groups at 1921 and 2015 cm−1. However, due to the presence of traces amounts of a paramagnetic impurity, we were unable to obtain a good 1H-NMR spectrum. In the 13C-NMR, however, resonances at 55.73 and 29.22 ppm can be attributed to the CH2 and CH carbons between the newly formed triazole and the COOH moiety. The peptide derivative 3A was isolated via preparative RP-HPLC purification. UPLC-MS analysis showed one peak in the chromatogram, with the m/z values assigned to the product with multiple charges. MALDI-TOF analysis also showed the expected peak at m/z 1289.6 corresponding to [M + H]+, but also the typical fragmentation of [M + H − Mn(CO)3]+. Such a fragmentation was already reported for similar biomolecules.30 As further confirmation of the identity of the compound, the IR spectrum of 3A showed the vibrations of the CO at 1927 and 2017 cm−1. When the Fc precursor B was used as a reaction substrate, lower yields were observed for both the amino acid and the peptide labelling reactions. This is probably due to the instability of Fc-methylene compounds.31,32 The harsh conditions of the click reaction (80 °C for 1 h) most probably led to partial decomposition of the ferrocene derivative. Nevertheless, products 2B and 3B could be isolated in 29% and 6.5% yields, respectively. For 2B, the HR ESI-MS analysis showed the [M − H]− peak at m/z 575.1391 (calculated value: 575.1387). In addition, the 13C-NMR revealed the carbon signals of the newly formed triazole moiety at 144.70 and 124.25 ppm. Furthermore, 2D experiments correlated the resonance at 124.25 ppm to the broad peak at 7.77 ppm in the 1H-NMR, which could be then assigned to the triazole CH. For 3B, MALDI-TOF analysis showed the most intense peak at m/z 1172.6, which can be attributed to [M + H]+.
Photoclick reaction
As a parallel approach to the one-pot click reaction, we investigated the applicability of the photoclick reaction to our metal precursors, to pursue a more biocompatible method for organometallic labelling. This procedure exploits the light-mediated formation of a reactive acyl-nitrene intermediate from a tetrazole group. This activated intermediate is then prone to 1,3-dipolar cycloaddition with a substituted alkene.23 This reaction belongs to the class of photo-mediated orthogonal reactions, which have been extensively exploited for in vivo applications due to their high selectivity and the lack of the toxic copper catalyst.20,21,33–35 The diphenyltetrazole group 7, which was synthesized as previously reported,36 was installed on the PNA backbone 8,37 to obtain a tetrazole-containing PNA monomer as a t-Bu ester 9 (Fig. 4). Removal of the t-Bu yielded compound 10, which can be inserted into a PNA or a peptide sequence by solid phase synthesis. The initial design of the system envisaged the use of a phenyl-allyloxy derivative as the alkene-reactive component. However, this molecule was found to be poorly reactive and only afforded the products in low yields. For these reasons, we decided to employ a maleimide group as alkene functionality. Maleimide derivatives of cymantrene and ferrocene (4A and 4B, Fig. 5) were therefore prepared and “photoclicked” with the protected PNA monomer 9 bearing the tetrazole reactive moiety. The Fc-containing compound 5B was isolated in 63% yield and fully characterized. The UV chromatogram of UPLC analysis showed one peak and the HR ESI-MS showed a peak at m/z 912.3041, corresponding to [M + H]+ (calculated: 912.3054). On the other hand, the Mn derivative 5A was isolated from the crude as a minor component (8.5%), whereas the main product of the reaction (40%) was still a product of photoclick lacking, however, the Mn(CO)3 moiety (m/z 891.5 [M − Mn(CO)3 + H]+). Comparison of the IR spectra confirmed the absence of the CO vibrations in the spectrum of the product isolated in higher amount, signals which are well visible in the IR spectrum of 5A (1927 and 2019 cm−1). This outcome is probably due to the known ability of cymantrenyl compounds to release CO upon light irradiation.38 Therefore, it is likely that the product of the photoclick decomposes during the reaction.
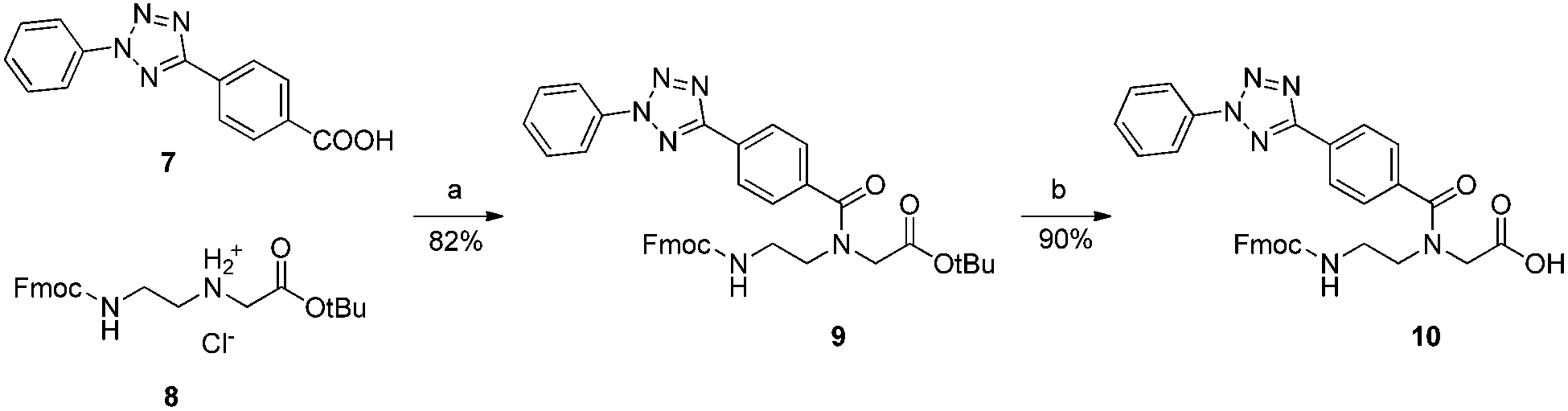 | ||
Fig. 4 Synthesis of the tetrazole-containing PNA monomer. (a) HATU, HOBt, DIPEA, DMF, o.n., RT. (b) DCM![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :TFA:TIPS 2:1:0.4, o.n., RT. :TFA:TIPS 2:1:0.4, o.n., RT. | ||
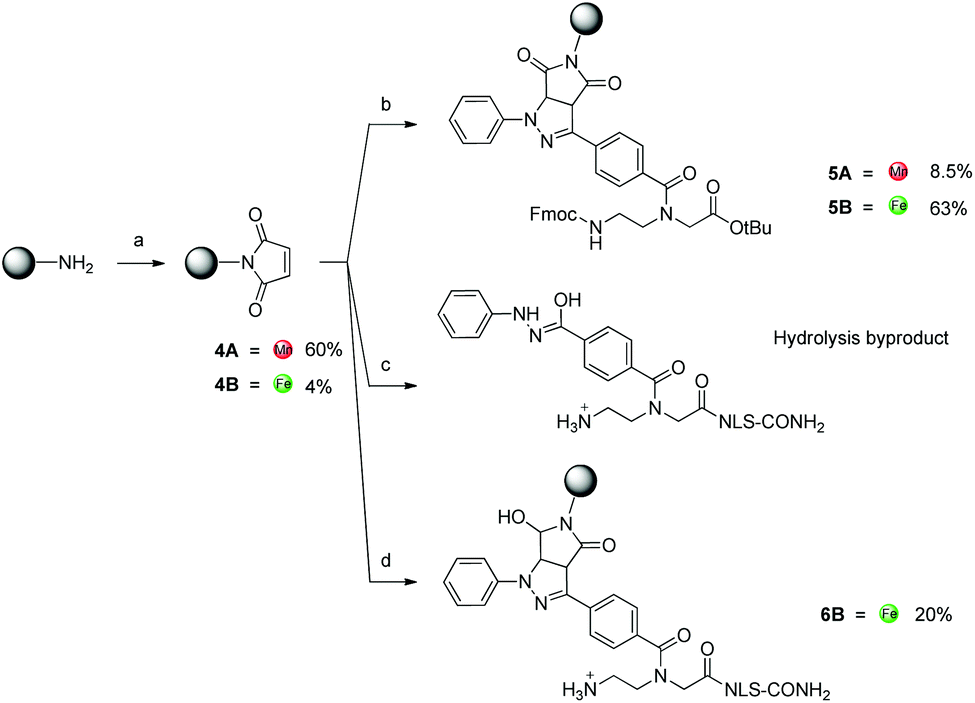 | ||
| Fig. 5 Photoclick reaction. (a) Maleic anhydride, AcOH, 8 h, reflux. (b) 9, ACN, 302 nm, 1 h, RT. (c) NLS-tetra, H2O or ACN:PBS (5:1), 302 nm, 30 min, RT. (d) NLS-tetra, DMSO, 302 nm, 1 h, RT. All amine groups in the peptide sequence are present as TFA salts. | ||
Motivated by the successful formation of 5B, we attempted the insertion of ferrocene-maleimide in a NLS peptide carrying the modified PNA monomer (NLS-tetra, Fig. 2), synthesized by solid phase synthesis.28 In a procedure reporting on the modification of a DNA strand with a maleimide functionalized dye via photoclick, this reaction was performed in PBS affording yields in the range of 8–34% depending on the conditions used, whilst the use of DMSO as solvent did not afford the desired product.39 Surprisingly, when we performed the reaction in water or ACN:PBS (5:1), after just 30 min irradiation NLS-tetra was found to have entirely turned into the hydrolysis byproduct of the acyl-nitrene intermediate (Fig. 5, ESI-MS (pos. detection mode): m/z [M + 3H]3+ 318.4, [M + 2H]2+ 476.9, [M + H]+ 952.6. UPLC r.t. (2.5 min): 0.3 min). By performing the reaction in DMSO and upon 1 h irradiation, we could obtain, next to the hydrolysis compound, the expected product 6B in 20% yield. MALDI-TOF analysis confirmed the identity of the compound, showing a peak at m/z 1229.6 [M + H]+.
PNA functionalization with photoclick
In order to assess the full potential of the photoclick synthetic procedure, we applied it to a PNA sequence. PNAs are DNA/RNA mimics which were first reported by Nielsen et al.40 They have gained significant interest owing to their ability to hybridize with DNA/RNA complementary strands with very high affinity, their proven stability to nuclease and protease degradation and their lack of unspecific interactions with proteins. Thanks to these characteristics, PNAs are very promising for several applications ranging from nanotechnology41,42 to in vivo antisense therapy43 and tumour pre-targeting.8 However, there is room for improvement of the potential of PNAs in biological applications, for example by increasing their low solubility in physiological environment or by equipping them with a probe for cellular localization. It is therefore important to develop techniques for PNA functionalization. In particular, substitution with metal complexes have been of great interest to exploit additional properties of the new PNA–metal complex conjugate.8,15,16,37,44–51 In this work, we applied photoclick to achieve the insertion of a ferrocene moiety into a PNA sequence that was previously applied as an antisense agent on hepatitis B virus.52 This sequence, which was synthesized via solid phase synthesis,53 was further modified with the insertion of the PNA monomer bearing the modified tetrazole and a cysteine for conjugation via Michael addition (PNA, Fig. 2). The photoclick reaction was performed several times to optimize the conditions and the structures of the isolated products are reported in Fig. 6. Having the PNA sequence as substrate, the reaction showed a similar behaviour to that reported previously with the DNA strand.39 When DMSO was used as solvent, the PNA starting material was converted to the hydrolyzed form of the acyl-nitrene intermediate 14. Therefore, in the second trial, the reaction was performed in water with 1 eq. of 4B. After 1 h irradiation, we could isolate all the products reported in Fig. 6. The photoclick product 11, containing a single Fc moiety, was isolated in 6.5% yield. The doubly Fc labelled PNA 12 was also formed in 8% yield, due to the occurrence of both photoclick and Michael addition. Furthermore 13, a PNA containing the hydrolyzed tetrazole and the ferrocene conjugated at the cys group via Michael addition, was isolated in 14% yield. Due to the presence of water, the main component of the mixture was found to be the unlabelled hydrolyzed product 14 (30%). Next to these components, two compounds with m/z 5380 and 5566, respectively were isolated but their structures were not elucidated. In the attempt to improve the labelling efficiency, increasing amounts of 4B were used (5 eq.). The yield of formation of the photoclick product 11 could be increased to 10%. Additionally, the double Fc containing 12 was isolated in 2.6% yield, the hydrolyzed intermediate 14 in 17% yield and 13 in 36% yield. The component with m/z 5566 was also formed. As became evident from the last reaction conditions used, it is possible to increase the formation of the photoclick product by increasing the amount of the Fc-maleimide starting material. The kinetic of the competing Michael reaction seems however to be faster than the one of the photoclick, therefore, higher yields could be obtained if the cysteine would not be present.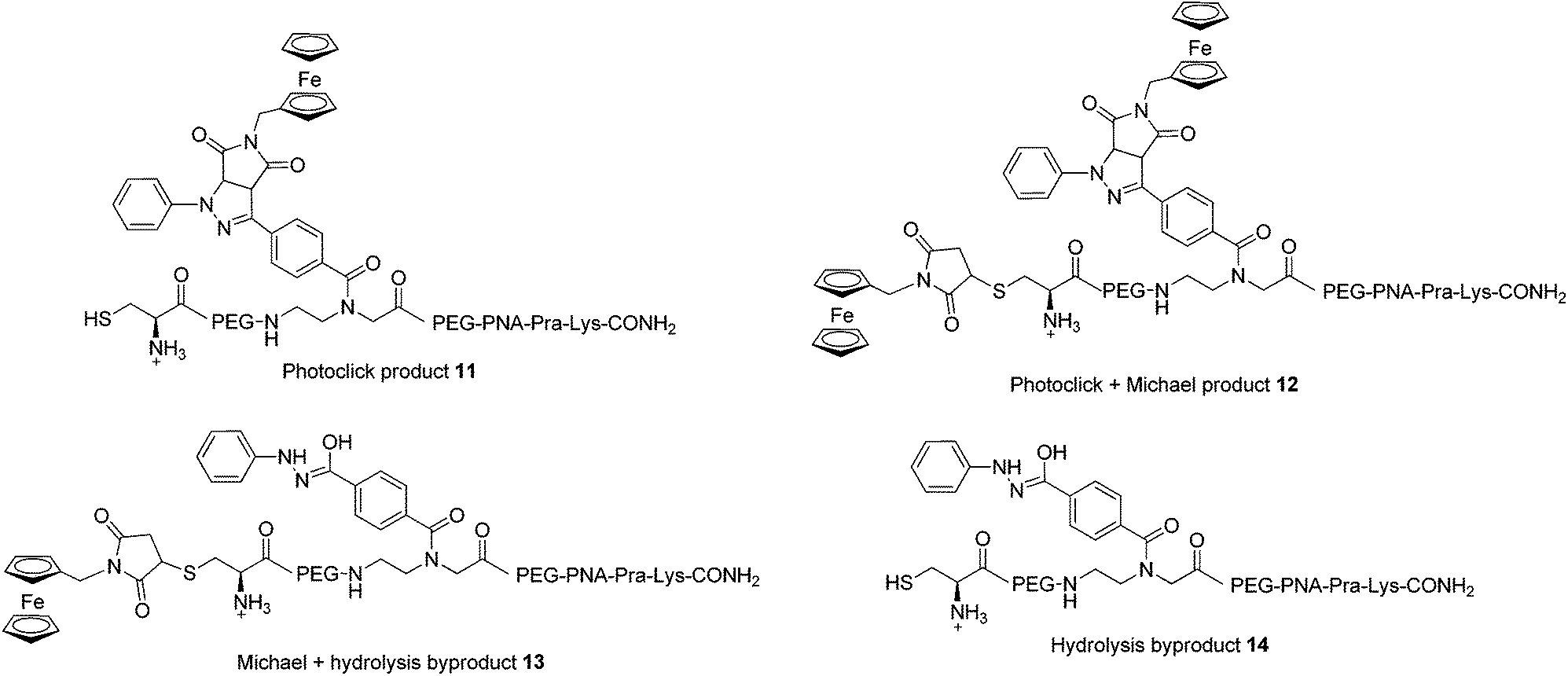 | ||
| Fig. 6 Products of the reaction between PNA and 4B upon 302 nm light irradiation. All amine groups in the PNA sequences are present as TFA salts. | ||
Experimental
Instruments and methods
All commercial chemicals were used without further purification. 1H and 13C NMR measurements were carried out on Bruker 400 and 500 spectrometers and referenced to residual solvent peaks. The abbreviations for the peak multiplicities are as follows: s (singlet), bs (broad singlet), d (doublet), dd (doublet of doublets), t (triplet), q (quartet), m (multiplet). UV spectra were recorded on a Varian Cary 100 spectrophotometer. Elemental microanalyses were performed on a LecoCHNS-932 elemental analyser. ESI-MS and R.P. UPLC-MS were performed using a Bruker Daltonics HCT 6000 mass spectrometer. R.P. UPLC-MS spectra were recorded on a Waters Acquity™ system equipped with a PDA detector and an auto-sampler. R.P. UPLC-MS was performed on an Acquity UPLC BEH C18 column (21.50 × 1.7 mm). The UPLC run (flow rate: 0.6 mL min−1) was carried out with a linear gradient of A (double distilled water containing 0.1% v/v formic acid) and B(acetonitrile): t = 0 min, 5% B; t = 0.25 min, 5% B; t = 1.5 min, 100% B; t = 2.5 min, 100% B. High resolution ESI-MS spectra were recorded using a Bruker ESQUIRE-LC quadrupole ion trap instrument. The matrix-assisted laser desorption/ionization time of flight mass spectrometry (MALDI-TOF) mass spectra were measured on a Bruker Daltonics Autoflex. The experiments were performed in reflector (RP) or linear (LP) mode using α-cyano-4-hydroxy-cinnamic acid on a Prespotted Anchor Chip (PAC HCCA) or sinapinic acid (SA) as the matrix. Preparative HPLC purification was carried out on a Varian ProStar system and an Agilent Zorbax 300 SB-C18 prep column (5 μm particle size, 300 Å pore size, 150 × 21.2 mm, flow rate: 20 mL min−1). The runs were performed with a linear gradient of A (double distilled water containing 0.1% v/v trifluoroacetic acid) and B (acetonitrile, Sigma-Aldrich HPLC grade). Microwave reactions were performed using a Biotage Initiator + Robot Eight instrument in 2.5 mL sealed microwave vials. UV irradiation at 302 nm were performed using a handheld UV lamp UVM-57, 6 Watt from UVP, Cambridge, UK. Samples were irradiated in a Starna GmbH 1.4 or 3.5 mL fluorescence quartz cuvette (width 1 cm) while stirring.Syntheses
A, B, 7, 8 were synthesized as previously described. All analytical data match with that reported in the literature.24,25,36,37:Hex (1:1) as the eluent to isolate the product (r.f. 0.65, 630 mg, 82%) as a yellowish solid. ESI-MS (pos. detection mode): m/z [M + Na]+ 667.2, [2M − H + Na]+ 1311.1. 1H-NMR δH (CDCl3, 500 MHz): 1.44 (s, 9H, tBu), 3.29 (bs, 1H, NHCH2CH2), 3.54 (bs, 2H, NCH2CO), 3.75 (bs, 1H, NHCH2CH2), 3.93 (bs, 1H, CH2CH2N), 4.14 (bs, 1H, CH2CH2N), 4.24 (m, 1H, FMOC–CHCH2), 4.29–4.40 (dd, 2H, CHCH2O), 5.68 (bs, 1H, NH), 7.29–7.33 (t, 2H, FMOC), 7.38–7.42 (t, 2H, FMOC), 7.51–7.63 (m, 7H, FMOC + Ph), 7.77–7.79 (d, 2H, FMOC), 8.16–8.29 (m, 4H, Ph). 13C-NMR (CDCl3, 126 MHz, rotamers observed): 28.10, 39.34, 47.27, 47.35, 47.59, 49.14, 67.14, 120.04, 120.11, 125.26, 125.31, 127.21, 127.32, 127.46, 127.83, 129.86, 136.94, 141.43, 144.09, 156.29, 156.90, 164.52, 168.98, 169.23, 171.90, 172.64. HR ESI-MS calc for C37H37N6O5 [M + H]+ 645.2028, found 645.2818, calc for C37H36N6O5Na [M + Na]+ 667.2639, found 667.2636.
:TFA:TIPS (2:1:0.4) solution. The mixture was stirred overnight at room temperature. The solvent was then removed at the rotary evaporator and the crude product purified by column chromatography on silica gel with eluent EtOAc:MeOH (10:3) to isolate the product as a white powder (r.f. 0.55, 320 mg, 90%). ESI-MS (pos. detection mode): m/z [M + H]+ 589.3, [2M + H]+ 1178.5. 1H-NMR δH (acetone-d6, 500 MHz): 3.34 (m, 1H, NHCH2CH2), 3.52 (m, 1H, NHCH2CH2), 3.58 (m, 1H, CH2CH2N), 3.73 (m, 1H, CH2CH2N), 4.19 (m, 1H, FMOC–CHCH2), 4.25 (s, 2H, NCH2COOH), 4.33–4.39 (dd, 2H, CHCH2O), 6.60 (bs, 1H, NH), 7.30–7.33 (t, 2H, FMOC), 7.39–7.42 (t, 2H, FMOC), 7.60–7.66 (m, 3H, Ph), 7.70–7.72 (m, 4H, FMOC + Ph), 7.86–7.88 (d, 2H, FMOC), 8.20–8.23 (m, 4H, Ph). 13C-NMR (acetone-d6, 126 MHz): 41.31, 47.42, 48.00, 50.53, 67.13, 120.78, 126.08, 127.63, 127.74, 127.94, 128.31, 128.54, 128.69, 130.90, 130.94, 137.78, 139.60, 142.15, 145.17, 156.98, 165.37, 171.28, 171.81. HR ESI-MS calc for C33H29N6O5 [M + H]+ 589.2194, found 589.2196.
:MeOH (100:2.5) as the eluent. The product was isolated as the first band (r.f. 0.67). The solvent was removed using a rotary evaporator to yield 16.6 mg of the expected product as a light brown oil (60%). 1H-NMR δH (CDCl3, 500 MHz): 1.79 (m, 2H, CH2CH2CH2), 2.18 (t, 2H, CpCH2CH2), 2.26 (t, 2H, CH2CH2CO), 3.45–3.47 (m, 2H, NHCH2CH2), 3.69–3.70 (m, 2H, CH2CH2NH), 4.63 (s, 4H, Cp), 5.81 (br s, 1H, NH), 6.73 (s, 2H, maleimide). 13C-NMR (CDCl3, 126 MHz): 26.68, 27.74, 35.85, 37.76, 39.17, 81.96, 106.32, 134.43, 171.10, 172.62, 225.33. ESI-MS (pos. detection mode): m/z [M + H+] 413.1. UPLC r.t. (2.5 min): 1.3 min.
:EtOAc 1:1) and UPLC (r.t. 1.35 min, m/z = 359.1) confirmed the full conversion of the amine group to the azide. The mixture was then transferred in a microwave reactor and the following reagents were added to it: Fmoc-L-Pra-OH (15.1 mg, 0.045 mmol, 1.0 eq.), TBTA (2.4 mg, 0.0045 mmol, 0.1 eq.) in 254 μL of MeOH, CuSO4 (0.143 mg, 0.0009 mmol, 0.02 eq.) in 15 μL of H2O and Na ascorbate (1.78 mg, 0.009 mmol, 0.2 eq.) in 154 μL of H2O and 0.5 mL of MeOH to help full dissolution. The mixture was refluxed in the MW oven for 1 h at 80 °C (60 W). The solvent was removed at the rotary evaporator and the mixture was purified by column chromatography on silica gel with DCM:MeOH (10:1) as the eluent to isolate the product (r.f. 0.27, 50%). Another batch was purified by preparative r.p. HPLC (linear gradient of A (double distilled water containing 0.1% v/v trifluoroacetic acid) and B (acetonitrile): t = 0–3 min 25% B, t = 33 min 100% B, t = 35 min 100% B, flow rate 20 mL min−1, detection 275 nm) to afford the same product. ESI-MS (neg. detection mode): m/z [M − H]− 692.1. UPLC r.t. (2.5 min): 1.40 min. HR ESI-MS calc for C34H31MnN5O8 [M − H]− 692.1559, found 692.1565. 1H-NMR not meaningful due to traces of paramagnetic impurities. 13C-NMR (MeOH-d4, 126 MHz): 27.91, 28.46, 29.23, 30.41, 55.73, 67.68, 82.85, 83.41, 107.49, 120.68, 125.88, 127.91, 128.54, 142.29, 144.97, 226.32. Some quaternary signals not observed.
:EtOAc 1:1) and UPLC (r.t. 1.60 min, m/z = 240.9) confirmed the full conversion of the amine group to azide. Then the mixture was transferred in a microwave reactor and the following reagents were added to it: Fmoc-L-Pra-OH (31.2 mg, 0.093 mmol, 1 eq.), TBTA (4.9 mg, 0.0093 mmol, 0.1 eq.) in 526 μL of MeOH, CuSO4 (0.297 mg, 0.00186 mmol, 0.02 eq.) in 30 μL of H2O and Na ascorbate (3.68 mg, 0.0186 mmol, 0.2 eq.) in 380 μL of H2O and 0.5 mL of MeOH to help full dissolution. The mixture was refluxed in MW oven for 1 h at 80 °C (60 W). After the reaction time, the solvent was removed using a rotary evaporator and the mixture was purified by column chromatography on silica gel with DCM:MeOH (10:1) as the eluent, to isolate the product (r.f. 0.52, 29%). Another batch was purified by preparative r.p. HPLC (linear gradient of A (double distilled water containing 0.1% v/v trifluoroacetic acid) and B (acetonitrile): t = 0–3 min 25% B, t = 33 min 100% B, t = 35 min 100% B, flow rate 20 mL min−1, detection 275 nm) to afford the same compound. ESI-MS (neg. detection mode): m/z [M − H]− 575.1, [2M − H]− 1151.3. UPLC r.t. (2.5 min): 1.55 min. HR ESI-MS calc for C31H27FeN4O4 [M − H]− 575.1387, found 575.1391. 1H-NMR δH (MeOH-d4, 500 MHz): 3.04–3.09 (m, 1H, triazCH2CH), 3.33 (below MeOH, m, 1H, triazCH2CH), 4.14–4.26 (m, 12H, Fc, OCH2CH, CH2CHFmoc), 4.49–4.51 (m, 1H, NHCHCOOH), 7.32–7.35 (m, 2H, Fmoc), 7.42–7.45 (m, 2H, Fmoc), 7.62–7.67 (m, 2H, Fmoc), 7.77 (bs, 1H, triaz), 7.85–7.87 (m, 2H, Fmoc). 13C-NMR (MeOH-d4, 126 MHz): 28.85, 48.27, 51.09, 55.13, 68.26, 121.16, 121.19, 124.25, 126.60, 126.61, 128.43, 129.03, 129.05, 142.67, 142.72, 144.70, 145.33, 145.37, 158.59, 174.59 (Fc signals below MeOH).
:EtOAc 1:1) and UPLC (r.t. 1.35 min, m/z = 359.1) confirmed the full conversion of the amine group to azide. Then the mixture was transferred in a microwave reactor and the following reagents were added to it: NLS-Pra as TFA salt (31.07 mg, 0.0224 mmol, 1 eq.) in 2.24 mL of H2O, TBTA (1.19 mg, 0.00224 mmol, 0.1 eq.) in 474 μL of MeOH, CuSO4 (0.071 mg, 0.00045 mmol, 0.02 eq.) in 11 μL of H2O and Na ascorbate (0.888 mg, 0.00448 mmol, 0.2 eq.) in 81 μL of H2O and 2.5 mL of MeOH to help full dissolution. The mixture was refluxed in MW oven for 1 h at 80 °C (60 W). After the reaction time, the solvent was removed using a rotary evaporator and the crude was purified by preparative r.p. HPLC (linear gradient of A (double distilled water containing 0.1% v/v trifluoroacetic acid) and B (acetonitrile): t = 0–1.48 min 5% B, t = 38 min 100% B, t = 40 min 100% B, flow rate 20 mL min−1, detection 275 nm). Chromatographic yield: 65%. ESI-MS (pos. detection mode): m/z [M + H]+ 1289.6, [M + 2H]2+ 645.8, [M + 3H]3+ 430.7. UPLC r.t. (2.5 min): 0.9 min. MALDI-TOF [M − Mn(CO)3 + H]+ 1151.7, [M − Mn(CO)3 + Na]+ 1173.7, [M − Mn(CO)3 + K]+ 1189.6, [M + H]+ 1289.6.
:EtOAc 1:1) and UPLC (r.t. 1.35 min, m/z = 240.9) confirmed the full conversion of the amine group to the azide. Then the mixture was transferred in a microwave reactor and the following reagents were added to it: NLS-Pra as TFA salt (17.6 mg, 0.0127 mmol, 1 eq.) in 760 μL of H2O, TBTA (0.672 mg, 0.00126 mmol, 0.1 eq.) in 71 μL of MeOH, CuSO4 (0.04 mg, 0.00025 mmol, 0.02 eq.) in 4 μL of H2O and Na ascorbate (0.50 mg, 0.00254 mmol, 0.2 eq.) in 44 μL of H2O and 1 mL of MeOH to help full dissolution. The mixture was refluxed in MW oven for 1 h at 80 °C (60 W). After the reaction time, the solvent was removed using a rotary evaporator and the crude was purified by preparative r.p. HPLC (linear gradient of A (double distilled water containing 0.1% v/v trifluoroacetic acid) and B (acetonitrile): t = 0–1.48 min 5% B, t = 38 min 100% B, t = 40 min 100% B, flow rate 20 mL min−1, detection 275 nm method). Chromatographic yield: 6.5%. ESI-MS (pos. detection mode): m/z [M − Fc − CH2 + 3H]3+ 325.7, [M + 3H]3+ 391.7, [M − Fc − CH2 + 2H]2+ 487.9, [M + 2H]2+ 586.9, [M + H]+ 1172.6. UPLC r.t. (2.5 min): 0.9 min. MALDI-TOF [M + H]+ 1172.6.
:MeOH (10:1). The yellow spot was isolated (r.f. 0.3) to yield the product and a yellow/green solid (2.3 mg, 63%). 1H-NMR not meaningful due to traces of paramagnetic impurities. 13C-NMR (CDCl3, 126 MHz): 28.27, 29.41, 29.83, 39.41, 47.37, 65.98, 67.18, 114.74, 120.18, 121.97, 125.39, 127.36, 127.93, 129.34, 132.07, 136.33, 141.47, 144.16, 169.30. Some quaternary signals not observed. ESI-MS (pos. detection mode): m/z [M]+ 911.4. HR ESI-MS calc for C52H50FeN5O7 [M + H]+ 912.3054, found 912.3041. UPLC r.t. (2.5 min): 1.8 min.
Conclusions
The functionalization of biologically relevant molecules with metal complexes is a very intriguing topic nowadays. The insertion of a metal fragment can strongly improve the properties of the system, from a therapeutic or diagnostic point of view as well as for the investigation of biological mechanisms. The use of copper-catalyzed azide–alkyne cycloaddition is deeply exploited for the functionalization of biomolecules, but some drawbacks limit its application for example in vivo. Therefore, the optimization of modified click procedure was extensively studied in the recent years. In this paper we present the use of modified click strategies (namely one-pot click and photoclick) to insert organometallic fragments in peptides and PNA sequences. One-pot click allows for the in situ conversion of an amine to the azide intermediate, which is then directly clicked with the alkynyl counterpart. Therefore, the isolation of the azide containing compound is not required, relieving the safety concerns linked to the manipulation of azide compounds. We obtained high labelling efficiencies using one-pot click on an amine derivative of cymantrene for derivatization of both a model alkynyl amino acid and a NLS peptide carrying this amino acid. When a ferrocene derivative was used as an amine substrate, yields were found to be lower. The harsh conditions used for one-pot click (1 h of microwave assisted reflux at 80 °C) might lead to the decomposition of the methylaminoferrocene precursor. We also investigated the application of metal-free photoclick to our metal-containing precursors, to establish a biocompatible strategy for organometallic derivatization of biomolecules. While the use of cymantrene as substrate for photoclick yielded mainly a by product of the light-mediated CO release from the metal core, the application of photoclick to a ferrocene-maleimide derivative allowed for fast and effective functionalization of both a tetrazole-derivatized PNA monomer and a NLS peptide containing this PNA residue. This technique was also used for Fc insertion into a 15-mer PNA sequence carrying the tetrazole group, with yields comparable to those of a previously reported photoclick application for dsDNA functionalization.39 Overall, we envisage the application of these very versatile techniques for the functionalization of biomolecules with other metal complexes. As a result, a wide range of metal fragments could be introduced into bioactive systems to prepare novel diagnostic tools useful to study cellular mechanisms. One could also imagine that novel therapeutic entities could be designed, for example, with the introduction of ruthenium complexes into targeting peptides to obtain selective compounds for traditional chemotherapy or photodynamic therapy. Overall, this study will expand the pool of metal-based probe-containing systems, which can be profitably applied in chemical biology or in medicinal chemistry.Acknowledgements
This work was financially supported by the Swiss National Science Foundation (Ambizione Fellowship No. PZ00P2_126404 as well as Professorships No. PP00P2_133568 and PP00P2_157545 to G.G), the University of Zurich (G.G), the Stiftung für wissenschaftliche Forschung of the University of Zurich (G.G.) and the Forschungskredit of the University of Zurich (Grant K-73532-01-01 to C.M). The authors thank Dr Thomas Fox for his help with the NMR experiments and Dr Phuc Ung for helpful discussions.References
- R. Mahato, W. Tai and K. Cheng, Adv. Drug Delivery Rev., 2011, 63, 659–670 CrossRef CAS PubMed.
- T. J. S. Binghe Wang and R. A. Soltero, Drug Delivery: Principles and Applications, John Wiley & Sons, Inc., Hoboken, 2005 Search PubMed.
- V. Stella, R. Borchardt, M. Hageman, R. Oliyai, H. Maag and J. Tilley, Prodrugs: Challenges and Rewards, Springer, New York, 2007 Search PubMed.
- G. Gasser, Inorganic Chemical Biology, Wiley-VCH Verlag GmbH & Co. KGaA, UK, 2014 Search PubMed.
- C. Preusch Peter, in Medicinal Inorganic Chemistry, American Chemical Society, 2005, vol. 903, pp. 15–29 Search PubMed.
- C. Mari, V. Pierroz, A. Leonidova, S. Ferrari and G. Gasser, Eur. J. Inorg. Chem., 2015, 2015, 3879–3891 CrossRef CAS.
- A. Leonidova, P. Anstaett, V. Pierroz, C. Mari, B. Spingler, S. Ferrari and G. Gasser, Inorg. Chem., 2015, 54, 9740–9748 CrossRef CAS PubMed.
- A. Leonidova, C. Foerster, K. Zarschler, M. Schubert, H.-J. Pietzsch, J. Steinbach, R. Bergmann, N. Metzler-Nolte, H. Stephan and G. Gasser, Chem. Sci., 2015, 6, 5601–5616 RSC.
- S. Jaracz, J. Chen, L. V. Kuznetsova and I. Ojima, Bioorg. Med. Chem., 2005, 13, 5043–5054 CrossRef CAS PubMed.
- J. S. Butler and P. J. Sadler, Curr. Opin. Chem. Biol., 2013, 17, 175–188 CrossRef CAS PubMed.
- V. Fernandez-Moreira, F. L. Thorp-Greenwood and M. P. Coogan, Chem. Commun., 2010, 46, 186–202 RSC.
- M. Meldal and C. W. Tornøe, Chem. Rev., 2008, 108, 2952–3015 CrossRef CAS PubMed.
- M. D. Best, Biochemistry, 2009, 48, 6571–6584 CrossRef CAS PubMed.
- W. Tang and M. L. Becker, Chem. Soc. Rev., 2014, 43, 7013–7039 RSC.
- G. Gasser, in Peptide Nucleic Acids, ed. P. E. Nielsen and D. H. Appella, Humana Press, 2013, vol. 1050, pp. 55–72 Search PubMed.
- G. Gasser, A. M. Sosniak and N. Metzler-Nolte, Dalton Trans., 2011, 40, 7061–7076 RSC.
- A. Leonidova, V. Pierroz, L. A. Adams, N. Barlow, S. Ferrari, B. Graham and G. Gasser, ACS Med. Chem. Lett., 2014, 5, 809–814 CrossRef CAS PubMed.
- S. Bräse, C. Gil, K. Knepper and V. Zimmermann, Angew. Chem., Int. Ed., 2005, 44, 5188–5240 CrossRef PubMed.
- P. Ostrovskis, C. M. R. Volla, M. Turks and D. Markovic, Curr. Org. Chem., 2013, 17, 610–640 CrossRef CAS.
- C. P. Ramil and Q. Lin, Curr. Opin. Chem. Biol., 2014, 21, 89–95 CrossRef CAS PubMed.
- M. A. Tasdelen and Y. Yagci, Angew. Chem., Int. Ed., 2013, 52, 5930–5938 CrossRef CAS PubMed.
- H. S. G. Beckmann and V. Wittmann, Org. Lett., 2007, 9, 1–4 CrossRef CAS PubMed.
- Y. Wang, C. I. Rivera Vera and Q. Lin, Org. Lett., 2007, 9, 4155–4158 CrossRef CAS PubMed.
- M. Patra, G. Gasser, M. Wenzel, K. Merz, J. E. Bandow and N. Metzler-Nolte, Organometallics, 2012, 31, 5760–5771 CrossRef CAS.
- P. D. Beer and D. K. Smith, Dalton Trans., 1998, 417–424 RSC.
- A. Vessières, M. Salmain, P. Brossier and G. Jaouen, J. Pharm. Biomed. Anal., 1999, 21, 625–633 CrossRef.
- M. Salmain, A. Vessières, P. Brossier, I. S. Butler and G. Jaouen, J. Immunol. Methods, 1992, 148, 65–75 CrossRef CAS PubMed.
- A. Leonidova, V. Pierroz, R. Rubbiani, J. Heier, S. Ferrari and G. Gasser, Dalton Trans., 2014, 43, 4287–4294 RSC.
- A. Leonidova, V. Pierroz, R. Rubbiani, Y. Lan, A. G. Schmitz, A. Kaech, R. K. O. Sigel, S. Ferrari and G. Gasser, Chem. Sci., 2014, 5, 4044–4056 RSC.
- W. Hu, K. Splith, I. Neundorf, K. Merz and U. Schatzschneider, J. Biol. Inorg. Chem., 2012, 17, 175–185 CrossRef CAS PubMed.
- G. Gasser, A. J. Fischmann, C. M. Forsyth and L. Spiccia, J. Organomet. Chem., 2007, 692, 3835–3840 CrossRef CAS.
- R. Jiang, Y. Zhang, Y.-C. Shen, X. Zhu, X.-P. Xu and S.-J. Ji, Tetrahedron, 2010, 66, 4073–4078 CrossRef CAS.
- R. K. V. Lim and Q. Lin, Acc. Chem. Res., 2011, 44, 828–839 CrossRef CAS PubMed.
- W. Song, Y. Wang, J. Qu, M. M. Madden and Q. Lin, Angew. Chem., Int. Ed., 2008, 47, 2832–2835 CrossRef CAS PubMed.
- Y. Wang, W. Song, W. J. Hu and Q. Lin, Angew. Chem., Int. Ed., 2009, 48, 5330–5333 CrossRef CAS PubMed.
- W. Song, Y. Wang, J. Qu, M. M. Madden and Q. Lin, Angew. Chem., Int. Ed., 2008, 47, 2832–2835 CrossRef CAS PubMed.
- G. Gasser, N. Huesken, S. D. Koster and N. Metzler-Nolte, Chem. Commun., 2008, 3675–3677 RSC.
- G. Dordelmann, T. Meinhardt, T. Sowik, A. Krueger and U. Schatzschneider, Chem. Commun., 2012, 48, 11528–11530 RSC.
- S. Arndt and H.-A. Wagenknecht, Angew. Chem., Int. Ed., 2014, 53, 14580–14582 CrossRef CAS PubMed.
- P. E. Nielsen, M. Egholm, R. H. Berg and O. Buchardt, Science, 1991, 254, 1497–1500 CAS.
- P. Anstaett, Y. Zheng, T. Thai, A. M. Funston, U. Bach and G. Gasser, Angew. Chem., Int. Ed., 2013, 52, 4217–4220 CrossRef CAS PubMed.
- P. Anstaett and G. Gasser, Chimia, 2014, 68, 264–268 CrossRef CAS PubMed.
- P. E. Nielsen, Curr. Opin. Mol. Ther., 2000, 2, 282–287 CAS.
- C. Mari, M. Panigati, L. D'Alfonso, I. Zanoni, D. Donghi, L. Sironi, M. Collini, S. Maiorana, C. Baldoli, G. D'Alfonso and E. Licandro, Organometallics, 2012, 31, 5918–5928 CrossRef CAS.
- T. Joshi, M. Patra, L. Spiccia and G. Gasser, Artif. DNA PNA XNA, 2013, 4, 11–18 CrossRef PubMed.
- G. Gasser, A. Pinto, S. Neumann, A. M. Sosniak, M. Seitz, K. Merz, R. Heumann and N. Metzler-Nolte, Dalton Trans., 2012, 41, 2304–2313 RSC.
- G. Gasser, A. M. Sosniak, A. Leonidova, H. Braband and N. Metzler-Nolte, Aust. J. Chem., 2011, 64, 265–272 CrossRef CAS.
- G. Gasser, K. Jaeger, M. Zenker, R. Bergmann, J. Steinbach, H. Stephan and N. Metzler-Nolte, J. Inorg. Biochem., 2010, 104, 1133–1140 CrossRef CAS PubMed.
- Z. Ma, F. Olechnowicz, Y. A. Skorik and C. Achim, Inorg. Chem., 2011, 50, 6083–6092 CrossRef CAS PubMed.
- D.-L. Popescu, T. J. Parolin and C. Achim, J. Am. Chem. Soc., 2003, 125, 6354–6355 CrossRef CAS PubMed.
- R. M. Franzini, R. M. Watson, G. K. Patra, R. M. Breece, D. L. Tierney, M. P. Hendrich and C. Achim, Inorg. Chem., 2006, 45, 9798–9811 CrossRef CAS PubMed.
- M. Robaczewska, R. Narayan, B. Seigneres, O. Schorr, A. Thermet, A. J. Podhajska, C. Trepo, F. Zoulim, P. E. Nielsen and L. Cova, J. Hepatol., 2004, 42, 180–187 CrossRef PubMed.
- Peptide Nucleic Acids: Methods and Protocols, ed. P. E. Nielsen and D. H. Appella, Humana Press, 2014 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5qi00270b |
| This journal is © the Partner Organisations 2016 |