
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Recent developments of iron pincer complexes for catalytic applications
Gerald
Bauer
and
Xile
Hu
*
Laboratory of Inorganic Synthesis and Catalysis, Institute of Chemical Sciences and Engineering, École Polytechnique Fédérale de Lausanne (EPFL), ISIC-LSCI, BCH 3305, 1015 Lausanne, Switzerland. E-mail: xile.hu@epfl.ch
First published on 1st February 2016
Abstract
Iron catalysis is attractive for organic synthesis because iron is inexpensive, abundant, and non-toxic. To control the activity and stability of an iron center, a large number of iron pincer complexes have been synthesized. Many such complexes exhibit excellent catalytic activity in a number of important organic reactions such as hydrogenation, hydrosilylation, dehydrogenation, and carbon–carbon bond forming reactions. In this review, recent examples of representative iron pincer catalysts are presented.
 Gerald Bauer | Gerald Bauer was born in 1984 in Tulln/Donau, Austria. He studied ‘Technical Chemistry’ at the University of Technology in Vienna with semesters at the UCT in Prague and the University of Waterloo, ON, Canada during his bachelor studies. Gerald received his M.Sc. in 2010 under the joint supervision of Prof. Karl Kirchner, UT Vienna, and Prof. Kazushi Mashima, University of Osaka. Shortly afterwards, he joined the lab of Prof. Xile Hu at the EPFL working on the catalytic applications and mechanistic investigation of iron pincer complexes in Kumada cross-coupling reactions, for which he was awarded his PhD in 2015. |
 Xile Hu | Xile Hu was born in 1978 in Putian, China. He received a B.S. degree from Peking University (2000) and a Ph.D. degree from the University of California, San Diego (2004; advisor: Prof. Karsten Meyer). He carried out a postdoctoral study at the California Institute of Technology (advisor: Prof. Jonas Peters) before joining the École Polytechnique Fédérale de Lausanne (EPFL) as a tenure-track assistant professor in 2007. He is currently an associate professor at the same institute. His research interests span from organometallic chemistry, synthetic methodologies, and reaction mechanisms to bio-mimetic and bio-speculated coordination chemistry to electrocatalysis and artificial photosynthesis. |
1. Introduction
Complexes of precious metals, particularly those from the platinum group, occupy a central place in homogeneous catalysis. However, the high cost and potential toxicity of precious metals make them less desirable for industrial applications. Thus, homogeneous catalysis using more abundant and cheaper first row transition metals has become a popular research theme. Iron catalysis is particularly desirable because iron is inexpensive, readily available, and non-toxic. In biological systems iron-containing enzymes are widely used as catalysts, and examples include cytochrome P-450, peroxidases, various oxygenases, and hydrogenases. While numerous reports of iron-catalyzed organic reactions are known,1,2 studies of well-defined iron catalysts are less common. Such studies, however, are essential for the in-depth understanding and further advancement of iron catalysis.This review focuses on the catalytic applications of iron pincer complexes.3,6–8 Pincer ligands, in general, are tridentate, 6-electron donor ligands that enforce a meridional coordination geometry on the metal center. Although there is no strict definition, a pincer ligand traditionally consists of a central benzene ring, which is 1,3-disubstituted with two chelating side arms (Fig. 1). Both the central atom (Dc) and the donors at the two arms (Ds) bind to the metal center, resulting in strong chelation.4,5,9–15 Replacement of the central aryl ring by a pyridinyl or diarylamine group leads to two major classes of variants of the original pincer ligands. In addition to their strong chelating ability and structural rigidity, the advantage of pincer ligands is their diversity. By modifying the central and side donors as well as the ligand backbone, it is possible to synthesize an almost endless number of ligands with varying electronic and steric properties.5 This diversity is very attractive for homogeneous catalysis where systematic studies of metal–ligand combinations are desired.
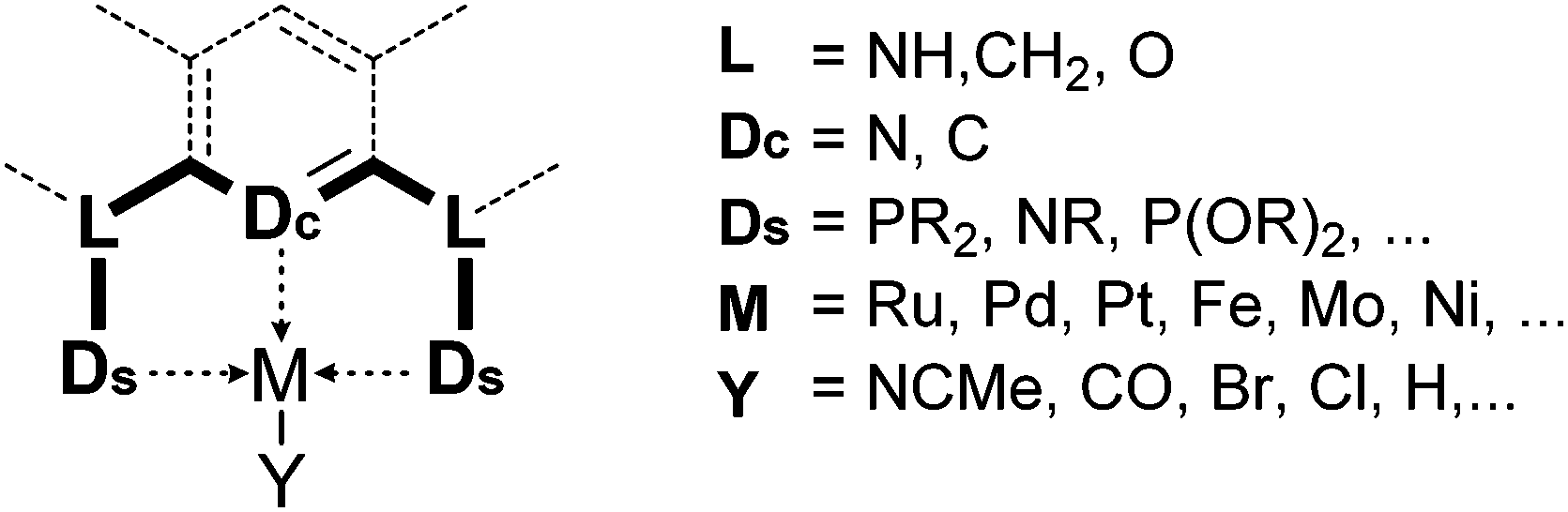 | ||
| Fig. 1 Generalized structure of pincer complexes.3–5 | ||
In this review, iron complexes of pincer ligands based on the following frameworks are discussed: 2,6-disubstituted pyridine, 1,3-disubstituted benzene, N,N-diarylamine, isoindoline and bis(phosphinoethyl)amine. Only catalysis by pre-formed iron complexes, but not in situ generated iron species, will be presented. The examples are selected to give a representative, but not comprehensive overview of the developments in the field. Mostly symmetrically substituted pincer ligands with a C2 or C2v symmetry are included. Unsymmetrically substituted ligands and terpyridine-type ligands are not treated. Pyridine diimines (PDI) might be considered as pincer ligands, and their iron complexes are very active for polymerization,16–19 hydrogenation, and other alkene, alkyne, and ketone functionalization reactions. Several excellent reviews covering these iron complexes have been published recently.20,21 Therefore, they will not be reviewed here.
2. Neutral pyridine-based PNP ligand systems and their applications in iron catalysis
The first iron PNP complexes were synthesized by Dahlhoff and Nelson in 1971 by reacting 2,6-bis(diphenylphosphinomethyl)pyridine with FeX2 (X = Cl, Br, I, NCS). The resulting complexes were in a high spin configuration and exhibited a 5-coordinate geometry with a slightly distorted square pyramidal structure.6 This ligand framework became popular, and in addition to the variance of the substituents on the phosphorus donors,5 the linker (L) that connects the phosphorus donors to the central pyridine ring could be modified to include not only CR2,6,22–31 but also NR5,32–39 and O (Fig. 2).40,41 To differentiate between the different linkers, these pincer ligands are abbreviated as “PLNLP”, where L = C, N, O, to represent CR2, NR, and O, respectively.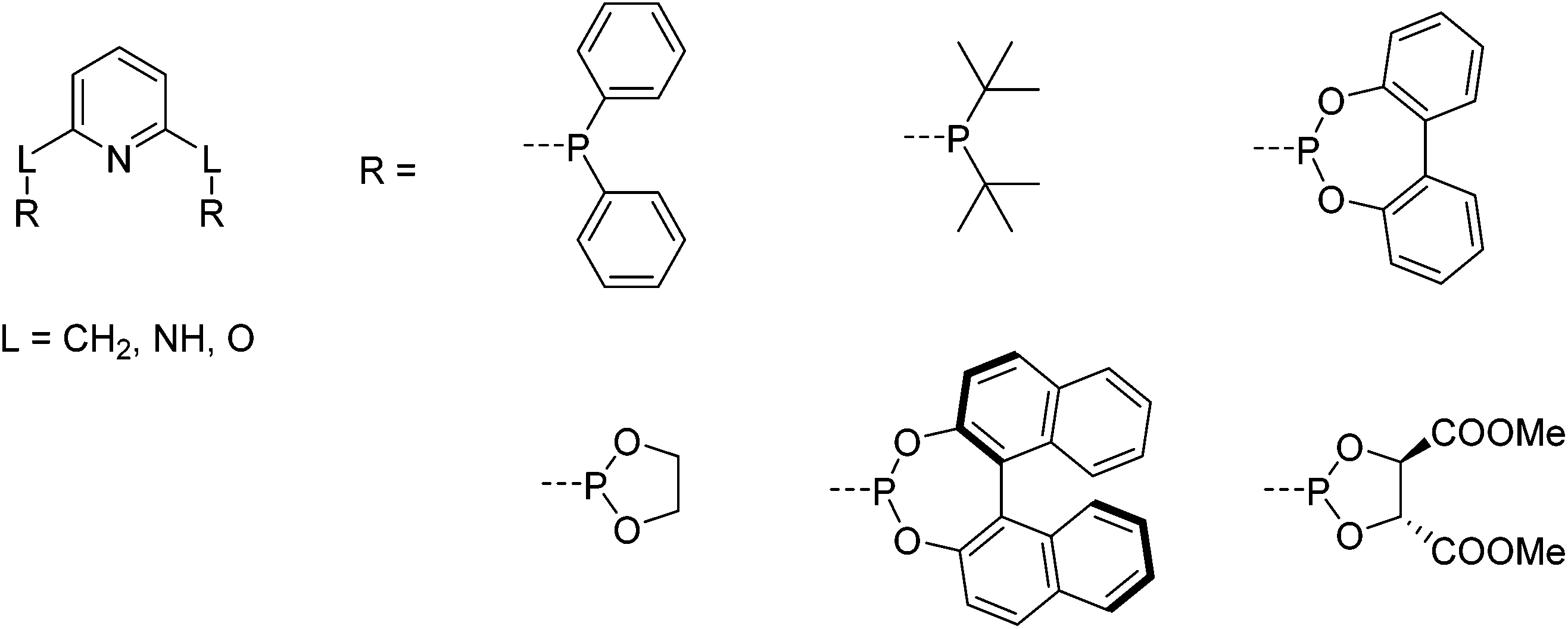 | ||
| Fig. 2 Structure of pyridine-based PNP ligands and selected chiral and achiral substituents on the phosphorus.5 | ||
Several groups studied the influence of linkers on the electronic density of the metal centre using the IR frequencies of M–CO bonds as a probe (Fig. 3).26,34,40 The CO stretching frequency of [(PNP)FeX2(CO)] (X = Cl, Br) decreases when the linker is changed from O to NH and to CH2. This result suggests that the electron donating character of the corresponding PNP ligands follows a similar order.
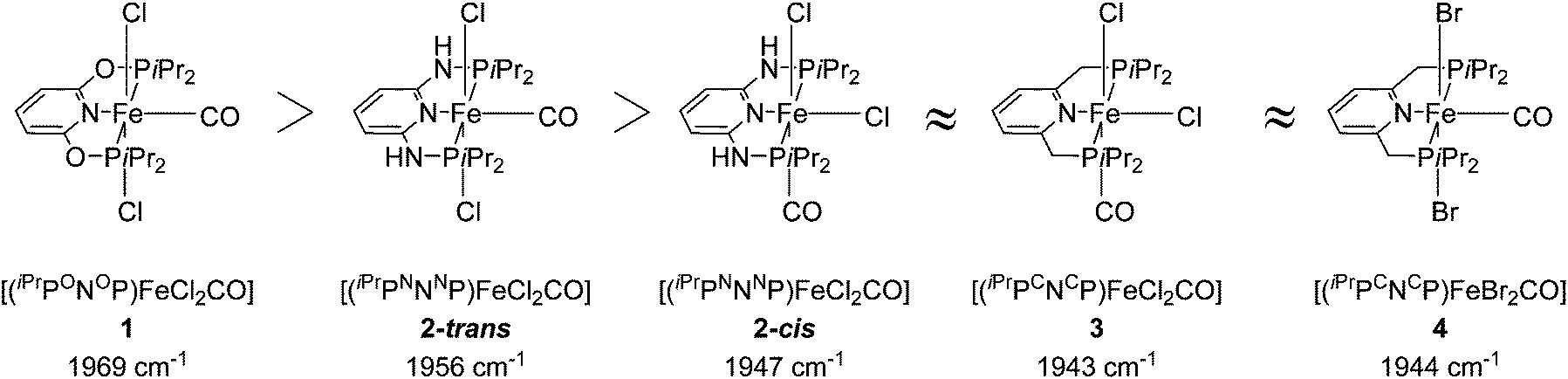 | ||
| Fig. 3 CO stretching frequencies for 1, 2-cis, 2-trans, 3 and 4.26,34,40 | ||
An interesting feature of these PNP pincer complexes is their metal–ligand cooperation. The slightly acidic CH2 and NH linkers are easily deprotonated under basic conditions, which causes a reversible dearomatization of the central pyridine ring.42 Further details will be discussed below.
2.1 Iron(II) PNNNP pincer complexes for the selective formation of 3-hydroxyacrylates from benzaldehydes and ethyl diazoacetates
Aldehydes react with ethyl diazoacetates (EDA) in the presence of Lewis acids (e.g. BF3, ZnCl2, AlCl3, SnCl2, GeCl2) to form β-keto esters.43 It was previously shown that the Lewis acid [(η5-Cp)Fe(CO)2THF](BF4) catalyzed the reaction of benzaldehydes with EDA to give β-hydroxy-2-aryl acrylates.44,45More recently Kirchner and co-workers employed cis-[(iPrPNNNP)Fe(CO)(CH3CN)2](X)2 (5-X; X = BF4−, BArF−) for the reaction of p-anisaldehyde with EDA (Fig. 4).33 These reactions produced selectively a hydroxyl acrylate (A, >80%) and only trace amounts of a β-keto ester (B). The two counter ions, BF4− and BArF−, gave similar results. A tentative mechanistic proposal is given in Fig. 5. The CO ligand exhibits a stronger trans effect than the nitrogen of the pyridine ring. Therefore the acetonitrile opposite to the carbonyl will be cleaved first. The dissociation of acetonitrile and successive coordination of an aldehyde gives intermediate 6. The nucleophilic attack of EDA on the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond gives the transition state 7, which upon elimination of N2 yields complex 8. Migration of the aryl substituent then leads to 9. The newly formed aldehyde is released and tautomerizes into the thermodynamically more stable ethyl 3-hydroxy-2-(-4-methoxyphenyl)acrylate.33
O bond gives the transition state 7, which upon elimination of N2 yields complex 8. Migration of the aryl substituent then leads to 9. The newly formed aldehyde is released and tautomerizes into the thermodynamically more stable ethyl 3-hydroxy-2-(-4-methoxyphenyl)acrylate.33
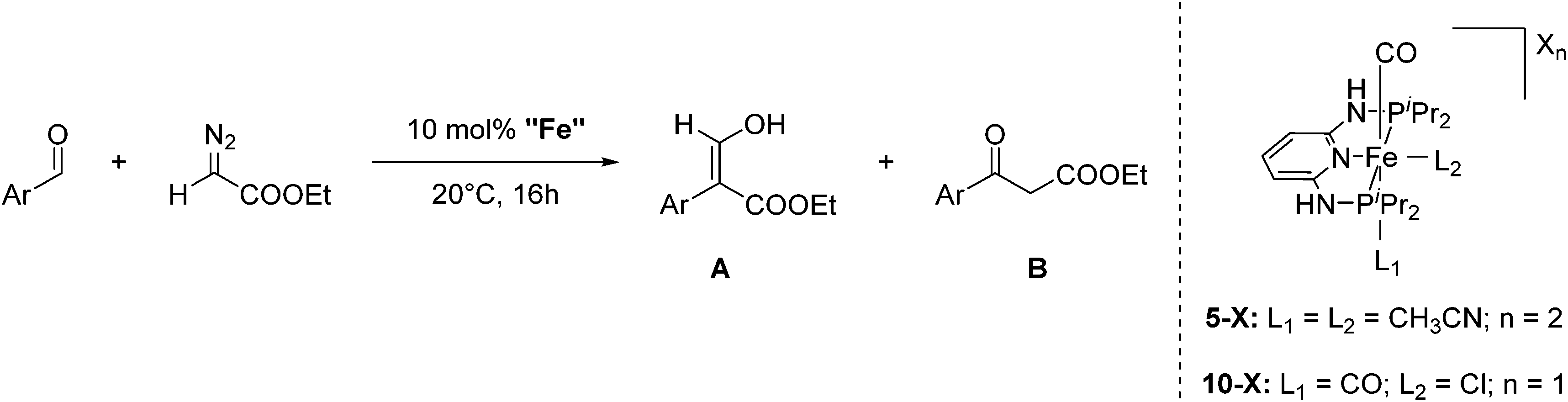 | ||
| Fig. 4 Reaction of p-anisaldehyde with EDA catalyzed by 5-X (X2 = BF4−, BArF) and 10-X (X = NO3−, CF3COO−, CF3SO3−, BF4−, PF6−, SbF6− and BArF−). | ||
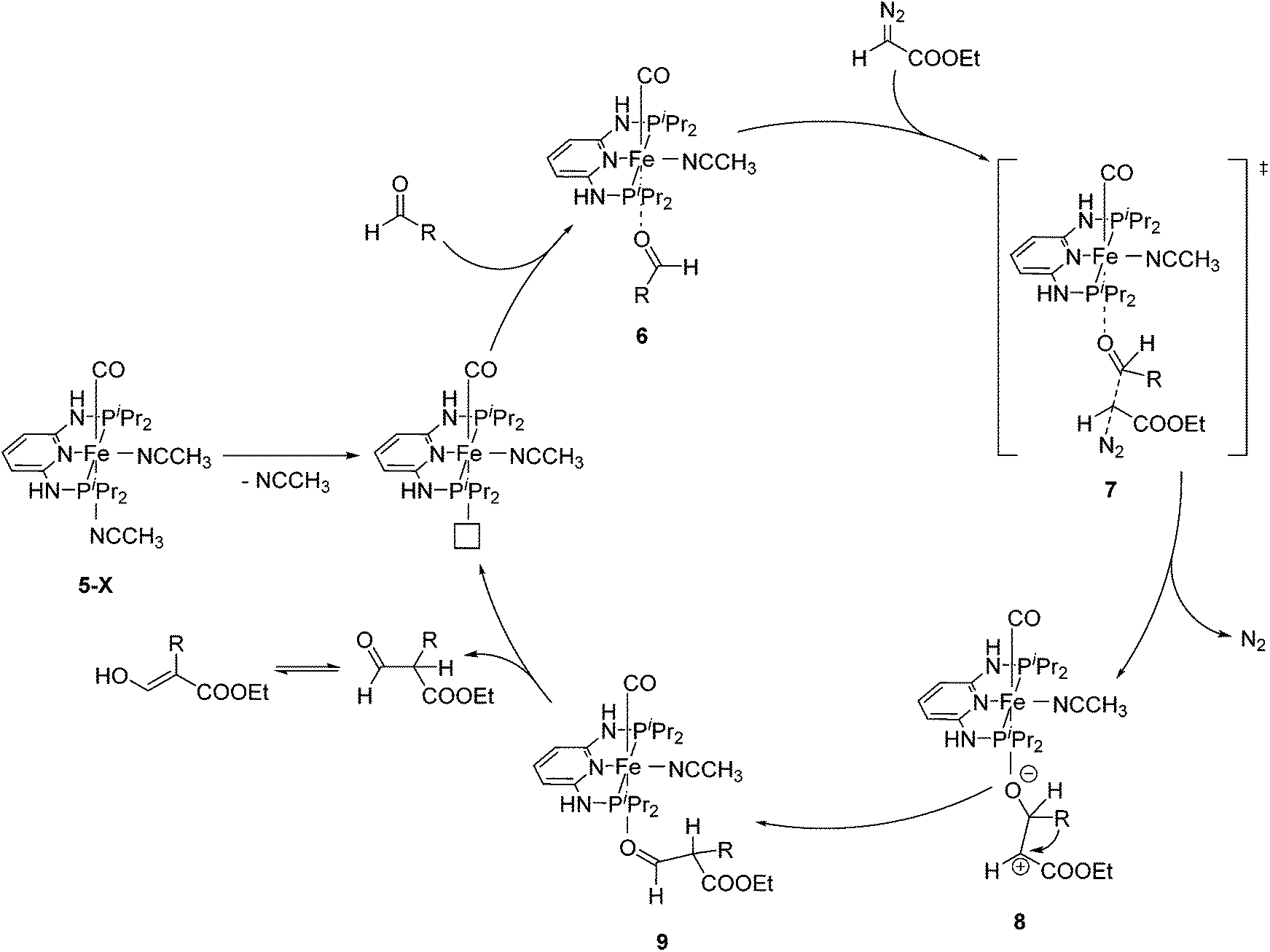 | ||
| Fig. 5 Mechanistic proposal for the formation of ethyl 3-hydroxy-2-(4-methoxyphenyl)acrylate from p-anisaldehyde and EDA. | ||
Kirchner and co-workers then examined the reaction using a series of mono-cationic dicarbonyl complexes trans-[(iPrPNNNP)Fe(CO)2Cl](X) (10-X, X = NO3−, CF3COO−, CF3SO3−, BF4−, PF6−, SbF6− and BArF−; Fig. 4).46 Complexes 10-X (X = NO3−, CF3COO−, CF3SO3−, SbF6−, BArF−) showed no activity. Complex 10-PF6 gave a 20% yield of the product while complex 10-BF4 gave 88% yield. These results are in strong contrast to previous results obtained using 5-BF4 and 5-BArF as catalysts, where the counter ion showed no influence.33 The reaction mechanism of the catalysis with 10-BF4 was investigated by DFT/B3LYP computations. It was found that the formation of ethyl 3-hydroxy-2-arylacrylates had a lower energy barrier compared to the formation of the β-keto ester. Hydrogen bonds between the acidic N–H of the ligand, the BF4 anion and the EDA (N–H⋯F–BF2–F⋯EDA) seemed to play an important role.46
2.2 Iron(II) PNP pincer complexes for catalytic hydrogenation
Chirik and co-workers reported that the dinitrogen cis-dihydride complex [(iPrPCNCP)Fe(H)2(N2)] (11, Fig. 6) catalyzed the hydrogenation of simple acyclic and cyclic alkenes.24 The hydrogenation of 1-hexene under 4 bar of H2 was achieved using a 0.3 mol% catalyst loading in three hours and with a conversion of more than 98%. The conversion for the hydrogenation of cyclohexene was only 10%. Milstein and co-workers developed a new iron pincer complex [(iPrPCNCP)FeH(CO)Br] (12, Fig. 6).26 The complex was active for the hydrogenation of ketones under mild conditions. The reactions typically proceeded in an ethanolic solution with 0.05 mol% of 12 under 4 bar of hydrogen pressure and temperatures of 26–28 °C. A maximum TON of 1880 was reached. A wide range of aromatic and aliphatic ketones could be hydrogenated. The catalysis was less selective for enone substrates such as E-4-phenylbut-3-en-2-one and cyclohex-2-enone, where the CC double bonds were preferably hydrogenated. Interestingly, for benzaldehyde only 36% of benzylic alcohol could be isolated after the hydrogenation. The low yield might be due to catalyst poisoning by benzoic acid, which was formed in small quantities via a Cannizzaro reaction. By lowering the catalyst loading to 0.025 mol%, increasing the loading of KOtBu (base) to 0.625% and running the reaction in an ethanol/NEt3 (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) solution at 40 °C under 30 bar H2, the yield for the hydrogenation of benzaldehyde could be increased to up to 99% and the TON was up to 4000. This modified protocol was applied for the hydrogenation of various aromatic and aliphatic aldehydes.31
:1) solution at 40 °C under 30 bar H2, the yield for the hydrogenation of benzaldehyde could be increased to up to 99% and the TON was up to 4000. This modified protocol was applied for the hydrogenation of various aromatic and aliphatic aldehydes.31
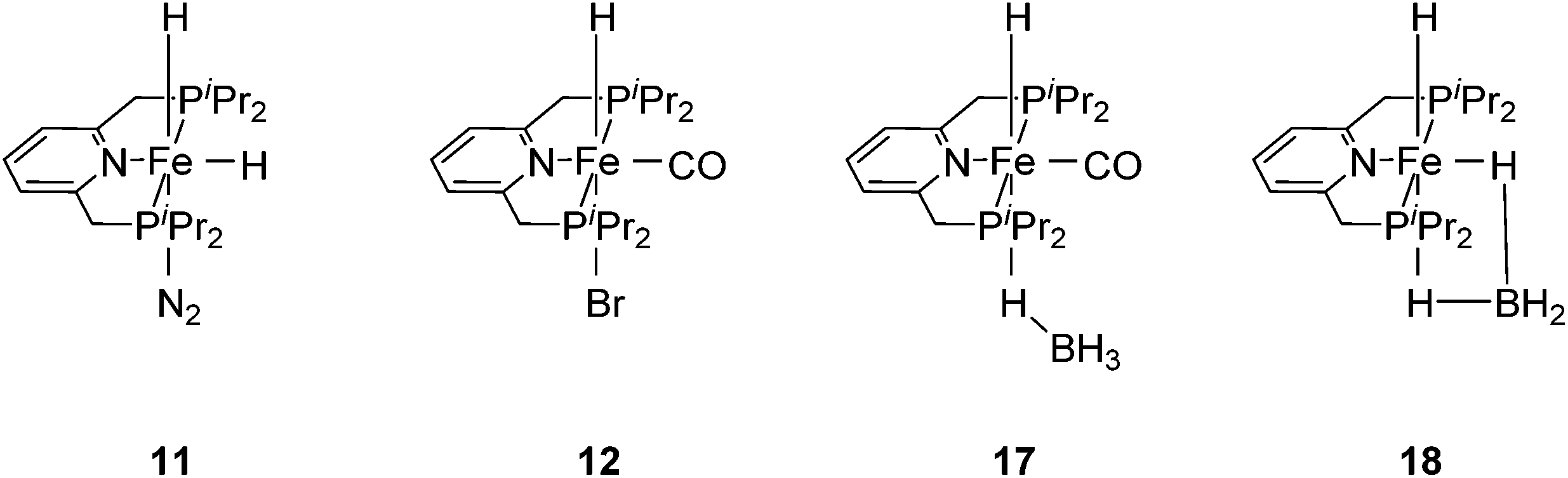 | ||
| Fig. 6 Structures of complexes 11, 12, 17 and 18. | ||
For the hydrogenation of ketones catalyzed by 12, the reactions were best run in ethanol; in THF or neat acetophenone no conversion was observed. Stoichiometric reactions revealed that the bridging methylene group of 12 was deprotonated by KOtBu to form the complex [(iPrPCNCP-H)FeH(CO)] (13, Fig. 7) containing a dearomatized pyridine ligand. It was proposed that the 5-coordinated 16-electron species 13 might be stabilized by the reversible addition of ethanol to afford the 6-coordinate species 13′. In the proposed mechanism, the coordination of ketone to 13 followed by insertion into the iron–hydride bond gave the alkoxide complex 14. The coordination of dihydrogen to 14 then gave intermediate 15. Heterolytic cleavage of the dihydrogen in 15 then generated either the hydrido alkoxide complex 16 or its dearomatized form 16′. The catalytic cycle was closed by elimination of the alcohol and regeneration of 13.26,47
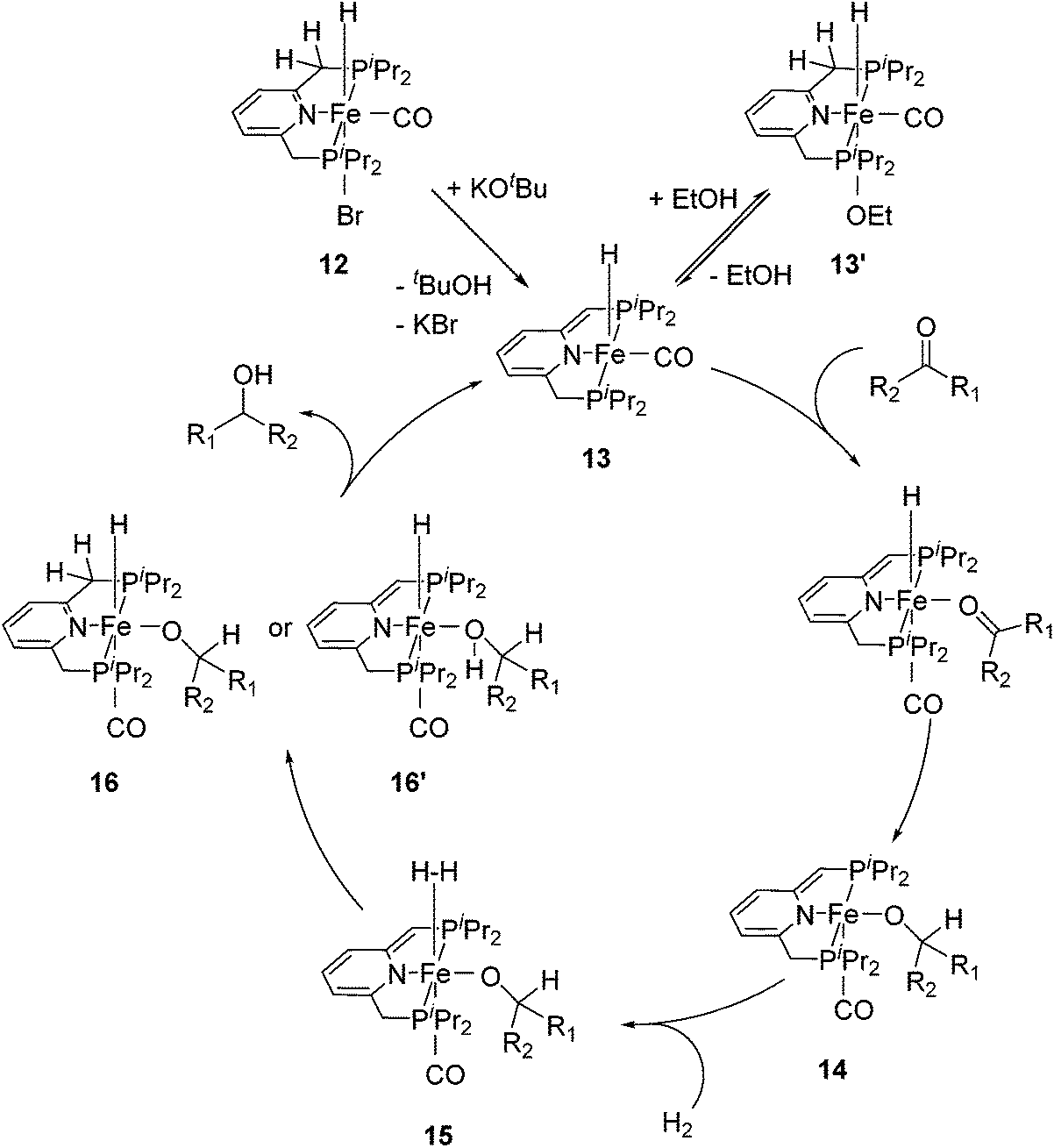 | ||
| Fig. 7 Proposed catalytic cycle for the hydrogenation reactions of ketones using [(iPrPCNCP)FeH(CO)Br] (12) as a pre-catalyst. | ||
The Milstein group further found that minor modifications of the [(iPrPCNCP)FeH(CO)Br] (12) catalyst led to base-free hydrogenation of ketones.28 They found that [(iPrPCNCP)FeH(CO)(η1BH4)] (17, Fig. 6) catalyzed the hydrogenation of acetophenone under 4 bar of hydrogen at 40 °C without the need for an additional base. Aromatic and aliphatic ketones could be readily reduced to the corresponding alcohols with yields of 53% to 99%. Interestingly, a related complex, [(iPrPCNCP)FeH(η2BH4)] (18, Fig. 6), was not active. In aprotic solvents such as benzene-d6 and toluene-d8, [(iPrPCNCP)FeH(CO)(η1BH4)] (17) loses BH3 to give trans-[(iPrPCNCP)Fe(H)2(CO)], which is in equilibrium with cis-[(iPrPCNCP)Fe(H)2(CO)]. Neither complex reacts with acetophenone, excluding its participation in the catalytic cycle.
Kirchner and co-workers recently synthesized a series of [(iPrPNNNP)FeH(CO)L]n (19-L) complexes with L = Br, CH3CN, pyridine, PMe3, SCN−, CO and BH4− and n = 0 and +1.39 The spacers between the phosphine and pyridine donors were NH and/or NMe. Complex 19a-Br (Fig. 8) was active in the hydrogenation of acetophenones under 5 bar hydrogen at 25 °C, with a catalyst loading of 1.0 mol% and 2.0 mol% KOtBu as the base. The hydrogenation only occurred in alcoholic solutions, with ethanol being the best solvent. Complex 19b-Br was completely inactive while complex 19c-Br had low activity. Changing the Br ligand of 19a-Br to CH3CN and BH4− resulted in similarly active catalysts. Changing the Br ligand to pyridine, PMe3, SCN−, CO, however, led to inactive complexes, because these ligands probably could not dissociate during catalysis. The turnover frequency (TOF) was up to 770 h−1. Interestingly, both 19a-Br and 19b-Br were active for the hydrogenation of aldehydes. Thus, 19b-Br is a chemoselective catalyst for the hydrogenation of aldehydes. The mechanism of hydrogenation catalyzed by 19a-Br was investigated by stoichiometric reactions and DFT computations.39 The results suggested that 19a-Br reacted with one equivalent of a base to afford the deprotonated 5-coordinated complex [(iPrPNNNP-H)FeH(CO)] (20; Fig. 9). An incoming ketone coordinated to the vacant coordination site on the complex and inserted into the iron–hydride bond to give the 5-coordinated alkoxide complex [(iPrPNNNP-H)Fe(OCHR1R2)(CO)] (21; OCHR1R2 = alkoxide). Hydrogen then bound to 21 to give [(iPrPNNNP-H)Fe(H2)(OCHR1R2)(CO)] (22). The heterolytic cleavage of hydrogen with the aid of the coordinated alkoxide led to the formation of an alcohol and regeneration of the hydride species [(iPrPNNNP-H)FeH(HOCHR1R2)(CO)] (23). Substitution of the alcohol by an incoming ketone closed the catalytic cycle. It was shown that the amine stays deprotonated throughout the catalytic cycle. The heterolytic cleavage of H2 involving protonation of the alkoxide ligand had a calculated barrier of 16.0 kcal mol−1, much lower than the 34.1 kcal mol−1 calculated for the heterolytic cleavage involving the deprotonated PNP ligand. The role of ethanol seemed to prevent the formation of unactive Fe dihydride species.
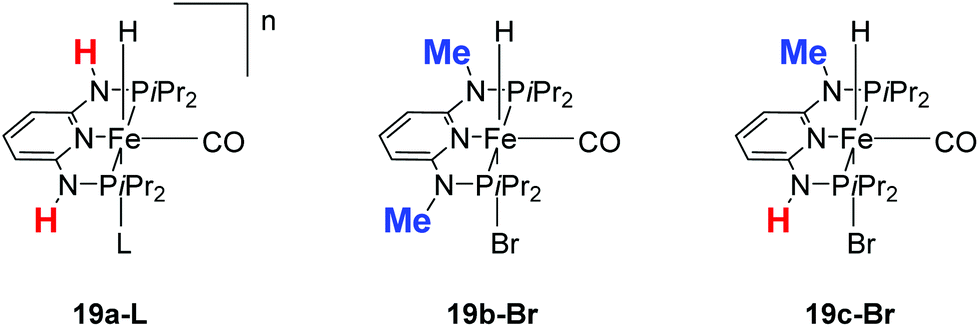 | ||
| Fig. 8 Structures of complexes 19a-L (L = Br, CH3CN, py, PMe3, SCN−, CO and BH4−; n = 0, +1), 19b-Br and 19c-Br. | ||
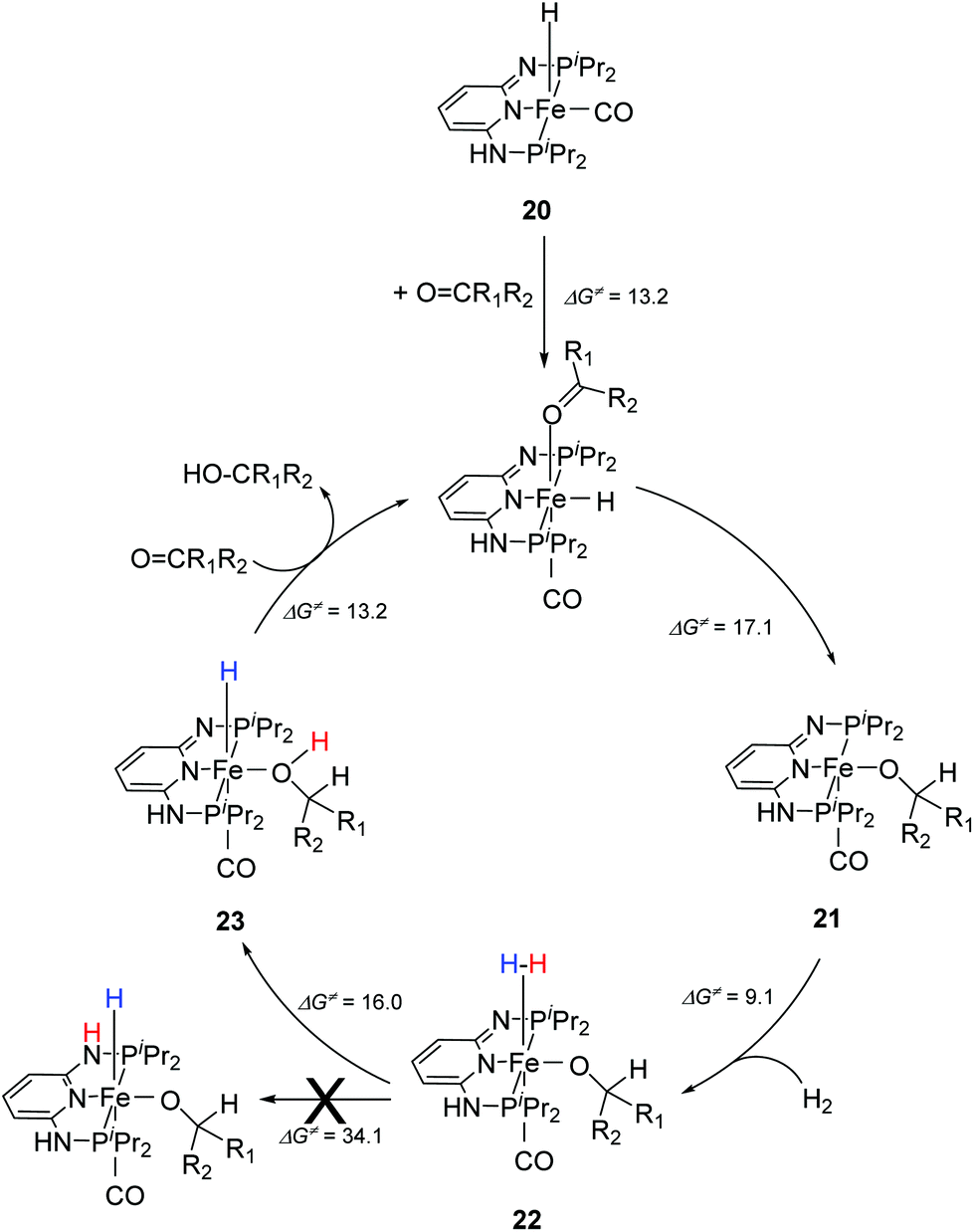 | ||
| Fig. 9 Proposed catalytic cycle for the hydrogenation reactions of ketones using [(iPrPNNNP)FeH(CO)Br] (19a-Br) as a pre-catalyst. | ||
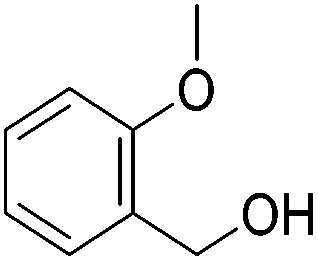
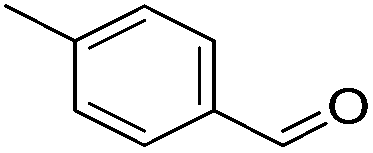
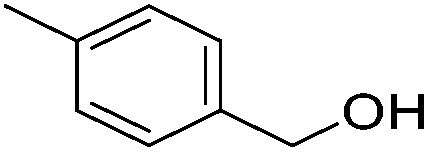
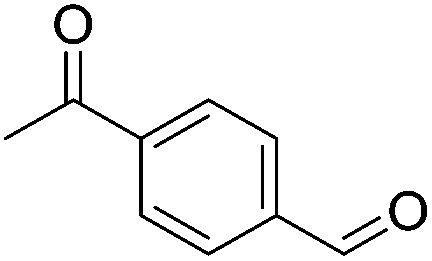
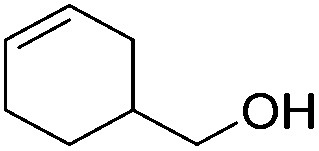
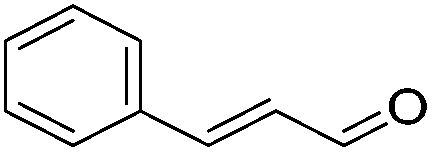
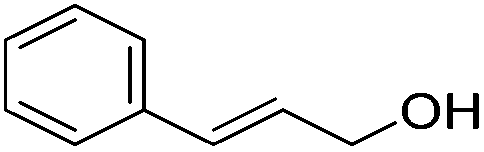
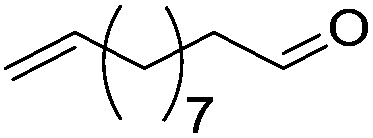
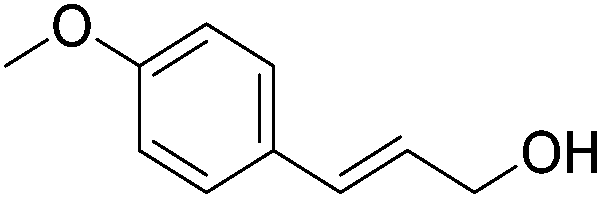
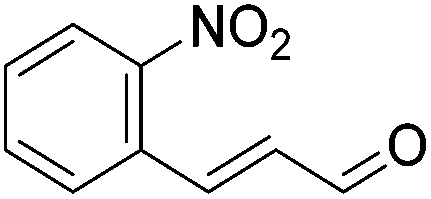
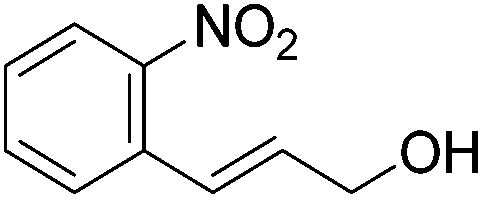
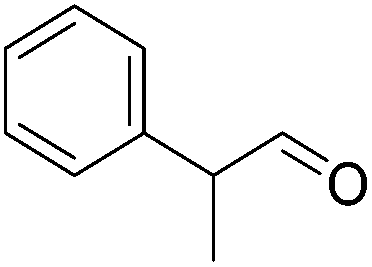
Hu and co-workers recently synthesized [(iPrPONOP)FeH(CO)L]n (24-L; L = CH3CN or Br and n = 0 or +1; Fig. 10) complexes, where L = CH3CN or Br and n = 0 or +1.41 The phosphinite donors are linked to the central pyridine ring by oxygen atoms, so a dearomatisation of the central pyridine ring upon deprotonation of the linker, as shown in the complexes of Milstein and Kirchner26,27,31,39 is not possible. Complex 24-Br is a chemoselective catalyst for the hydrogenation of aldehydes. The scope of the reaction was probed for a range of different aliphatic and aromatic aldehydes, using 10 mol% of 24-Br in methanol under 8 bar hydrogen at room temperature (Table 1). Importantly, functional groups such as terminal and internal alkenes, α,β-unsaturated aldehydes and keto were tolerated. The addition of 10 mol% HCOONa promoted the reaction. The catalyst loading and the H2 pressure could be reduced to 5 mol% and 4 bar, respectively (Table 2). While 24-CH3CN was not active for hydrogenation under the conditions shown in Table 1, it was active when HCOONa was used as an additive.41
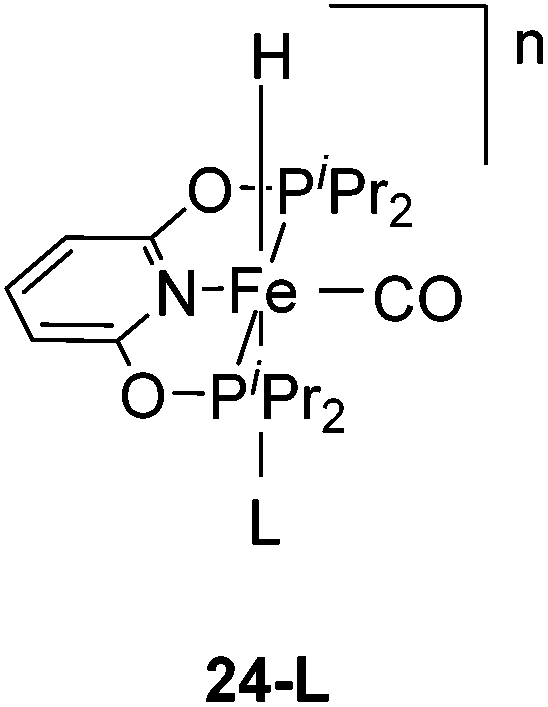 | ||
| Fig. 10 Structure of [(iPrPONOP)FeH(CO)L]n (24-L; L = CH3CN or Br and n = 0 or +1). | ||
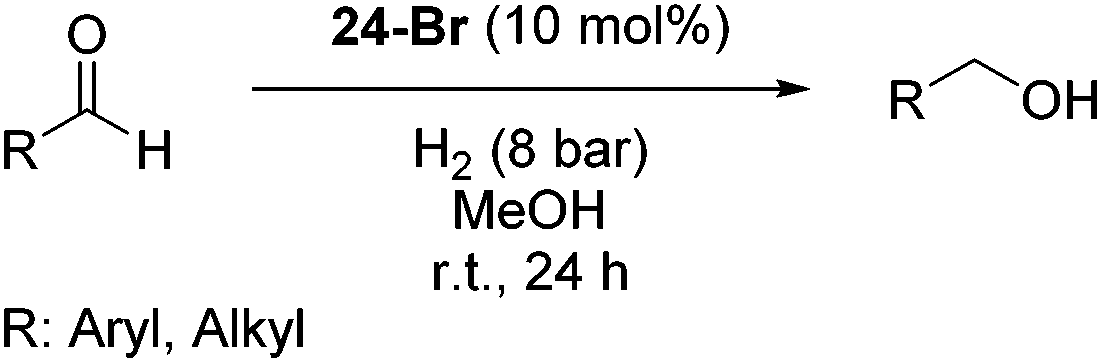
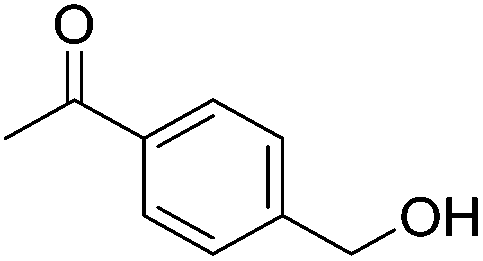
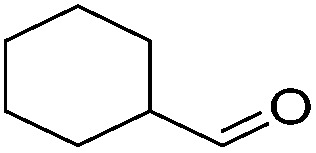
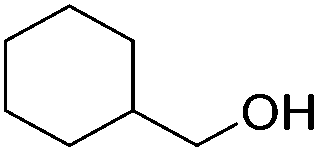
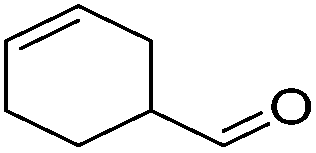
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Aldehyde | Product | Yielda [%] | Yielda [%] | ||
| Cond. A | Cond. B | |||||
| 24-Br | 24-CH3CN | 24-Br | 24-CH3CN | |||
| a Yield determined by GC (isolated yield). b Not accessible. | ||||||
| 1 |

|
|
80 (75) | 75 | 98 (91) | 95 |
| 2 |
|
|
83 (80) | 94 | 98 (93) | 94 |
| 3 |

|
|
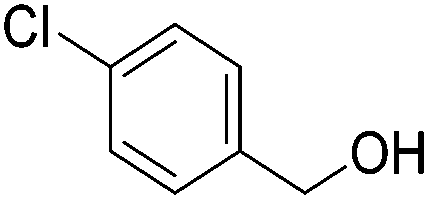
86 (81) | 76 | 98 (91) | 95 |
| 4 |
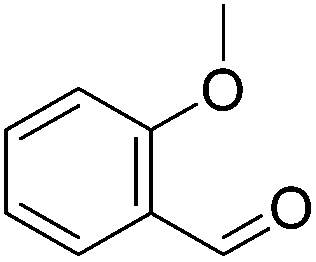
|
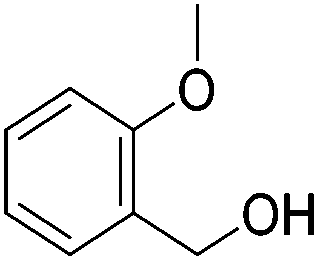
|
92 (88) | 72 | 97 (92) | 85 |
| 5 |
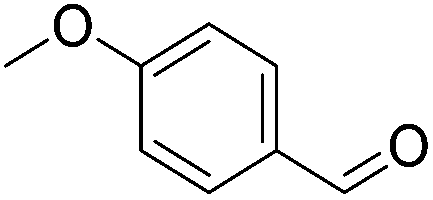
|
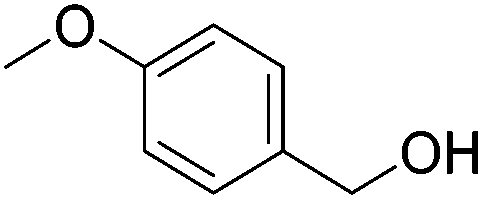
|
56 (50) | 53 | 93 (89) | 86 |
| 6 |
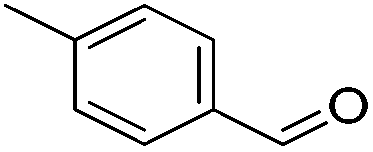
|
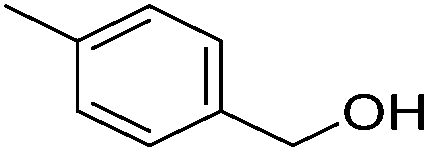
|
95 (87) | 76 | 99 (92) | 96 |
| 7 |
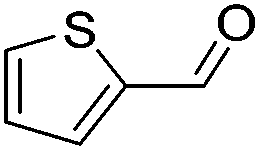
|
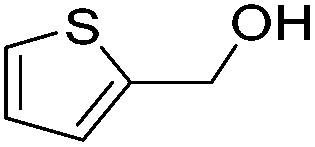
|
71 | 57 | 82 | 79 |
| 8 |

|
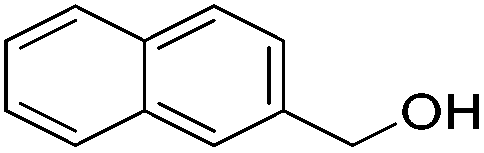
|
66 (62) | 76 | 99 (93) | 96 |
| 9 |
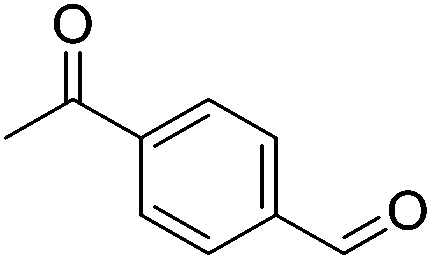
|

|
84 (76) | 82 | n/ab | n/ab |
| 10 |
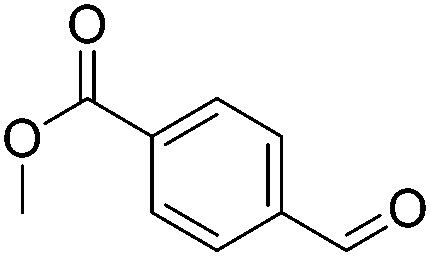
|
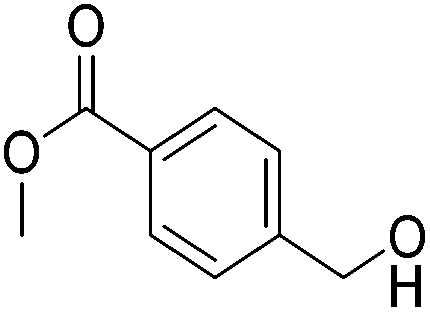
|
87 (83) | 92 | 97 (87) | 95 |
| 11 |
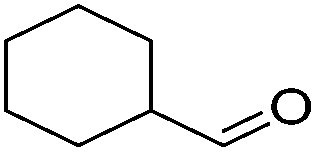
|
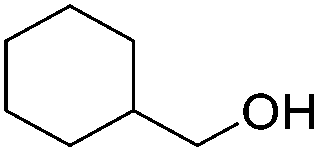
|
84 | 90 | 97 | 84 |
| 12 |
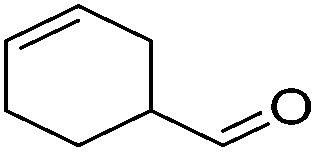
|
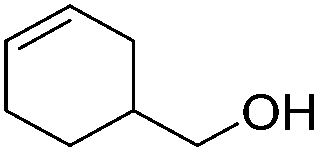
|
58 | 65 | 98 | 94 |
| 13 |
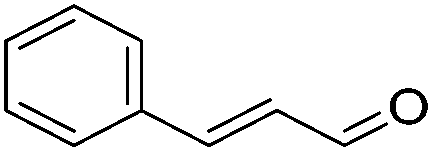
|
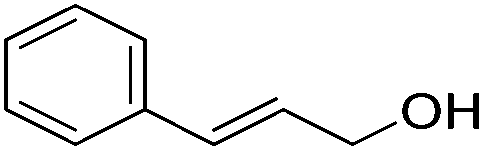
|
65 (60) | 78 | 85 (76) | 72 |
| 14 |
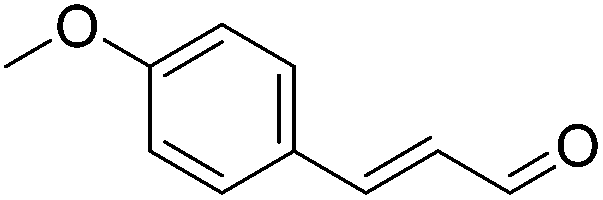
|
|
(64) | (67) | (72) | (68) |
| 15 |
|
|
70 (63) | 64 | 78 (65) | 69 |
| 16 |
|
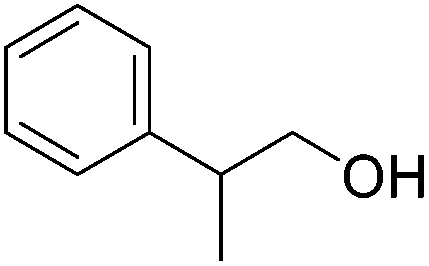
|
60 | 68 | 95 | 90 |
| 17 |
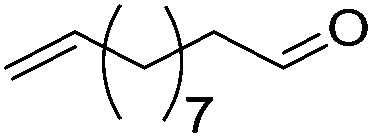
|
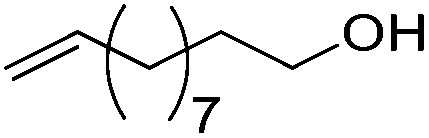
|
75 | 83 | 95 | 75 |
The promotion by HCOONa encouraged the study of 24-L for transfer hydrogenation of aldehydes using sodium formate as the hydrogen donor. Indeed, both catalysts were active and chemoselective for this reaction (Table 2).41
Milstein and co-workers showed that trans-[(tBuPCNCP)Fe(H)2CO] (25; Fig. 11) catalyzed the hydrogenation of sodium bicarbonate.27 A TON of up to 788 and a TOF of up to 156 h−1 were obtained. The yields of sodium formate were around 40%. NMR and IR studies suggested that carbon dioxide inserted into one iron hydride bond to give [(tBuPCNCP)Fe(H)(η1-OOCH)CO] (26). The structure of this compound was confirmed by X-ray analysis. The formate ligand in 26 was easily replaced by water to form [(tBuPCNCP)Fe(H)(H2O)CO] (27). A catalytic cycle was proposed (Fig. 12), in which the coordination of hydrogen to 27 gives [(tBuPCNCP)Fe(H)(H2)CO] (28). An incoming hydroxide deprotonated the hydrogen via heterolytic cleavage (29). This might involve the deprotonation of the PNP ligand forming the dearomatized complex 29′. Either way, after elimination of water the starting dihydride complex 25 was regenerated.27 Complex 25 was also an active catalyst for the decomposition of formic acid, with a TON of up to 100000.29
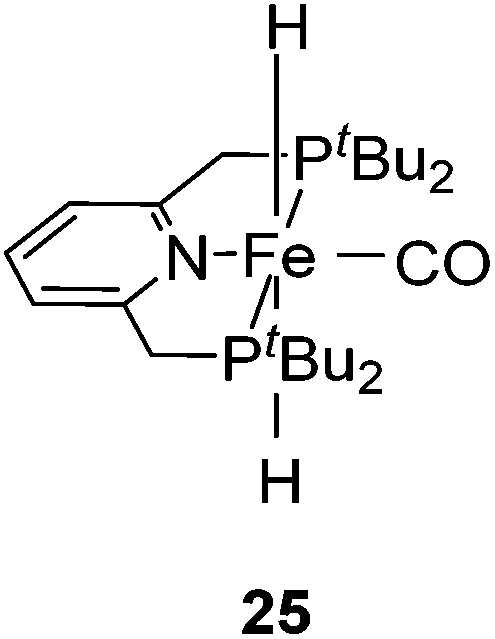 | ||
| Fig. 11 Structure of trans-[(tBuPCNCP)Fe(H)2CO] (25). | ||
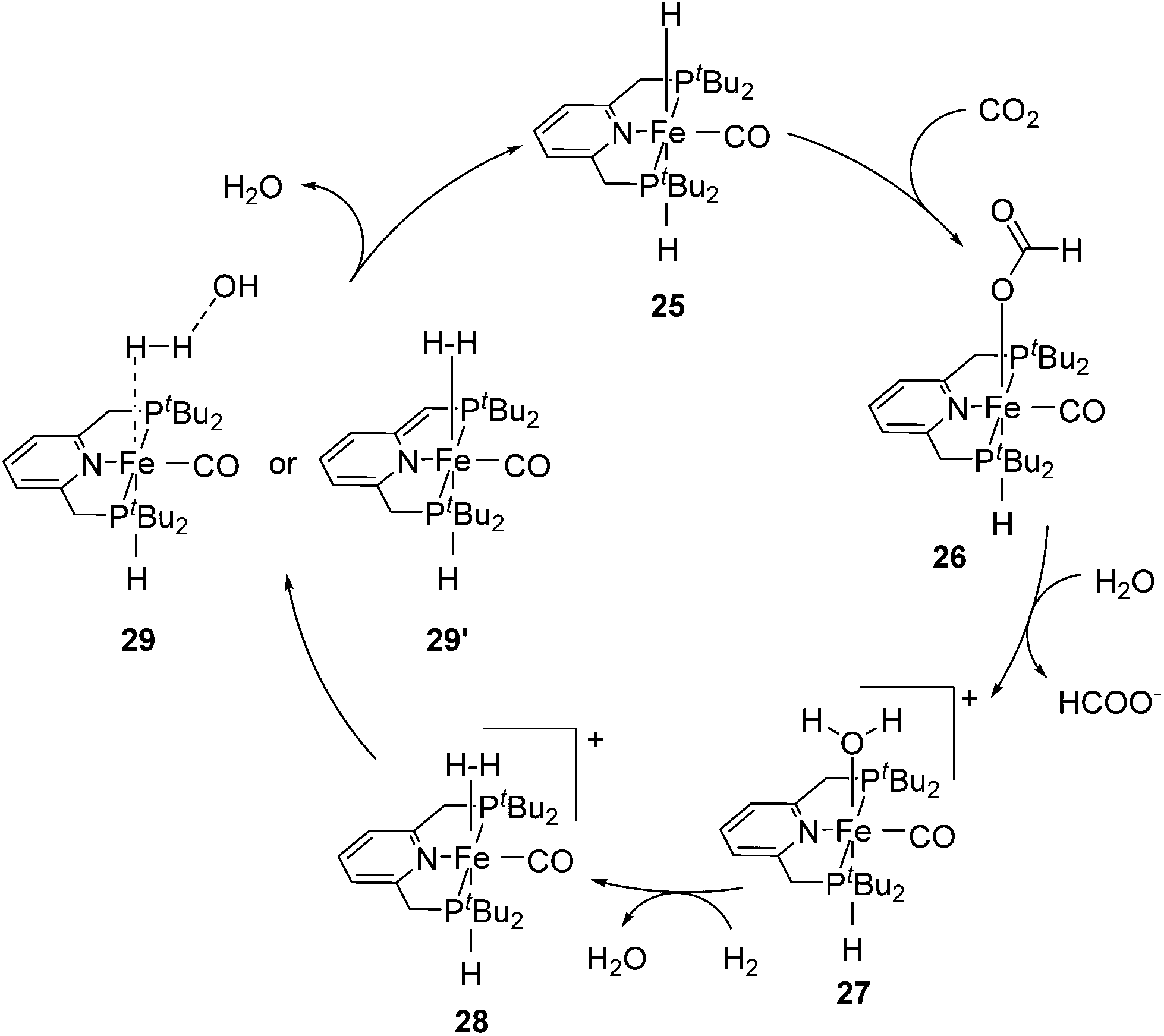 | ||
| Fig. 12 Proposed mechanism for the hydrogenation of carbon dioxide catalyzed by trans-[(tBuPCNCP)Fe(H)2(CO)] (25). | ||
3. Monoanionic benzene-based PCP ligand systems
The synthesis and application of PCP iron complexes are less reported than their PNP counterparts. These complexes are mostly formed via cyclometalation reactions. To promote the iron–carbon bond formation, iron precursors in a low oxidation state or with basic ligands such as alkoxides and alkyl groups are used.48–50Creaser and Kaska reported the synthesis of [(MePCCCP)Fe(H)(dmpe)] (30, Fig. 13).51 Treating the MePCCCP ligand with hydrated ferrous chloride in an ethanolic solution resulted in the precipitation of a white polymeric compound [(MePCCCP)FeCl2]n (A). This polymeric compound was converted to 30 in the presence of dmpe (1,2-bis(dimethylphosphino)ethane) and 30% sodium amalgam. The structure and composition of 30 were confirmed by NMR and IR. The insertion of a Fe ion into the C–H bond of benzene was proposed to take place when the Fe(II) ion was reduced to Fe(0).
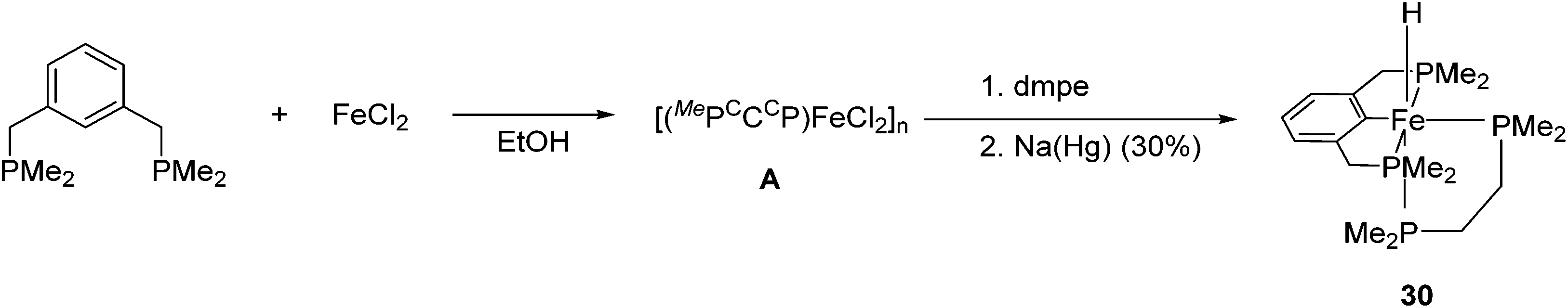 | ||
| Fig. 13 Synthesis of [(MePCCCP)Fe(H)(dmpe)] (30). | ||
Guan and co-workers synthesized [(RPOCOP)FeH(PMe3)2] (31-R; R = iPr, Ph; Fig. 15)52,53 by cyclometalation of the RPOCOP ligand with Fe(0)(PMe3)4. 31-iPr catalyzed the hydrosilylation of various aryl aldehydes by (EtO)3SiH. Aryl ketones were less reactive under similar reaction conditions. In order to gain some mechanistic insights, stoichiometric reactions were probed. Benzaldehyde did not react with 31-iPr, excluding its insertion into the Fe–H bond as a catalytic step. The reaction of 31-iPr with CO for 24 hours at room temperature gave quantitatively [(iPrPOCOP)FeH(PMe3)(CO)] (32), where the CO ligand was trans to the hydride. This result was expected because the hydride had a strong trans-effect, which made the trans PMe3 more labile than the cis one. Complex 32 slowly isomerized to the thermodynamically more stable complex [(iPrPOCOP)FeH(PMe3)(CO)] (32′) where CO was cis to the hydride, but the reaction took more than 7 days at 60 °C. The reaction of deuterium-labelled C6H5CDO with a mixture of Ph2SiD2 and 31-iPr (1:1:1) led to the formation of Ph2SiD(OCD2C6H5) and Ph2Si(OCD2C6H5)2 in a ratio of 4:1. No deuterium incorporation in 31-iPr was detected. This result further excluded the involvement of the hydride in 31-iPr in the hydrosilylation reaction. Based on the above results Guan and co-workers proposed the following mechanism (Fig. 14).52
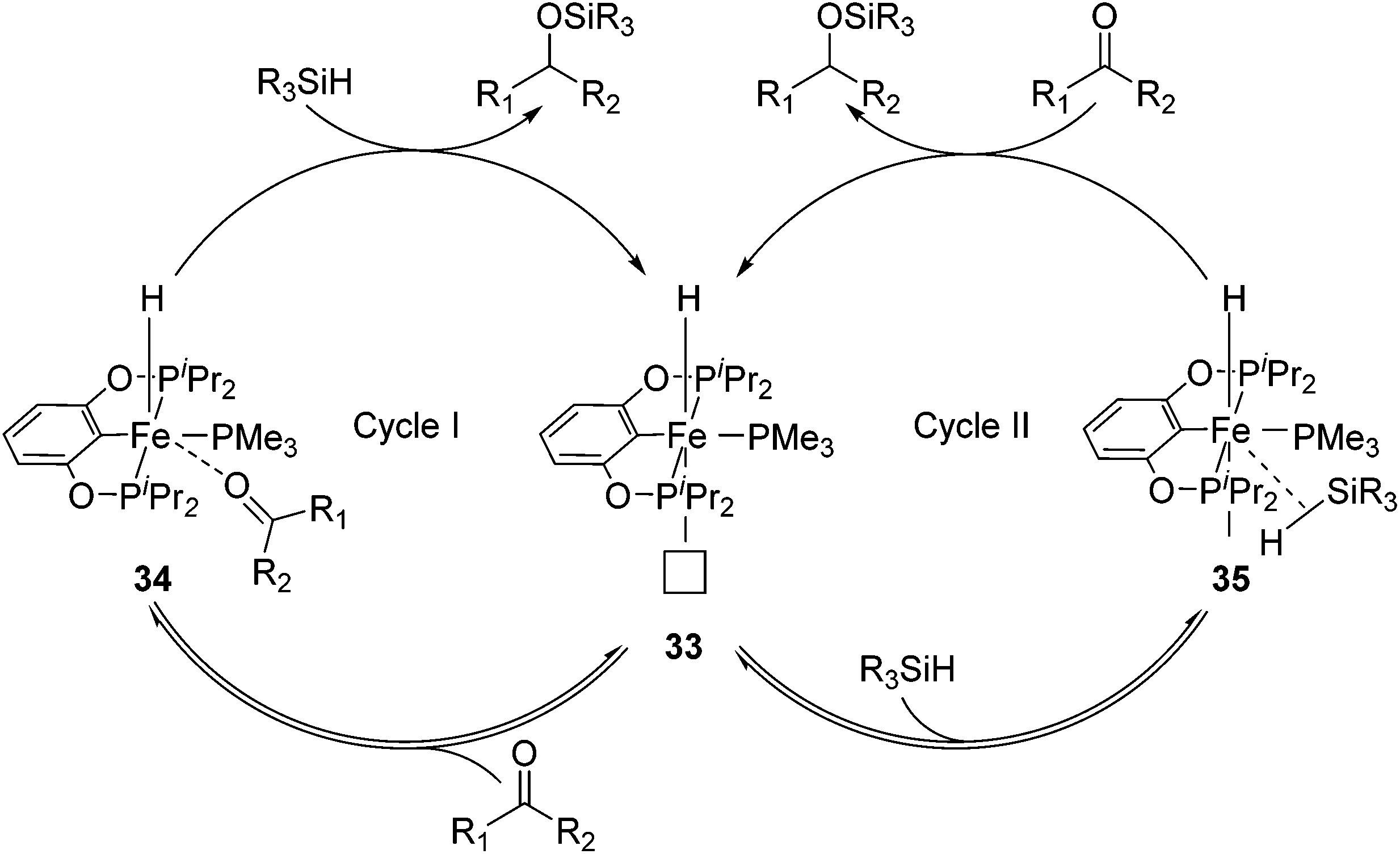 | ||
| Fig. 14 Proposed mechanism for the hydrosilylation of carbonyl compounds catalyzed by 31-iPr. | ||
The PMe3 in 31-iPrtrans to the hydride dissociated to give a vacant coordination site (33, Fig. 14). The isomerisation in which the remaining PMe3 ligand moved to a position trans to the hydride was too slow to be catalytically relevant. An incoming carbonyl compound might coordinate to the open site at first before being reduced by silane (34, Fig. 14; cycle I). Alternatively, silane might first coordinate to the vacant site, get activated, and then reduce the carbonyl compound (35, Fig. 14; cycle II).52 The details of these steps were not clear. Wang et al. reported a DFT study on the mechanism of this hydrosilylation reaction, favouring cycle I. They found that the carbonyl-coordinated compound underwent an isomerization to move PMe3 to a position trans to the hydride. This was followed by the insertion of the carbonyl compound into the Fe–H bond, giving an iron alkoxide complex. The cycle was closed by σ-bond metathesis with silane, yielding silyl ether and regenerating the iron hydride species.54 The computed mechanism, however, was at odds with the experimental findings, which excluded the insertion of benzaldehyde into the Fe–hydride of 31-iPr as a relevant step.52
Guan and co-workers also used 31-iPr for the decomposition of NH3BH3 (AB) to give H2 (Fig. 15).55 The reactivity of 31-iPr could be increased by replacing the two PMe3 ligands with PMe2Ph, giving the complex [(iPrPOCOP)Fe(H)(PMe2Ph)2] (36). Complex 36, which showed an increased activity in the beginning of the reaction, was less stable than 31-iPr during the reaction. A further attempt was undertaken to increase the electron density of the metal center, by adding a methoxy group para to the Fe–C bond (37, Fig. 15). Complex 37 was indeed more active than 31-iPr and 36, and more stable than 36.55
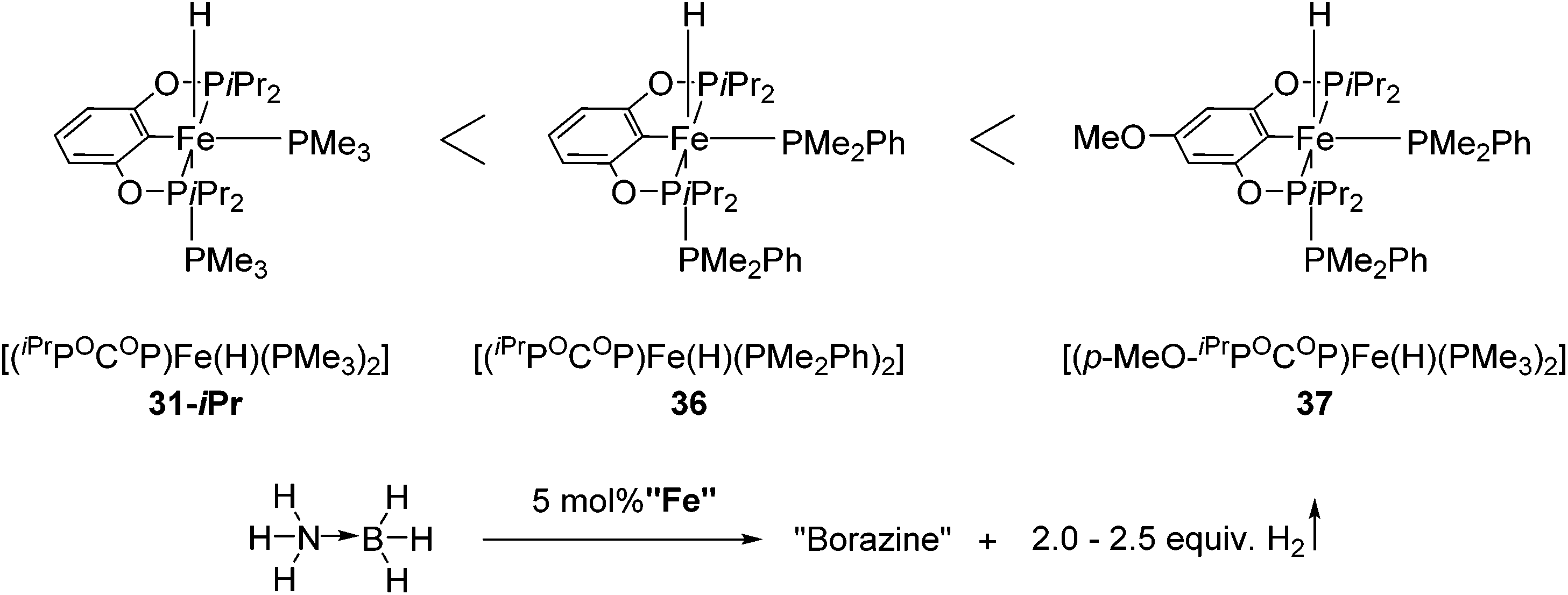 | ||
| Fig. 15 Activity of Fe–POCOP complexes 31-iPr, 36 and 37 for the decomposition of amine borane. | ||
Recently Guan and co-workers prepared cationic complexes [(iPrPOCOP)Fe(PMe3)(CO)](BF4) (38) and cis-[(iPrPOCOP)Fe(CO)2](BF4) (39) by the hydride abstraction of [(iPrPOCOP)FeH(PMe3)(CO)] (40) and [(iPrPOCOP)FeH(CO)2] (41) using a Brønsted acid such as HBF4·Et2O.5638 and 39 were able to activate hydrogen in the presence of Hünig's base to give back the neutral hydride complexes 40 and 41, respectively. 38 and 39 were active catalysts for the hydrosilylation of benzaldehyde and acetophenone, with increased activity over the corresponding neutral complexes, 40 and 41.56
4. Pyridine- and phenyl bis(oxazolinyl) pincer systems
Iron pyridine bis(oxazoline) (pybox) complexes (Fig. 16) have been widely used in catalytic reactions. In the majority of the cases, these complexes were formed in situ by combining iron salts and pybox ligands. The reactions studied included cyclizations,57–60 additions,61–65 the kinetic resolution of racemic sulfoxides,66 and hydrosilylation of aromatic ketones.67 The active species were unclear, so they were not further discussed.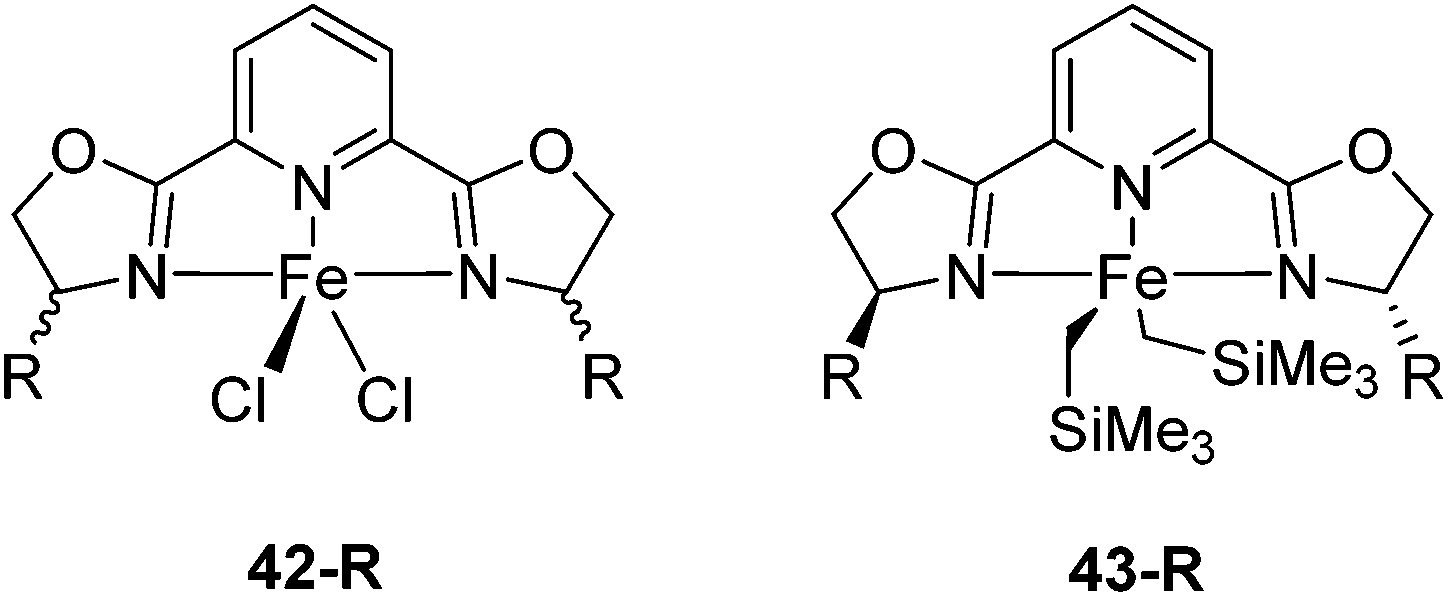 | ||
| Fig. 16 Structures of complexes 42-R (R = Me2, Ph2, iPr(S), tBu(S), Ph(S)) and 43-R (R = iPr(S), tBu(S), Bn(S), iBu(S)). | ||
The groups of Huang and Nomura employed pre-made [(pybox-R)FeCl2] 42-R (R = Me2, Ph2, iPr(S)) complexes as catalysts for the polymerisation of ethylene.68–70 Redlich and Hossain prepared a series of [(pybox-R)FeCl2] (42-R; R = iPr(S), tBu(S), Ph(S)) complexes and used them for the cyclopropanation of imines (Fig. 17).45 In the presence of one equivalent of the halide abstractor, AgSbF6 (relative to catalyst), 42-R (R = iPr, tBu) catalyzed the reaction to yield 47% of the cis-aziridine with ee's below 50%. The trans-aziridine and β-amino-α,β-unsaturated esters were side products.
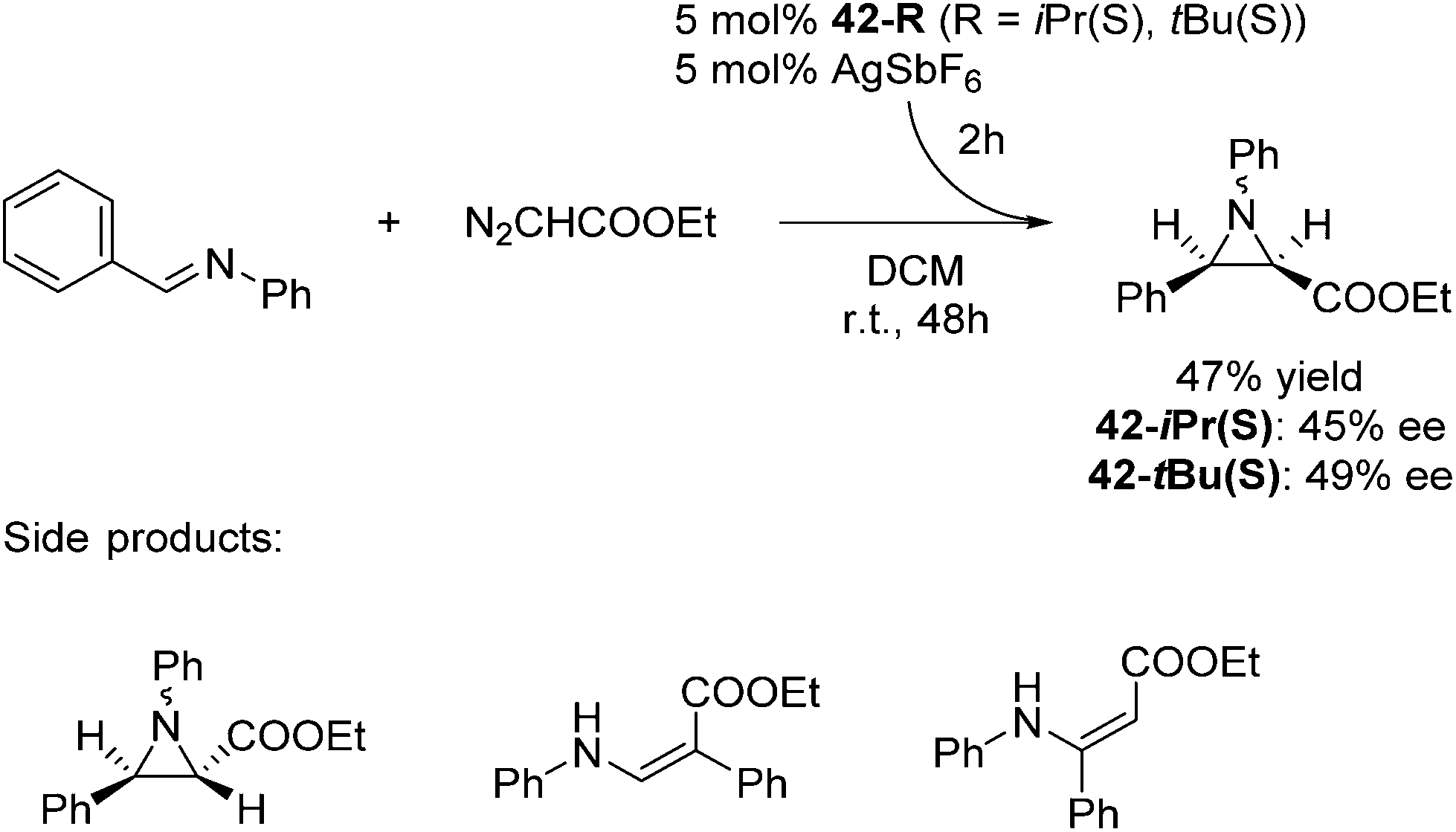 | ||
| Fig. 17 Cyclopropanation catalyzed by complexes 42-R (R = iPr(S), tBu(S)). | ||
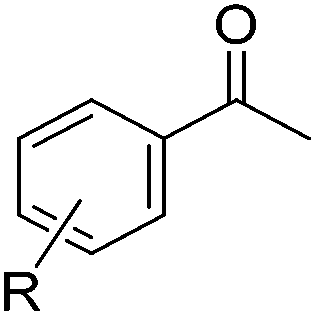
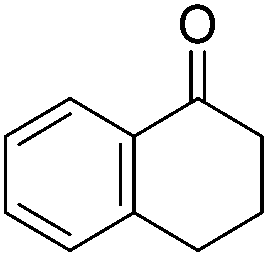
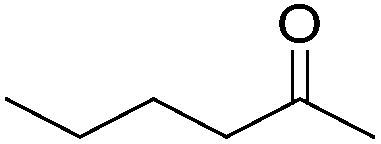
Chirik and co-workers studied enantioselective hydrosilylation of ketones using chiral iron pybox complexes71 [(pybox-R)Fe(CH2SiMe3)2] (43-R; R = iPr(S), tBu(S), indane(1R,2S), Bn(S), iBu(S); Fig. 16). The results in Table 3 show that the catalysts were generally active, and the reactions have high yields and a broad functional group tolerance. Bulky substrates (2,4,6-trimethylacetophenone and 2,6-dimethyl-4-tBu-acetophenone), however, could not react. The enantioselectivity was low in all cases. Nishiyama and co-workers synthesized iron complexes of a phenyl-bis(oxazoline) (phebox) ligand (44-R, Fig. 18). The synthesis involved the reaction of Fe2(CO)9 with (phebox-R)Br (R = Me2, iPr(S)), probably via oxidative addition of the C–Br bond at the Fe center (Fig. 18).72
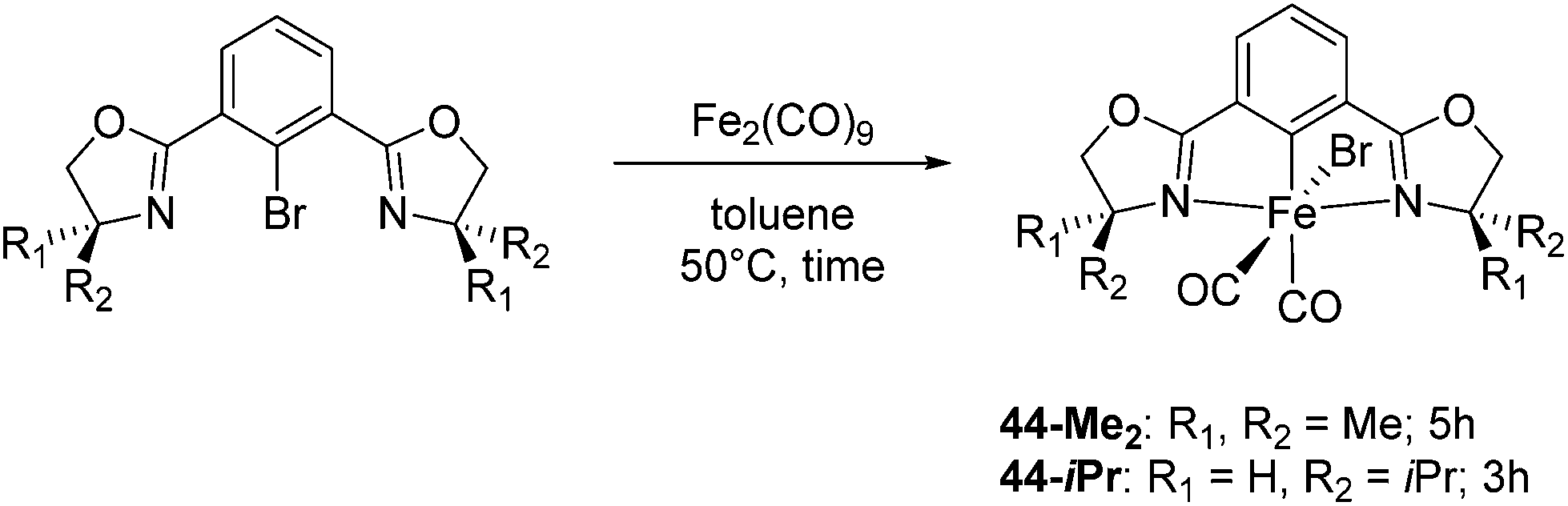 | ||
| Fig. 18 Reaction scheme for the complexation of [(phebox-R)FeBr(CO)2] (44-R; R = Me2, iPr(S)). | ||
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Compound | R = iPr(S) | iPr(S)b | tBu(S) | Bn(S) | iBu(S) |
| a Conversion [%] (ee [%]). b The catalyst was activated by the addition of 0.95 equivalents of B(C6F5)3. | ||||||
| 1 |
|
80 (49) | 99 (54) | 99 (30) | 99 (18) | 99 (8) |
| R = H | ||||||
| 2 | R = 4-tBu | 99 (23) | 99 (13) | 99 (25) | 99 (25) | 99 (11) |
| 3 | R = 4-OMe | 99 (5) | 55 (25) | 99 (3) | 99 (29) | 97 (5) |
| 4 | R = 4-CF3 | 99 (20) | 99 (20) | 99 (12) | 99 (21) | 99 (12) |
| 5 | R = 3,5-(CF3)2 | 99 (6) | 99 (6) | 72 (12) | 99 (6) | 99 (4) |
| 6 | R = 2,4-(OMe)2 | 99 (32) | 99 (32) | 99 (50) | 99 (32) | 99 (22) |
| 7 | R = 2,4,6-Me3 | <1 | 1 (30) | 2 (5) | 4 (44) | 2 (26) |
| 8 | R = 2,6-Me2-4-tBu | <1 | <1 | 1 (30) | <1 | <1 |
| 9 |
|
99 (30) | 99 (41) | 60 (2) | 93 (22) | 25 (7) |
| 10 |
|
58 (10) | 99 (11) | 99 (25) | 46 (17) | 46 (17) |
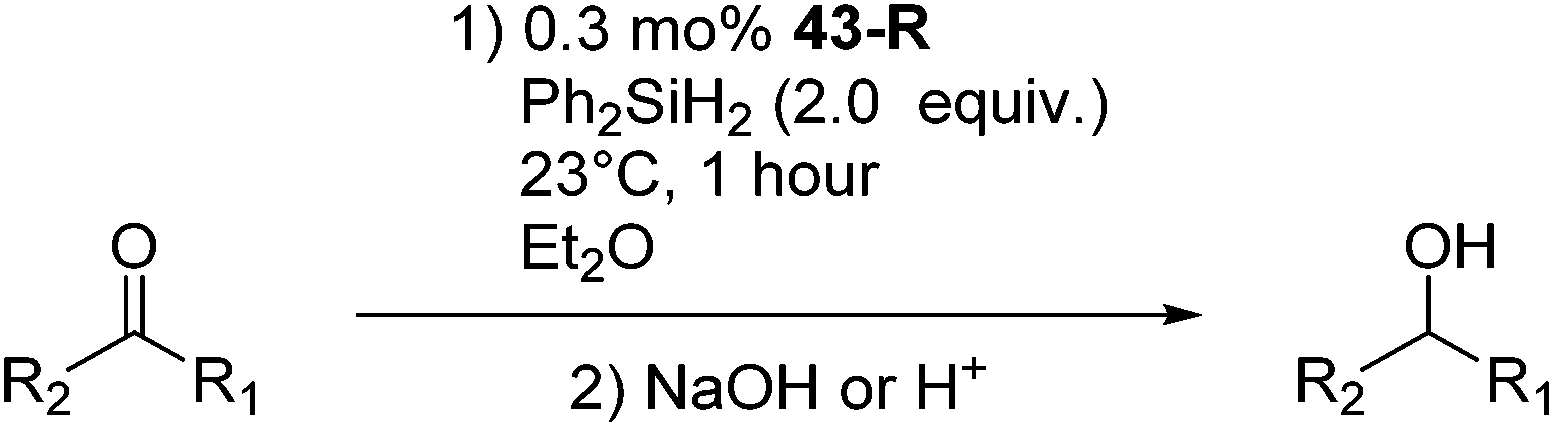
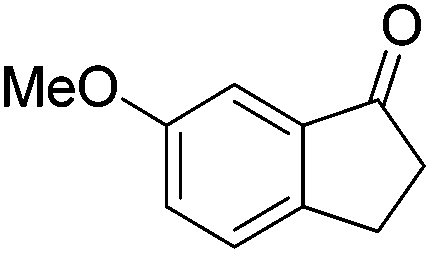
The chiral complex 44-iPr(S) catalyzed hydrosilylation of aromatic ketones. The reduction of 4-acetylbiphenyl in the presence of 2 mol% 44-iPr(S) and 2 mol% of Na(acac) in hexane at 50 °C gave 1-(4-biphenyl)ethanol in a 99% yield and 66% ee for the R-configuration. The reduction of 2-acetylanthracene and 2-acetylnaphthalene gave 49 and 53% ee, respectively. However, the reduction of 1-acetyl-4-methoxybenzene and 1-acetyl-4-methylbenzene had low enantioselectivity (21–38%).72
5. Pyridine bis(N-heterocyclic carbene) pincer systems
Bedford and co-workers showed that the iron(II) pyridyl bis(carbene) pincer complex, 45-Br2 (Fig. 19) was an excellent catalyst for the cross coupling of alkyl halides with p-tolyl Grignard reagents (Table 4).73 While chlorocyclohexane (Table 4, entry 1) was unreactive, bromocyclohexane and 4-bromo-1-methylcyclohexane could be coupled in 94 and 89% yields (Table 4, entries 2 and 3). 1-Bromooctane was coupled in a 71% yield.73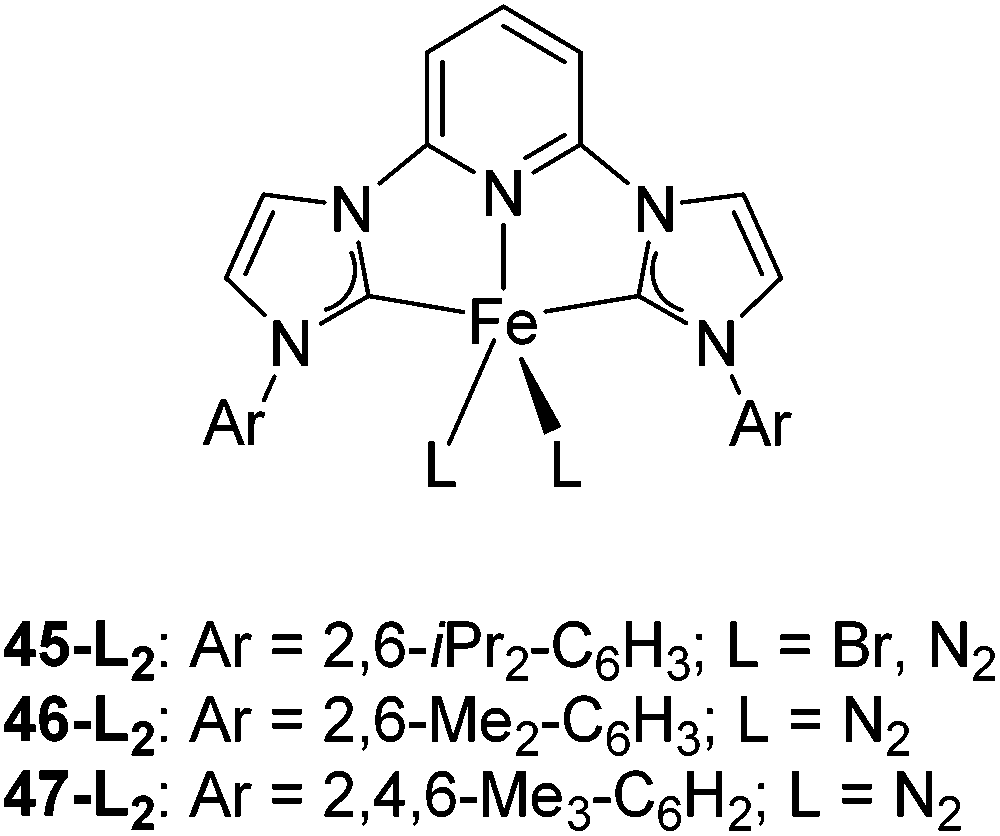 | ||
| Fig. 19 Structures of 45-L2 (L = Br, N2), 46-(N2)2 and 47-(N2)2. | ||
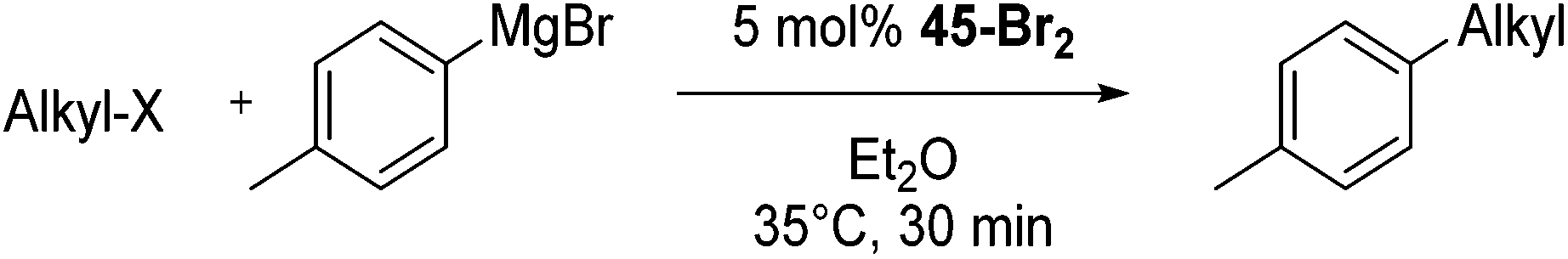
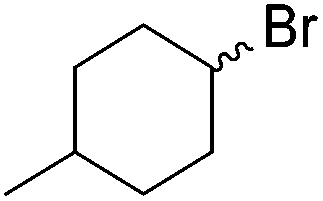
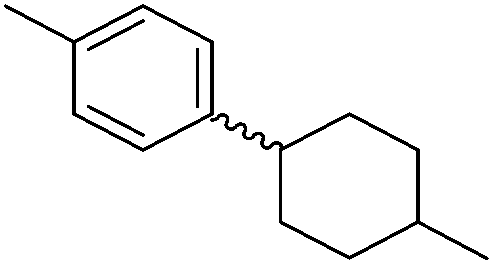
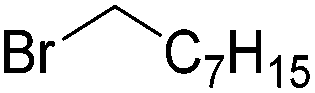
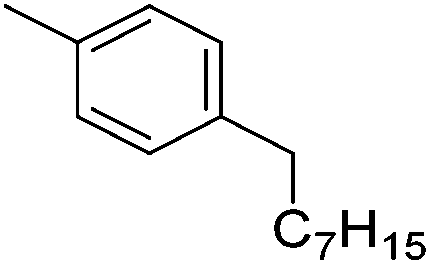
Chirik and co-workers synthesized the iron(0) complex 45-(N2)2 (Fig. 19) and employed it for the hydrogenation of ethyl 3-methyl-2-butenoate. A complete conversion was obtained after 1 hour.74 The results prompted the examination of analogous complexes bearing sterically less demanding aryl substituents, 46-(N2)2 and 47-(N2)2 (Fig. 19).
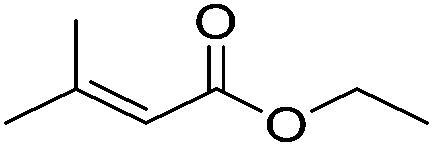
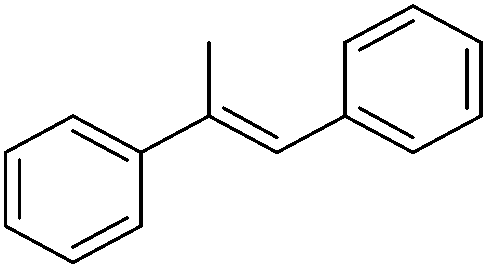
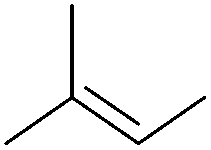
Table 5 compares the activity of complexes 45-(N2)2–47-(N2)2 in the hydrogenation of various internal alkenes (5 mol% catalyst, 4 atm H2, room temperature). The hydrogenation of ethyl 3-methyl-2-butenoate using 46-(N2)2 or 47-(N2)2 as a catalyst was less efficient as compared to that using 45-(N2)2 as a catalyst, due to competing deactivation pathways.75 However, for other acyclic trisubstituted olefins, 46-(N2)2 and 47-(N2)2 are as active as 45-(N2)2. They are even more active for the hydrogenation of cyclic trisubstituted olefins. A cyclic tetrasubstituted olefin could be hydrogenated using 46-(N2)2 or 47-(N2)2, but acyclic tetrasubstituted olefins could not be hydrogenated.74
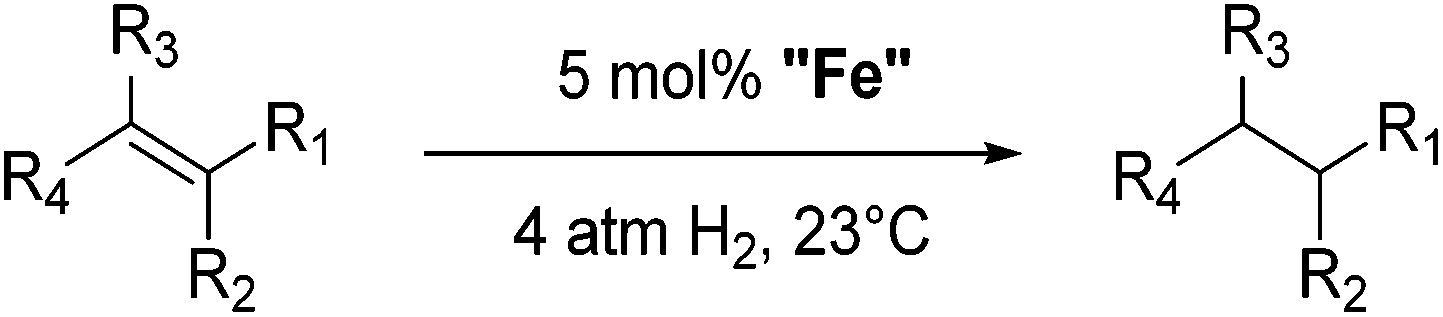
6. N,N-Diphenylamino-based pincer systems
Milstein and co-workers prepared iron complexes of an acridine ligand [(AcrPNP)FeBr2] (48) and [(HAcrPNP)Fe(CH3CN)(μ2-CH3CHNBH3)] (49) (Fig. 20, AcrPNP = 4,5-bis(diphenylphospino)acridine, HAcrPNP = 4,5-bis(diphenylphospino)-9H-acridine-10-ide).76 Interestingly, in complex 49 the acridine is in the reduced form and the μ2-CH3CHNBH3 ligand originates from the insertion of acetonitrile into BH4−.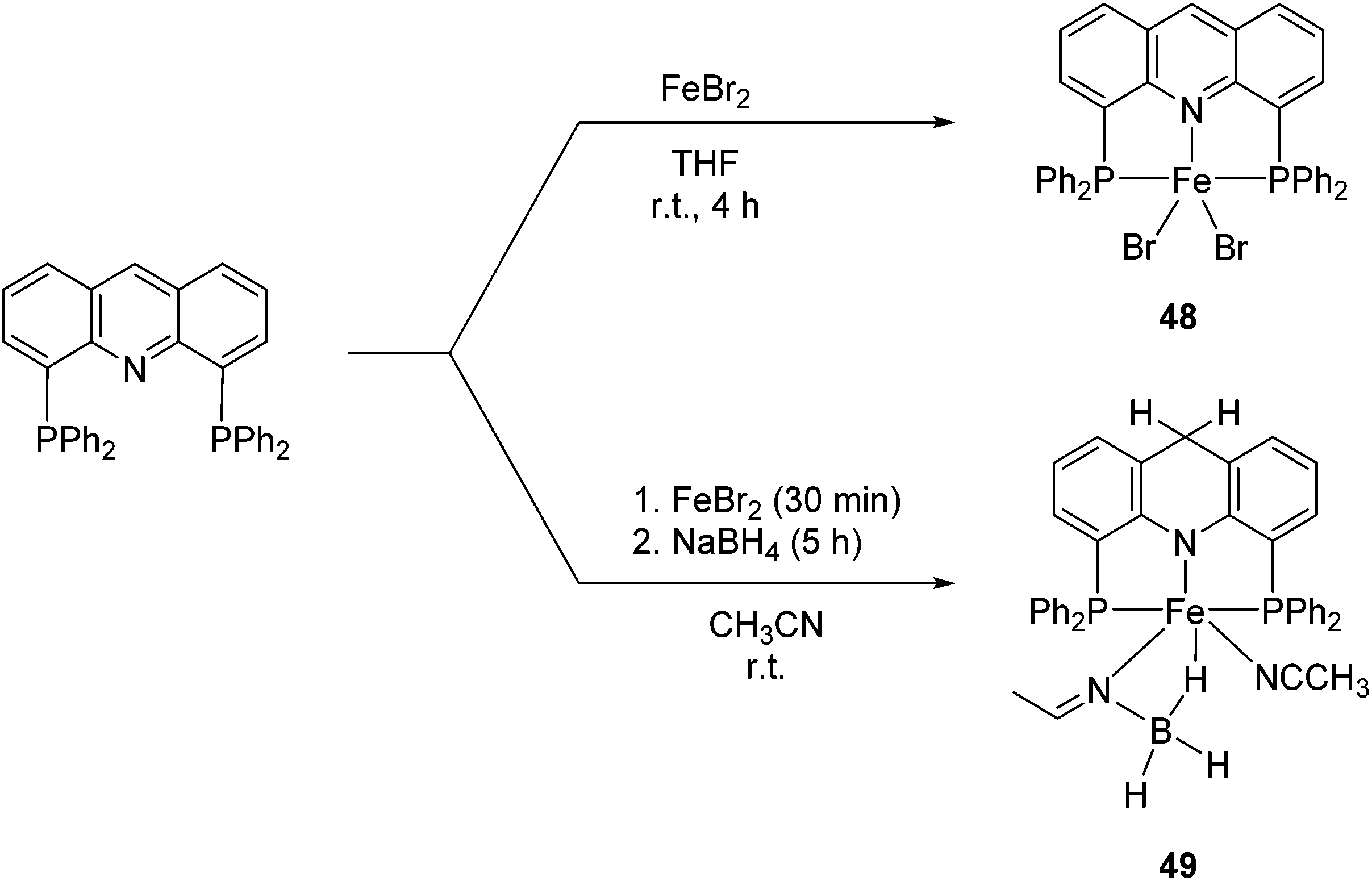 | ||
| Fig. 20 Synthesis of 48 and 49. | ||
48 and 49 were employed for hydrosilylation of internal alkynes. While 48 was not active, 49 was an efficient and selective catalyst (Table 6). The yields were generally between 85 to 100%, and the reactions were E-selective. A high functional group tolerance was obtained: ether, ester, ketone, halide and nitrile substituents remained intact during the reduction. A terminal alkyne, phenylacetylene, could also be reduced.76
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Alkyne | 49 [mol%] | Time [h] | Pressure [bar] | Yield [%] |
E:Z |
| a The yield in parentheses is the corresponding alkane product [%]. | ||||||
| 1 |
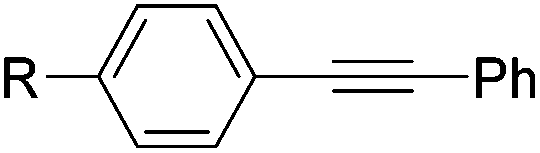
|
0.6 | 12 | 4 | 99 | 100:0 |
| R = H | ||||||
| 2 | R = OMe | 0.6 | 36 | 4 | 99 | 99:1 |
| 3 | R = COCH3 | 2 | 65 | 4 | 99 | 64:36 |
| 4 | R = COOEt | 2.2 | 22 | 4 | 89 (11)a | 99:1 |
| 5 | R = CN | 4 | 72 | 10 | 94 | 99:1 |
| 6 | R = Cl | 2.2 | 23 | 10 | 87 (13)a | 99:1 |
| 7 |
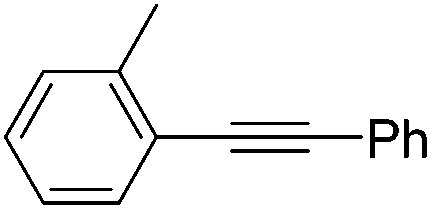
|
2.5 | 70 | 10 | 99 | 99:1 |
| 8 |
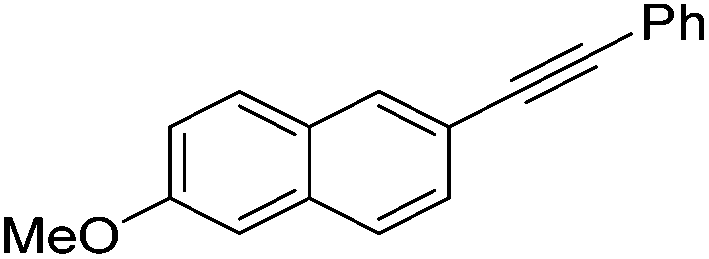
|
4 | 21 | 4 | 98 (2)a | 100:0 |
| 9 |
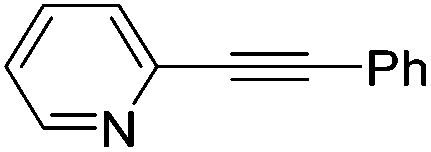
|
2 | 48 | 4 | 70 (30)a | 61:39 |
| 10 |
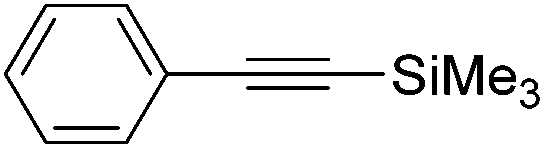
|
1.7 | 17 | 4 | 76 (24)a | 99:1 |
| 11 |
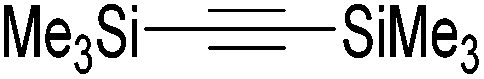
|
1 | 30 | 4 | 85 | 100:0 |
| 12 |
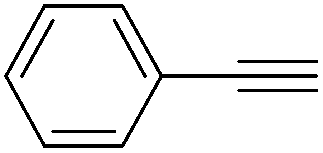
|
0.6 | 11 | 4 | 99 | — |
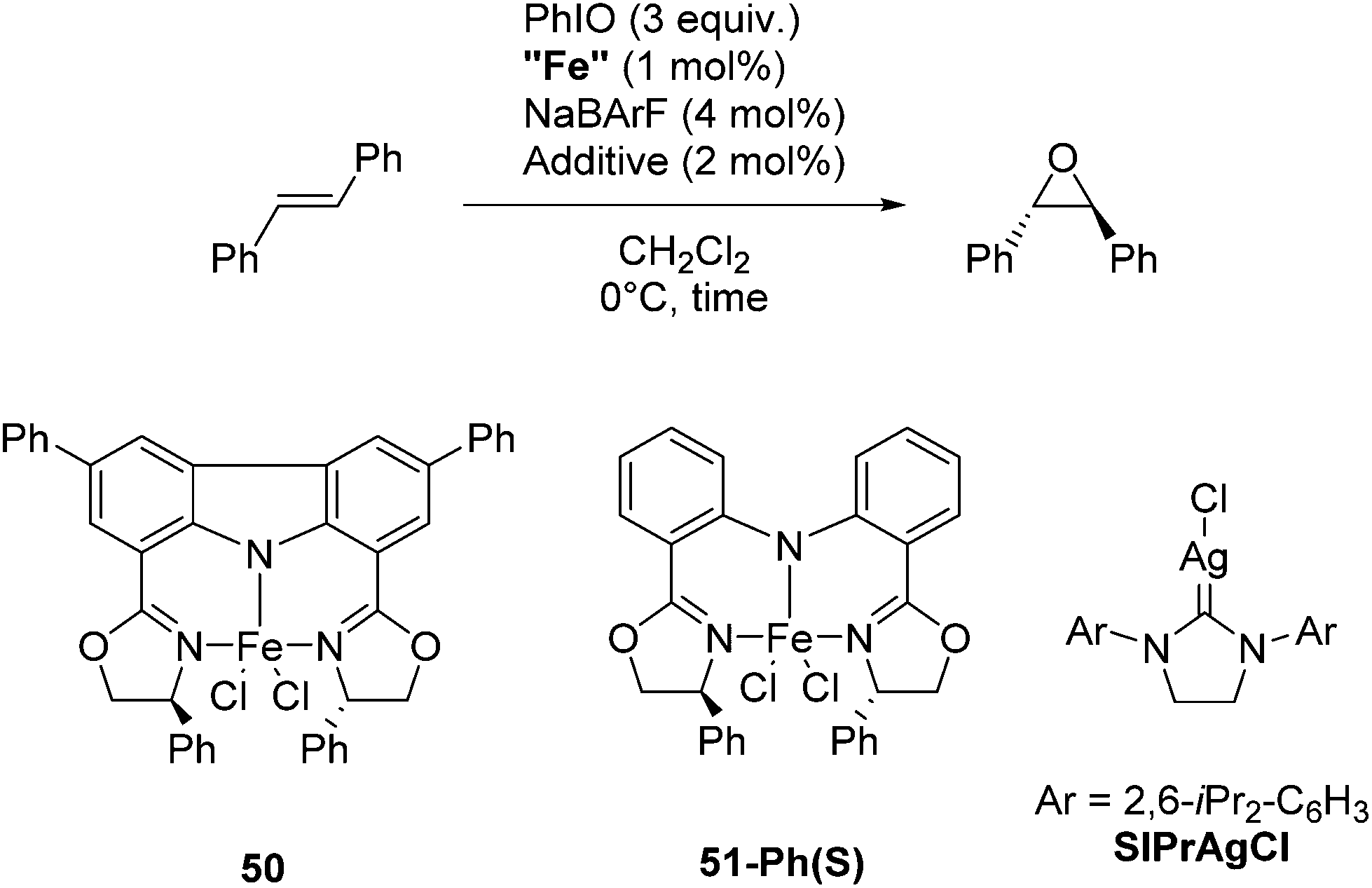
Niwa and Nakada developed a carbazole-based tridentate bis(oxazoline) ligand (CarBox-R) that displayed porphyrin-like properties in the iron catalyzed epoxidation reaction of (E)-olefins.77 In the presence of 4 mol% NaBArF, [(CarBox-Ph)FeCl2] (50, 1 mol%) catalyzed the epoxidation of trans-stilbene using iodosobenzene as an oxidising reagent (Table 7). The reaction gave a 35% yield with an ee of 83%. In the absence of NaBArF no reaction was observed. When SIPrAgCl (SIPr = N,N′-bis(2,6-iPr2-C6H3)-4,5-dihydroimidazol-2-ylidene) was used as an additive the yield and ee could be increased to 55% and 88%, respectively. The reaction with the structurally similar [(bopa-Ph)FeCl2] (51-Ph(S); vide infra) gave no yield. The investigation of the substrate scope showed the broad utility of this reaction. Various substituted trans-stilbenes and cinnamyl alcohol derivatives reacted to give the corresponding (S,S) epoxides in high enantioselectivity.77
|
|
|||||
|---|---|---|---|---|---|
| Entry | Complex | Additive | Time [min] | Yield [%] | ee [%] |
| 1 | 50 | None | 30 | 35 | 83 |
| 2 | 50 | None | 60 | Trace | n/a |
| 3 | 50 | SIPrAgCl | 60 | 55 | 88 |
| 4 | 51 | SIPrAgCl | 60 | Trace | n/a |
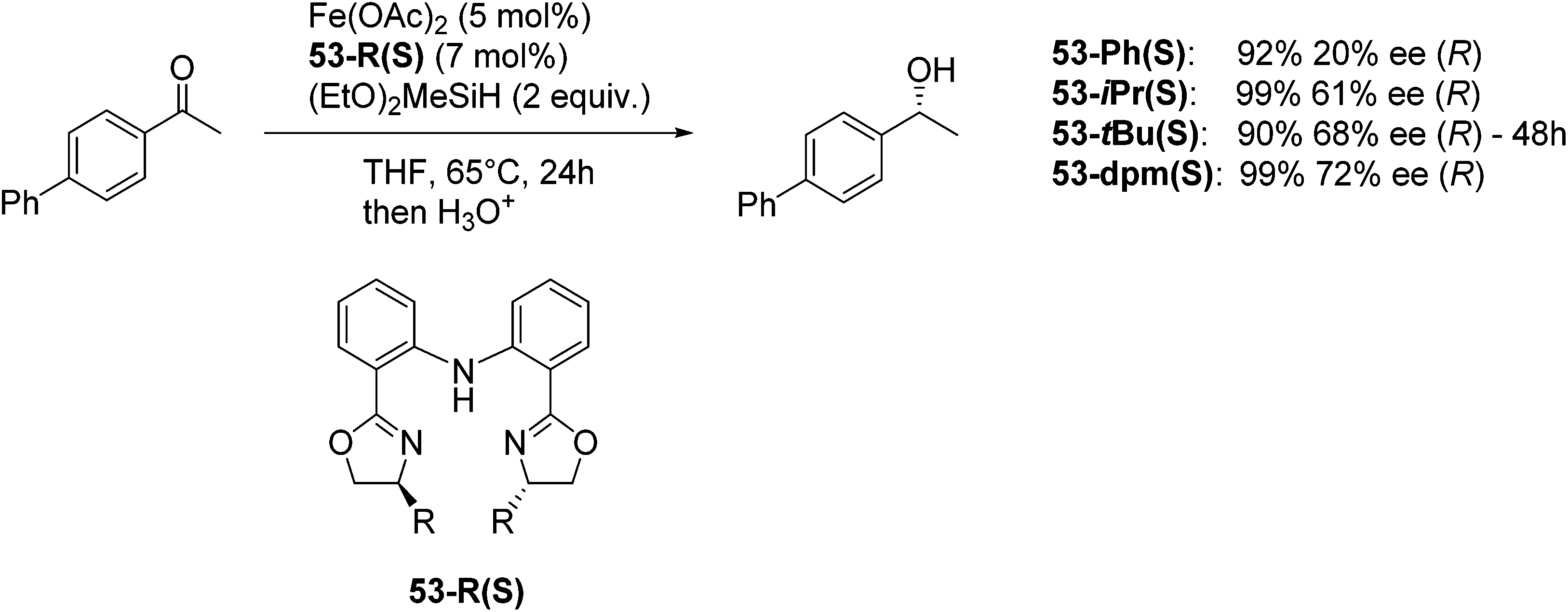
Nishiyama and co-workers studied the enantioselective hydrosilylation of ketones catalyzed by iron bis(oxazolinylphenyl)amino complexes (bopa-R; 53-R(S); R = dpm (CH(Ph)2), iPr, tBu, Ph, Bn). The reduction of 4-acetylbiphenyl by (EtO)2MeSiH was used as test reaction. The best results were obtained with 53-dpm(S), with a 99% yield and a 72% ee (Fig. 21). The scope of the reaction was then further probed (Table 8; condition A). The yields were usually high and a range of functional groups were tolerated. The enantioselectivities were modest to good. The R-enantiomer was the major product.78 Attempts to isolate the active complex by mixing iron(II) acetate with 53-R(S) were unsuccessful. However, they were able to isolate [(bopa-iPr)FeIIICl2] (51-iPr(S)) from a refluxing mixture of FeCl2 and 53-iPr(S) in THF under air. Interestingly, 51-iPr(S) was inactive for the hydrosilylation of 4-acetylbiphenyl under the previously described conditions.
 | ||
| Fig. 21 Enantioselective hydrosilylation using the in situ formed iron catalyst. | ||
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Ketone | Condition A | Condition B | ||||
| Yielda [%] | ee [%] | Abs. config | Yielda [%] | ee [%] | Abs. config | ||
| a All reported yields were of the isolated product. b Reaction time was 96 hours. c Reaction time was 48 hours. | |||||||
| 1 |
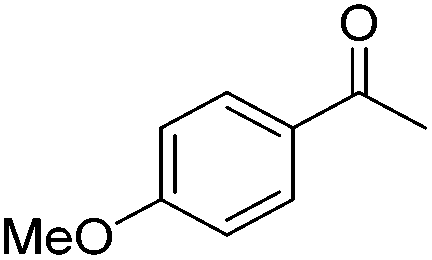
|
98 | 78 | R | 99 | 63 | S |
| 2 |
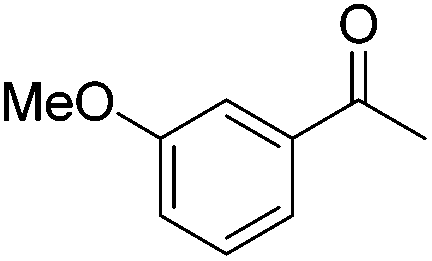
|
99 | 54 | R | 99 | 76 | S |
| 3 |
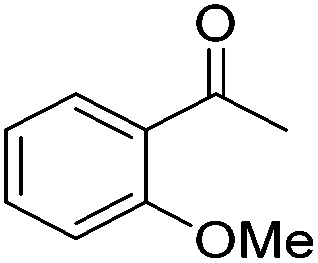
|
95 | 50 | R | 99 | 36 | S |
| 4 |
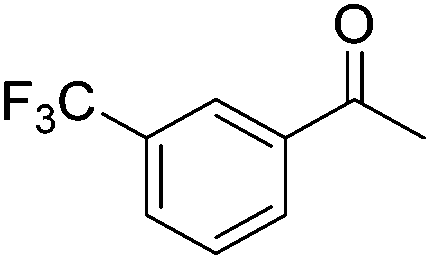
|
99 | 40 | R | 99 | 55 | S |
| 5 |
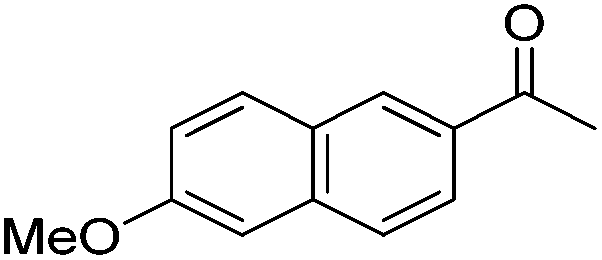
|
99 | 71 | R | 99 | 82 | S |
| 6 |
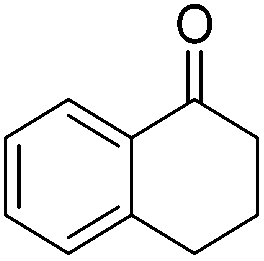
|
99 | 85 | R | 99 | 80 | S |
| 7 |
|
97 | 90 | R | 99 | 95 | S |
| 8 |
|
93 | 86 | R | 99 | 81 | S |
| 9 |
|
97c | 89 | R | 99b | 95 | S |
| 10 |
|
94 | 35 | S | 99 | 33 | S |
| 11 |
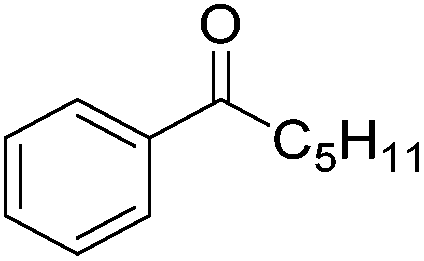
|
88 | 58 | R | 98 | 1 | S |
| 12 |
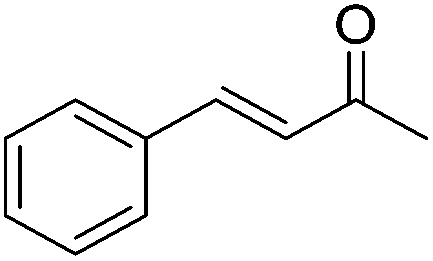
|
57 | 15 | R | 87 | 60 | S |
| 13 |
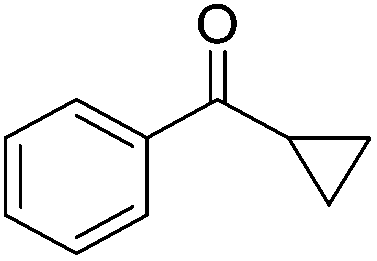
|
40b | 18 | S | 40b | 32 | S |
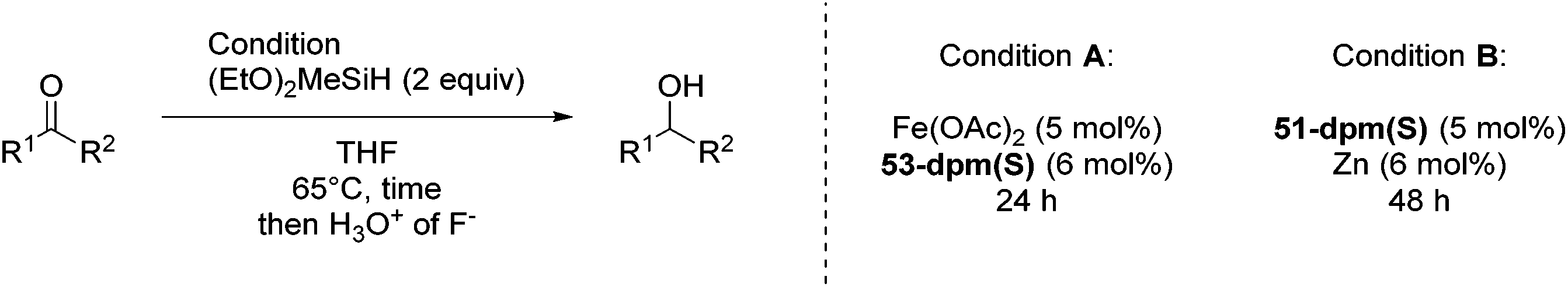
In a subsequent study, Nishiyama and co-workers showed that 51-R could be activated by the addition of 6 mol% Zn. The model substrate, 4-acetylbiphenyl, could be reduced in a 98% yield with a 65% ee.79 But in this reaction the S-enantiomer was the major product, which is opposite to the reaction using the mixture of Fe(OAc)2 with a ligand. The origin of the change in selectivity was unclear. The new reaction conditions were also general for the enantioselective hydrosilylation of ketones (Table 8; condition B).
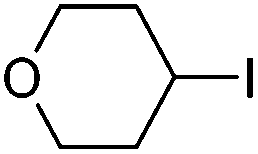
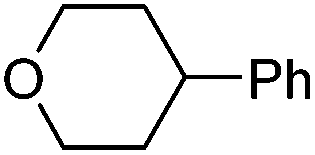
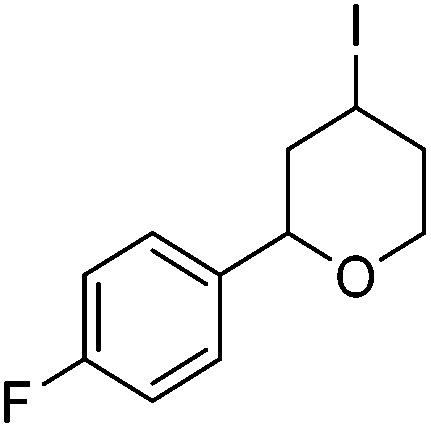
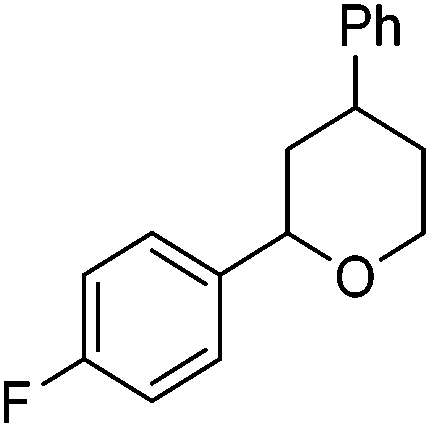
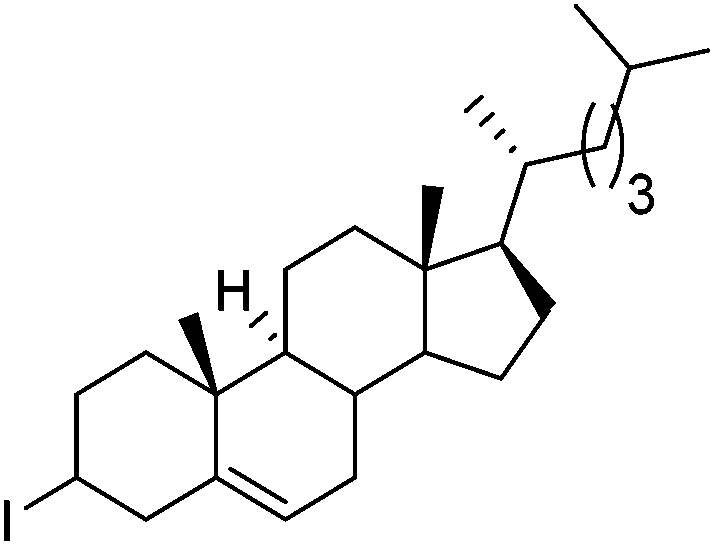
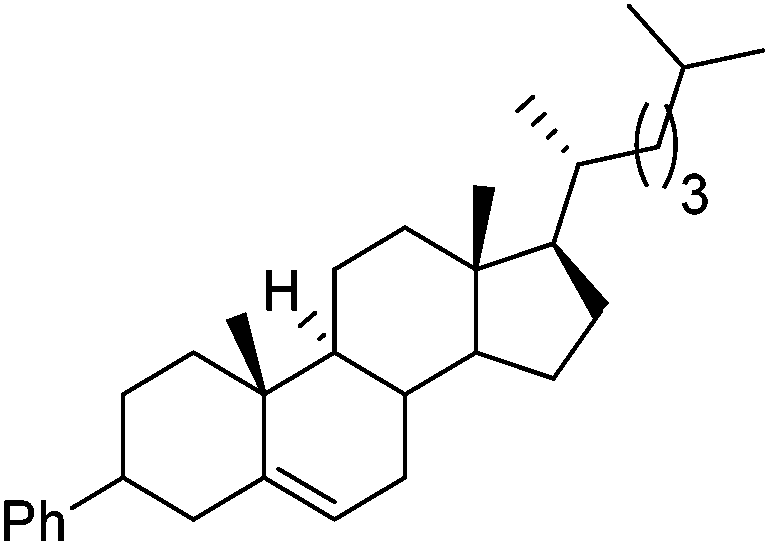
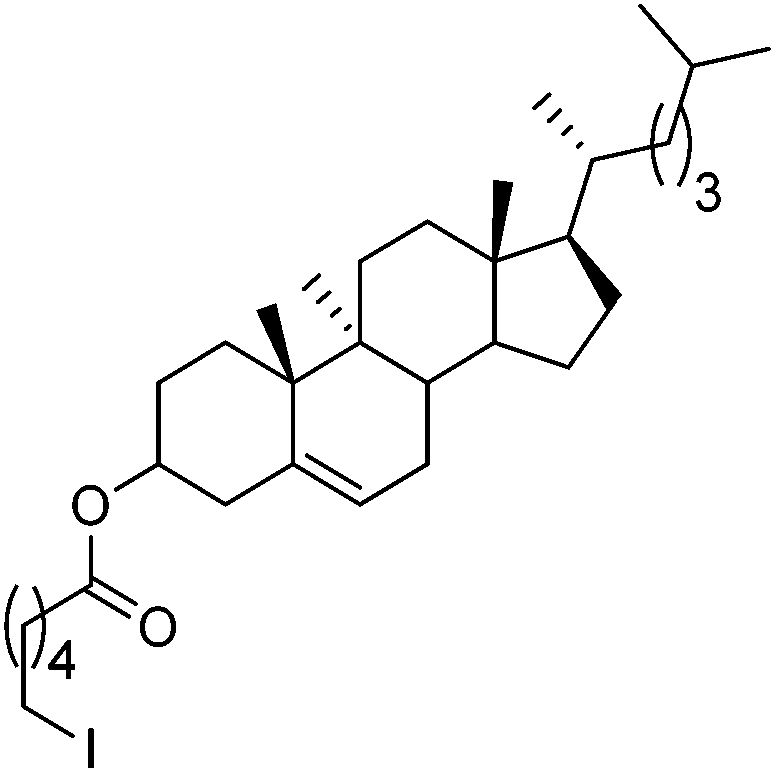
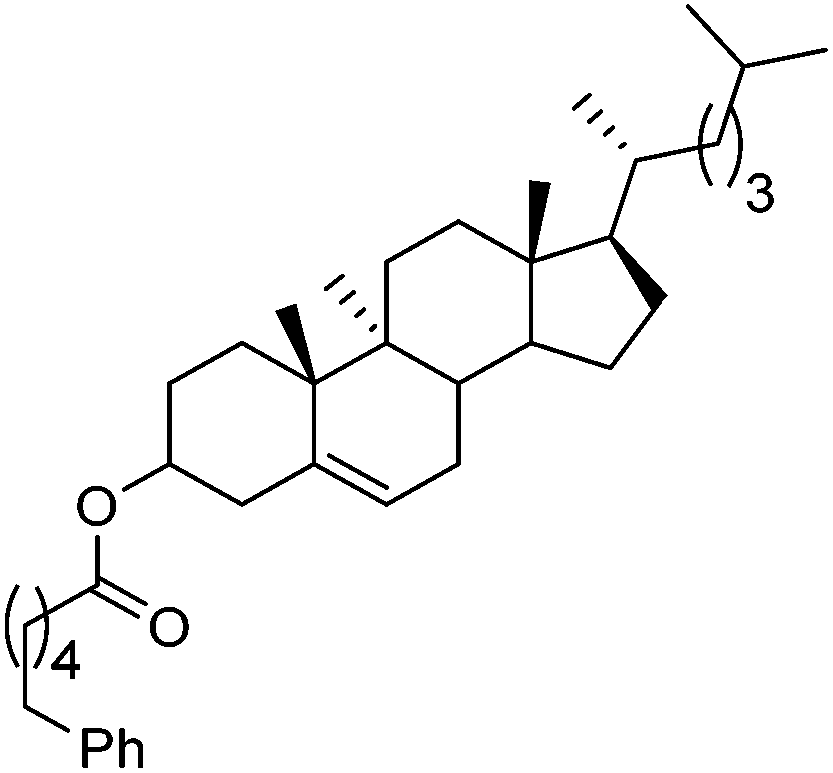
Hu and co-workers recently reported that complex 51-R (R = tBu(S), Ph(R)) was an excellent catalyst for the Kumada cross coupling reaction of non-activated alkyl halides (Table 9).80,81 The protocol tolerates a range of functional groups, including carbamates (entry 11), N-heterocycles (entries 2 and 4), Boc-protected piperidine (entry 10), tetrahydropyran (entries 7 and 8), base-sensitive ester (entries 3 and 9) and ketone (entry 4). Natural product-derived compounds, including 3-iodocholestene, cholestenyl-6-iodohexanoate and menthyl-6-iodohexanoate (Table 9, entries 12–14) were also coupled in moderate to good yields. The enantioselectivity for the coupling of 3-iodobutylbenzene, 2-iodobutylbenzene and 2-iodopropylbenzene, however, was low. Only an ee of up to 20% could be obtained.80
|
|
|||
|---|---|---|---|
| Entry | Halide | Product | Yielda (%) |
|
a Isolated yields at 100% conversion.
b Reaction at −40 °C.
c Use of 51-Ph(R) as a catalyst.
d 25% starting material was recovered.
e Mixture of diastereoisomers: 66:34 (d.r. for RX = 91:9).
f Mixture of diastereoisomers: 52:48 (d.r. for RX = 81:19).
g Mixture of stereoisomers: 81:19 (d.r. for RX = 100:0).
|
|||
| 1 |
|
|
92b,c |
| 82c | |||
| 2 |
|
|
83 |
| 3 |
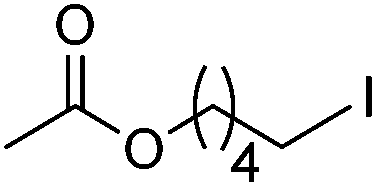
|
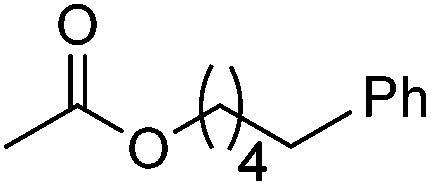
|
83 |
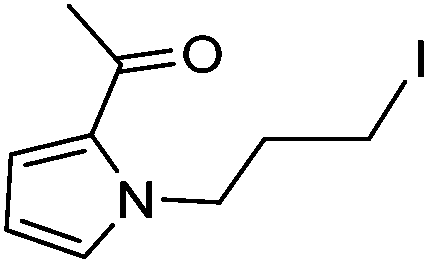
| 4 |
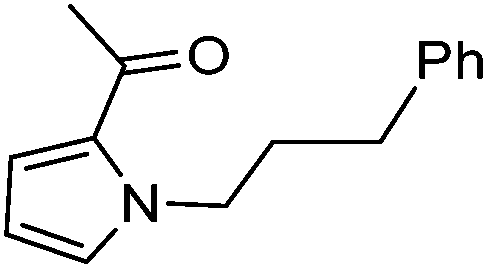
|
|
57 (25)d |
| 5 |
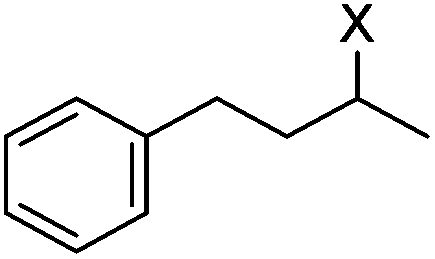
|
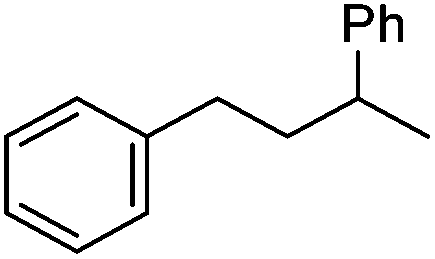
|
95b,c (X = I) |
| 88c (X = I) | |||
| 93c (X = Br) | |||
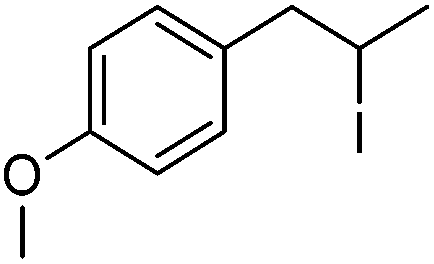
| 6 |
|
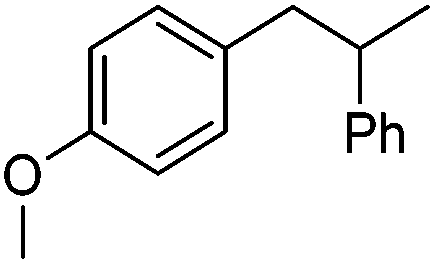
|
90 |
| 7 |
|
|
75 |
| 8 |
|
|
92e |
| 9 |
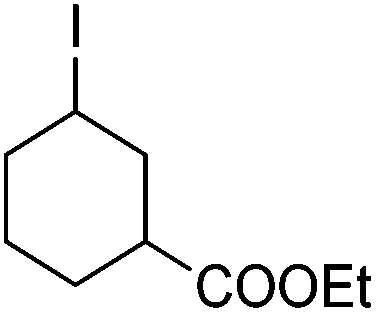
|
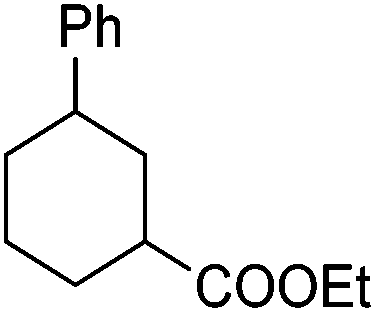
|
71f |
| 10 |
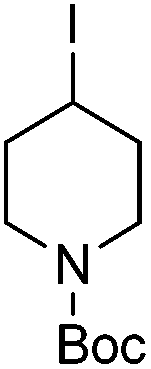
|
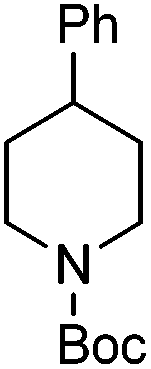
|
88 |
| 11 |
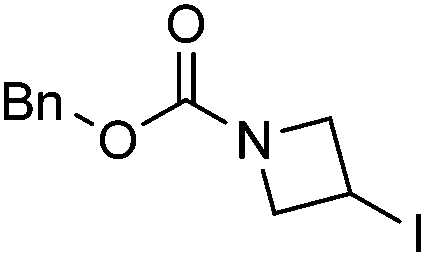
|
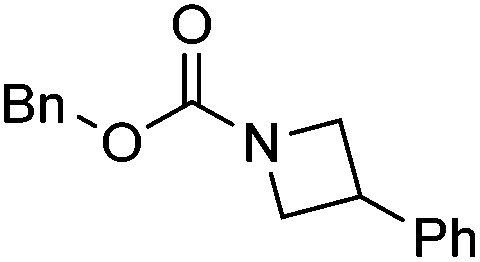
|
65 |
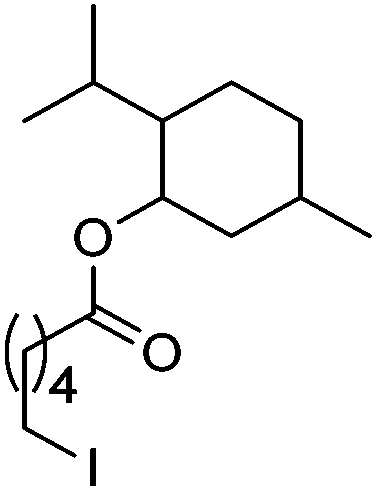
| 12 |
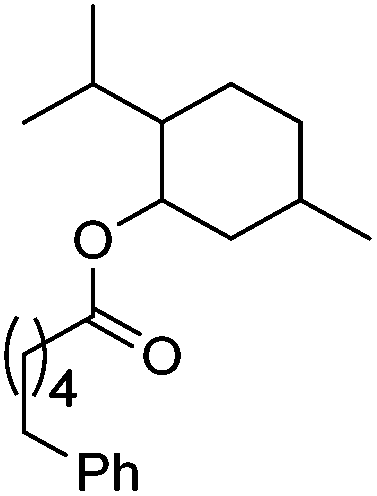
|
|
83 |
| 13 |
|
|
53g |
| 14 |
|
|
85 |
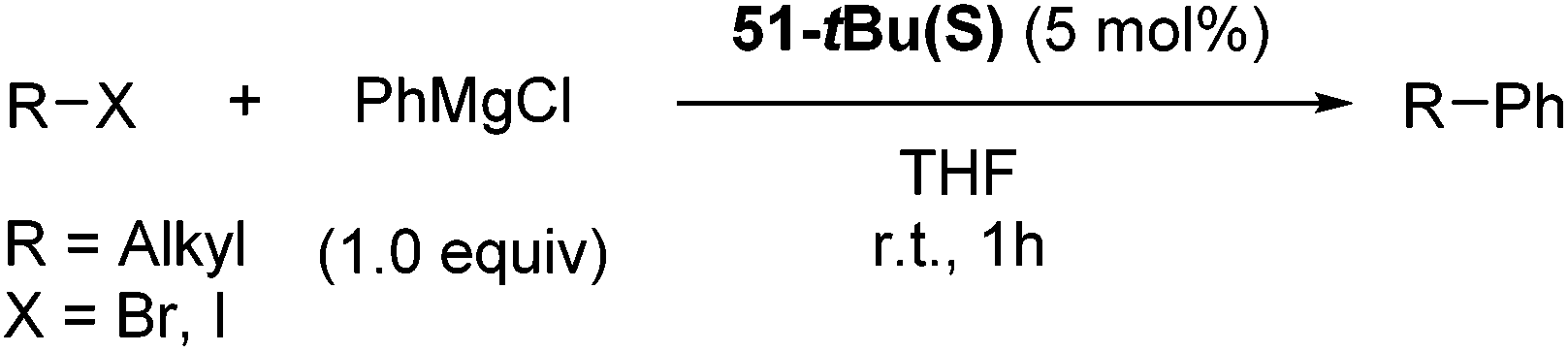
7. 2,5-Disubstituted pyrrolidine-based pincer ligands
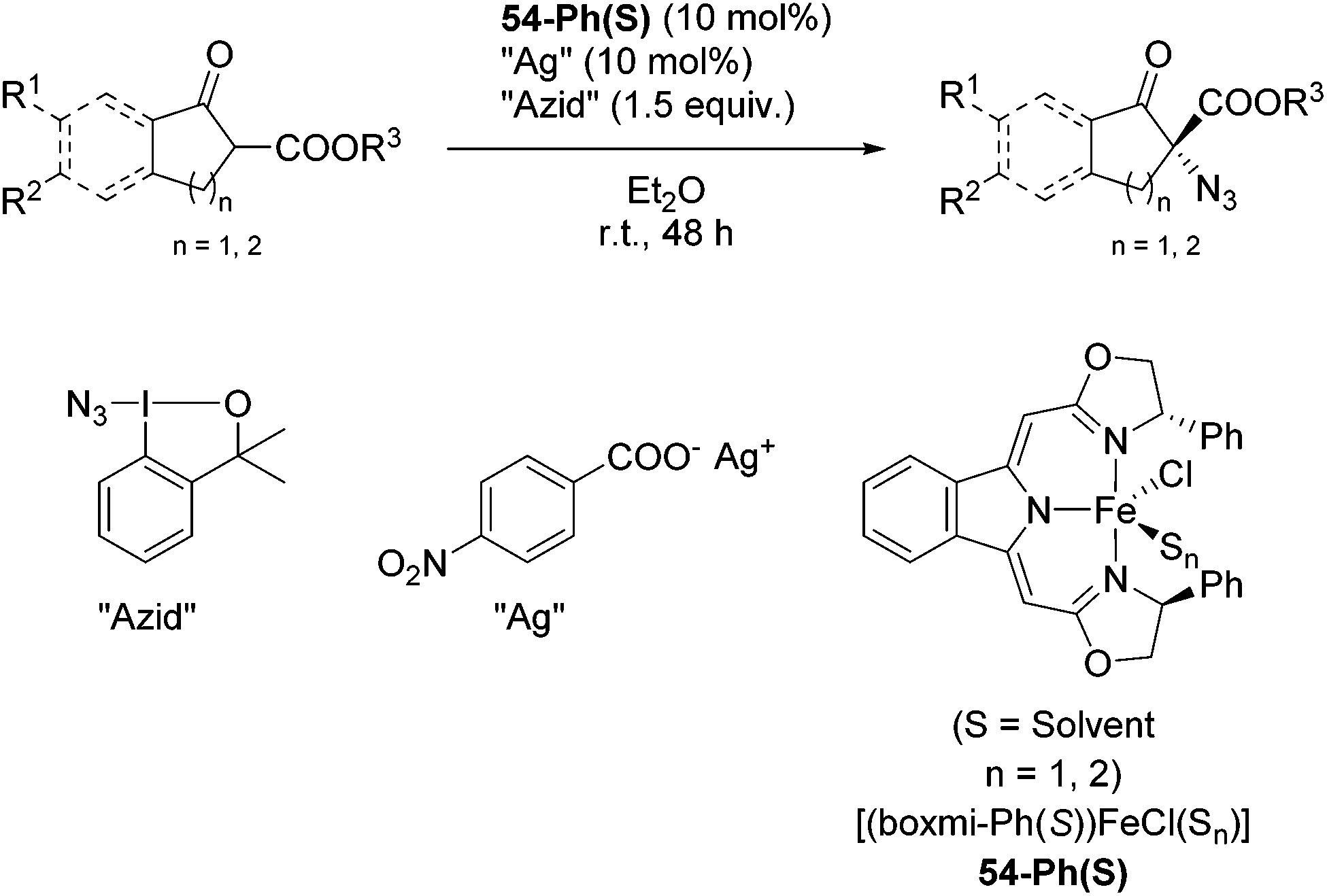
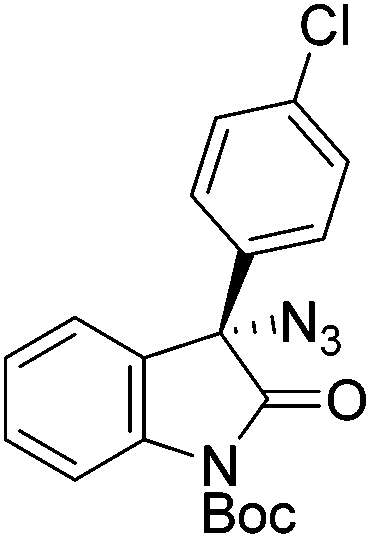
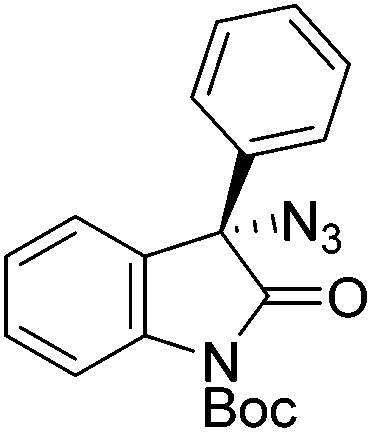
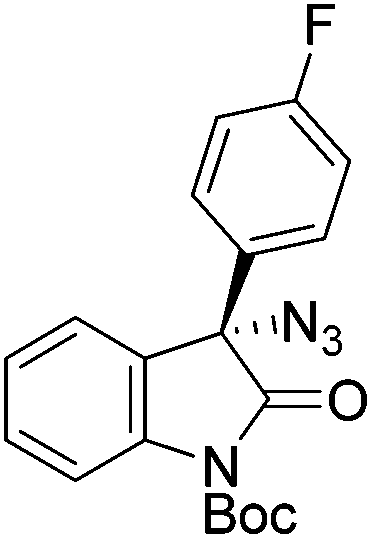
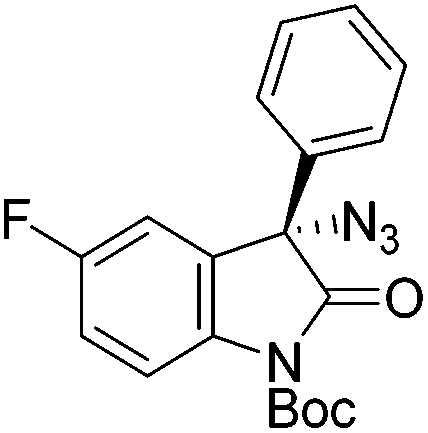
The group of Gade developed a new class of pincer ligands based on bis(oxazolinylmethylidene)isoindolines (boxmi).Complex [(boxmi-Ph(S))FeCl(Sn)] (54-Ph(S); S = solvent; n = 1 or 2) was an active catalyst for the azidation of cyclic β-keto esters and 1-indanones (Table 10).82 The indanone derived tert butyl β-keto esters could be converted in high yields and high enantioselectivities (90–93%) were achieved for many substrates. It was shown that the bulky tert butyl substituent on the ester is crucial for higher enantioselectivities (compare entry 7 with entry 9). Substituted cyclopentenone derivatives could also be converted in high enantioselectivity.82
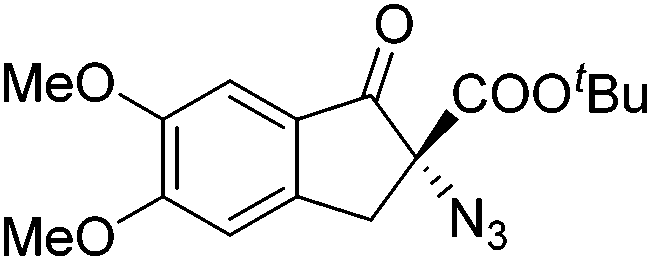
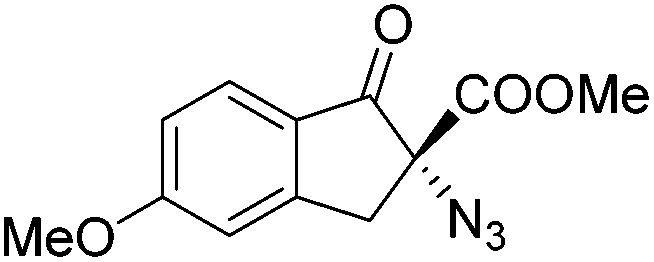
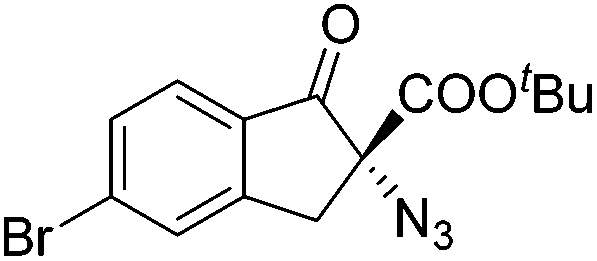
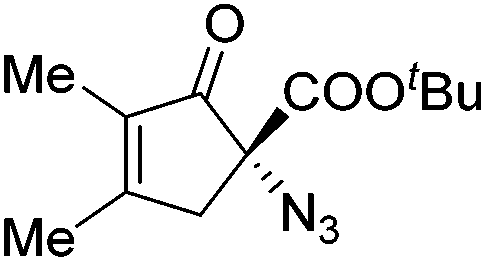
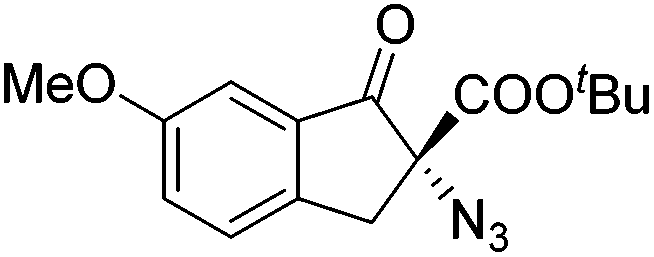
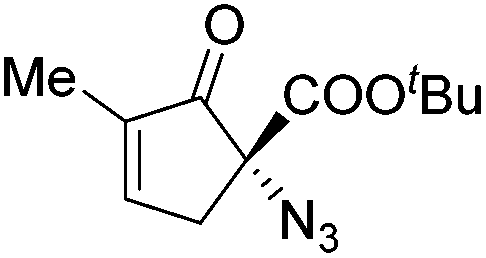
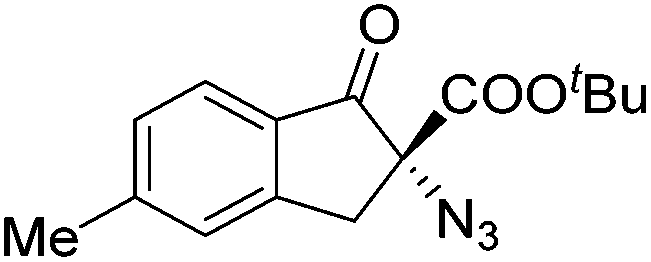
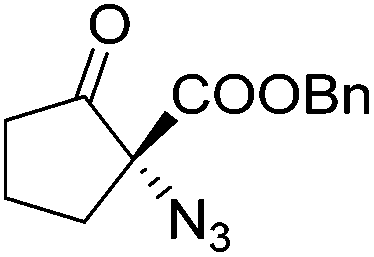
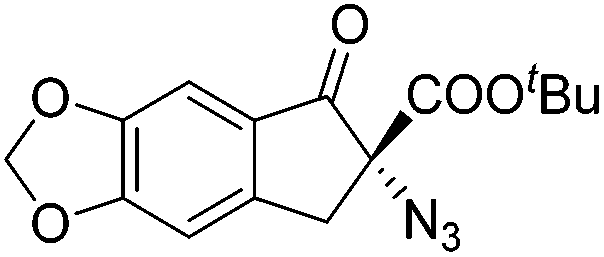

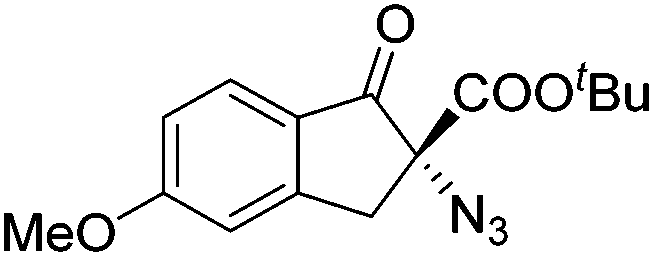
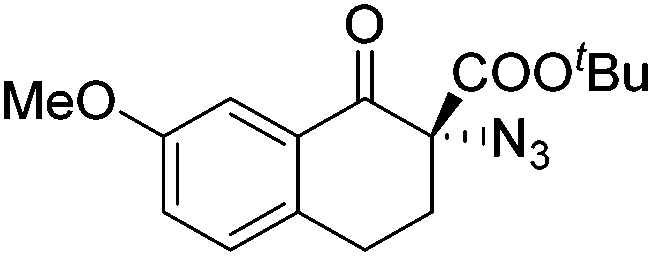
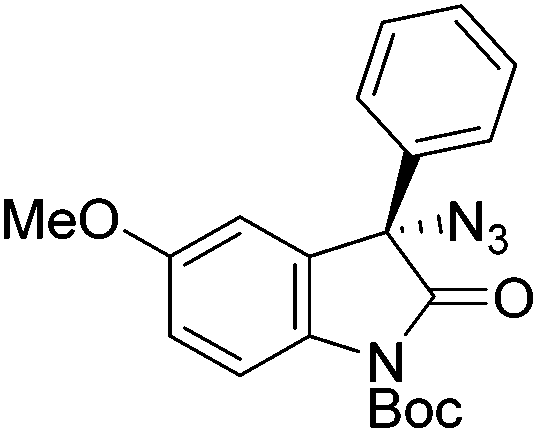
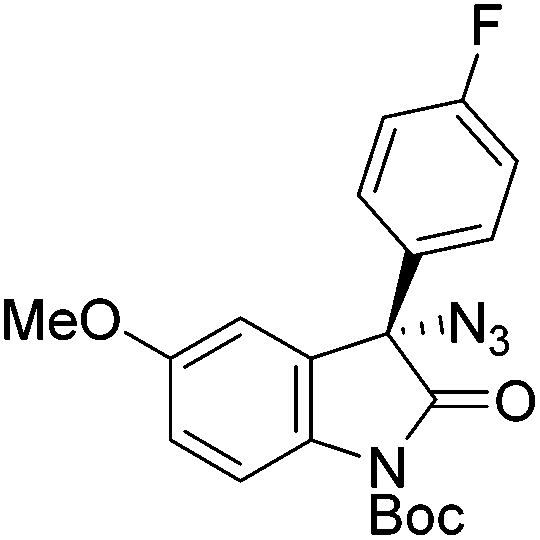
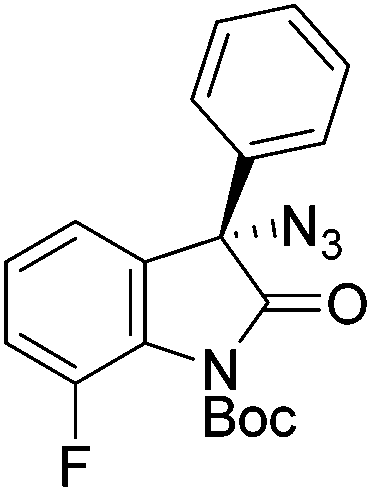
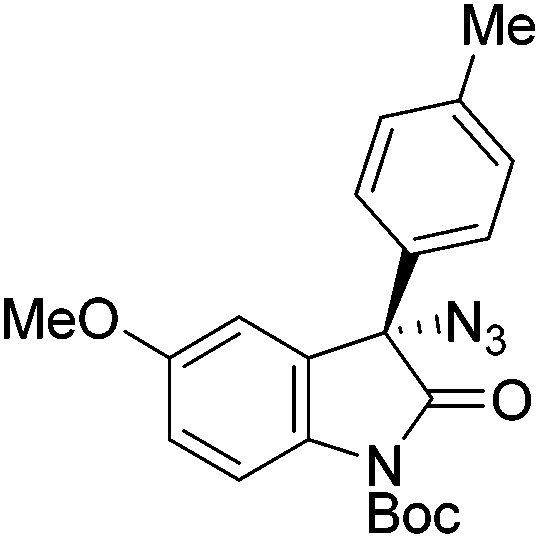
Gade and co-workers then applied this catalytic system for the azidation of 3-aryloxindoles. The previously optimized conditions gave lower ee's. The selectivity could be increased by the in situ formation of the catalytically active species, which was generated by mixing 10 mol% Fe(OOCEt)2 with 12 mol% 55-Ph(S) in diethylether at room temperature. Under the optimized reaction conditions the reactions of various 3-aryloxindoles were successful. The yields were between 85 and 90% and the ee were between 87 and 94% (Table 11).82
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Product | Yielda [%] | eeb [%] | Entry | Product | Yielda [%] | eeb [%] |
| a All reported yields were of the isolated products. b The absolute configuration was determined by a crystal structure of an 1,2,3-triazole which was obtained by click reaction of the azide compound, entry 3 with (3-bromophenyl)acetylene. The configuration was confirmed as the R-enantiomer. | |||||||
| 1 |
|
87 | 91 | 5 |
|
85 | 94 |
| 2 |
|
85 | 91 | 6 |
|
89 | 90 |
| 3 |
|
90 | 91 | 7 |
|
85 | 91 |
| 4 |
|
85 | 94 | 8 |
|
89 | 87 |
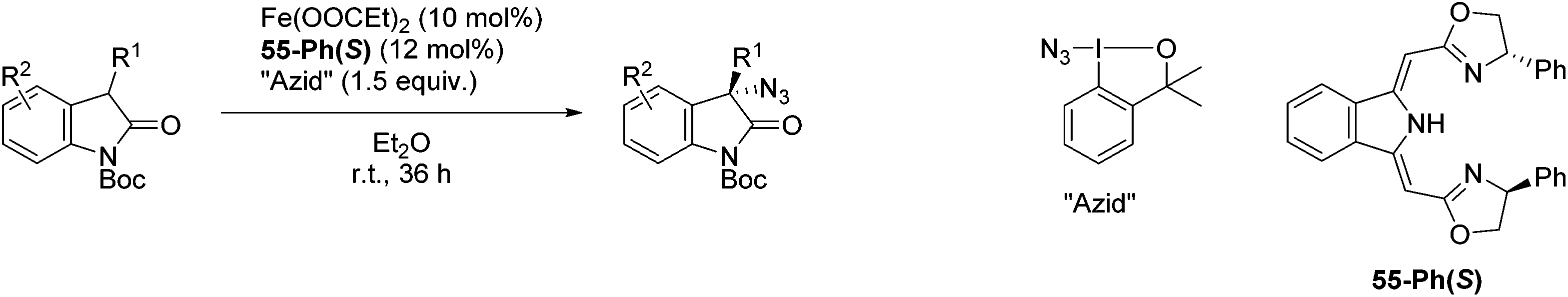
Gade and co-workers then showed that [(boxmi-Ph(R))Fe(OAc)] (56-Ph(R)) and [(boxmi-Ph(R)Fe((S)-OCHCH3Ph)] (57-Ph(R)) catalyzed the enantioselective hydrosilylation of aryl ketones (Fig. 22).83 The latter complex was employed for the hydrosilylation of various substrates (Table 12).
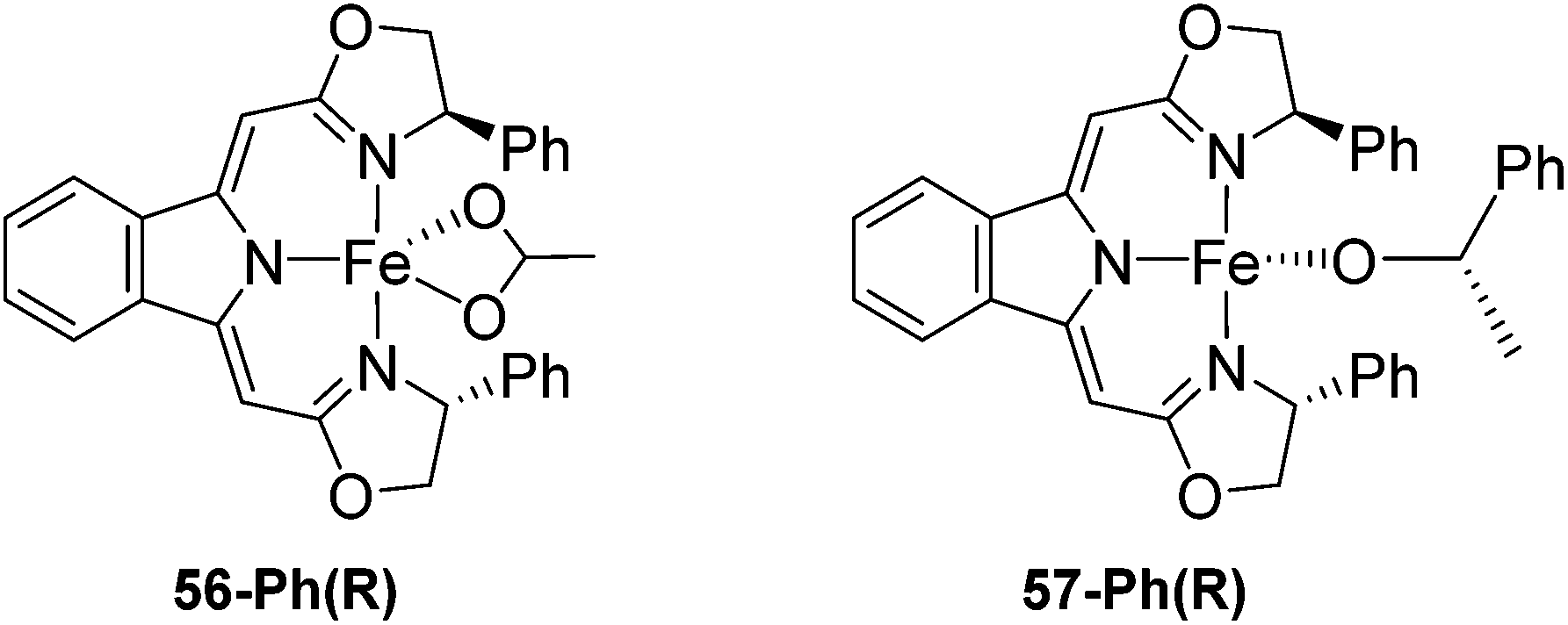 | ||
| Fig. 22 Structures of complexes [(boxmi-Ph(R))Fe(OAc)] (56-Ph(R)) and [(boxmi-Ph(R)Fe((S)-OCHCH3Ph)] (57-Ph(R)). | ||
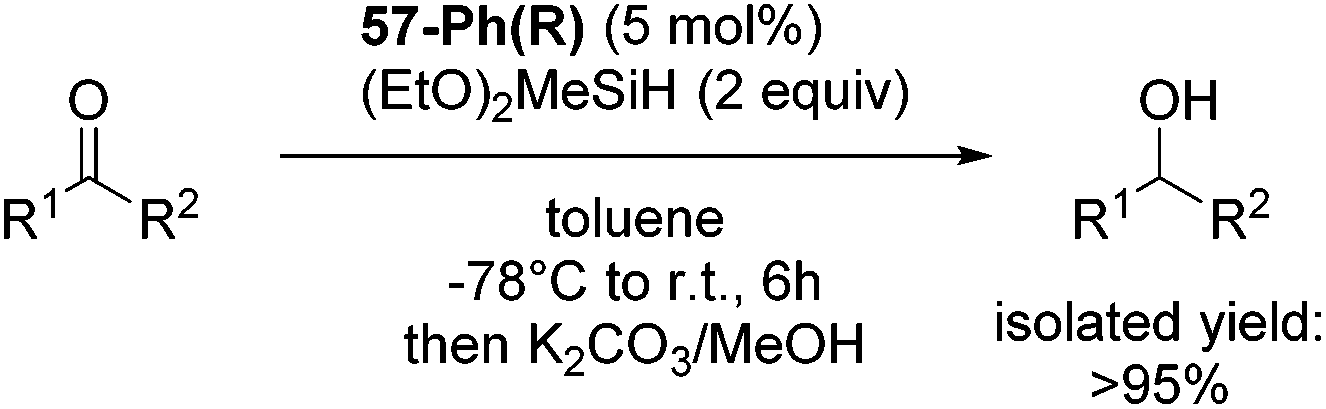
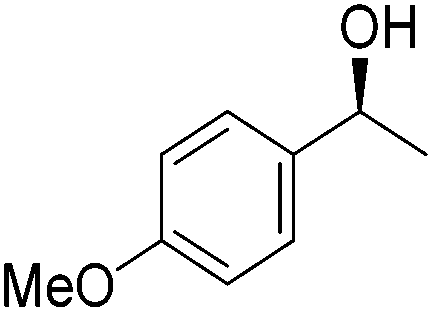
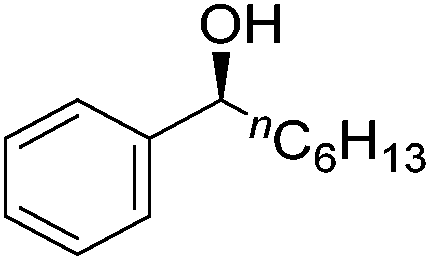
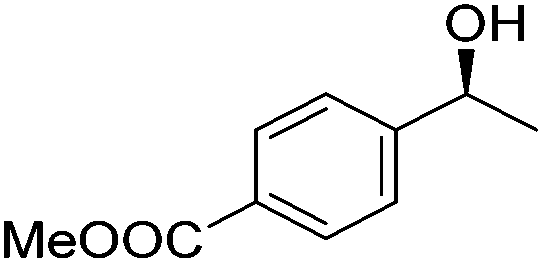
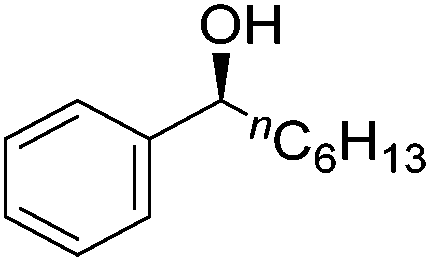
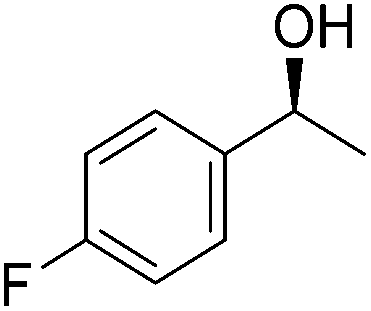
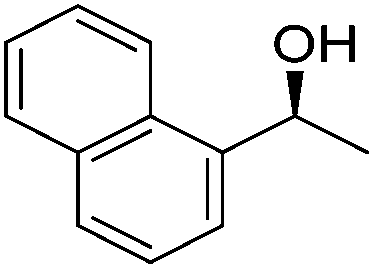
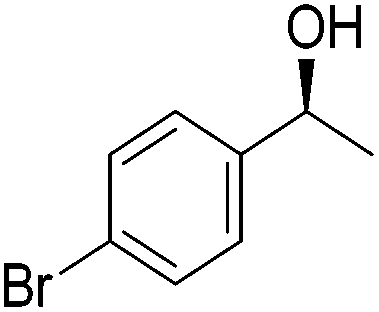
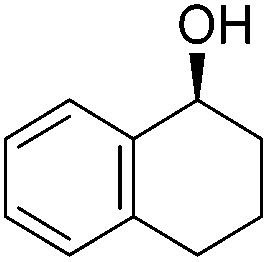
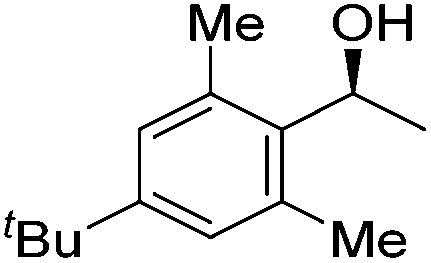
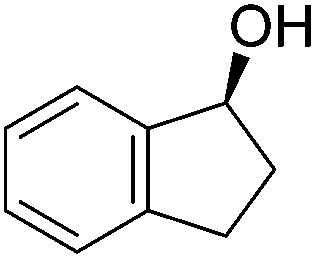
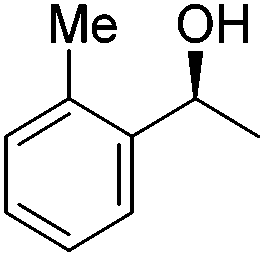
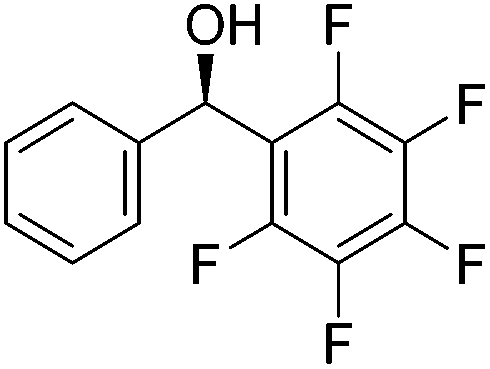
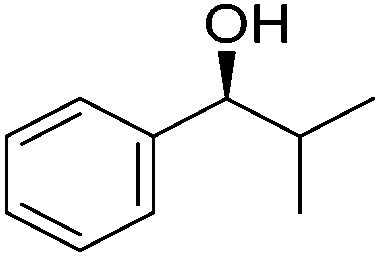
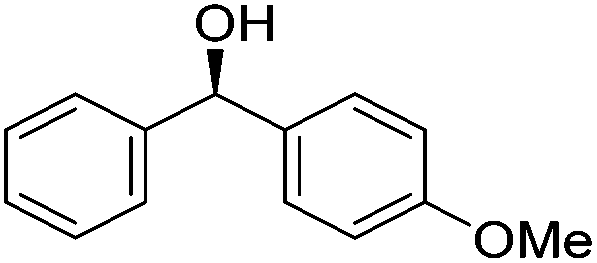
Good yields and selectivity were achieved for many substituted acetophenones. Different substituents had almost no influence on the outcome of the reaction. Changing the alkyl substituent of acetophenone to a more sterically demanding isopropyl group decreased the ee to 73% (entry 8). The use of diaryl ketones decreased the selectivity (entries 15 and 16). An inner sphere mechanism was proposed. Activation of the pre-catalyst gave an iron alkoxy complex, which undergoes σ-bond metathesis with silane to give the silyl ether product and the iron hydride species. The prochiral ketone coordinates to the hydride complex and subsequently inserts into the metal hydride bond to regenerate the iron alkoxy complex.83
8. Bis(phosphinoethyl)amino pincer ligands
Although pincer ligands were mostly based on rigid backbones such as arene, pyridine, and diarylamine, ligands based on more flexible alkyl linkers have also been reported. The P(CH2)2NH(CH2)2P ligand, also known as MACHO,84 enforces a meridional coordination geometry on many transition metal ions. Their iron complexes have found many catalytic applications.3,85–95Schneider and co-workers synthesized a series of different Fe(II) and Fe(0) complexes supported by RP(CH2)2NH(CH2)2P (R = cy (C6H11), iPr; Fig. 23).85 The complexes [Fe(RP(CH2)2NH(CH2)2P)Cl2(CO)] (58-R; R = cy, iPr) were prepared by treating the ligand with FeCl2 under a CO atmosphere. Both 58-cy and 58-iPr show a low spin configuration. The Fe(0) complexes, [Fe(RP(CH2)2NH(CH2)2P)(CO)2] (59-R; R = cy, iPr) were prepared by mixing Fe(CO)5 with the ligand under UV irradiation. Both 59-cy and 59-iPr are in a low spin configuration. 59-R reacted with CH2Cl2 to give 58-R. 59-iPr reacted with HCl to give the cationic hydride species, [Fe(iPrP(CH2)2NH(CH2)2P)H(CO)2]Cl (60-iPr). NMR spectroscopy suggested that both cis and trans CO isomers were formed. The neutral hydride complexes [Fe(RP(CH2)2NH(CH2)2P)H(CO)Cl] (61-R) were obtained either by the reaction of 58-R with nBu4NBH4 in acetonitrile at room temperature or by UV irradiation of the cationic complex 60-R. The reaction of 58-iPr with an excess amount of NaBH4 (10 equiv.) generated the borohydride complex [Fe(iPrP(CH2)2NH(CH2)2P)H(CO)(η1-HBH3)] (62-iPr). High resolution X-ray crystallography and NOESY experiments of 62-iPr revealed an intramolecular hydrogen bond between the terminal hydrides of the η1-HBH3 and the N–H of the ligand. Protonation of 62-iPr with 2,6-lutidinium tetraphenylborate gave the dimeric complex [(Fe(iPrP(CH2)2NH(CH2)2P)(CO))2(μ2, η1:η1-H2BH2)](BPh4) (63-iPr). This compound is a group 8 metal complex with a μ2, η1:η1-H2BH2 ligand.85
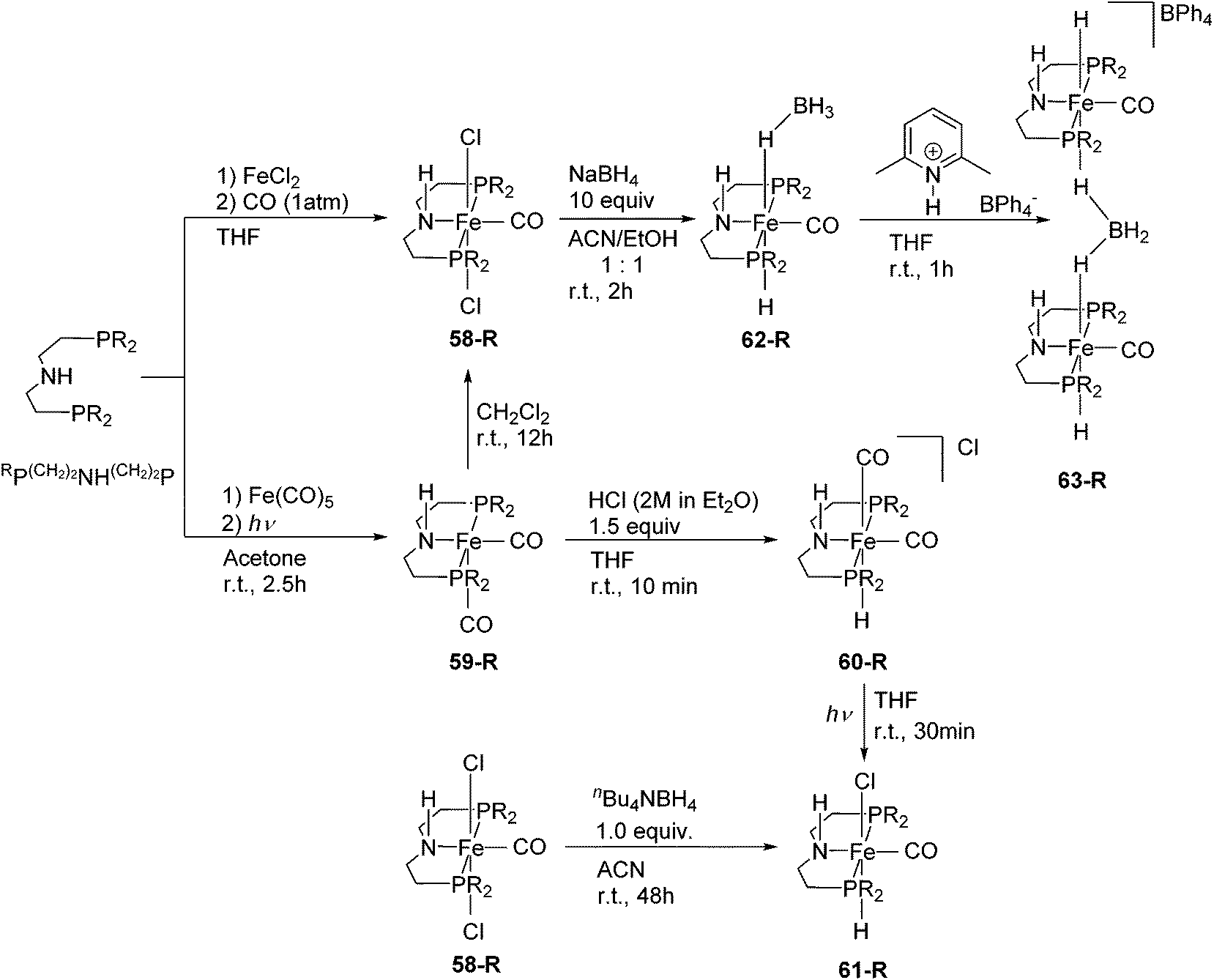 | ||
| Fig. 23 Synthesis and reactivity of iron complexes of RP(CH2)2NH(CH2)2P. | ||
8.1 Hydrogenation of esters and nitriles
The groups of Guan and Beller independently reported the hydrogenation of esters to alcohols catalyzed by 62-iPr.53,86,87 Both aliphatic and aromatic esters were hydrogenated in moderate to high yields. Aromatic esters showed higher reactivity compared to the aliphatic esters. For a substrate containing both CC and CO groups, methyl cinnamate, hydrogenation of both groups was obtained.
Guan and co-workers then reported the hydrogenation of CE-1270, a methyl ester derived from coconut oil.86 The substrate consisted of methyl laurate (C12, 73%), methyl myristate (C14, 26%) and trace amounts of C10 and C16 methyl esters (∼1%). The hydrogenation was carried out under neat conditions under 52 bar hydrogen pressure at 135 °C, using 1 mol% 62-iPr as the catalyst. The reaction gave a full conversion after three hours, giving a combined GC yield of 99% for fatty alcohols. In an upscale experiment (100 g), the yield was only 25%, suggesting catalyst degradation. Lowering the temperature to 115 °C increased the yield to 40%.86 Complex 62-iPr was then further employed in the hydrogenation of neat coconut oil, without transforming it to the corresponding fatty acid methyl esters. A yield of 12% was reached after 23 hours.92
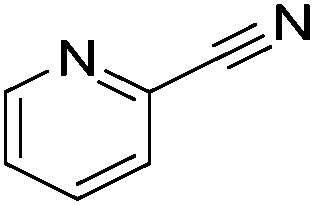
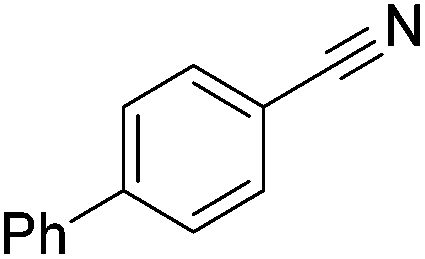
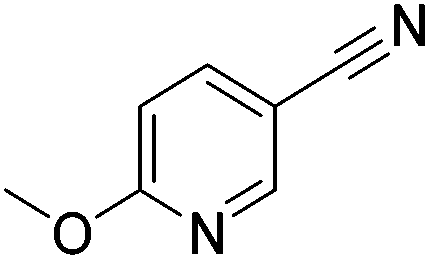
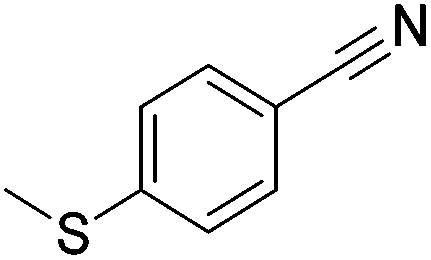
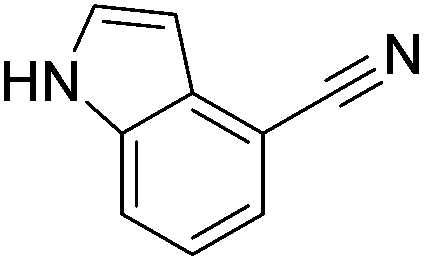
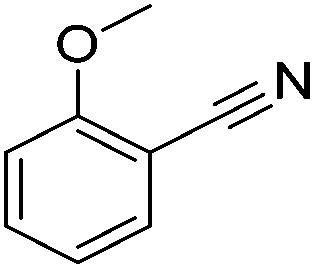
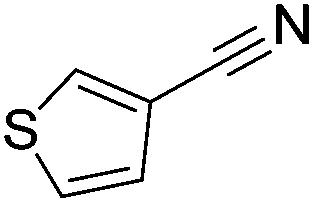
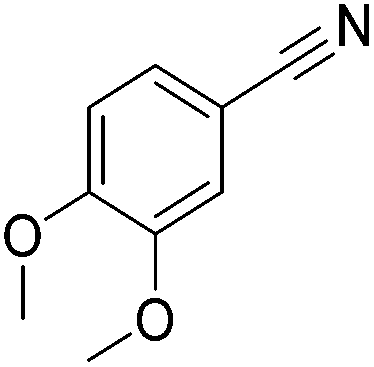
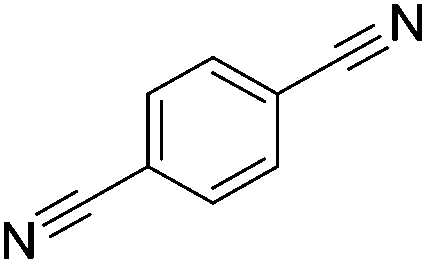
Beller and co-workers reported the hydrogenation of nitriles and dinitriles using 62-iPr as the catalyst.91 The catalyst showed good chemoselectivity, tolerating various functional groups (Table 13).
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Substrate | T [°C] | Yielda [%] | Entry | Substrate | T [°C] | Yielda [%] |
| a Isolated yields at full conversion (primary amines were isolated as HCl-salts). b GC-yield with hexadecane as the internal standard. c 10% transesterification product (–COOiPr) product were observed. d Reaction was quenched after 20 min. | |||||||
| 1 |
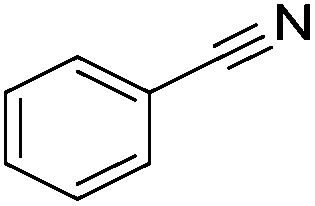
|
70 | 97b | 13 |
|
100 | 80 |
| 2 |
|
70 | 99 | 14 |
|
130 | 92 |
| 3 |
|
100 | 81 | 15 |
|
100 | 40b |
| 4 |
|
70 | 84 | 16 |
|
100 | 58b |
| 5 |
|
70 | 92 | 17 |
|
130 | 75c |
| 6 |
|
70 | 98 | 18 |
|
100 | 93 |
| 7 |
|
100 | 99 | 19 |
|
100 | 70 |
| 8 |
|
100 | 71 | 20 |
|
100 | 93 |
| 9 |
|
100 | 81 | ||||
| 10 |
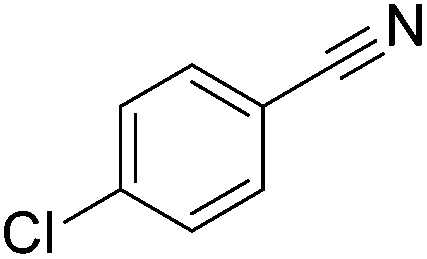
|
130 | 78 | 21 |
|
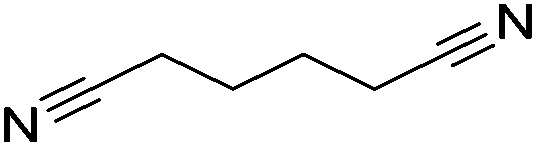
100 | 95 |
| 100 | 83d | ||||||
| 11 |
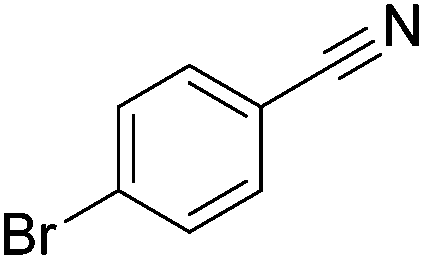
|
100 | 88 | 22 |
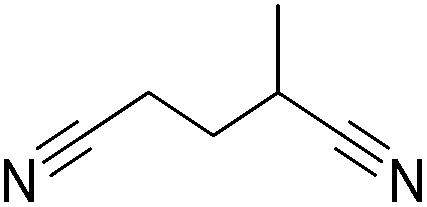
|
100 | 41 |
| 12 |

|
100 | 92 | 23 |
|
100 | 85 |
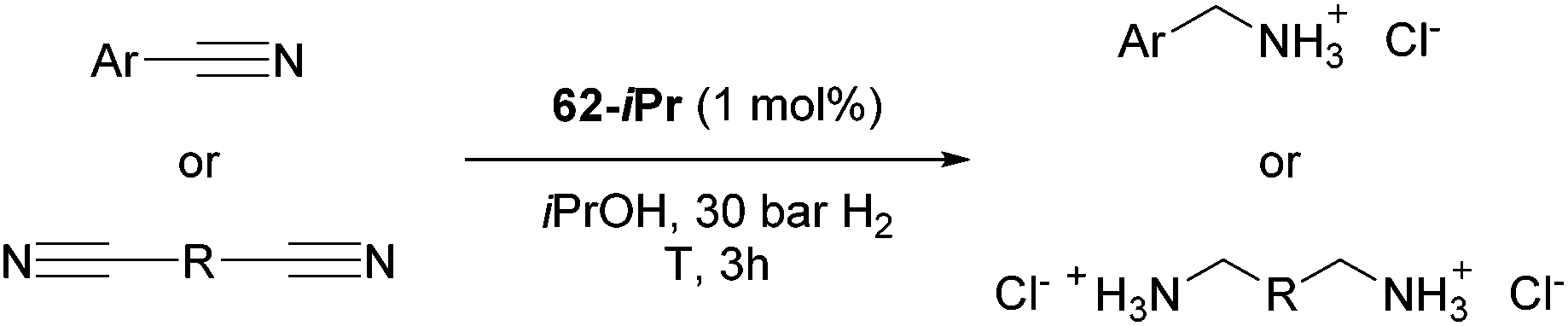
Aryl nitriles with electron-donating groups could be reduced at a lower temperature than aryl nitriles with electron-withdrawing groups. The method could be employed for the hydrogenation of more challenging aliphatic nitriles.
8.2 Acceptorless dehydrogenation of alcohols, formic acid and N-heterocycles
Beller and co-workers showed that 62-iPr and [Fe(iPrP(CH2)2NH(CH2)2P)H(CO)Br] (64-iPr; Fig. 24), were active catalysts for the dehydrogenation of methanol to give hydrogen and CO2 in the presence of a base (8 M KOH) at 91 °C. The TON was up to 10000 after 46 hours. In order to improve the stability of 62-iPr in the reaction, an additional ligand (up to 5 equivalents) was added. The life time of 62-iPr could be improved from 43–66 h to 5 days.3 Hazari and co-workers95 showed that complex [Fe(RP(CH2)2N(CH2)2P)H(CO)] 65-R (R = iPr, cy; Fig. 24) catalyzed the base-free dehydrogenation of methanol in water. Methyl formate was obtained as a side product, suggesting the incomplete dehydrogenation of methanol. Following the reaction by NMR revealed the formation of [Fe(cyP(CH2)2NH(CH2)2P)H(CO)(OOCH)] (66-cy; Fig. 24), which took the catalyst away from the catalytic cycle. In the presence of a Lewis acid the accumulation of 66-cy could be prevented. Thus, Lewis acids were used to promote the dehydrogenation of methanol. A TON of up to 51000 could be reached.95 In an analogous study, LiBF4 was used to promote the dehydrogenation of formic acid catalyzed by 65-R and 66-R (R = iPr, cy). They obtained the best results using 0.0001 mol% of 66-iPr in a 2.2% solution of formic acid in dioxane at 80 °C. 10 mol% of LiBF4 was used as the additive. The reaction finished after 9.5 hours giving a TON of 983642 with a TOF of 196728 h−1.89
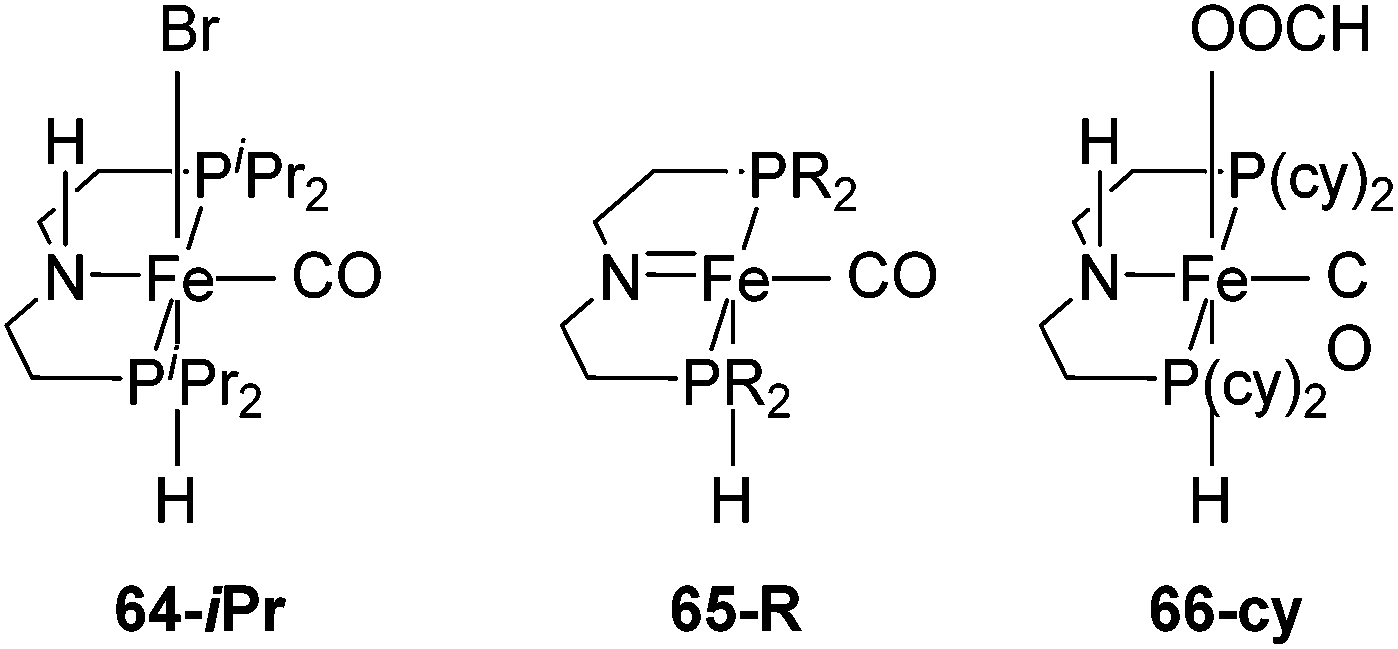 | ||
| Fig. 24 Structure of complexes [Fe(iPrP(CH2)2NH(CH2)2P)H(CO)Br] (64-iPr) and [Fe(RP(CH2)2N(CH2)2P)H(CO)] 65-R (R = iPr, cy). | ||
The groups of Schneider and Jones further reported the acceptorless dehydrogenation of different primary and secondary alcohols using 62-iPr as the catalyst.90 The formed H2 was removed from the reaction medium by a steady flow of N2, in order to shift the reaction equilibrium. Secondary benzylic alcohols (entries 1–8) were oxidized to the corresponding acetophenone derivatives in good to excellent yields (65–92%). The protocol also tolerates a range of different functional groups (MeO, Me, NO2 and halides). For substrates with electron withdrawing groups (entries 4–6) the catalyst loading could be lowered to 0.1 mol%. An aliphatic cyclohexanol (entry 10) was oxidized to cyclohexanone in a 64% yield. Primary alcohols and diols were also used as substrates. Benzylic alcohol formed benzyl benzoate exclusively (entry 9). The chemoselectivity of secondary over primary alcohol was tested using 1,2-propandiol and 1,3-butanediol (entries 13 and 14). In both cases the secondary hydroxyl group was oxidized before the primary group. The diols 1,2-benzenedimethanol and 1,5-pentanediol (entries 11 and 12) gave the corresponding lactones in 96 and 59% yield. Furthermore, it was shown that the reactions could be reversible, and the hydrogenation of ketones was achieved with 62-iPr as the catalyst (Table 14).93
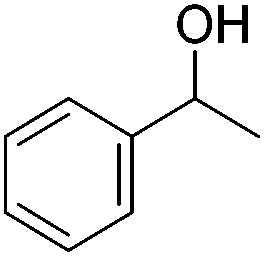
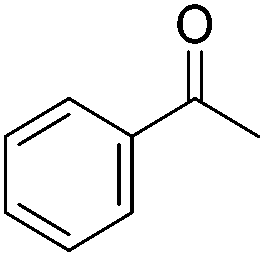
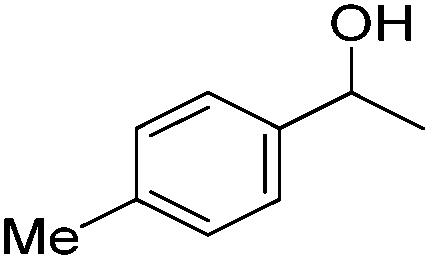
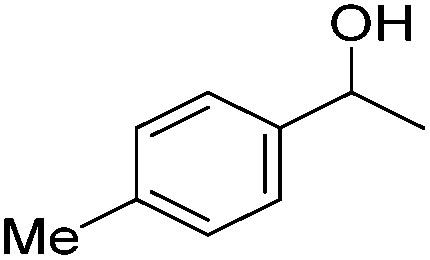
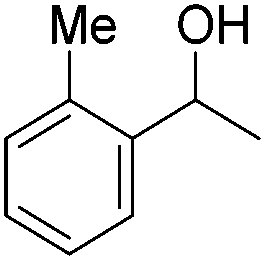
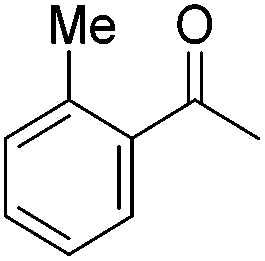
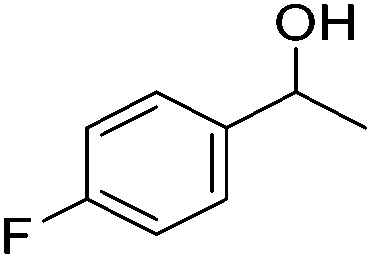
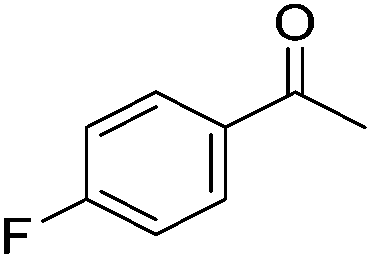
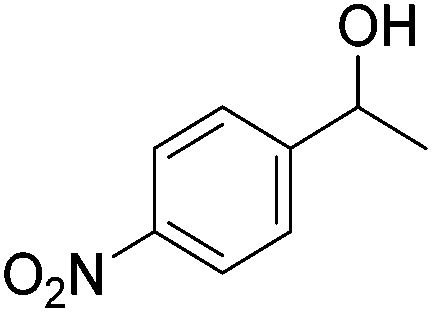
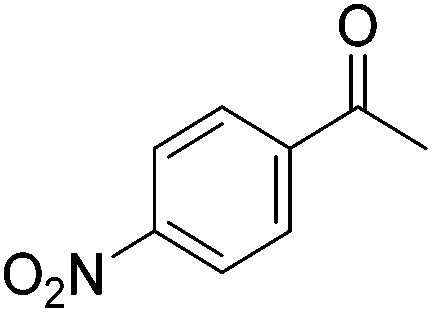
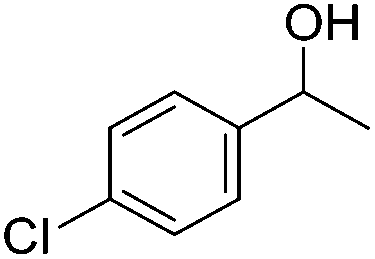
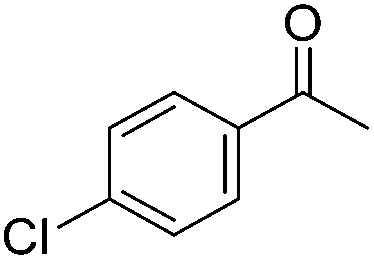
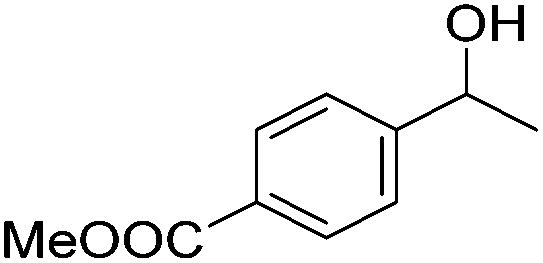
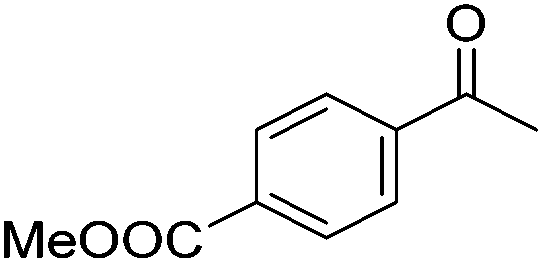
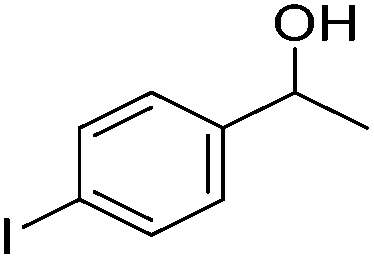
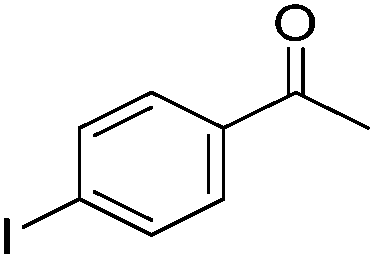
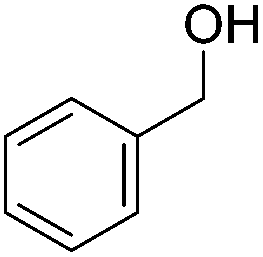
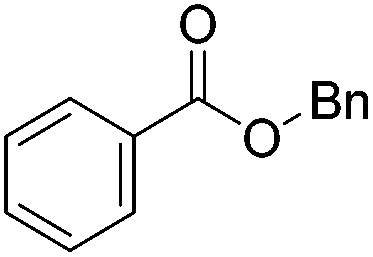
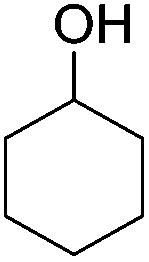
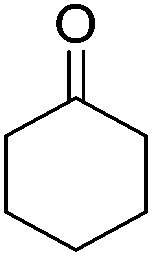
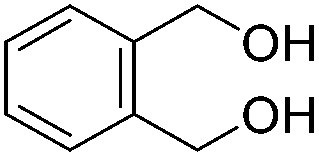
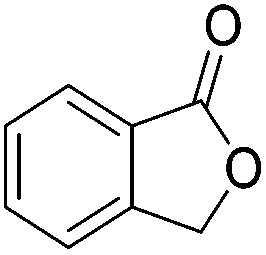

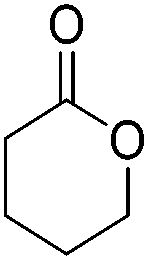
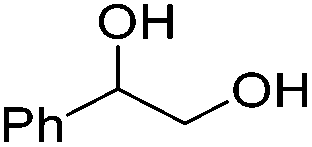
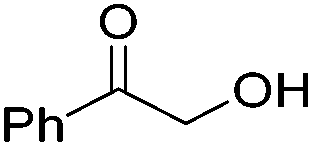
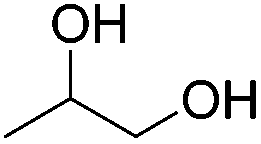
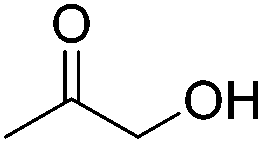
Beller and co-workers further studied the dehydrogenation of diol substrates to give lactones catalyzed by 62-iPr.94 In contrast to the conditions described by Schneider and Jones.90 (toluene, reflux, 1 mol% 62-iPr), their reactions took place in tert-amyl alcohol at 150 °C with 0.5 mol% 62-iPr and 10 mol% K2CO3. The conditions were applied to 18 different aromatic and aliphatic diols which could be converted to the corresponding 5-membered lactones in good to excellent yields. Also 6-membered and 7-membered lactones can be synthesized. The scope was further extended to 9 different α,ω-amino alcohols giving the corresponding lactams in good to excellent yields.
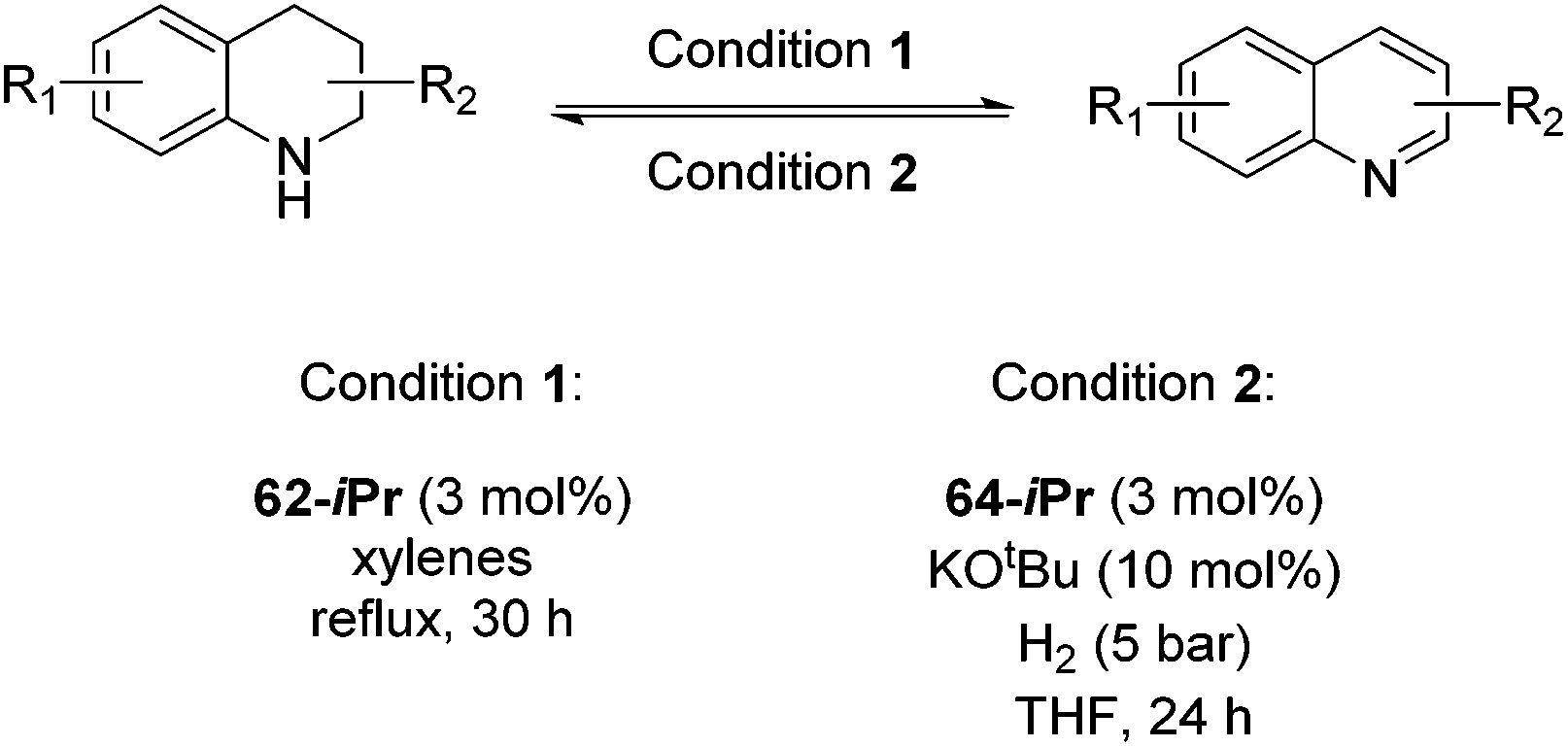
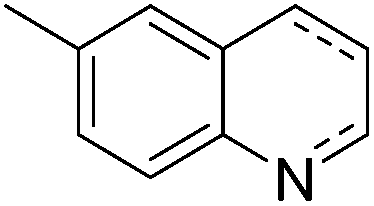
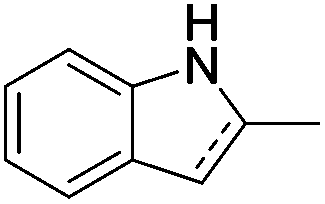
Jones and co-workers applied complexes 62-iPr, 64-iPr and 65-iPr for the acceptorless dehydrogenation of saturated N-heterocycles to their aromatic counter parts and molecular H2.88 The method was applied to a range of different 1,2,3,4-tetrahydroquinolines (Table 15, condition 1, entries 1–6). 2,6-Dimethylpiperidine and 2-methylindoline could be oxidized as well (entries 7 and 8). Partial dehydrogenation products were not observed. Remarkably, pyridine and quinoline products, which were potential ligands for iron, did not influence the reaction. It was observed that in the presence of 5 bar of H262-iPr completely lost its catalytic activity for the dehydrogenation reaction, which suggested that this complex catalyzed the reverse reaction, the hydrogenation of unsaturated heterocycles in the presence of H2. This was indeed true, and conditions were found for the hydrogenation of many such unsaturated heterocycles (Table 15, condition 2).

9. Summary and outlook
As described above, many new classes of iron pincer complexes have been recently synthesized and applied for organic synthesis. The reactivity of these complexes depends critically on the nature of the pincer ligands. The large number of successful applications underscores the advantages of pincer ligands, namely their diversity, modularity, and strong chelating ability. These examples also suggest that pincer ligands and iron catalysis are a perfect match for one another.While iron pincer complexes are often used for hydrogenation, hydrosilylation, and related dehydrogenation reactions, their applications in other reactions including epoxidation, aziridation, and C–C bond-forming reactions are emerging. Given the many benefits of iron catalysis in organic synthesis, it is clear that there is a huge amount of unexplored potential of iron pincer complexes in catalysis. Until now, enantioselective catalysis using chiral iron pincer complexes remains scarce. This state will likely change rapidly in the near future.
References
- B. Plietker, Iron Complexes in Organic Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, 2008 Search PubMed.
- B. Plietker, Iron Catalysis - Fundamentals and Applications, Springer, Berlin Heidelberg, 2011 Search PubMed.
- E. Alberico, P. Sponholz, C. Cordes, M. Nielsen, H.-J. Drexler, W. Baumann, H. Junge and M. Beller, Angew. Chem., Int. Ed., 2013, 52, 14162–14166 CrossRef CAS PubMed.
- G. Van Koten, in Organometallic Pincer Chemistry, ed. G. Van Koten and D. Milstein, Springer, Berlin Heidelberg, 2013, vol. 40 Search PubMed.
- D. Benito-Garagorri and K. A. Kirchner, Acc. Chem. Res., 2008, 41, 201–213 CrossRef CAS PubMed.
- W. V. Dahlhoff and S. M. Nelson, J. Chem. Soc. A, 1971, 2184 RSC.
- C. J. Moulton and B. L. Shaw, Dalton Trans., 1976, 1020 RSC.
- G. Van Koten, Pure Appl. Chem., 1989, 61, 1681–1694 CrossRef CAS.
- B. Rybtchinski and D. Milstein, Angew. Chem., Int. Ed., 1999, 38, 870–883 CrossRef.
- C. M. Jensen, Chem. Commun., 1999, 2443–2449 RSC.
- M. Albrecht and G. van Koten, Angew. Chem., Int. Ed., 2001, 40, 3750–3781 CrossRef CAS.
- A. Vigalok and D. Milstein, Acc. Chem. Res., 2001, 34, 798–807 CrossRef CAS PubMed.
- M. E. van der Boom and D. Milstein, Chem. Rev., 2003, 103, 1759–1792 CrossRef CAS PubMed.
- D. Milstein, Pure Appl. Chem., 2003, 75, 445–460 CrossRef CAS.
- J. T. Singleton, Tetrahedron, 2003, 59, 1837–1857 CrossRef CAS.
- B. L. Small, M. Brookhart and A. M. A. Bennett, J. Am. Chem. Soc., 1998, 120, 4049–4050 CrossRef CAS.
- B. L. Small and M. Brookhart, J. Am. Chem. Soc., 1998, 120, 7143–7144 CrossRef CAS.
- G. J. P. Britovsek, V. C. Gibson, B. S. Kimberley, P. J. Maddox, S. J. McTavish, G. A. Solan, A. J. P. White and D. J. Williams, Chem. Commun., 1998, 849–850 RSC.
- G. J. P. Britoek, M. Bruce, V. C. Gibson, B. S. Kimberley, P. J. Maddox, S. Mastroianni, S. J. McTavish, C. Redshaw, G. A. Solan, S. Stromberg, A. J. P. White and D. J. Williams, J. Am. Chem. Soc., 1999, 121, 8728–8740 CrossRef.
- P. J. Chirik, in Catalysis without Precious Metals, Wiley-VCH Verlag GmbH & Co. KGaA, 2010, pp. 83–110 Search PubMed.
- P. J. Chirik, in Pincer and Pincer-Type Complexes, Wiley-VCH Verlag GmbH & Co. KGaA, 2014, pp. 189–212 Search PubMed.
- P. Giannoccaro, G. Vasapollo, C. F. Nobile and A. Sacco, Inorg. Chim. Acta, 1982, 61, 69–75 CrossRef CAS.
- J. Zhang, M. Gandelman, D. Herrman, G. Leitus, L. J. W. Shimon, Y. Ben-David and D. Milstein, Inorg. Chim. Acta, 2006, 359, 1955–1960 CrossRef CAS.
- R. J. Trovitch, E. Lobkovsky and P. J. Chirik, Inorg. Chem., 2006, 45, 7252–7260 CrossRef CAS PubMed.
- E. M. Pelczar, T. J. Emge, K. Krogh-Jespersen and A. S. Goldman, Organometallics, 2008, 27, 5759–5767 CrossRef CAS.
- R. Langer, G. Leitus, Y. Ben-David and D. Milstein, Angew. Chem., Int. Ed., 2011, 50, 2120–2124 CrossRef CAS PubMed.
- R. Langer, Y. Diskin-Posner, G. Leitus, L. J. W. Shimon, Y. Ben-David and D. Milstein, Angew. Chem., Int. Ed., 2011, 50, 9948–9952 CrossRef CAS PubMed . The same group also reported a PNP–Fe complex with a new pyrazine-based pincer ligand and applied it for the hydrogenation of CO2, see: R. Langer, Y. Diskin-Posner, G. Leitus, L. J. W. Shimon, Y. Ben-David and D. Milstein, Inorg. Chem., 2015, 54, 4526 CrossRef PubMed.
- R. Langer, M. A. Iron, L. Konstantinovski, Y. Diskin-Posner, G. Leitus, Y. Ben-David and D. Milstein, Chem. – Eur. J., 2012, 18, 7196–7209 CrossRef CAS PubMed.
- T. Zell, B. Butschke, Y. Ben-David and D. Milstein, Chem. – Eur. J., 2013, 19, 8068–8072 CrossRef CAS PubMed.
- T. Zell, Y. Ben-David and D. Milstein, Angew. Chem., Int. Ed., 2014, 53, 4685–4689 CrossRef CAS PubMed.
- T. Zell, Y. Ben-David and D. Milstein, Catal. Sci. Technol., 2015, 5, 822–826 CAS.
- D. Benito-Garagorri, E. Becker, J. Wiedermann, W. Lackner, M. Pollak, K. Mereiter, J. Kisala and K. A. Kirchner, Organometallics, 2006, 25, 1900–1913 CrossRef CAS.
- D. Benito-Garagorri, J. Wiedermann, M. Pollak, K. Mereiter and K. A. Kirchner, Organometallics, 2007, 26, 217–222 CrossRef CAS.
- D. Benito-Garagorri, L. G. Alves, M. Puchberger, K. Mereiter, L. F. Veiros, M. J. Calhorda, M. D. Carvalho, L. P. Ferreira, M. Godinho and K. A. Kirchner, Organometallics, 2009, 28, 6902–6914 CrossRef CAS.
- D. Benito-Garagorri, L. G. Alves, L. F. Veiros, C. M. Standfest-Hauser, S. Tanaka, K. Mereiter and K. A. Kirchner, Organometallics, 2010, 29, 4932–4942 CrossRef CAS.
- B. Bichler, C. Holzhacker, B. Stöger, M. Puchberger, L. F. Veiros and K. A. Kirchner, Organometallics, 2013, 32, 4114–4121 CrossRef CAS PubMed.
- B. Bichler, M. Glatz, B. Stöger, K. Mereiter, L. F. Veiros and K. A. Kirchner, Dalton Trans., 2014, 43, 14517–14519 RSC.
- M. Glatz, B. Bichler, M. Mastalir, B. Stöger, M. Weil, K. Mereiter, E. Pittenauer, G. Allmaier, L. F. Veiros and K. A. Kirchner, Dalton Trans., 2015, 44, 281–294 RSC.
- N. Gorgas, B. Stöger, L. F. Veiros, E. Pittenauer, G. Allmaier and K. A. Kirchner, Organometallics, 2014, 33, 6905–6914 CrossRef CAS.
- W. S. DeRieux, A. Wong and Y. Schrodi, J. Organomet. Chem., 2014, 772, 60–67 CrossRef PubMed.
- S. Mazza, R. Scopelliti and X. Hu, Organometallics, 2015, 34, 1538–1545 CrossRef CAS.
- T. Zell and D. Milstein, Acc. Chem. Res., 2015, 48, 1979–1994 CrossRef CAS PubMed.
- C. R. Holmquist and E. J. Roskamp, J. Org. Chem., 1989, 54, 3258–3260 CrossRef CAS.
- S. J. Mahmood and M. M. Hossain, J. Org. Chem., 1998, 63, 3333–3336 CrossRef CAS.
- M. Redlich, S. J. Mahmood, M. F. Mayer and M. M. Hossain, Synth. Commun., 2000, 30, 1401–1411 CrossRef CAS.
- L. G. Alves, G. Dazinger, L. F. Veiros and K. A. Kirchner, Eur. J. Inorg. Chem., 2010, 493160–493166 Search PubMed.
- G. Bauer and K. A. Kirchner, Angew. Chem., Int. Ed., 2011, 50, 5798–5800 CrossRef CAS PubMed.
- M. Albrecht, Chem. Rev., 2010, 110, 576–623 CrossRef CAS PubMed.
- P. Bhattacharya and H. R. Guan, Comments Inorg. Chem., 2011, 32, 88–112 CrossRef CAS.
- A. A. Kulkarni and O. Daugulis, Synthesis, 2009, 4087–4109 CAS.
- C. S. Creaser and W. C. Kaska, Inorg. Chim. Acta, 1978, 30, L325–L326 CrossRef CAS.
- P. Bhattacharya, J. A. Krause and H. R. Guan, Organometallics, 2011, 30, 4720–4729 CrossRef CAS.
- S. Chakraborty, P. Bhattacharya, H. Dai and H. Guan, Acc. Chem. Res., 2015, 48, 1995–2003 CrossRef CAS PubMed.
- W. Wang, P. Gu, Y. Wang and H. Wei, Organometallics, 2014, 33, 847–857 CrossRef CAS.
- P. Bhattacharya, J. A. Krause and H. R. Guan, J. Am. Chem. Soc., 2014, 136, 11153–11161 CrossRef CAS PubMed.
- P. Bhattacharya, J. A. Krause and H. R. Guan, Organometallics, 2014, 33, 6113–6121 CrossRef CAS.
- D. F. Lu, C. L. Zhu, Z. X. Jia and H. Xu, J. Am. Chem. Soc., 2014, 136, 13186–13189 CrossRef CAS PubMed.
- H. Usuda, A. Kuramochi, M. Kanai and M. Shibasaki, Org. Lett., 2004, 6, 4387–4390 CrossRef CAS PubMed.
- M. Kawatsura, K. Kajita, S. Hayase and T. Itoh, Synlett, 2010, 1243–1246 CrossRef CAS.
- M. Nakanishi, A. F. Salit and C. Bolm, Adv. Synth. Catal., 2008, 350, 1835–1840 CrossRef CAS.
- A. Sharma and J. F. Hartwig, Nature, 2015, 517, 600–604 CrossRef CAS PubMed.
- J. Jankowska, J. Paradowska and J. Mlynarski, Tetrahedron Lett., 2006, 47, 5281–5284 CrossRef CAS.
- J. Jankowska, J. Paradowska, B. Rakiel and J. Mlynarski, J. Org. Chem., 2007, 72, 2228–2231 CrossRef CAS PubMed.
- M. Kawatsura, Y. Komatsu, M. Yamamoto, S. Hayase and T. Itoh, Tetrahedron Lett., 2007, 48, 6480–6482 CrossRef CAS.
- M. Kawatsura, Y. Komatsu, M. Yamamoto, S. Hayase and T. Itoh, Tetrahedron, 2008, 64, 3488–3493 CrossRef CAS.
- J. Wang, M. Frings and C. Bolm, Chem. – Eur. J., 2014, 20, 966–969 CrossRef CAS PubMed.
- H. Nishiyama and A. Furuta, Chem. Commun., 2007, 760–762 RSC.
- T. Chen, L. M. Yang, D. R. Gong and K. W. Huang, Inorg. Chim. Acta, 2014, 423, 320–325 CrossRef CAS.
- Y. Imanishi and K. Nomura, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 4613–4626 CrossRef CAS.
- K. Nomura, W. Sidokmai and Y. Imanishi, Bull. Chem. Soc. Jpn., 2000, 73, 599–605 CrossRef CAS.
- A. M. Tondreau, J. M. Darmon, B. M. Wile, S. K. Floyd, E. Lobkovsky and P. J. Chirik, Organometallics, 2009, 28, 3928–3940 CrossRef CAS.
- S. Hosokawa, J. Ito and H. Nishiyama, Organometallics, 2010, 29, 5773–5775 CrossRef CAS.
- R. B. Bedford, M. Betham, D. W. Bruce, A. A. Danopoulos, R. M. Frost and M. Hird, J. Org. Chem., 2006, 71, 1104–1110 CrossRef CAS PubMed.
- R. P. Yu, J. M. Darmon, J. M. Hoyt, G. W. Margulieux, Z. R. Turner and P. J. Chirik, ACS Catal., 2012, 2, 1760–1764 CrossRef CAS PubMed.
- R. J. Trovitch, E. Lobkovsky, M. W. Bouwkamp and P. J. Chirik, Organometallics, 2008, 27, 6264–6278 CrossRef CAS.
- D. Srimani, Y. Diskin-Posner, Y. Ben-David and D. Milstein, Angew. Chem., Int. Ed., 2013, 52, 14131–14134 CrossRef CAS PubMed.
- T. Niwa and M. Nakada, J. Am. Chem. Soc., 2012, 134, 13538–13541 CrossRef CAS PubMed.
- T. Inagaki, L. T. Phong, A. Furuta, J. Ito and H. Nishiyama, Chem. – Eur. J., 2010, 16, 3090–3096 CrossRef CAS PubMed.
- T. Inagaki, A. Ito, J. Ito and H. Nishiyama, Angew. Chem., Int. Ed., 2010, 49, 9384–9387 CrossRef CAS PubMed.
- G. Bauer, C. W. Cheung and X. Hu, Synthesis, 2015, 1726–1732 CAS.
- G. Bauer, M. D. Wodrich, R. Scopelliti and X. Hu, Organometallics, 2015, 34, 289–298 CrossRef CAS.
- Q.-H. Deng, T. Bleith, H. Wadepohl and L. H. Gade, J. Am. Chem. Soc., 2013, 135, 5356–5359 CrossRef CAS PubMed.
- T. Bleith, H. Wadepohl and L. H. Gade, J. Am. Chem. Soc., 2015, 137, 2456–2459 CrossRef CAS PubMed.
- W. Kuriyama, T. Matsumoto, O. Ogata, Y. Ino, K. Aoki, S. Tanaka, K. Ishida, T. Kobayashi, N. Sayo and T. Saito, Org. Process Res. Dev., 2012, 16, 166–171 CrossRef CAS.
- I. Köhne, T. J. Schmeier, E. A. Bielinski, C. J. Pan, P. O. Lagaditis, W. H. Bernskoetter, M. K. Takase, C. Würtele, N. Hazari and S. Schneider, Inorg. Chem., 2014, 53, 2133–2143 CrossRef PubMed.
- S. Chakraborty, H. Dai, P. Bhattacharya, N. T. Fairweather, M. S. Gibson, J. A. Krause and H. Guan, J. Am. Chem. Soc., 2014, 136, 7869–7872 CrossRef CAS PubMed.
- S. Werkmeister, K. Junge, B. Wendt, E. Alberico, H. Jiao, W. Baumann, H. Junge, F. Gallou and M. Beller, Angew. Chem., Int. Ed., 2014, 53, 8722–8726 CrossRef CAS PubMed.
- S. Chakraborty, W. W. Brennessel and W. D. Jones, J. Am. Chem. Soc., 2014, 136, 8564–8567 CrossRef CAS PubMed.
- E. A. Bielinski, P. O. Lagaditis, Y. Zhang, B. Q. Mercado, C. Würtele, W. H. Bernskoetter, N. Hazari and S. Schneider, J. Am. Chem. Soc., 2014, 136, 10234–10237 CrossRef CAS PubMed.
- S. Chakraborty, P. O. Lagaditis, M. Förster, E. A. Bielinski, N. Hazari, M. C. Holthausen, W. D. Jones and S. Schneider, ACS Catal., 2014, 4, 3994–4003 CrossRef CAS.
- C. Bornschein, S. Werkmeister, B. Wendt, H. Jiao, E. Alberico, W. Baumann, H. Junge, K. Junge and M. Beller, Nat. Commun., 2014, 5, 4111 CAS.
- N. T. Fairweather, M. S. Gibson and H. Guan, Organometallics, 2015, 34, 335–339 CrossRef CAS.
- P. J. Bonitatibus Jr., S. Chakraborty, M. D. Doherty, O. Siclovan, W. D. Jones and G. L. Soloveichik, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 1687–1692 CrossRef PubMed.
- M. Pena-Lopez, H. Neumann and M. Beller, ChemCatChem, 2015, 7, 865–871 CrossRef CAS.
- E. A. Bielinski, M. Forster, Y. Zhang, W. H. Bernskoetter, N. Hazari and M. C. Holthausen, ACS Catal., 2015, 5, 2404–2415 CrossRef CAS.
| This journal is © the Partner Organisations 2016 |