
Recent advances in iron-catalysed cross coupling reactions and their mechanistic underpinning
T. L.
Mako
and
J. A.
Byers
*
Department of Chemistry, Boston College, Chestnut Hill, Massachusetts, USA. E-mail: jeffery.byers@bc.edu
First published on 18th February 2016
Abstract
Advances in iron-catalysed cross coupling from 2010–2015 are critically reviewed. In addition to a description of the systems that have emerged since 2010, the significant mechanistic work carried out to understand the mechanisms of these transformations will be discussed. An emphasis will be placed on mechanistic studies that utilize in situ reaction monitoring and the tools used to achieve this goal.
 J. A. Byers and T. L. Mako | Teresa L. Mako earned her undergraduate degree at the University of Rhode Island in Chemistry and Forensic Chemistry, graduating Summa Cum Laude in 2013. Under the direction of Prof. Mindy Levine, Mako studied cyclodextrin-mediated energy transfer from polycyclic aromatic hydrocarbons to squaraine fluorphores. She is now pursuing a doctoral degree as a member of the Byers group at Boston College where her research focuses on the study of iron catalyzed coupling protocols as well as the development of new ligand systems to promote iron catalysis. Jeffery A. Byers attended Washington University in St Louis, where he received his B.A. in Chemistry in 2000. In 2007, Jeff obtained his Ph. D. from the California Institute of Technology under the guidance of Prof. John E. Bercaw. After a postdoctoral position in the laboratory of Prof. Timothy F. Jamison at the Massachusetts Institute of Technology, Jeff started his independent career at Boston College in the summer of 2011, where he is currently an Assistant Professor of Chemistry. The research carried out in the Byers group centers around fundamental transition metal chemistry and its application in chemical catalysis pertaining to the synthesis of new biodegradable polymers, energy storage, and cross coupling reactions. |
Introduction
Organic chemists have utilized metal-catalysed cross coupling reactions to assemble many natural products, pharmaceutically active compounds, and novel polymeric materials from relatively simple starting materials. To recognize these powerful transformations, the Nobel Prize in Chemistry was awarded to Richard Heck, Ei-ichi Negishi, and Akira Suzuki in 2010. Although these methods have been enormously useful in synthetic organic chemistry and materials science, most methods involve the use of palladium- or nickel-based catalysts, which are toxic, expensive, or both. Moreover, most cross coupling reactions have largely been relegated to substrates containing sp2- or sp-hybridized carbons. Reactions involving sp3-hybridized substrates are uncommon, although some excellent progress has been made recently.1–3 These drawbacks have limited the use of cross coupling reactions on scale for the synthesis of many pharmaceutically active agents and in general for their use in the agrochemical industry.In this review article, cross coupling reactions will be discussed that utilize catalysts based on the earth abundant and inexpensive metal iron. In addition to the environmental and economic advantages, the possibility for one or two electron redox events provides access to different classes of substrates compared to the most commonly used palladium catalysts. Nevertheless, since the first report of an iron-based catalyst for cross coupling appeared in 1971 from Kochi and co-workers,4 the progress towards a practical and general protocol for iron-based cross coupling catalysis has not been as rapid as for the development of analogous palladium and nickel-based catalysis. In part, this progress has been stymied by a lack of mechanistic understanding for these reactions. However, in recent years, the area of iron-based cross coupling catalysis has enjoyed a renaissance that is due in part to the emergence of detailed mechanistic studies aimed towards understanding these processes.
This review is divided into two parts. The first is dedicated towards establishing the state-of-the-art in iron catalysed cross coupling reactions by recounting the progress made in this field since 2010. While significant contributions have certainly been made prior to 2010, we direct the interested readers to the several excellent reviews that have described this chemistry elsewhere.3,5–8 For organizational purposes, this part of the review is arranged by the type of cross coupling reaction using names commonly referred to in the palladium cross coupling literature. In the second part of the review, we describe several key mechanistic features of the most thoroughly studied catalytic systems. A challenging aspect of iron-catalysed cross coupling reactions is that there is not one unified mechanistic manifold that is sufficient to describe all cross coupling reactions. Some of the studies reviewed here were carried out prior to 2010, but they are presented because they are the most thoroughly studied systems and they are illustrative of the mechanistic complexity that exists within the field. It is not our intent to exhaustively cover all mechanistic proposals, but rather to recount the best studied systems so as to highlight the key experiments and physical techniques that have been used.
Kumada–Tamao–Corriu
By far the types of cross coupling reactions that are most commonly catalysed by iron are reactions involving Grignard reagents as the transmetalating nucleophile. Effective methods have been developed for the cross coupling of aryl, alkyl, and alkynyl Grignard reagents with aryl, vinyl, and alkyl halides. These reactions are organized by the type of nucleophilic Grignard reagent used followed by the corresponding electrophile.Aryl Grignard
In recent years, few examples of aryl–aryl coupling have surfaced. Ligand design is a prominent aspect of a report published by Duong and co-workers12 that was inspired by work carried out by Nakamura9 wherein an iron(III) trifluoride catalyst supported by an N-heterocyclic carbene (NHC) formed from the imidazolinium salt N,N′-(2,6-diisopropylphenyl)dihydroimidazolium chloride 1, was utilized for aryl–aryl cross coupling. The authors proposed that, like fluoride ligands, alkoxide ligands can enhance selectivity by inhibiting the formation of the ferrate complex that leads to the undesired homocoupled product. Unlike fluoride ligands, however, alkoxide ligands are strong π-donors that can be modified to fine tune the iron centre. In fact, the authors find that aryl–aryl coupling is promoted by their ligand system utilizing a wide variety of NHC ligands. Aryl chlorides were shown to couple readily, however aryl bromides and iodides were not explored. Electron rich and electron deficient substrates were tolerated as well as sterically demanding aryl chlorides.
Also similar to the aforementioned work by Nakamura and co-workers,9 coupling of aryl pseudohalides (tosylates and sulfamates) with aryl Grignard reagents was shown by Cook and co-workers in the presence of FeF3 and 2, an NHC precursor, N,N′-(2,6-diisopropylphenyl) imidazolium chloride, which resulted in moderate yields of desired product (Scheme 1b).13ortho Substitution, as well as strongly electron donating and withdrawing groups of both coupling partners led to a decrease in yield. An example of vinyl–aryl cross coupling using a vinyl substrate in conjugation with an aromatic system was also shown. Coupling of vinyl tosylate 3 led to a majority of desired product with a prominent vinyl homocouple side product (Scheme 1c).
 | ||
| Scheme 1 (a) The coupling of aryl and heteroaryl chlorides with aryl Grignard reagents in the presence of catalytic amounts of Fe(OtBu)3 and imidazolinium salt 1.12 (b) The coupling of aryl tosylates and sulfamates with aryl Grignard reagents in the presence of a catalytic amount of FeF3 and imidazolium salt 2.13 (c) Coupling of conjugated vinyl tosylate 3.13 | ||
Iron-catalysed aryl–aryl cross coupling reactions have been particularly successful when N-heteroaromatic halide electrophiles are used as the substrate. For example, Chen and co-workers were able to produce desired biaryl coupling products in moderate yields when catalysed by a dicationic iron(II) supported by a bidentate pyrimidine NHC ancillary ligand.14 Another instance of N-heteroaromatic halide coupling was published by the Knochel group in which iron halide salts and careful solvent selection were enough to produce the desired product in excellent yields (Scheme 2).15 They were able to enhance reaction performance by replacing the traditional Grignard reagent with a lithium–chloride adduct.
 | ||
| Scheme 2 The coupling of lithiated aryl Grignard reagents with N-heteroaromatic halides in the presence of catalytic amounts of FeBr3.15 | ||
Knochel and co-workers also discovered that an isoquinoline additive accelerates the coupling of N-heteroaromatic halides with aryl Grignard reagents via ligation to the iron tribromide salt.16 Without the additive, 40% yield of the desired product was formed from the coupling of 2-chloropyridine with phenyl magnesium chloride. In the presence of isoquinoline, 92% of the product was formed. This work was expanded to include the cross coupling of heteroaromatic electrophiles with heteroaromatic nucleophiles.17
 | ||
| Scheme 3 Stereoselective coupling of vinyl chlorides with phenyl magnesium chloride in the presence of catalytic amount of Fe(acac)3 and with NMP as an additive.19 | ||
 | ||
| Scheme 4 (a) FeCl2(dppbz)2 and (b) FeCl2(SciOPP) complexes are commonly used to catalyse Kumada couplings. (c) The use of FeCl2(dppbz)2 complex 4 to facilitate the coupling of aryl Grignard reagents with benzyl chlorides.21 | ||
As previously mentioned, ligand design is a prominent aspect of many current efforts in iron-catalysed cross coupling reactions. Readily available iron precursors, such as Fe(acac)3 or FeCl3, are commonly active as catalysts for cross coupling, but improvements in the system via ligand design are still desirable. In 2011, Yamaguchi, Asami, and co-workers used a β-aminoketonato ligand as a nitrogen-containing analogue of the acetylacetonato (acac) ligand for the purpose of better stabilizing coordinatively unsaturated metal centres (Scheme 5).22 The use of iron(III) dichloride complex 5 allowed for the coupling of aryl Grignard reagents with cyclohexyl bromide in good yields, albeit without tolerating sterically hindered Grignard reagents. The substrate scope was not extensively explored, but cyclohexyl iodide was an equally competent electrophile as cyclohexyl bromide, and cyclohexyl chloride was inferior. An acyclic secondary alkyl bromide was less productive than its cyclic analogue, while a primary alkyl bromide led to a moderate yield of coupled product. The tertiary alkyl bromide, t-butyl bromide, resulted in no desired product.
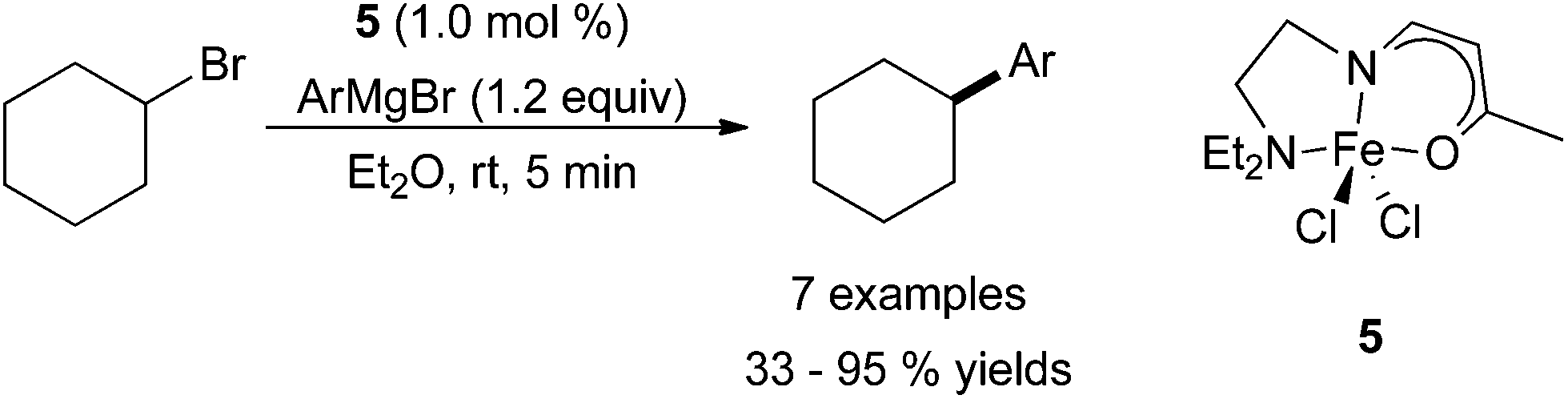 | ||
| Scheme 5 The coupling of aryl Grignard reagents with cyclohexyl bromide in the presence of β-aminoketonato iron dichloride complex 5.22 | ||
Denmark and Cresswell developed a method to effect the cross coupling of aryl Grignard reagents with alkyl aryl-sulfones or alkyl aryl-thioethers (Scheme 6).23 Optimal conditions were revealed to be 20% Fe(acac)3, 3 equivalents of a Grignard reagent, and 8 equivalents of TMEDA additive, which afforded moderate yields of cross coupled product. The reaction was limited to electron rich aromatic Grignard reagents that were void of heteroatoms and was completely inactive for alkyl, alkenyl, and alkynyl Grignard reagents. Aryl-sulfone electrophiles were much more active in this system than aryl-thioethers, and the aryl group of the thioether was found to require a nitrogen atom in the aromatic system to afford the highest yields. While secondary alkyl electrophiles were active under these conditions, primary and tertiary alkyl electrophiles were not examined. The Bedford group have subsequently suggested that the role of TMEDA in similar reactions is to sequester iron so as to make it unreactive to unproductive side reactions rather than to act as an ancillary ligand during cross coupling as speculated by Denmark and Cresswell.24
 | ||
Scheme 6 The coupling of aryl Grignard regents with secondary alkyl aryl-sulfones and aryl-thioethers in the presence of a catalytic amount of Fe(acac)3 and a stoichiometric excess of TMEDA. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Coupled with 2-OiPrPhMgBr. bCoupled with PhMgBr.23 Coupled with 2-OiPrPhMgBr. bCoupled with PhMgBr.23 | ||
Nakamura and co-workers discovered systems that were capable of carrying out the cross coupling of primary and secondary alkyl chlorides in the presence of either the well-defined iron dichloride SciOPP catalyst, 6, or a complex generated in situ from imidazolium salt 2 and FeCl3. Utilizing SciOPP ligands, primary and secondary alkyl halides underwent cross coupling with aryl magnesium halides resulting in good yields of the desired product (Scheme 7a).25 These studies demonstrated that the SciOPP ligands are superior to dppbz ligands for this system, and that an additional equivalent of ligand relative to the catalyst was required for maximum yield. Using imidazolium salt 2 (Scheme 7b), moderate to good yields were seen with primary and secondary chlorides.26 Sterically hindered Grignard reagents, such as mesityl magnesium bromide, led to trace amounts of desired product, but electron withdrawing fluoro- and electron donating methoxy-substituents were well tolerated. Impressively, the tertiary alkyl chloride, adamantyl chloride, underwent cross coupling with both systems reported by Nakumura in 81% and 90% yields for the SciOPP and NHC supported systems, respectively. Unfortunately, the reaction was not universal for coupling tertiary alkyl chlorides as the cross coupling of phenyl magnesium bromide and t-butyl chloride led to 12% yield of the cross coupling product for the iron-NHC catalysed system with remainder of the alkyl halide presumably being converted to either isobutene or isobutane. In general, coupling of tertiary alkyl halides is challenging and an effective and versatile method for coupling this important class of substrates still requires development.
 | ||
| Scheme 7 (a) The coupling of alkyl halides and aryl Grignard reagents in the presence of an FeCl2(SciOPP) catalyst 6 and additional ligand, requiring the slow addition of the Grignard reagent.25 (b) A similar system to a in which imidazolium salt 2 forms the active species in situ with FeCl3.26 | ||
Similar to what is seen in palladium and nickel-catalysed cross coupling reactions,3 some iron-catalysed cross coupling reactions favour alkyl iodides or alkyl bromides compared to alkyl chlorides; alkyl fluorides are usually unreactive. However, in 2012, Deng and co-workers developed conditions for the successful cross coupling of primary alkyl fluorides with aryl magnesium bromides in the presence of an NHC ligand (Scheme 8).27 In order to accomplish this challenging transformation, a dinuclear iron complex, 7, was used as the pre-catalyst. Although effective for the cross coupling of primary alkyl fluorides, secondary and tertiary alkyl fluorides and aryl fluorides were found to be inactive. Nevertheless, this report is promising for the development of iron-catalysed cross coupling reactions involving alkyl fluorides.
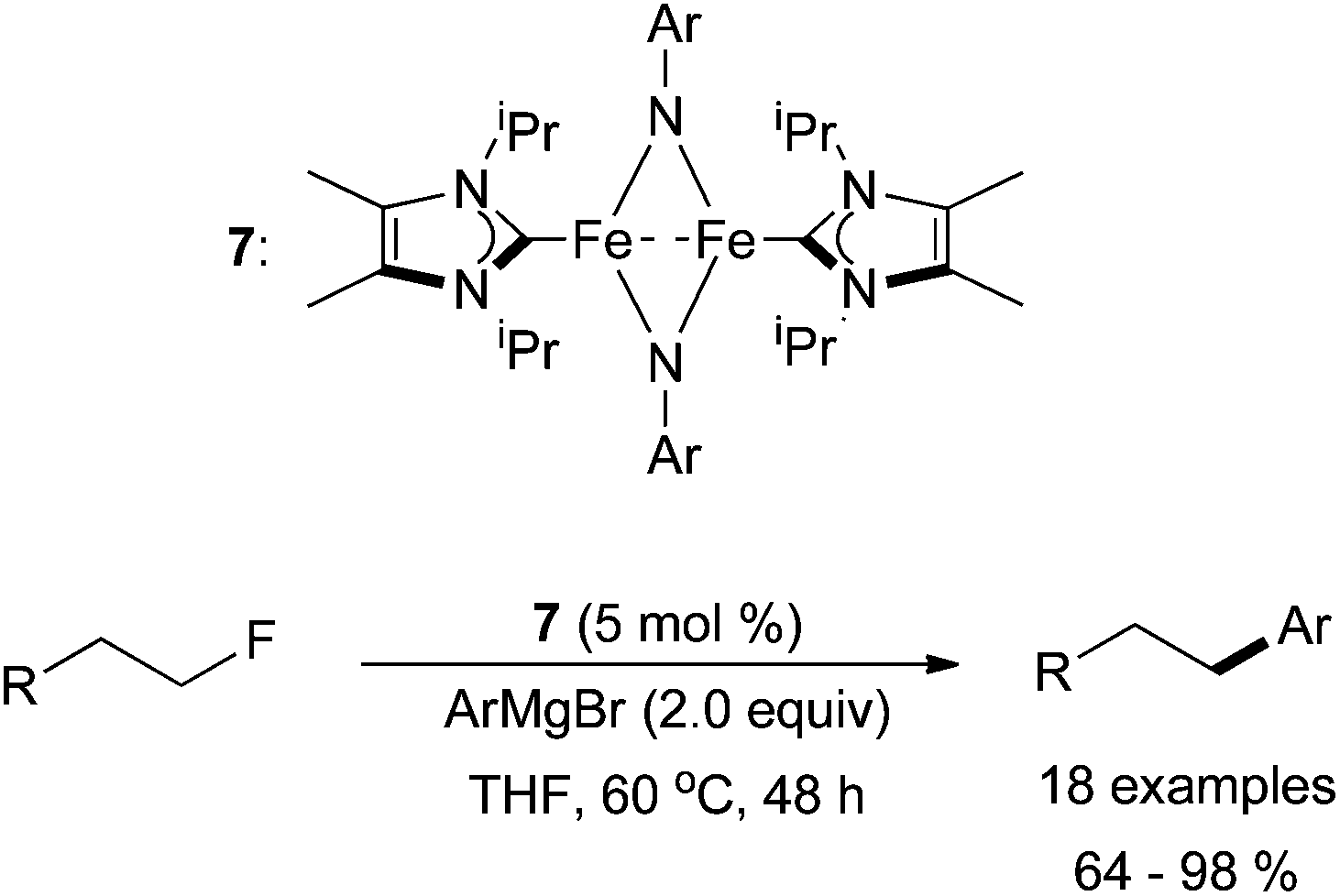 | ||
| Scheme 8 The coupling of primary alkyl fluorides with aryl magnesium bromide reagents catalysed by the dinuclear complex 7.27 | ||
Fürstner and co-workers have utilized stoichiometric reactions to investigate the tendency for sterically encumbered aryl Grignard reagents to be inactive in cross coupling reactions.28 Using iron dichloride complex 8, the group discovered that a bisarylated bisphosphine iron catalyst 9 is generated in situ upon reaction with an aryl Grignard reagent (Scheme 9a). This proceeded without the production of [Mes3Fe]−, which the Neidig group has shown leads to unproductive side reactions such as homocoupling of the Grignard reagent.29 Using this knowledge, the group was able to develop a protocol to couple a variety of sterically hindered aryl Grignard reagents with primary alkyl bromides and iodides (Scheme 9b). Sterically demanding neopentyl iodide electrophiles undergo the cross coupling reaction to give good yields of cross coupling products, but employing secondary alkyl halides led to elimination rather than cross coupling. Alkyl iodides performed better than bromide, and alkyl chlorides were not reported. The use of alkyl tosylates required an iodide source to convert the tosylate into an alkyl iodide in situ. The Fürstner group demonstrated the usefulness of this method for the synthesis of 10, a useful precursor to the angular tricyclic core of the silphiperfolene family of natural products, such as 11, via an elegant intramolecular arene–alkene meta-photocycloaddition reaction (Scheme 9c).
 | ||
| Scheme 9 (a) Supposed active species bis(mesityl) iron bis(phosphine) 9 is formed from treatment of an iron dichloride bis(diphosphine) complex 8. (b) Coupling of primary alkyl halides with aryl Grignard reagents in the presence of 8. (c) Abridged total synthesis of sliphiperfol-6-ene, 11, using the developed reactivity to form the crucial precursor 10.28 | ||
An interesting report by Rueping and co-workers utilized iron-catalysed cross coupling to functionalize azetidines, a common functionality in pharmaceutically active molecules (Scheme 10).30 Using Fe(acac)3, protected 3-iodiazetidines were coupled with aryl, vinyl, and methyl magnesium bromides in moderate yields.
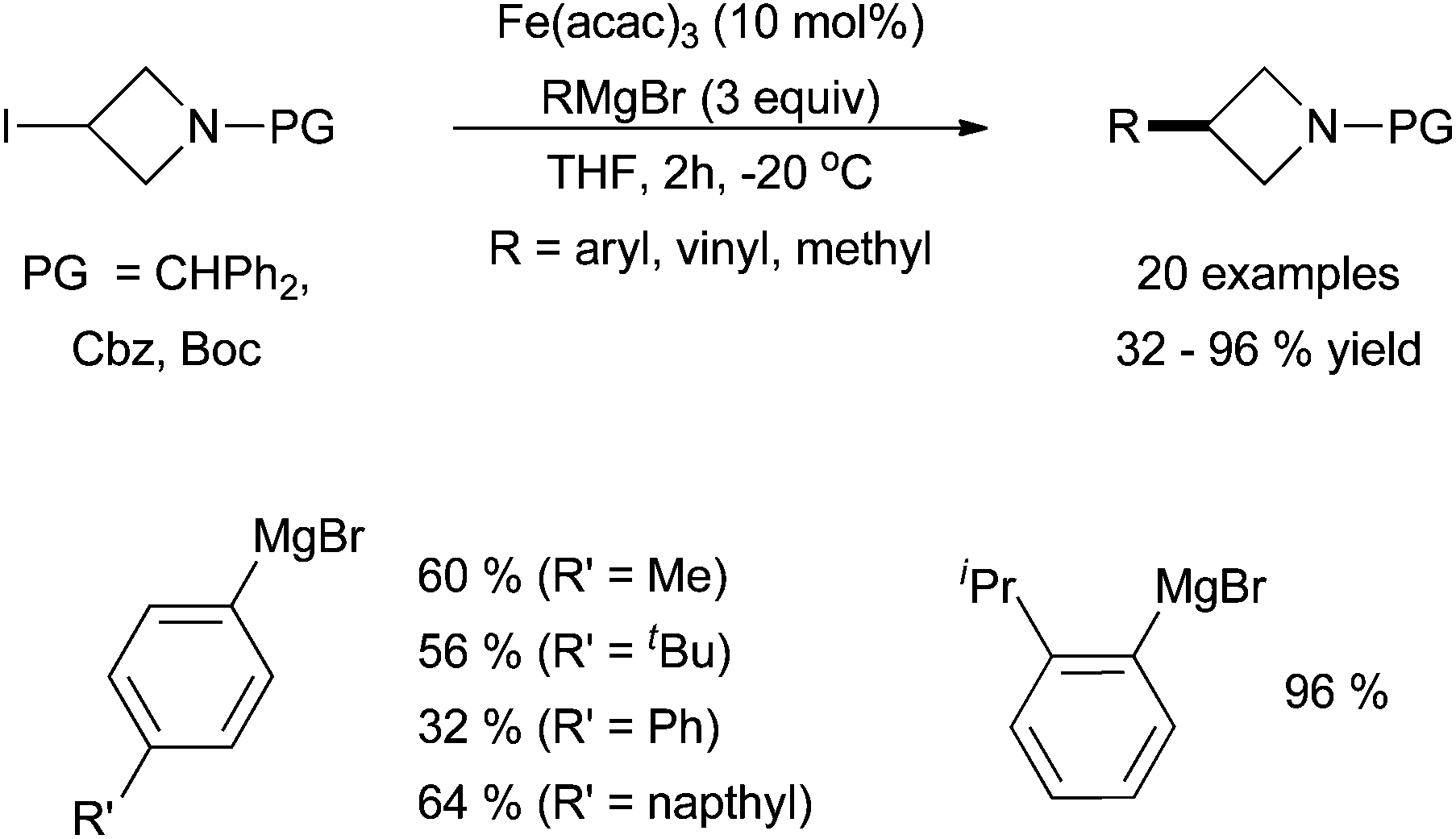 | ||
| Scheme 10 The coupling of 3-iodiazetidines with aryl, vinyl, and methyl Grignard reagents in the presence of catalytic amounts of Fe(acac)3.30 | ||
Jacobi von Wangelin and co-workers have found that the cross coupling of aryl halides with alkyl halides proceeds in the presence of Mg0, TMEDA and FeCl3.31 In this system, the identity of the Grignard reagent generated in situ is unclear, since the alkyl and aryl halides are exposed to magnesium simultaneously. Moderate yields were observed for this system, but iron-based catalysts were superior to other metal catalysts examined. By switching the metal salt to NiCl2, the yields decreased by half, and when Cu, Pd, Mn, or Cr were used, only trace amounts of desired product were seen.
Enantioselective cross coupling reactions catalysed by iron are very rare. In a preliminary communication, the Nakamura group had discovered that α-bromoesters undergo cross coupling with aryl Grignard reagents to yield racemic products using Fe(acac)3 as the catalyst.32 Later, they revealed that chiral bisphosphine ligand 12 and Fe(acac)3 could be used to carry out the stereoconvergent coupling of α-chloroesters (13) with aryl Grignard reagents to obtain good yields of the cross coupled product in moderate stereoselectivities (Scheme 11).33 Although only moderate enantioselectivity was achieved, the authors were able to enhance the optical purity of the products through ester hydrolysis and subsequent crystallization with octylamine (Scheme 11b). The reaction tolerated aryl Grignard reagents that contained electron donating and electron withdrawing groups, although strongly electron withdrawing Grignard reagents such as 3,4,5-trifluorophenylmagnesium bromide led to significantly lower yields. Moreover, sterically constrained ortho-substituted Grignard reagents were not tolerated except when the ortho-substitution was an extended aromatic system. With respect to the alkyl halide, it was found that substrates with bulky esters (R′ = tBu, theptyl) were superior to maximize yields. Nickel and palladium metal precursors more commonly used as catalysts for cross coupling reactions did not produce the desired product under the reaction conditions shown in Scheme 11a.
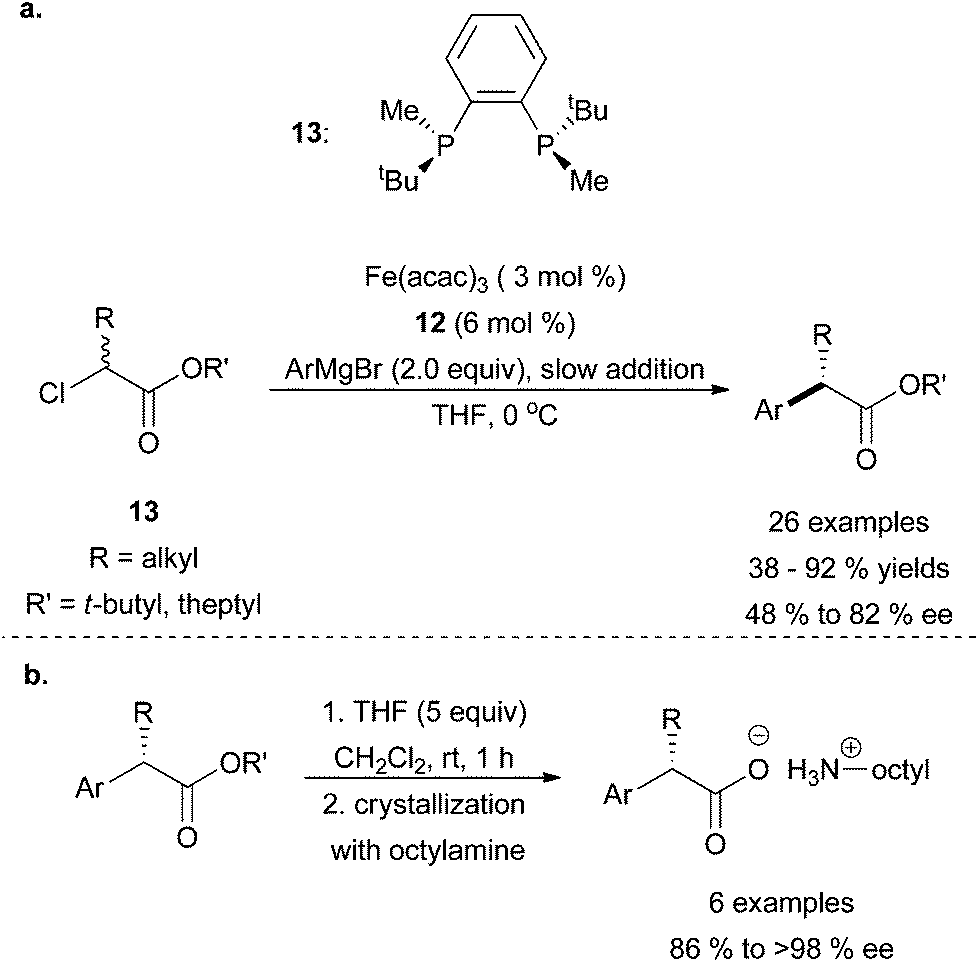 | ||
| Scheme 11 (a) The stereoconvergent coupling of α-chloroesters (13) with aryl Grignard reagents in the presence of catalytic amounts of Fe(acac)3 and chiral ligand 12. (b) Crystallization of coupled product with octylamine to enhance % ee of the compounds.33 | ||
Hu and co-workers presented the use of iron pincer complex 14 in an attempt to afford enantiomerically enriched products (Scheme 12).34 However, the highest enantiomeric excess reached was only 19% for prochiral substrates, and the highest diastereomeric ratio was 66:34 when chiral substrates were used. The coupling procedure was shown to be very general, successfully coupling primary and secondary alkyl bromides and iodides, and tolerant of ester, ether, and ketone functionalities. Late stage cross-coupling of compounds derived from natural products was also shown to be effective. Unfortunately, the scope of the Grignard reagent was not examined. Hu also used a similar catalyst for extensive mechanistic studies,35 which will be discussed below.
 | ||
| Scheme 12 The coupling of primary and secondary alkyl halides with aryl Grignard reagents in the presence of complex 14.34 | ||
In 2011, Knochel and co-workers published a diastereoselective coupling of enantiomerically enriched cyclic aryl iodides achieved by using bulky protecting groups positioned adjacent to the coupling site.36 Although catalytic amounts of iron dichloride provided good diastereoselectivities, high yields were only obtained in the presence of stoichiometric amounts of iron and 4-fluorostyrene as an additive, which was present to encourage reductive elimination.
Alkyl Grignard
Perry and co-workers have reported the use of imidazolium salt 2 to promote good yields for the cross coupling of aryl chlorides with both primary and secondary alkyl Grignard reagents when the Grignard reagent was present in excess (Scheme 13a).37 As this research focused on the cross coupling of unactivated aryl chlorides, only moderate substitution of the electrophile was examined. When cyclohexyl magnesium bromide was employed, moderate cross coupling yields were obtained. However, when acyclic secondary alkyl Grignard reagents were used, a mixture of branched and linear cross coupling products were observed, which resulted from chain walking that occurred as a result of β-hydride elimination and reinsertion prior to reductive elimination. The amount of chain walking could be controlled with a large excess of Grignard reagent (6 equivalents), albeit at the expense of yield.
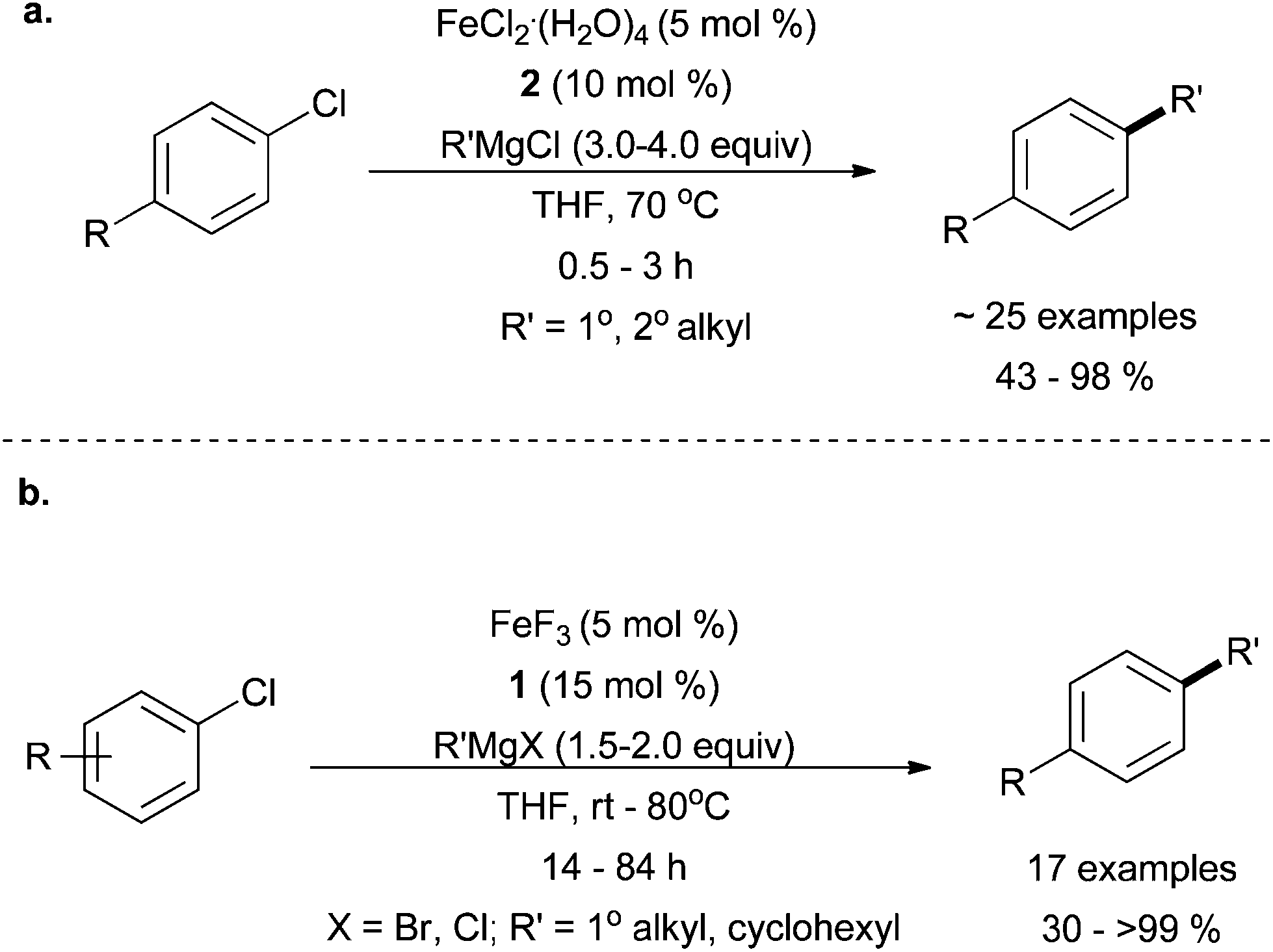 | ||
| Scheme 13 The coupling of primary and secondary alkyl Grignard reagents in the presence of (a) FeCl2·(H2O)4 and 237 and (b) FeF3 and 1.38 | ||
In a related system, Nakamura and co-workers used the imidazolinium salt 1 in combination with iron trifluoride to carry out the cross coupling of methyl, primary alkyl, and secondary alkyl Grignard reagents (with or without hydrogens that can undergo β-hydrogen elimination), with aryl halides in good yields (Scheme 13b).38 As was the case with the Perry system, chain walking was also prevalent for secondary alkyl Grignard reagents.
The groups of Z.-X. Wang and X.-C. Wang have both studied the use of aryl pseudo halides in place of traditional aryl halides. Z.-X. Wang and co-workers carried out the cross coupling of aryltrimethylammonium triflates, 15, with alkyl Grignard reagents in high yields (Scheme 14).39 The process tolerated electron rich and electron poor aryltrimethylammonium triflates and primary alkyl Grignard reagents. Acyclic secondary alkyl Grignard reagents were compatible nucleophiles, but cyclohexyl magnesium halides led to modest yields of cross coupled products. The authors do not comment on the propensity for the secondary alkyl Grignard reagent to undergo chain walking. Interestingly, the use of a well-defined ligand system detracted from the process, while addition of N-methyl pyrrolidinone (NMP) as a minor co-solvent gave the most promising results. It was found that 99.9% pure Fe(acac)3 gave similar results as 98% pure Fe(acac)3. Moreover, the addition of 0.1 mol% copper to reactions catalysed by the 99.9% pure Fe(acac)3 had no effect. These results were consistent with an iron-catalysed process rather than a copper catalysed process.
 | ||
| Scheme 14 The coupling of aryl trimethylammonium triflates (15) with alkyl Grignard reagents in the presence of Fe(acac)3 with NMP as an additive.39 | ||
X. C. Wang40 and co-workers described iron-catalysed cross coupling reactions between N-heterocyclic aryl tosylates and alkyl Grignard reagents that was focused toward the synthesis of biologically active molecules (Scheme 15). Their efforts were specifically directed toward the coupling of ethyl 4-methyl-6-aryl-2-(tosyloxy) pyrimidine-5-carboxylate, 16, with various alkyl and aryl Grignard reagents. With the use of iron trichloride and NMP additive, conditions first reported by Cahiez and Aveissian,41 moderate yields of cross coupling products were achieved with both primary and secondary alkyl Grignard reagent. The group briefly evaluated the system with other pyridine and pyrimidine based scaffolds, but these efforts resulted in low yields of the cross-coupled product.
 | ||
| Scheme 15 The coupling of N-heterocyclic aryl tosylates (16) with alkyl Grignard reagents in the presence of FeCl3 with NMP as an additive.40 | ||
Garg and co-workers found that aryl sulfamates and carbamates were suitable pseudohalides for cross coupling partners with alkyl Grignard reagents in the presence of FeCl2 and the imidazolinium salt N,N′-(2,4,6-trimethylphenyl)dihydroimidazolium chloride, 17 (Scheme 16).42 It was found that electron donating substituents on the electrophile led to an increased yield of desired product when a carbamate was used. ortho-Substitution was tolerated, even in the case of a bulky TMS group, and N-heterocycles were tolerated. Interestingly, the reaction required a sub-stoichiometric amount of methylene chloride to attain higher yields. The rationale for this additive was not provided. Primary and secondary alkyl Grignard reagents were tolerated.
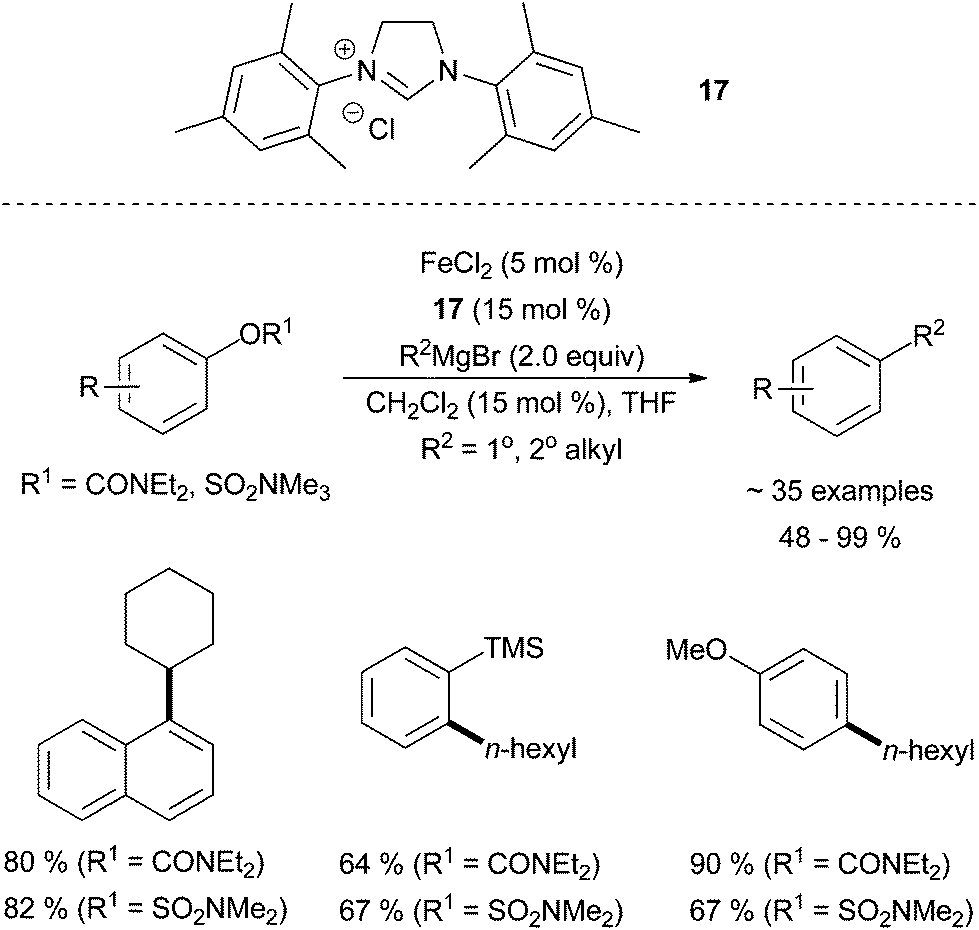 | ||
| Scheme 16 The coupling of aryl sulfamates and carbamates with primary and secondary alkyl Grignard reagents in the presence of FeCl2 and 17.42 | ||
The Cook group discovered that the same system they found to facilitate cross coupling of aryl Grignard reagents with aryl and vinyl tosylates in the presence of 2 and FeF3 (Scheme 1b and c) can also effect the cross coupling of aryl sulfamates with methyl magnesium bromide (Scheme 17a).13 The authors expanded the scope of this system by utilizing NHC ligands to couple aryl sulfamates and tosylates with alkyl Grignard reagents in the presence of FeF3·3H2O and 1 (Scheme 17b).43 Although only moderate yields were achieved, electron withdrawing and electron donating groups were tolerated, as well as para- and meta-substitution of the aromatic electrophiles. The use of secondary rather than primary alkyl magnesium chlorides led to reduced yields, but the catalysts were still active for product formation. Once again, the secondary alkyl Grignard cross coupling partners were prone to chain walking (Scheme 17c). Interestingly, isopropyl magnesium chloride underwent cross coupling with [1,1′-biphenyl]-4-yl dimethylsulfamate, 18, to give predominately the branched product (65% yield, 6.5:1 b:l) while the analogous aryl tosylate, 19, led to the production of mostly linear product (50% yield, 1:10 b:l). Moreover, the selectivity for the coupling of the sulfamate 18 could be reversed to favor the linear product (1:5 b:l) if FeCl3 was used as the iron source instead of FeF3·3H2O. While the authors provided no explanation for the difference in b:l for aryl sulfamates and aryl tosylates, they hypothesized that the high b:l observed for FeF3 was due to the affinity of fluoride for iron, which can bind to open coordination sites that would be otherwise available for β-hydride elimination.
 | ||
| Scheme 17 (a) The coupling of an aryl sulfamate with methyl magnesium bromide in the presence of FeF3 and imidazolium salt 2.13 (b) The coupling of primary and secondary alkyl Grignard reagents in the presence of an imidazolinium salt 1 and FeF3·H2O.43 (c) The isomerisation from branched to linear that occurs during the coupling of iPrMgCl with aryl pseudo halides. When aryl sulfamate 18 was used, the branched product dominated in a 6.5:1 ratio over linear. If aryl tosylate 19 was used, the linear product became the major product in a 10:1 ratio.43 | ||
In 2013, Jacobi von Wangelin and co-workers demonstrated that aryl chlorides bearing olefin substituents could increase the reaction yield by acting as a directing group for the metal centre.44 The reaction was found to be highly chemoselective for aryl chlorides that were positioned ortho to the olefin-directing group (Scheme 18a). An impressive demonstration of the chemoselectivity afforded by an ortho-vinyl directing group was the greater than 15:1 selectivity for production of the cross coupled product from reaction with the aryl chloride moiety in preference to the aryl bromide moiety present in compound 20 (Scheme 18b). Over 30 examples of olefin assisted cross coupling reactions were reported, all in moderate to good yield. Cyclic Grignard reagents were tolerated well, but no cross coupling reactions were reported for acyclic secondary alkyl Grignard reagents.
 | ||
| Scheme 18 (a) The coupling of aryl chlorides with n-decylmagnesium bromide can be facilitated by the presence of an adjacent vinyl group when catalysed by Fe(acac)3 with NMP as an additive. (b) Coupling of 20 occurs preferentially at the aryl chloride moiety that has an adjacent vinyl group rather than the aryl bromide even though aryl bromides couple more readily in most systems.44 | ||
A research group out of Genentech Inc. pursued a similar strategy for the chemoselective cross coupling of alkyl Grignard reagents with N-heteroaromatic dihalides as coupling partners.45 Based on the substitution pattern of the dihalides, chemoselective cross coupling was observed, allowing for one-pot sequential and selective addition of different alkyl Grignard reagents to a single aryl electrophile. N-heteroaromatic dichlorides with a variety of substitution patterns and electronic profiles were studied, the majority of which demonstrated selectivities that were 20:1 and yields that were synthetically useful. While the authors do not comment on the source of chemoselectivity, selectivity was often observed for aryl halides proximal to potential directing groups.
 | ||
| Scheme 19 The cross coupling of unsaturated cyclic esters with methyl magnesium bromide via ring opening oxidative addition in the presence of catalytic amounts of Fe(acac)3. aSolvent: Et2O/toluene (1:1).46 | ||
In 2010, Tanabe and co-workers reported that enol tosylates are competent cross coupling partners, undergoing smooth reaction with methyl, butyl, and phenyl magnesium bromides with good yields and in a stereoretentive manner.47 The Jacobi von Wangelin group has also described iron-catalysed cross coupling of vinyl acetates with alkyl Grignard reagents (Scheme 20).48 Under FeCl2/NMP conditions, moderate to good yields were produced in over 30 examples. Primary and secondary alkyl Grignard reagents were tolerated, as well as a variety of substitution patterns of the electrophile, although electron-withdrawing groups tended to decrease the yield.
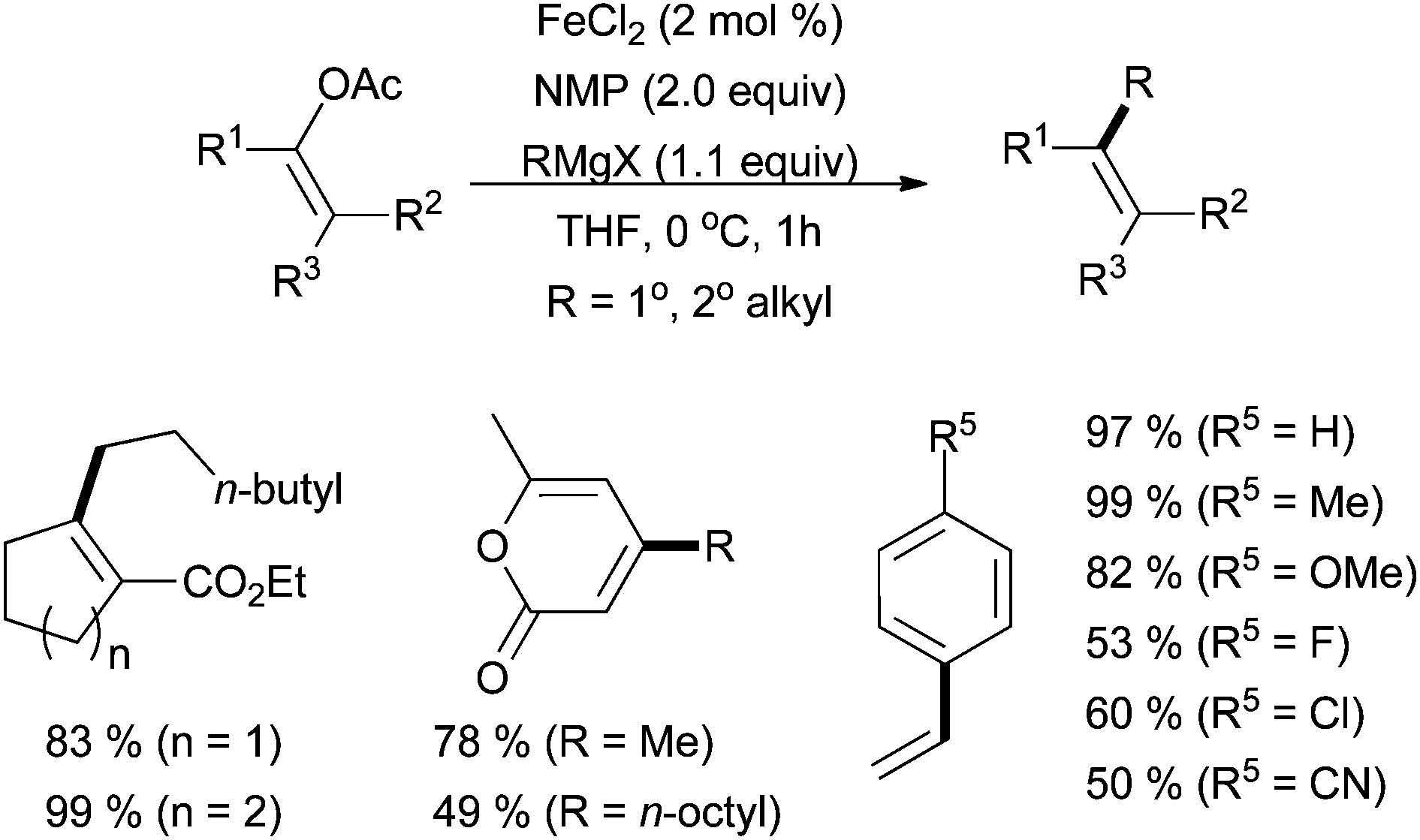 | ||
| Scheme 20 The coupling of vinyl acetates with alkyl Grignard reagents in the presence of NMP and catalytic amounts of FeCl2.48 | ||
Cahiez and co-workers attempted to find a simple ligand system to replace NMP, which is a common additive in many cross coupling reactions between vinyl halides and alkyl Grignard reagents.49 NMP is a known reproductive toxin,50 and would be impractical for industrial and pharmaceutical applications. The group found that iron(II) thiolates are competent catalysts for the coupling of vinyl halides with primary alkyl Grignard reagents providing yields similar to those seen with iron salts and NMP. The coupling of secondary and even tertiary alkyl Grignard reagents in this system is possible, but the latter required small amounts of NMP to proceed with high yields.
 | ||
| Scheme 21 The coupling of alkyl iodides with alkyl magnesium bromides in the presence of catalytic amounts of Fe(OAc)2 and imidazolium salt 21 with representative examples.52 | ||
The only other report that describes alkyl–alkyl cross coupling appeared in 2012 from Ghosh and co-workers, who reported iron-catalysed cross coupling of alkyl and aryl Grignard reagents with polychlorinated solvents, such as dichloromethane, chloroform, and carbon tetrachloride.53 In each case, multiple coupling products were produced. The best results are obtained from the coupling of chloroform with phenyl magnesium bromine, in which triphenylmethane is produced in 96% yield.
Alkynyl Grignard
Thus far, there are only two examples of alkyl–alkynyl coupling via iron catalysis. The first, by Nakamura, describes the cross coupling of alkyl chlorides, bromides and iodides with alkynyl magnesium bromides in the presence of the bisphosphine ligand SciOPP at elevated temperatures.54 Twelve examples were reported in good yields. Hu and co-workers subsequently developed a mild and phosphine free example of alkyl–alkynyl Kumada coupling (Scheme 22).55 Sixty examples are reported with a wide variety of substitution patterns and functional group tolerance, many proceeding in good yields. The electrophile substrate scope includes primary and secondary alkyl iodides and bromides. Whereas the Nakamura system only reports the cross coupling of cyclic alkyl halides, Hu reports competent cross coupling with secondary alkyl halide substrates that are cyclic or acyclic. The substrate scope with respect to the alkynyl Grignard reagents was also broad with tolerance of Grignard reagents that contain ethers, esters, amides, piperidines, carbazoles, and silanes.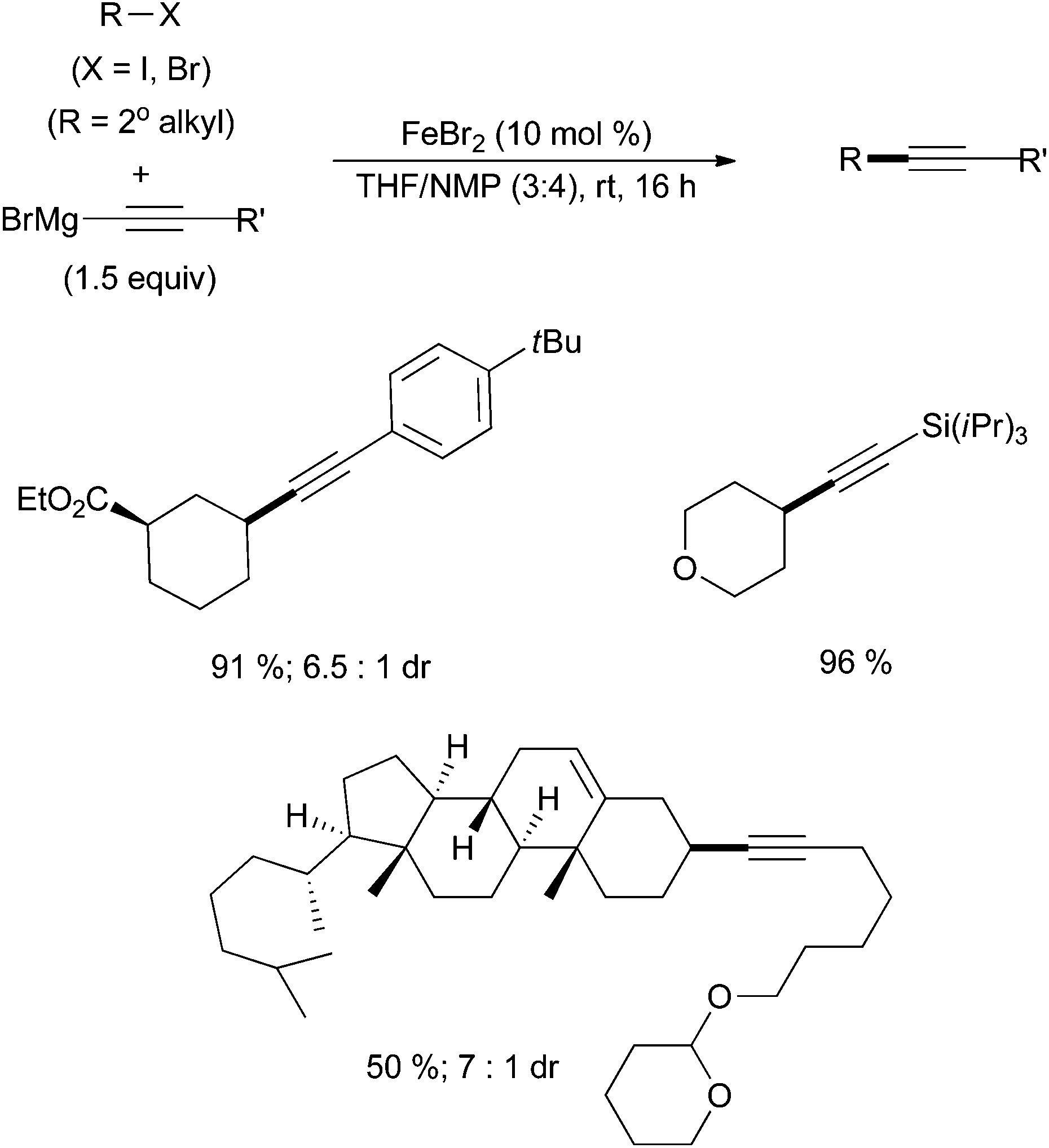 | ||
| Scheme 22 The coupling of alkyl halides with alkynyl magnesium bromides in the presence of catalytic amounts of FeBr2 and with NMP as an additive.55 | ||
Negishi
Despite the preponderance of iron-catalysed Kumada-type cross coupling reactions, several reports of iron-catalysed cross coupling reactions with organozinc reagents have emerged in the past five years. Nakamura successfully coupled alkyl halides with vinyl trimethylsilylmethyl zinc species to give the desired product in high yields and in a stereoretentive fashion (Scheme 23).56 The reactions were highly chemoselective and tolerant of a variety of electron withdrawing groups installed on the vinyl zinc reagents. No aryl zinc or alkyl zinc nucleophiles were reported. Primary and secondary alkyl chlorides, bromides, and iodides were all tolerated, but no tertiary electrophiles were reported.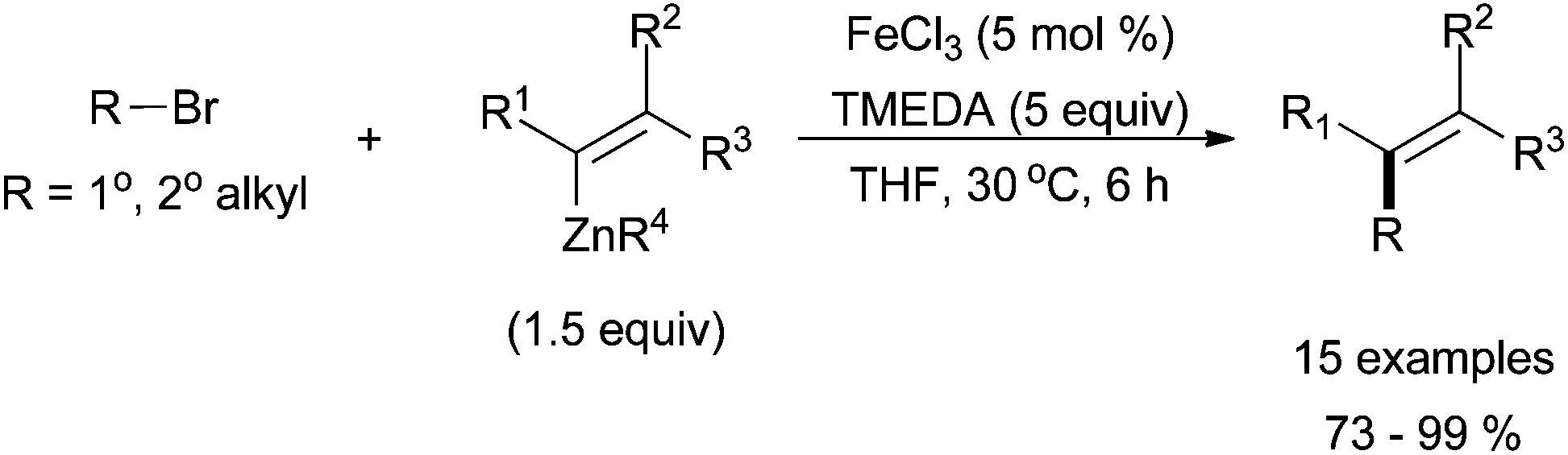 | ||
| Scheme 23 The coupling of primary and secondary alkyl halides with vinyl zinc species in a stereoretentive manner catalysed by FeCl3 and with TMEDA as an additive.56 | ||
A system developed by Qing and co-workers demonstrated that the commercially available bis(diphenylphosphino)propane (dppp) was an effective ligand for the coupling of alkyl halides bearing β-fluorides with aryl Grignard reagents to synthesize gem-difluoromethylenated compounds (Scheme 24).57 For this system, a zinc additive is required for the reaction to form desired product. Although diphenyl zinc is most likely formed from the reaction of zinc dichloride with phenyl magnesium bromide, it was not studied as a transmetalation reagent in this account.
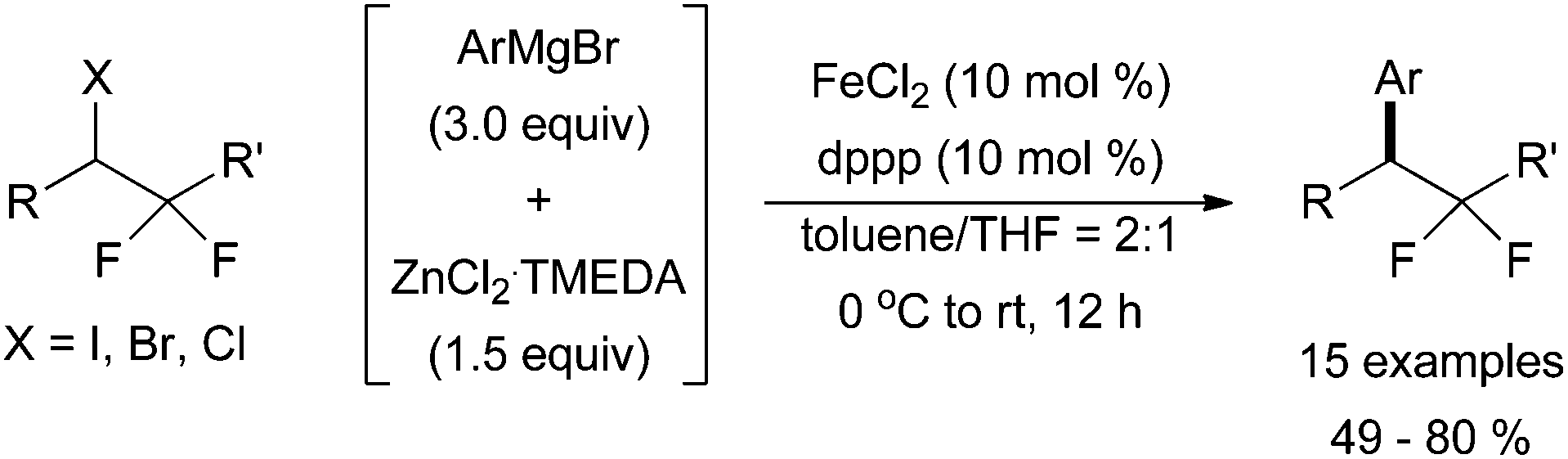 | ||
| Scheme 24 The coupling of alkyl halides containing two geminal fluorine atoms with an in situ generated aryl zinc species in the presence of catalytic amounts of FeCl2 and dppp.57 | ||
Bedford disclosed the cross coupling of benzyl bromides and chlorides with diaryl zinc species in good yields using the well-defined FeCl2(dppbz)2 species 22 (Scheme 25).58 A variety of substitution in the para position of the benzyl electrophile was tolerated, but few substrates with ortho substituents were examined and only para-substituted nucleophiles were studied. Whereas Nakamura used iron(III) trichloride with a TMEDA additive, Bedford's work required the use of iron dichloride complexes 22, presumably to combat the facile homocoupling of aryl and benzyl substrates. Further reports examined the role of bisphosphine ligation, and revealed that the commercially available bis(diphenyl-phosphino)ethane (dppe) as a competent ligand for iron, in the forms of complexes 23 and 24. High yields of desired product were seen with complex 23 especially when one additional equivalent of ligand was present and when the complex was reduced to an iron(I) species prior to the cross coupling reaction.59 This pre-catalyst proved to be competent to catalyse Negishi cross coupling reactions with diaryl zinc reagents as the nucleophiles and benzylic chlorides and bromides or a secondary alkyl bromide as the electrophiles. The FeCl(dppe)2 complex 24 was shown to be an excellent catalyst with no need for additional dppe ligand or pre-reduction.
 | ||
| Scheme 25 (a) The use of complex 22 in the coupling of benzyl bromides and diaryl zinc reagents.58 (b) A similar system to a in which complexes made from commercially available ligands, 23 and 24, are shown to be proficient catalysts for the aforementioned reactivity.59 | ||
Suzuki–Miyaura
Due to their ease in synthesis and handling, organoboron transmetalating reagents are arguably the most useful to employ industrially in cross coupling reactions. Despite their usefulness, efficient protocols for iron-catalysed Suzuki cross coupling reactions have only begun to emerge in the last decade, and all of the known protocols involve particularly activated borate transmetalating reagents and the presence of Zn or Mg additives.60 In 2009, Bedford and co-workers reported that the well-defined iron complex 22 was a competent catalyst for Suzuki–Miyaura cross coupling reactions with tetraphenyl borates in the presence of (tolyl)2Zn (Scheme 26).61 It was hypothesized that the zinc additive acts to stabilize the active iron species to prevent catalyst decomposition, although no evidence was provided to support this hypothesis. The optimal boron-containing cross coupling partner was found to be sodium tetraphenyl borate. Interestingly, in the absence of the borate and with stoichiometric amounts of the zinc reagent, Negishi cross coupling proceeds with the ditolyl zinc compound acting as the transmetalation partner. | ||
| Scheme 26 The coupling of benzyl bromides with sodium tetraphenyl borates in the presence of a diaryl zinc reagent and complex 22.61 | ||
A drawback of the Bedford system is that three of the four aromatic equivalents present in the transmetalating reagent serve as spectators in the cross coupling reactions. To address this limitation, Nakamura and co-workers have published several accounts utilizing activated borate species such as 25, which can be accessed by treating aryl boronates with butyl lithium.62 Alkyl–aryl cross coupling was accomplished in good yields using the iron SciOPP catalyst 26. Interestingly, cross coupling only proceeded in high yields with magnesium dibromide as an additive (Scheme 27a). The precise role of the additive is unknown, but it was hypothesized that it accelerated the inherently slow transmetalation step. Aryl borates with electron donating and electron withdrawing functional groups were tolerated although sterically hindered aryl borates were not examined. Ketone and aldehyde functionalities were tolerated, and alkyl bromides were superior to alkyl chloride as coupling partners. Secondary and primary alkyl halides reacted readily; tertiary substrates were not examined.
 | ||
| Scheme 27 (a) The coupling of primary and secondary alkyl chlorides with activated aryl borate 25, in the presence of 26.62 (b) Vinyl borates 27,63 alkynyl borates 28,64 and alkyl borates 2965,66 can also act as the nucleophilic coupling partner under these and/or similar conditions. | ||
Although the borate 25 contained both an aryl and alkyl group that could potentially participate in transmetalation, transfer of the alkyl group was never observed. However, by varying the transmetalating reagent, alkynyl, alkyl, and vinyl transfer could be achieved (Scheme 27b). Vinyl borate 27 was successful under a system similar to that of the aryl borates.63 Remarkably, the cross-coupling involving alkynyl borate 2864 and alkyl borate 2965,66 did not require the magnesium dibromide additive to form the desired product. In order for 29 to undergo efficient cross-coupling, an ancillary ligand with a larger bite angle was required, which is why iron complexes containing Xantphos instead of SciOPP were more effective catalysts for cross coupling reactions with 28.
Bedford and co-workers published a system similar to the 2010 report by Nakamura, but using commercially available bisphosphine ligands dppe and dppp.67 The group demonstrated that the borate reagent did not have to be synthesized a priori and could be generated in situ with the addition of tert-butyl lithium to the reaction mixture containing a boronate. Excellent yields were presented for the coupling of alkyl and benzyl bromides, but the analogous chlorides produced much lower yields. An additional report was published by the group in which it was found that 2-halobenzylhalides coupled at both the benzylic and aromatic positions, presumably because the initial coupling of the benzyl halide allowed for coordination of the iron catalyst, which could then activate the aryl halide.68 While these recent publications from the Nakamura and Bedford groups mark significant advances towards a practical iron-catalysed Suzuki–Miyaura cross coupling protocol, further advances are still needed so that Suzuki–Miyaura cross coupling reactions can proceed with less activated boron-containing transmetalating reagents.
Sonogashira
The first report of an iron-catalysed Sonogashira reaction was by Bolm and co-workers in 2008 (Scheme 28).69 In this publication, the group successfully coupled aryl iodides with aryl alkynes and silyl alkynes in good yields, albeit with a limited substrate scope. The use of alkyl-substituted alkynes, aryl bromides, and aryl tosylates led to diminished yields. However, most substitution on the aryl iodide was tolerated. | ||
| Scheme 28 The coupling of aryl iodides with aryl alkynes and silyl alkynes in the presence of catalytic amounts of FeCl3 with DMEDA and Cs2CO3 additives.69 | ||
Following this initial report, the groups of Tsai70 and Chen71 have done significant work to advance the field. Tsai and co-workers reported a creative aqueous system using iron trichloride and cationic bipyridyl ligand, 30 (Scheme 29).70 The conditions were optimized using phenylacetylene and iodobenzene, and then the system was expanded to use masked terminal alkynes, 4-aryl-2-methlbut-3-yn-2-ols, 31. By treating 31 with base, an alkynyl anion is formed in situ via an elimination reaction. With this method, they were able to couple aryl iodides with the protected alkynes in moderate yields. Various electron-withdrawing groups in the para-position of the aryl iodide were tolerated, although the yield was enhanced in the presence of electron withdrawing groups and decreased in the presence electron donating groups. Moderate steric hindrance of the aryl iodide was also tolerated, but 4-alkyl-2-methlbut-3-yn-2-ols were not tolerated. The presence of the zinc additive along with the required aqueous conditions led to the deiodination of the aryl iodide, which was the major reaction by-product. Finally, Chen and co-workers used an iron catalysed Sonogashira for the synthesis of 2-arylbenzo[b]furans supporting moderate functionality of the substrates in adequate yields.71
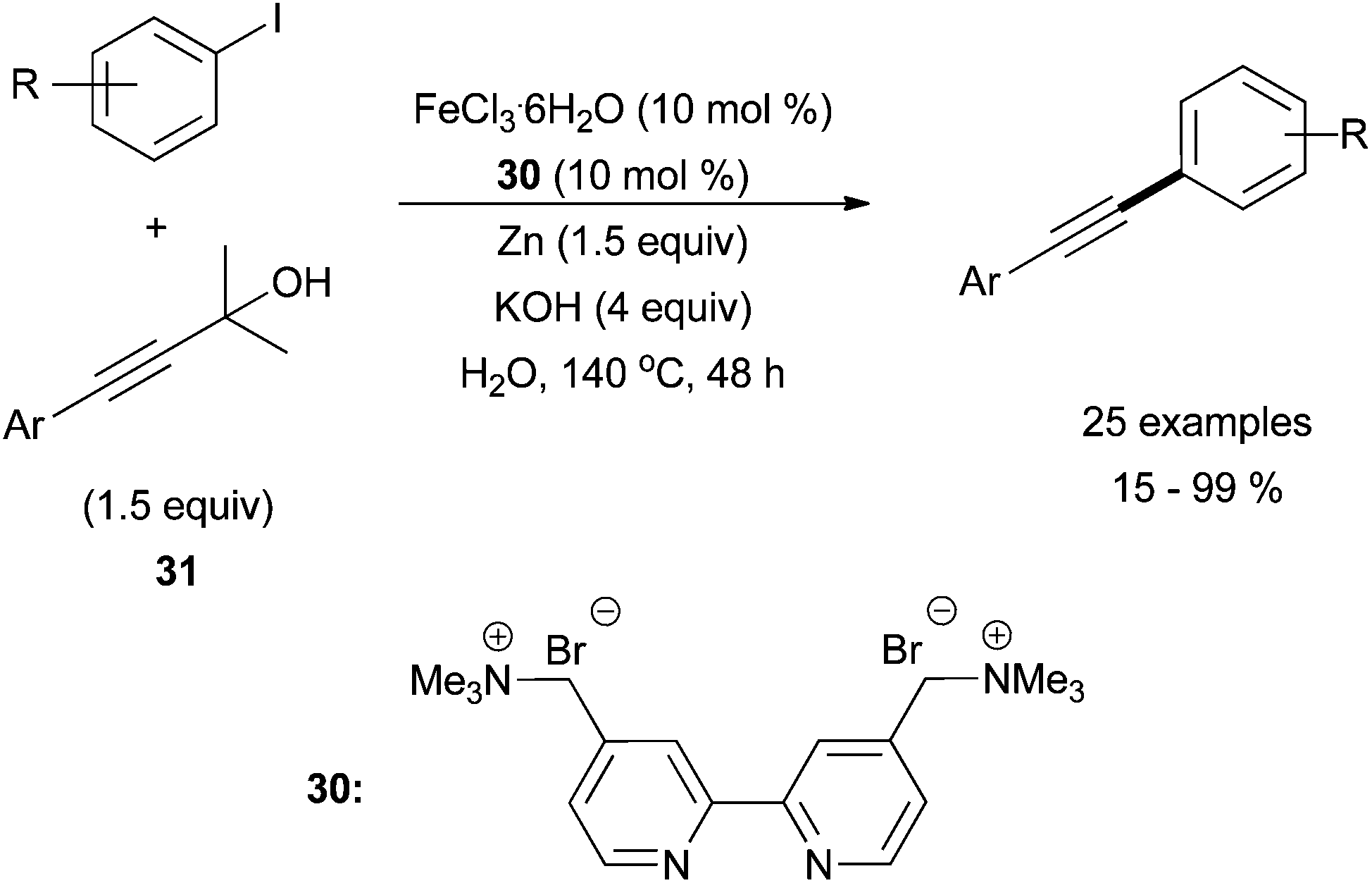 | ||
| Scheme 29 The coupling of aryl iodides with protected terminal alkynes, 31, in the presence of catalytic amounts of FeCl3·6H2O and bipyridyl ligand 30, with Zn and KOH additives.70 | ||
Heck
The first reported instance of iron-catalysed Heck reactions was presented by Vogel and co-workers in 2008, which utilized iron dichloride as the catalyst.72 Since then, only two further reports have been published. Azizi and co-workers utilized a silica supported acac ligand that, upon metalation with iron, formed the heterogeneous iron catalyst 32. This species was efficient for the coupling of aryl iodides with olefins to produce the desired cross coupled product in 70–85% yields (Scheme 30).73 They successfully cross coupled a variety of substituted aryl iodides, containing both electron donating and electron withdrawing functionalities, with styrene, methyl methacrylate, and methacrylate. The reaction required potassium carbonate as a base, polyethylene glycol as a solvent, and elevated temperatures. The heterogeneous catalyst could be isolated and reused in multiple reactions before a decrease in yield was seen. Furthermore, aryl iodides were more efficient electrophiles than aryl bromides. | ||
| Scheme 30 The coupling of aryl iodides and terminal olefins via a silica supported heterogeneous iron catalyst 32.73 | ||
The second recent report of iron-catalysed Heck coupling was inspired by previous evidence for arylation of styrenes using a stoichiometric quantity of iron species 33 (Scheme 31a).74 Mankad and coworkers discovered that this system could be manipulated for catalytic applications by employing UV irradiation to drive the catalytic reaction (Scheme 31b).75 By using the anionic complex 34, the group was successful in achieving an iron-catalysed Heck reaction that was E-selective. With an excess of olefin, and under UV irradiation to induce CO dissociation, the desired cross coupling product was produced in moderate yields along with an olefin migration product. The reaction was limited to benzyl chlorides, which were selectively activated in the presence of aryl chlorides. Styrene was the only alkene reportedly used in the cross coupling reaction, and an excess of it was required to achieve high yields of cross coupled products.
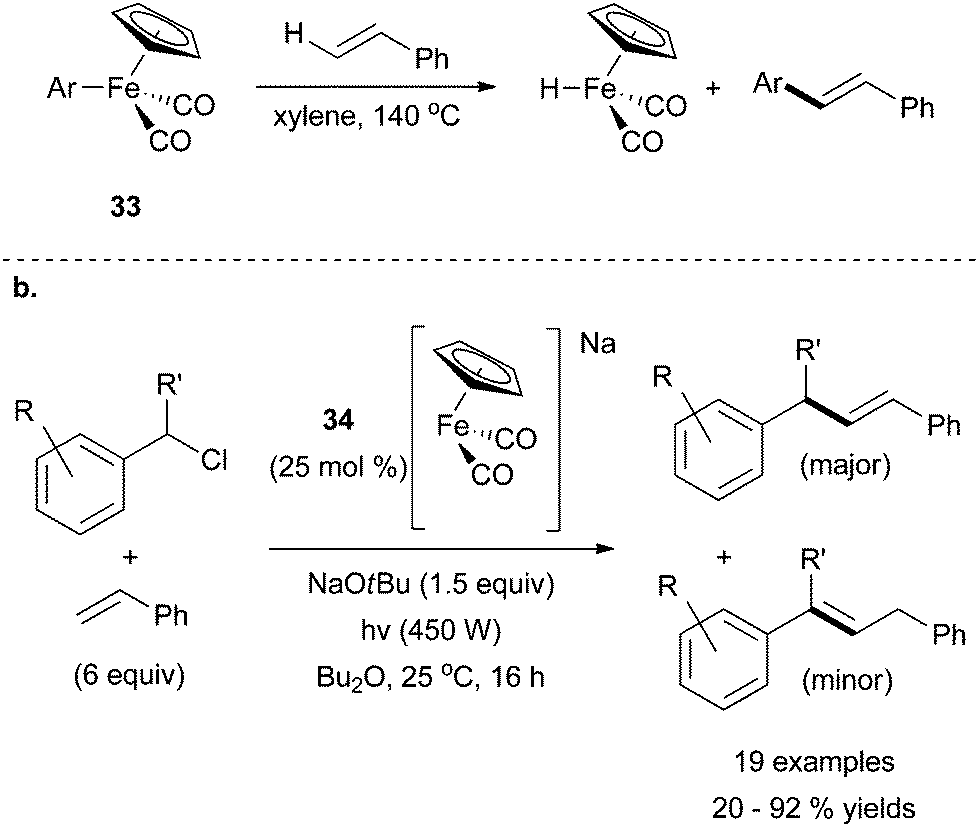 | ||
| Scheme 31 (a) Arylation of styrene using complex 33 to transfer an aryl ligand to the terminal olefin.74 (b) A modification of 33 for catalytic purposes in which benzyl chlorides were successfully coupled with styrene in the presence of 34 in an E-selective fashion.75 | ||
Additional variations
In addition to utilizing the more common metalloids as transmetalating reagents, there have been some significant advances in iron-catalysed cross coupling reactions that involve transmetalating reagents that are less commonly employed in palladium or nickel-catalysed cross coupling reactions. In 2010, Nakamura and co-workers evaluated the effectiveness of in situ generated phenylaluminate complexes prepared with various ratios of Grignard reagents. They discovered that the ratio of the Grignard reagent impacts their ability to participate in cross coupling reactions.76 Subsequently, the group employed iron SciOPP complex 26 in a screen of Mg, Zn, B, and Al transmetalation reagents for the coupling of a hydroxylated primary alkyl chloride with a phenyl nucleophile (Scheme 32).77 It was reported that an anionic tetraphenylaluminate complex with a magnesium chloride counter cation provided a 91% yield of desired product. Other transmetalating reagents such as PhMgBr and Ph2B(pin)Li, led to extremely low yields despite being useful transmetalating reagents elsewhere (see above). Additionally, Nakamura has employed in situ generated organoaluminium reagents for iron-catalysed cross coupling that were formed as a result of hydro- or carboalumination of alkynes.78 | ||
| Scheme 32 A screen of aryl nucleophiles for the coupling of alkyl chlorides in the presence of 26. It was determined that the phenylaluminate complex shown in entry 6 was the best metal nucleophile for this reaction a2.0 equivalents of PhMgBr used.77 | ||
Knochel and co-workers coupled benzyl manganese chlorides with aryl and heteroaryl electrophiles.79 Fourteen examples were reported, all of which led to product in moderate yields. The manganese reagents were generated in situ by first forming the benzyl Grignard reagent before transmetalation with lihum chloride adduct of manganese dichloride. Similarly, aryl magnesium bromides combined with a copper chloride additive to generate organocuprates in situ were used to carry out cross coupling reactions with alkynylbromides.80 The identity of the organocuprate was not determined in the report, and evidence of its existence was not provided. Liu and co-workers have developed a biaryl cross coupling using titanium additives.81
Bedford and co-workers have also studied alternative metal nucleophiles in which they screen group 13 metals as transmetalation agents with iron complex 22.66 Effective cross coupling reactions were mostly reported for the coupling of benzyl chlorides with aryl nucleophiles. It was found that boron, gallium, and thallium-based nucleophiles required the presence of a zinc co-catalyst to yield the cross-coupling product, whereas aluminium and indium provided good yields without the zinc additive. A subsequent substrate scope revealed that although aluminium and indium provided similar activities for most substrates, only the aluminium nucleophiles were able to undergo cross coupling reactions with alkyl halides.
Iron-catalysed examples of C–N and C–S bond forming Buchwald–Hartwig type reactions have also been pursued. In 2012, the Nakamura group achieved C–N bond formation in the absence of a copper co-catalyst by using diarylamine Grignard reagents, 35, as coupling partners for arylbromides in the presence of a stoichiometric amount of lithium salt (Scheme 33).82 A moderate range of electron donating and withdrawing groups were tolerated on the electrophile, and it was suggested that an iron(II) diamide dimer 36 is the in situ generated pre-catalyst. The purity of the iron salts was determined by Inductively Coupled Plasma Mass Spectrometry (ICP-MS) and Inductively Coupled Plasma Atomic Emission Spectroscopy (ICP-AES). It was found that ppm level amounts of Pd, Cu, Co, and Ni were present in the iron salts. Control experiments were therefore run with PdCl2, CuCl2 and CoCl2, showing low catalytic activity. Nickel salts had previously been shown to lead to lower product yields in this system compared to the iron salts.
 | ||
| Scheme 33 The coupling of aryl bromides with diarylaminomagnesium bromide reagent 35 in the presence of catalytic amounts of FeCl2 with LiBr as an additive; thought to proceed via36.82 | ||
A similar sulfonation route was then published by Lee and co-workers in which vinyl halides were coupled with alkyl and aryl thiols in the presence of iron trichloride and Xantphos.83 Moderate yields were achieved and a variety of thiols were shown to couple efficiently. The iron source used was of high purity (99.99%), so there was little reason to believe that trace amounts of other metals were the source of the catalytic activity.
Very recently, Hu and co-workers reported a one-pot synthesis of phenothiazines from 1,2-dihaloarenes via C–S coupling with an aryl thiol and subsequent C–N coupling with an aryl amide adjacent to the thiol in the presence of iron dichloride and phenathrone, 37 (Scheme 34).84 Substitution of the 1,2-dihaloarenes, was not tolerated and coupling of 1,2-dichlorobenzene was inefficient; bromoarene and iodoarene coupling sites were preferred. Moderate substitution of the N-(2-mercaptophenyl) acetamide nucleophile was tolerated. The report did not examine whether the amine analogue was a competent electrophile.
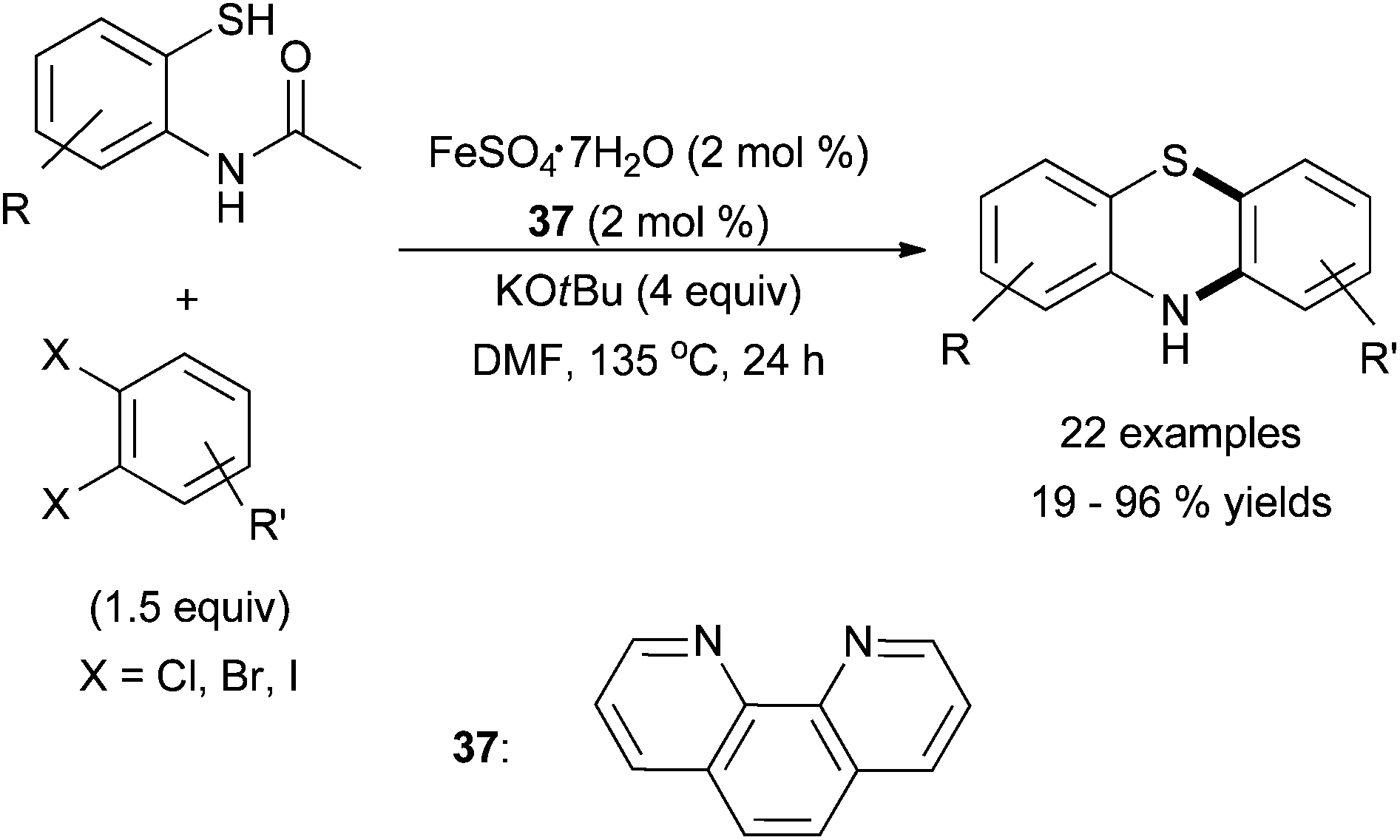 | ||
| Scheme 34 The synthesis of phenothiazines via a cascade C–S C–N coupling in the presence of an iron salt and ligand 37.84 | ||
Mechanistic considerations
While there have been several useful iron-catalyzed cross coupling protocols that have been established, a longstanding challenge in the field has been the development of a firm mechanistic understanding of these processes. Many early experiments relied on analyzing product distributions and employing classical physical organic techniques. Mechanistic proposals were often made based on the identity and oxidation state of the starting pre-catalysts and the characterization of stable species by X-ray crystallography or EPR spectroscopy. These studies suggested the likely existence of several reaction pathways whose operation depended on the identity of metal pre-catalyst, the cross coupling reagents, and the conditions employed. In recent years, a diverse set of in situ spectroscopic techniques have emerged as valuable tools to better evaluate these catalyst systems.Before discussing the various mechanisms in iron-catalysed cross coupling reactions, it is important to mention that several processes have been scrutinized for not being catalysed by iron.85–87 Careful control experiments have revealed that copper or palladium impurities in iron sources can sometimes be the catalytically active species. While relevant to all iron-catalysed processes, the reactions that have demonstrated evidence for such behavior has been iron-catalysed aryl–aryl cross coupling reactions,85 and iron-catalysed Buchwald–Hartwig type cross coupling reactions.86,87 A number of measures have been taken to ensure that iron is the active metal for catalysis including carrying out elemental analysis on iron sources and testing trace metals found from such analyses for their catalytic competency as well as carrying out catalytic reactions using 99.99% pure iron sources.
Although redox-neutral substitution reactions akin to reactions occurring in cuprate chemistry have been proposed as a possible mechanistic pathway in iron-catalysed cross coupling reactions,88 there is little experimental data that supports such pathways. Most mechanistic proposals with experimental backing follow three steps: transmetalation of the nucleophile, oxidative cleavage of the electrophile, and reductive coupling to form the product. The mechanistic diversity for iron-catalysed processes exists in three aspects: (1) the oxidation states of the metal in reactive intermediates, (2) the oxidative cleavage step occurring by a concerted oxidative addition mechanism (Scheme 35a), by an oxidative addition mechanism involving sequential one electron processes (Scheme 35a), by C–X bond homolysis resulting in one electron oxidation of the metal and formation of a carbon centered radical (Scheme 35b), or by a bimolecular oxidative addition mechanism (Scheme 35c), and (3) reductive coupling occurring by a concerted reductive elimination process or from rapid radical recombination of a carbon centered radical and an organometallic radical.
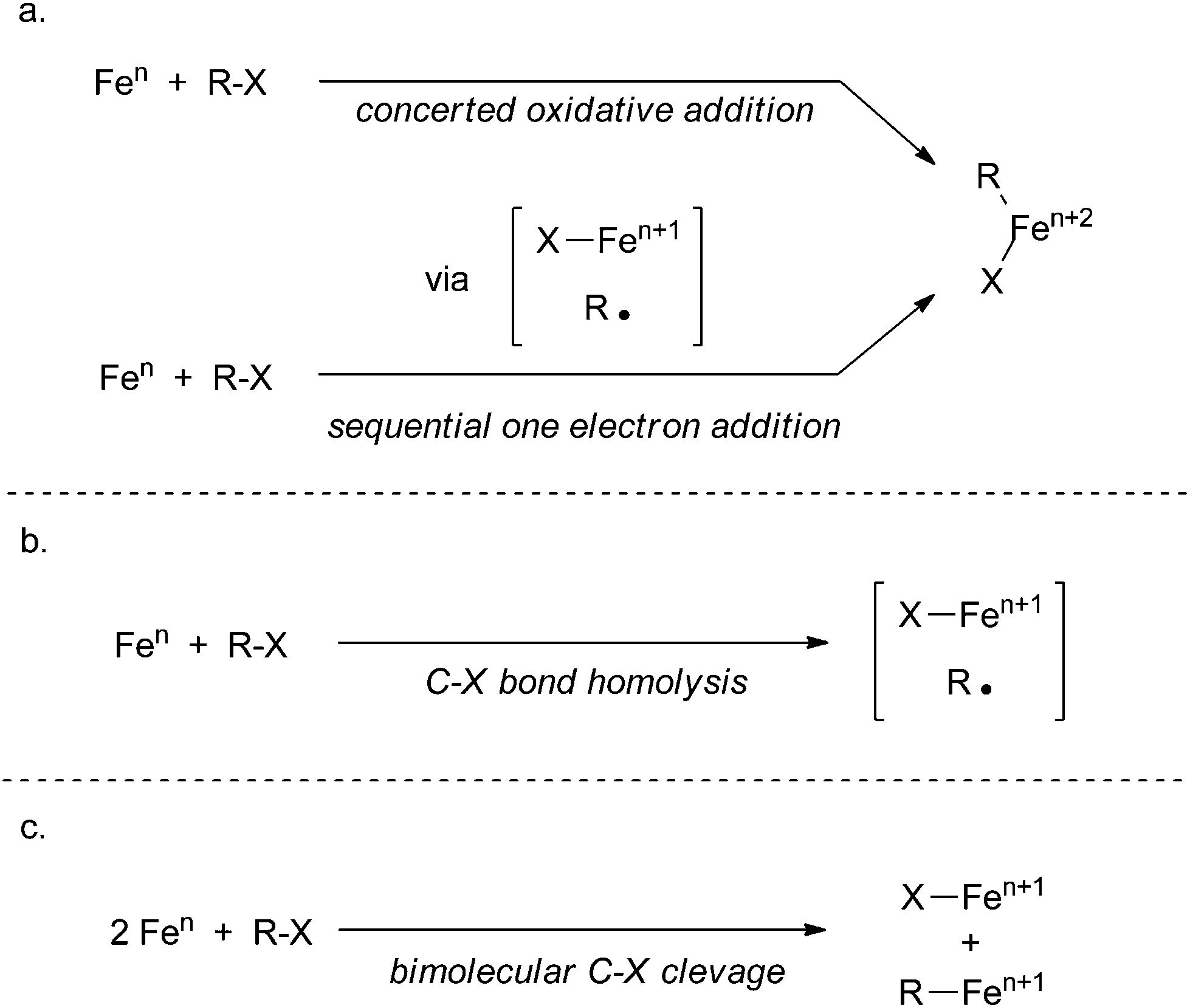 | ||
| Scheme 35 The proposed possibilities for the oxidative cleavage step of iron-catalysed cross coupling, including: (a) concerted oxidative addition or sequential one electron additions; (b) C–X bond homolysis resulting in a free carbon-centred radical; or (c) an oxidation facilitated by two metal centres. | ||
Alkyl nucleophile–aryl or vinyl electrophile
In the original reports that described the cross coupling of methyl Grignard reagents with alkenyl bromides, Kochi and co-workers proposed a cycle that involves an iron(I)/iron(III) redox couple that proceeds similarly to the canonical mechanism accepted for palladium cross coupling reactions (Scheme 36).89,90 The proposal for the existence of an iron(I) intermediate was made in large part from the formation of ethane and the observation of an S = 1/2 species observed by EPR spectroscopy upon reacting iron(III) trichloride with methyl magnesium bromide. From these studies, it was revealed that the role of the methyl Grignard reagent was twofold: it first served to reduce the iron(III) precursor to an iron(I) active catalyst and then it serves as the transmetalating reagent that is necessary for catalyst turnover. Fürstner was the first to obtain structural evidence that reduction of iron(III) to iron(II) occurs upon addition of methyl anion to iron(III) trichloride. Using X-ray crystallography, the iron(II) homoleptic compound 38 was isolated from reaction of methyl lithium with iron(III) trichloride in diethyl ether (Scheme 37a).91 In stoichiometric reactions, this species was found to be competent for forming cross coupling products with alkenyl triflates but incompetent for cross coupling with aryl bromides. | ||
| Scheme 36 Proposed mechanism for iron-catalysed cross coupling of alkenyl halides and methyl Grignard reagents.89,90 | ||
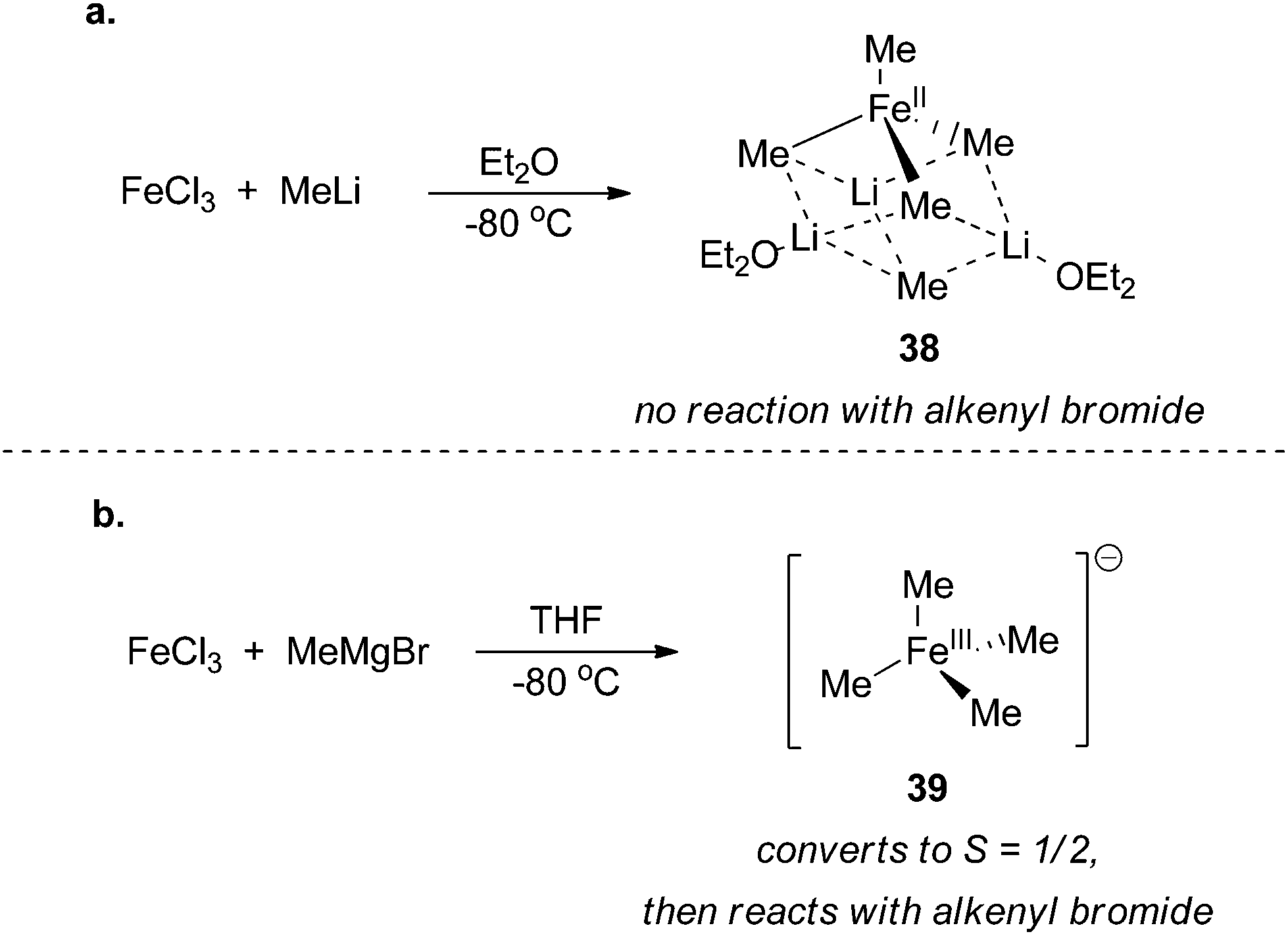 | ||
| Scheme 37 Homoleptic iron complexes isolated from reacting FeCl3 with (a) methyl lithium in diethyl ether91 and (b) methyl magnesium bromide in tetrahydrofuran.92 | ||
Recently, Neidig and co-workers isolated and characterized a different homoleptic anionic tetramethyl iron(III) complex 39 that forms from mixing iron(III) trichloride with methyl magnesium bromide at −40 °C (Scheme 37b).92 Using low temperature in situ EPR spectroscopy, the distorted square planar complex was shown to be an intermediate spin S = 3/2 iron(III) complex that undergoes clean transition to an S = 1/2 complex with concomitant production of ethane. These observations are consistent with Kochi's proposal for the formation of an iron(I) complex. Interestingly, addition of alkenyl halides prior to the formation of the S = 1/2 species led to no reaction even after prolonged reaction times, which suggests that it is an intermediate en route to the S = 1/2 iron(I) complex that is the presumed catalyst for the reaction. While the precise identity of the iron(I) complex that forms upon in situ reduction has remained elusive, the authors demonstrated by spin counting of the EPR spectrum that the iron(I) complex is the major iron species that exists during the cross coupling reaction. Moreover, the species was also shown to be catalytically competent and kinetically relevant for cross coupling. At the current stage of understanding, it is not clear whether the redox reactions occur by a two-electron process or by sequential one-electron processes.
In addition to iron(I) intermediates being involved in the cross coupling of vinyl halides and alkyl Grignard reagents, there have been alternative proposals involving iron complexes with even lower oxidation states of iron.91,93 The most experimental support for such proposals have emerged from the Fürstner group, who have put forth an Fe(-II)/Fe(0) cycle (Scheme 38).91 Notably, efficient cross coupling of aryl halides with Grignard reagents is only observed if the Grignard reagent contains hydrogen atoms that can undergo β-hydride elimination upon binding to iron. Based on reactivity originally observed by Bogdanovic,94,95 iron complexes were proposed to be reduced to oxidation states lower than iron(I) when treated with ethyl magnesium bromide. To support this claim, the group synthesized the iron(-II) olefin complex 40, which proved to be a particularly efficient pre-catalyst for cross coupling reactions between aryl halides and alkyl Grignard reagents (Scheme 38b). It is based on this reactivity difference that the authors favor an iron(-II)/iron(0) catalytic cycle.
 | ||
| Scheme 38 (a) Proposed catalytic cycle for iron-catalysed cross coupling of alkyl Grignard reagents and aryl halides involving an Fe(-II)/Fe(0) redox cycle. (b) Fe(-II) complex that is unusually active for Kumada-type coupling reactions.91 | ||
Since this report, the existence of an iron(-II)/iron(0) redox couple for the cross coupling of aryl halides and alkyl Grignard reagents has received some scrutiny.7 Using a series of iron(-I) anthracene complexes, Wolf and co-workers demonstrated that it is the coordinating ability of the ligands rather than the initial oxidation state of the metal that is most important for active cross coupling reactions.96 Werner and Bauer determined that species formed upon reacting Fe(acac)3 with phenyl magnesium bromide were capable of undergoing cross coupling between aryl chlorides and hexyl magnesium bromide.97 Based on X-ray absorption near edge spectroscopy (XANES) and X-ray absorption fine structure (XAFS) measurements, the authors concluded that the major iron species present during the reaction are iron nanoparticles with an average oxidation state of iron(II), which the authors attribute to geometric averaging of iron(I) and iron(III) sites in iron nanoparticles. Notably absent was any evidence for iron complexes in oxidation states lower than iron(I). Nevertheless, it is important to note that XAFS and XANES spectroscopy provide an averaged picture for all iron species present during cross coupling reactions. Therefore, despite the undoubtable existence of iron nanoparticles in these experiments, their mere presence does not necessitate catalysis by nanoparticles nor rule out a mechanism involving low valent iron complexes that exist in quantities undetectable by XANES and XAFS.
Norrby and co-workers have provided some insight into the mechanism for oxidative addition using linear free energy relationships (e.g. Hammett plots) for electronically different aryl triflate electrophiles in cross coupling reactions catalyzed by Fe(acac)3 in THF/NMP.98 The researchers found that relative reaction rates correlated well with the normal σ-values with reaction rates being significantly faster for electron deficient aryl triflates. The positive ρ-value obtained (ρ = +3.8) suggested significant negative charge buildup in the rate determining step, which is more consistent with a classic three-centered oxidative addition rather than a radical mediated process. Presumably, reductive elimination likely occurs by a similar mechanism although no experiments have been devoted to understanding the reductive elimination pathway.
Aryl nucleophile–alkyl electrophile
There are two mechanisms that are commonly proposed for iron-catalysed cross coupling reactions between aryl nucleophiles and alkyl electrophiles. The first involves an iron(I)/iron(II)/iron(III) redox couple that is similar to the catalytic cycle proposed by Kochi for the cross coupling of vinyl halides and methyl magnesium bromide with the exception that the oxidative addition occurs by two sequential one electron processes (Scheme 39).8,24,29,59,65,104 The second mechanism deviates somewhat from mechanisms traditionally proposed for cross coupling reactions, but its existence is supported by significant experimental evidence (Scheme 40).25,29,32,62,99,100 In this mechanism, originally proposed by Nakumura,62 the transmetalation of an aryl Grignard reagent to an iron(II) catalyst precursor occurs without reduction of iron to yield an iron(II) aryl-chloride species 41 and/or an iron(II) bis-aryl species 42 (if iron(III) precursors are utilized, rapid reduction to iron(II) occurs prior to transmetalation). These compounds undergo halide abstraction from the alkyl halide, which results in the liberation of an alkyl radical and the formal oxidation of the metal to form the iron(III) complex 43. Next, the alkyl radical recombines with 43 in the C–C bond forming step to produce the cross coupled product and liberate the iron(II) intermediate 44. Finally, 44 undergoes transmetalation with an equivalent of aryl Grignard reagent to regenerate 41 (or 42). | ||
| Scheme 39 Common mechanism proposed for iron-catalysed coupling of aryl nucleophiles and alkyl electrophiles. | ||
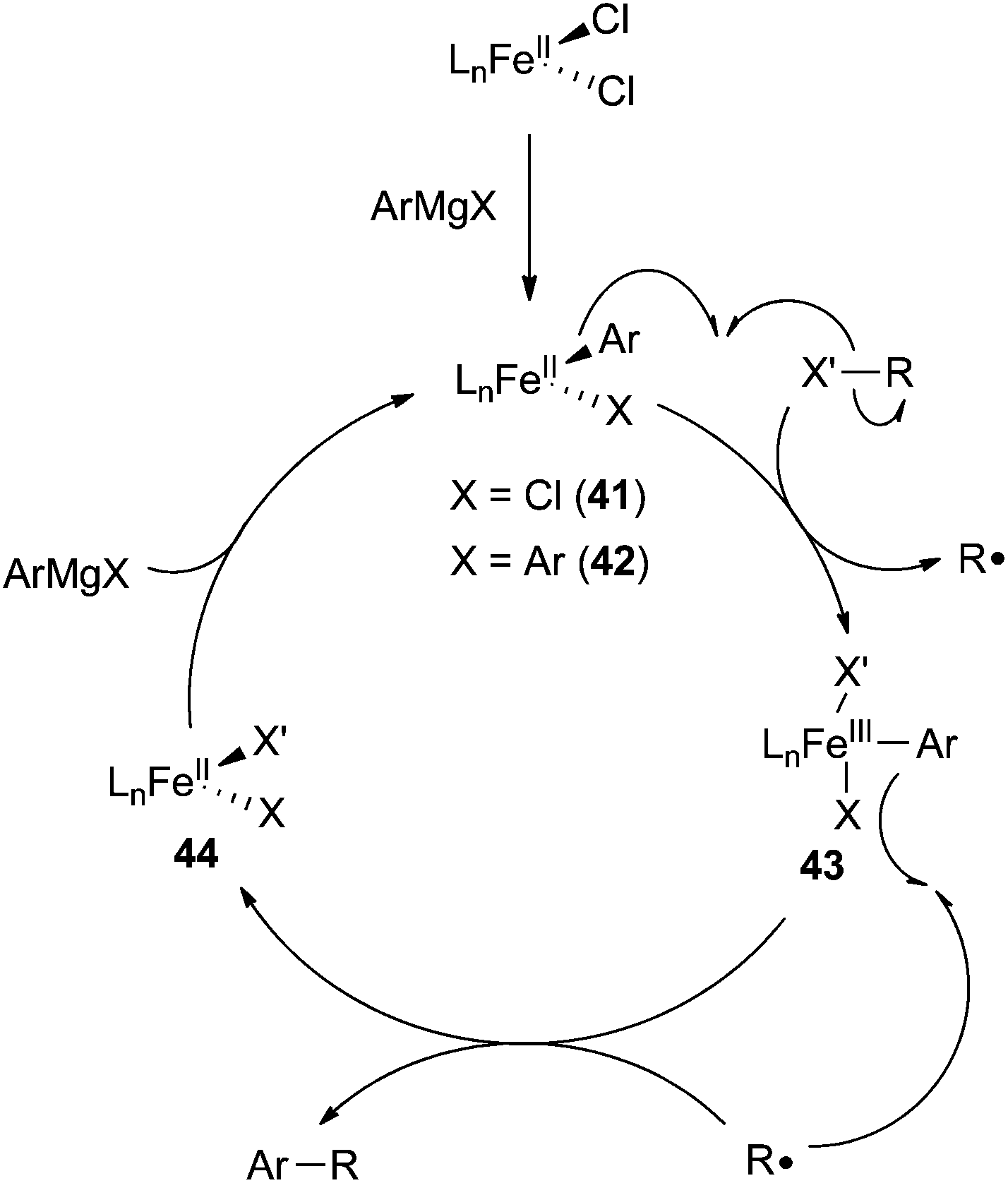 | ||
| Scheme 40 Common mechanism proposed for iron-catalysed coupling of aryl nucleophiles and alkyl electrophiles. | ||
Mechanistic support for carbon centered radical intermediates during the oxidative addition step are numerous.8,25–28,32,52,55–57,62–65,67,78,91,100 Several groups have utilized radical clock alkyl halide substrates, which undergo ring opening or ring closing reactions prior to cross coupling (Scheme 41).25,27,28,32,52,55–57,62–65,67,78 Although these outcomes are consistent with the existence of a carbon-centered radical intermediate, the radical clock substrates typically used are not entirely suitable for probing radical intermediates in organometallic chemistry. This is because ring opening and/or ring closing can occur via well-established two electron processes such as migratory insertion and β-elimination reactions. Additionally, palladium systems have shown this same reactivity although they are well known to proceed via two-electron processes.2,101 It is therefore useful to carry out additional experiments to support outcomes from radical clock substrates. For example, Fürstner91 and Nakumura26 have utilized enantiomerically enriched or diastereomerically pure alkyl halides, which undergo racemization as a result of C–X bond hemolysis. Additionally, the Norrby group have utilized linear free energy relationships that correlate well with radical reactions,8 and the Tonzetich group have utilized radical scavengers that inhibit cross coupling reactions.100 The existence of an alkyl radical intermediate is important because it suggests a mechanism for oxidative cleavage that occurs by one electron events rather than a concerted two-electron process akin to oxidative addition in palladium cross coupling reactions. The one-electron events can occur as two sequential oxidations of the iron center (e.g.Scheme 39) or via the formation of the free radical intermediate (e.g.Scheme 40).
 | ||
| Scheme 41 Radical clock substrates that implicate radical intermediates in iron-catalysed coupling reactions with aryl nucleophiles and alkyl electrophiles. | ||
An open question in the cross coupling of alkyl halides with aryl nucleophiles is whether the oxidation state of the iron complex that is capable of undergoing oxidative addition is always iron(II) or if a lower oxidation state of iron is the catalytically active species in some cases. On the basis of higher reactivity of an iron(-II) complex compared to iron(I), iron(II) or iron(III) complexes, the Fürstner group proposed a catalytic cycle based on an iron(-II)/iron(0) couple.91 Cahiez and co-workers have also proposed an iron(0)/iron(I)/iron(II) cycle.102 Although not definitively ruled out, there is minimal experimental support for these mechanisms and in situ X-ray spectroscopic studies on similar systems are inconsistent with the formation of these low valent species.97,103
Evidence for the iron(I)/iron(III) pathway shown in Scheme 39 has been put forth by Norrby and Bedford.8,24,29,59,65,104 The existence of an iron(I) intermediate was originally surmised from the observation that early on in the cross coupling reaction, one-half equivalent of biphenyl is produced per iron(II) precursor.8,104 This observation is consistent with the reduction of the metal by one electron to form an iron(I) complex. The Bedford group later demonstrated that in the absence of alkyl halide the formation of (dppbz)2FeAr (45) resulted (Scheme 42a).104 They also found conditions for synthesizing (dppbz)2FeBr (46). Both complexes were found to be catalytically competent although only complex 46 was kinetically competent with the rates observed during catalytic cross coupling reactions. Based on this observation, the Bedford group concluded that complex 45 was off the catalytic cycle. Similar results were found for related cross coupling reactions involving dppe as the ancillary ligand.67
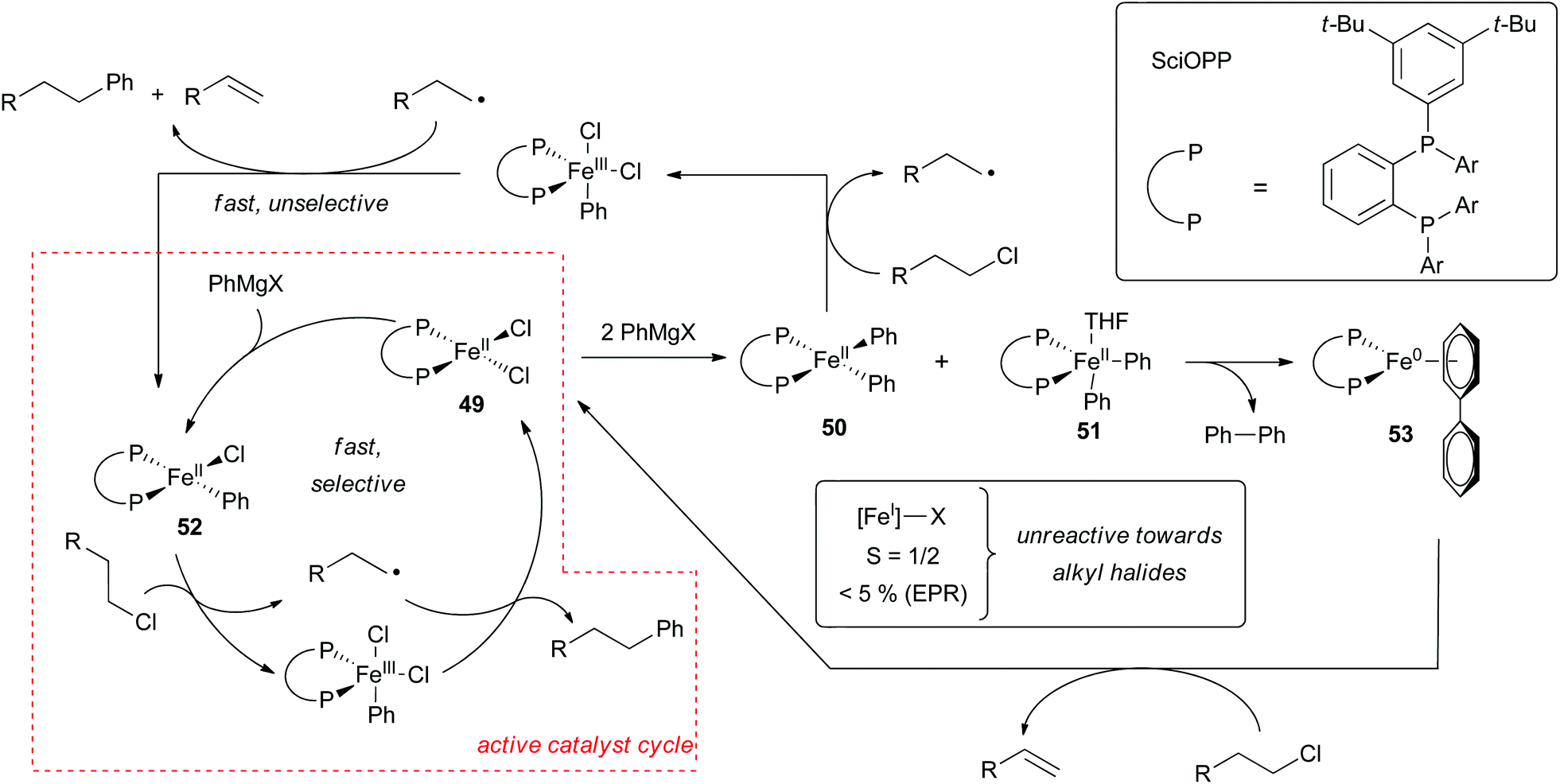 | ||
| Scheme 42 Mechanistic findings for the cross coupling of phenyl magnesium bromides and alkyl electrophiles catalysed by FeCl2(SciOPP).29,99 | ||
Further support for the intermediacy of an iron(I) compound was obtained by detection of the S = 1/2 complex (dppe)2FeCl (47) by in situ EPR spectroscopy carried out during Suzuki–Miyura cross coupling reactions (Scheme 42b).67 Based on kinetic studies, the Bedford group determined that the five coordinate iron(I) complex (dppe)2FeCl (47) was in equilibrium with the three coordinate iron(I) (dppe)FeCl (48) complex, which was surmised to be the complex responsible for oxidative addition that proceeds by halide abstraction and rapid radical rebound to form an iron(III) organometallic intermediate. Subsequent transmetalation followed by reductive elimination completes the catalytic cycle (Scheme 39). Despite significant effort devoted to identifying an iron(I) intermediate in these reactions, little experimental support has been obtained for the existence of an iron(III) alkyl intermediate.
Therefore, reasonable alternatives for the reductive coupling step, such as the one discussed below, are possible.
Despite some evidence for an iron(I)/iron(II)/iron(III) redox cycle, several systems containing sterically and electronically different ligands appear to proceed by the mechanism shown in Scheme 40.25,29,32,62,99,100 In a very thorough study, the Neidig group used a combination of in situ Mössbauer, EPR, and magnetic circular dichroism (MCD) spectroscopies to demonstrate that the resting state in Kumada and Suzuki type cross coupling reactions utilizing the Nakamura SciOPP ligands are iron(II) compounds (Scheme 43).29,99 The key experiments used to establish this fact involved rapid freeze trapping of in situ formed iron species using isotopically pure 57Fe. Analysis of reactions between (SciOPP)57FeCl2 (49) and two equivalents of PhMgBr by Mössbauer and MCD spectroscopies revealed the presence of two high spin iron(II) organometallic species, 50 and 51 as the major species in solution. As observed by Bedford and Norrby, analysis of the reaction mixture by EPR spectroscopy revealed the presence of an S = 1/2 iron(I) species. However, quantification of the paramagnetic compound by spin counting in the EPR spectrum revealed that only 5% of the total iron was iron(I), the remainder of the iron presumably being integer spin and EPR silent iron(II) species. Similar quantifications of EPR spectra in reaction mixtures were not carried out by the Bedford group. Addition of an excess of alkyl halides to the reaction mixture led to the rapid formation of the cross coupled product and an alkene byproduct with the concomitant conversion of 50 and 51 to (SciOPP)FeCl2 (49). Interestingly, the small amount of iron(I) species remained unreacted, thereby suggesting that the iron(I) complex is off cycle.
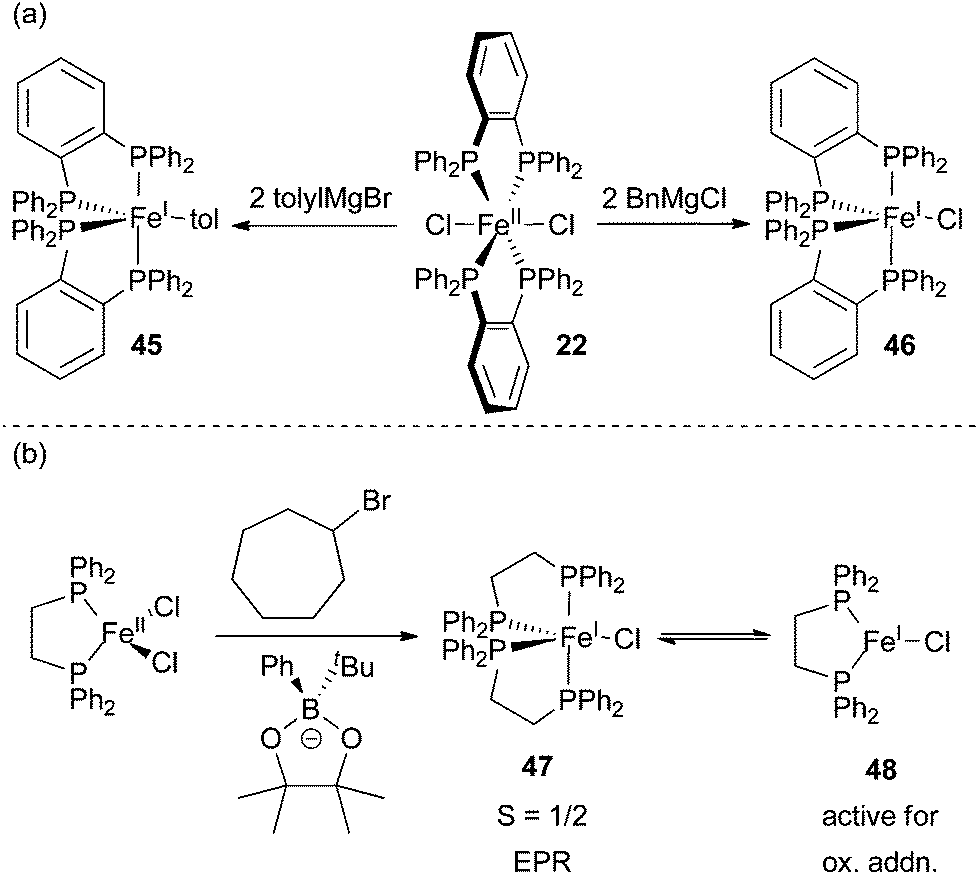 | ||
| Scheme 43 Evidence for iron(I) intermediates in the iron-catalysed cross coupling of aryl nucleophiles and alkyl electrophiles. (a) Reaction of 22 with different Grignard reagents will form different organometallic species.104 (b) The formation of S = 1/2 species 47.67 | ||
The Neidig group also demonstrated that (SciOPP)FePhCl (53), formed by adding one equivalent of PhMgBr to 49, was a superior catalyst for the formation of cross coupled product without the simultaneous formation of alkene byproducts. A common requirement in many iron-catalyzed Kumada-type cross coupling reactions is the slow addition of the Grignard reagent, which is presumably needed to form species analogous to 49 and avoid the production of alkene byproducts that result as a consequence of forming species analogous to 50 and 51. In a subsequent report, the Neidig group demonstrated that slow addition of the borates utilized for Suzuki cross coupling is not required because transmetalation is significantly slower and the formation of the bisarylated species 50 and 51 is avoided.99
Prolonged reaction of 50 and 51 or heating of the catalytic reaction mixture led to the formation of the iron(0) complex 53 as the major species in solution. This species is presumably formed from the reductive elimination of biphenyl from 50 and 51. Exposure of the electrophile to 53 led to the production of the cross coupled product, but at a rate significantly slower than the catalytic reaction and with the production of the alkene byproduct as the major product in the reaction. This led the researchers to conclude that compound 53 is off cycle. Importantly, the authors note that quenching of the organometallic species 50 and 51 led to the production of biphenyl. Thus, the authors concluded that estimation of iron oxidation states by quantifying the amount of biphenyl produced can be misleading.
While the studies carried out by the Neidig group may not be generally applied to other systems, they clearly reveal the importance in evaluating and quantifying reaction mixtures in situ because the formation of several off cycle, low valent iron species is likely. Furthermore, the iron speciation can change in the presence and absence of alkyl electrophile.
In addition to these commonly proposed mechanisms, there is an additional reaction mechanism that has some experimental support. In 2015, Hu and co-workers carried out mechanistic investigations for an iron-catalyzed cross coupling reaction between alkyl halides and aryl Grignard reagents using iron catalyst 54 (Scheme 44).35 Using kinetic studies, the group determined that the cross coupling reaction was first order in the aryl Grignard reagent, second order in catalyst, and zero order in alkyl halide electrophile. Radical clock substrates and stereochemical probes suggested the intermediacy of a free radical. The group ruled out a rapid rebound mechanism by carrying out cross coupling reactions for alkenyl halide radical clock substrate 55 at different catalyst concentrations (Scheme 44a). Higher selectivity for the linear product 56 as opposed to the cyclic product 57 was observed with increasing catalyst concentration, which is inconsistent with a radical rebound mechanism where the selectivity is expected to be insensitive to catalyst concentration due to both the radical rebound and the radical cyclization reactions being effectively zero order in catalyst.
 | ||
| Scheme 44 (a) Radical clock probe with substrate 55 and catalyst 54. (b) Proposed bimetallic oxidative addition mechanism for iron-catalysed cross coupling of alkyl halides and aryl magnesium bromides.35 | ||
Based on these studies, the group proposed a mechanism for cross coupling that involved a bimetallic oxidative addition (Scheme 44b). The kinetic studies were most consistent with a bimolecular mechanism for transmetalation that precedes oxidative addition, which was proposed to occur from the iron(II) biaryl “ate” complex 58 rather than the neutral iron(II) substrate 59. Liberation of the free radical followed by radical recombination with a second equivalent of 58 results in the iron(III) complex 60 that can undergo reductive elimination to give the cross coupled product and iron(I) species 61. To complete the catalytic cycle, the authors speculate that the iron(I) complex 61 reacts with the iron(III) product 62 that results from free radical formation so as to generate two iron(II) species in a comproportionation reaction. While this mechanism is consistent with the experimental data, the authors cannot rigorously rule out other possibilities, such as a unimolecular oxidative cleavage step involving radical recombination with iron(III) species 62 akin to the reaction mechanism in Scheme 40. Further experimentation is needed to rule out such possibilities.
Alkyl nucleophile–alkyl electrophile
Due in part to the paucity of systems, mechanistic studies involving the cross coupling of alkyl nucleophiles and alkyl halides are lacking. The most thorough study presented thus far is from the Cárdenas group, who have utilized iron(II) acetate combined with N-heterocyclic carbenes to effect alkyl–alkyl cross coupling reactions (Scheme 45).52 The reaction shares mechanistic features of the aryl nucleophile/alkyl electrophile cross coupling reactions (Scheme 39), namely the ring opening of radical clock substrates and the observation of an S = 1/2 species by EPR spectroscopy. This species was deduced to be an iron(I) intermediate from the amount of product formed from homocoupling of the nucleophile. For these reasons, the authors concluded that one of the two catalytic cycles shown in Scheme 45 were most likely. Interestingly, mechanistic studies from Tonzetich and co-workers for a very similar system (the two groups used slightly different N-heterocyclic carbene ligands and the Cárdenas group generated their iron complexes in situ) is more consistent with an iron(II)/iron(III) reaction mechanism analogous to the mechanism proposed by Nakamura (Scheme 40).100 These studies focused on isolating iron complexes that were presumed intermediates and demonstrating their catalytic competency. Considering the limited data presented thus far, it is difficult to ascertain whether there is a unified mechanism for alkyl/alkyl cross coupling reactions or whether these systems are particularly sensitive to subtle changes in reaction conditions.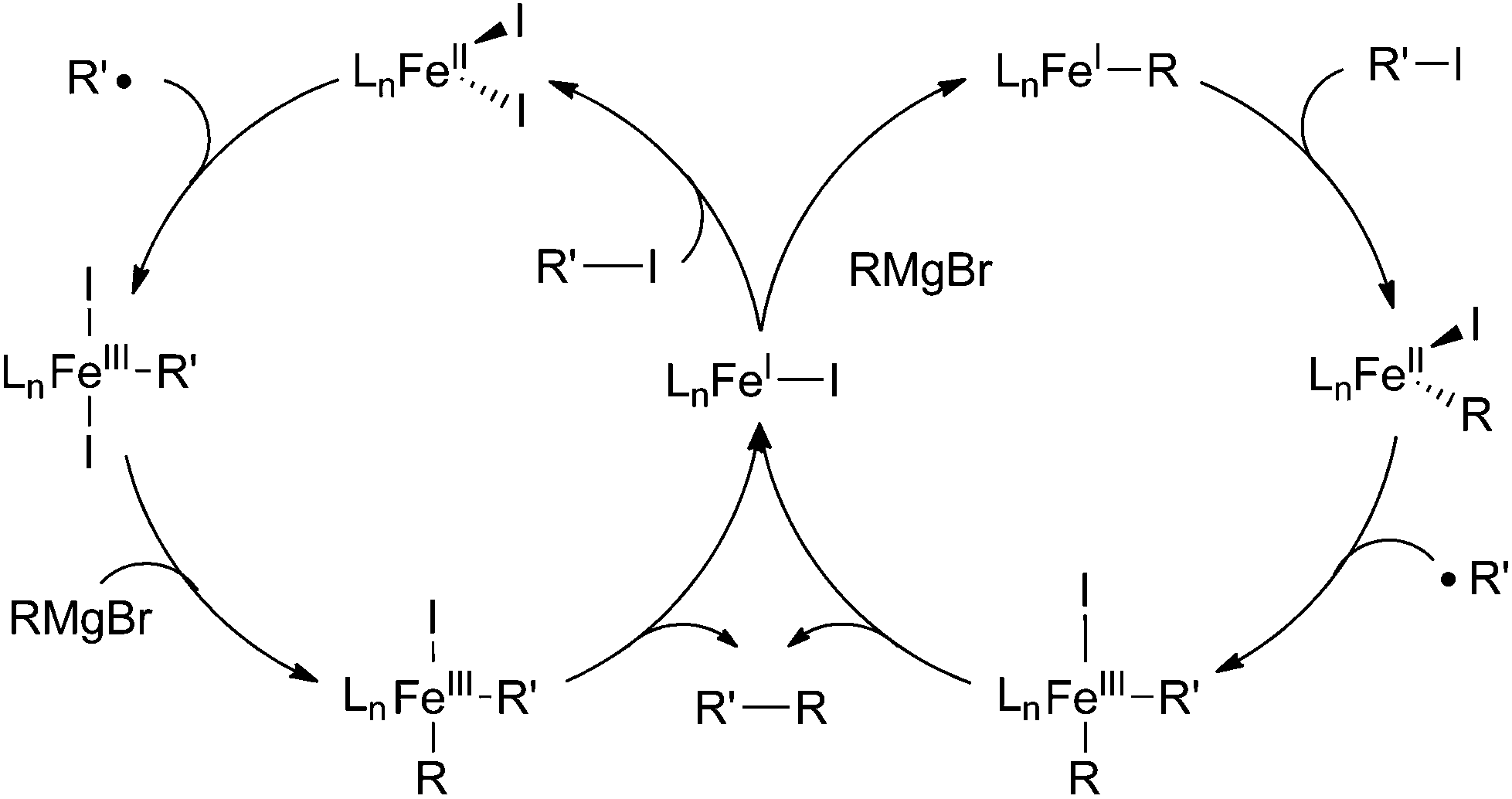 | ||
| Scheme 45 Proposed mechanism for the iron-catalysed cross coupling of alkyl Grignard reagents with alkyl iodides.52 | ||
Mechanistic insights pertaining to transmetalation
The majority of mechanistic investigations for iron-catalysed cross coupling reactions have been carried out for Kumada-type reactions. While the development of cross coupling protocols that utilize transmetalating reagents other than Grignard reagents is still in its infancy, there has been some effort directed toward understanding the key transmetalation step that distinguishes the types of cross coupling reactions.In a particularly relevant study, Ingleson and coworkers studied stoichiometric reactions between NHC iron complex 63 and aryl borate 64 (Scheme 46a).105 These studies revealed that 64 undergoes transmetalation with iron to selectively deliver methoxide in preference to aryl, which reflects the greater oxophilicity of iron compared to palladium. When the authors remove this competition by utilizing borate 65, transmetalation to iron was observed when reacted with (dppe)FeCl2 (23) or (dppbz)FeCl2 (22) at elevated temperatures (Scheme 46b).106 Aryl transmetalation at room temperature using 65 was not observed, but when the authors utilized the borate (tBu)(Ph)B(pin) instead, transmetalation to 23 occurred readily at room temperature and in the absence of any additives. This finding is significant because (tBu)(Ph)B(pin) was utilized in catalytic cross coupling reactions by Bedford,67 but such reactions required the presence of MgBr2 to proceed in high yields.
 | ||
| Scheme 46 (a) Transmetalation of borate 64 to NHC-iron complex 63.105 (b) transmetalation of borate 65 to iron complexes 22 and 23.106 (c) catalytic cross coupling using borate 65 facilitated by zincate 67 and catalysed by iron complex 23.106 | ||
Interestingly, when Ingleson employed his borate 65 in cross coupling reactions with alkyl halides, the desired cross coupled product was only observed when zincate 67 was used an additive (Scheme 46c). In the absence of such additives, the homocoupled product derived from borate 65 was the only product observed. These results highlight the sophistication of iron-catalysed cross coupling reactions and they emphasize the need for more studies such as these so that new developments may occur.
Conclusions
Since the early 2000s, the field of iron-catalysed cross coupling has experienced a renaissance period, which has resulted in the development of many useful alternatives to palladium-catalysed cross coupling reactions. Moreover, iron-catalysed reactions that are difficult to catalyse with palladium have also emerged. Nevertheless, established methods that utilize iron catalysts for cross coupling reactions have thus far been largely relegated to basic transmetalation reagents, such as Grignard reagents. While these reactions demonstrate remarkable chemoselectivity due to extremely fast cross coupling kinetics, this limitation is likely the reason why iron-based catalysts have not found as wide spread use as catalysts based on palladium and nickel. In order to advance the field further, more general methods are needed that involve iron catalysts and less basic coupling partners. Better mechanistic understanding of the various pathways available to iron-catalysed cross coupling reactions are also needed to achieve this goal. Moreover, there are very few examples of iron-catalysed cross coupling reactions that proceed enantioselectively, which is an area that will become more important as methods continue to develop for the cross coupling of sp3-hybridized coupling partners. Despite the significant challenges that remain, recent developments in the field including productive collaborations between organic and inorganic chemists are encouraging for the development of cross coupling reactions involving iron-based catalysts.Notes and references
- J. Terao and N. Kambe, Acc. Chem. Res., 2008, 41, 1545 CrossRef CAS PubMed.
- N. Kambe, T. Iwasaki and J. Terao, Chem. Soc. Rev., 2011, 40, 4937 RSC.
- R. Jana, T. P. Pathak and M. S. Sigman, Chem. Rev., 2011, 111, 1417 CrossRef CAS PubMed.
- M. Tamura and J. K. Kochi, J. Am. Chem. Soc., 1971, 93, 1487 CrossRef CAS.
- (a) C. Bolm, J. Legros, J. Le Paih and L. Zani, Chem. Rev., 2004, 104, 6217 CrossRef CAS PubMed; (b) A. Fürstner and R. Martin, Chem. Lett., 2005, 34, 624 CrossRef; (c) S. Ito and M. Nakamura, Organomet. News, 2007, 83 CAS; (d) W. M. Czaplik, M. Mayer, J. Cvengros and A. Jacobi von Wangelin, ChemSusChem, 2009, 2, 396 CrossRef CAS PubMed; (e) W. M. Czaplik, M. Mayer, S. Grupe and A. Jacobi von Wangelin, Pure Appl. Chem., 2010, 82, 1545 CrossRef CAS; (f) P. Knochel, T. Thaler and C. Diene, Isr. J. Chem., 2010, 50, 547 CrossRef CAS; (g) T. Mesganaw and N. K. Garg, Org. Process Res. Dev., 2013, 17, 29 CrossRef CAS; (h) E. Nakamura, T. Hatakeyama, S. Ito, K. Ishizuka, L. Ilies and M. Nakamura, Org. React., 2014, 83, 1 CAS; (i) R. B. Bedford and P. B. Brenner, Top. Organomet. Chem., 2015, 50, 19 CrossRef; (j) O. M. Kuzmina, A. K. Steib, A. Moyeux, G. Caheiz and P. Knochel, Synthesis, 2015, 1696 CAS.
- B. D. Sherry and A. Fürstner, Acc. Chem. Res., 2008, 41, 1500 CrossRef CAS PubMed.
- R. B. Bedford, Acc. Chem. Res., 2015, 48, 1485 CrossRef CAS PubMed.
- A. Hedström, Z. Izakian, I. Vreto, C.-J. Wallentin and P.-O. Norrby, Chem. – Eur. J., 2015, 21, 5946 CrossRef PubMed.
- (a) T. Hatakeyama and M. Nakamura, J. Am. Chem. Soc., 2007, 129, 9844 CrossRef CAS PubMed; (b) T. Hatakeyama, S. Hashimoto, K. Ishizuka and M. Nakamura, J. Am. Chem. Soc., 2009, 131, 11949 CrossRef CAS PubMed.
- (a) M. Talifer, N. Xia and A. Oualli, Angew. Chem., Int. Ed., 2007, 46, 934 CrossRef PubMed; (b) X. Wu and C. Darcel, Eur. J. Org. Chem., 2009, 5753 Search PubMed; (c) D. Guo, H. Huang, Y. Zhou, J. Xu, H. Jian, K. Chen and H. Liu, Green Chem., 2010, 12, 276 RSC.
- I. Thomé, A. Nijs and C. Bolm, Chem. Soc. Rev., 2012, 41, 979 RSC.
- Y.-Y. Chua and H. A. Duong, Chem. Commun., 2014, 50, 8424 RSC.
- T. Agrawal and S. P. Cook, Org. Lett., 2014, 16, 5080 CrossRef CAS PubMed.
- Y. Zhang, B. Liu, H. Y. Wu and W. Z. Chen, Chin. Sci. Bull., 2012, 57, 2368 CrossRef CAS.
- O. M. Kuzmina, A. K. Steib, D. Flubacher and P. Knochel, Org. Lett., 2012, 14, 4818 CrossRef CAS PubMed.
- O. M. Kuzmina, A. K. Steib, J. T. Markiewicz, D. Flubacher and P. Knochel, Angew. Chem., Int. Ed., 2013, 52, 4945 CrossRef CAS PubMed.
- O. M. Kuzmina, A. K. Steib, S. Fernandez, W. Boudot, J. T. Markiewicz and P. Knochel, Chem. – Eur. J., 2015, 21, 8232 CrossRef PubMed.
- M. Sova, R. Frlan, S. Gobec, G. Stavber and Z. Časar, Appl. Organomet. Chem., 2015, 29, 528 CrossRef CAS.
- R. N. Shakhmaev, A. S. Sunagatullina and V. V. Zorin, Russ. J. Org. Chem., 2014, 50, 322 CrossRef CAS.
- W. M. Czaplik, M. Mayer and A. Jacobi von Wangelin, ChemCatChem, 2001, 3, 135 CrossRef.
- S. Kawamura and M. Nakamura, Chem. Lett., 2013, 42, 183 CrossRef CAS.
- Y. Yamaguchi, H. Ando, M. Nagaya, H. Hinago, T. Ito and M. Asami, Chem. Lett., 2011, 40, 983 CrossRef CAS.
- S. E. Denmark and A. J. Cresswell, J. Org. Chem., 2013, 78, 12593 CrossRef CAS PubMed.
- R. B. Bedford, P. B. Brenner, E. Carter, P. M. Cogswell, M. F. Haddow, J. N. Harvey, D. M. Murphy, J. Nunn and C. H. Woodall, Angew. Chem., Int. Ed., 2014, 53, 1804 CrossRef CAS PubMed.
- T. Hatakeyama, Y. Fujiwara, Y. Okada, T. Itoh, T. Hashimoto, S. Kawamura, K. Ogata, H. Takaya and M. Nakamura, Chem. Lett., 2011, 40, 1030 CrossRef CAS.
- S. K. Ghorai, M. Jin, T. Hatakeyama and M. Nakamura, Org. Lett., 2012, 14, 1066 CrossRef CAS PubMed.
- Z. Mo, Q. Zhang and L. Deng, Organometallics, 2012, 31, 6518 CrossRef CAS.
- C. Sun, H. Krause and A. Fürstner, Adv. Synth. Catal., 2014, 356, 1281 CrossRef CAS.
- S. L. Daifuku, M. H. Al-Afyouni, B. E. R. Snyder, J. L. Kneebone and M. L. Neidig, J. Am. Chem. Soc., 2014, 136, 9132 CrossRef CAS PubMed.
- D. Parmar, L. Henkel, J. Dib and M. Rueping, Chem. Commun., 2015, 51, 2111 RSC.
- W. M. Czaplik, M. Mayer, S. Grupe and A. Jacobi von Wangelin, Pure Appl. Chem., 2010, 82, 1545 CrossRef CAS.
- M. Jin and M. Nakamura, Chem. Lett., 2011, 40, 1012 CrossRef CAS.
- M. Jin, L. Adak and M. Nakamura, J. Am. Chem. Soc., 2015, 137, 7128 CrossRef CAS PubMed.
- G. Bauer, C. W. Cheung and X. Hu, Synthesis, 2015, 1726 CAS.
- G. Bauer, M. D. Wodrich, R. Scopelliti and X. Hu, Organometallics, 2015, 34, 289 CrossRef CAS.
- A. K. Steib, T. Thaler, K. Komeyama, P. Mayer and P. Knochel, Angew. Chem., Int. Ed., 2011, 50, 3303 CrossRef CAS PubMed.
- M. C. Perry, A. N. Gillett and T. C. Law, Tetrahedron Lett., 2012, 53, 4436 CrossRef CAS.
- R. Agata, T. Iwamoto, N. Nakagawa, K. Isozaki, T. Hatakeyama, H. Takaya and M. Nakamura, Synthesis, 2015, 1733 CAS.
- W.-J. Guo and Z.-X. Wang, Tetrahedron, 2013, 69, 9580 CrossRef CAS.
- X. Chen, Z.-J. Quan and X.-C. Wang, Appl. Organomet. Chem., 2015, 29, 296 CrossRef CAS.
- G. Cahiez and H. Avedissian, Synthesis, 1998, 1199 CrossRef CAS.
- A. L. Silberstein, S. D. Ramgren and N. K. Garg, Org. Lett., 2012, 14, 3796 CrossRef CAS PubMed.
- T. Agrawal and S. P. Cook, Org. Lett., 2013, 15, 96 CrossRef CAS PubMed.
- S. Gülak, T. N. Gieshoff and A. Jacobi von Wangelin, Adv. Synth. Catal., 2013, 355, 2197 CrossRef.
- S. Malhotra, P. S. Seng, S. G. Koenig, A. J. Deese and K. A. Ford, Org. Lett., 2013, 15, 3698 CrossRef CAS PubMed.
- C. Sun and A. Fürstner, Angew. Chem., Int. Ed., 2013, 52, 13071 CrossRef CAS PubMed.
- H. Hishikado, H. Nakatsuji, K. Ueno, R. Nagase and Y. Tanabe, Synlett, 2010, 14, 2087 Search PubMed.
- D. Gärtner, A. L. Stein, S. Grupe, J. Arp and A. Jacobi von Wangelin, Angew. Chem., Int. Ed., 2015, 54, 10545 CrossRef PubMed.
- G. Cahiez, O. Gager, J. Buendia and C. Patinote, Chem. – Eur. J., 2012, 18, 5860 CrossRef CAS PubMed.
- Reprotoxic Category 2, R61, Official Journal of the European Union, December 31, 2008, European regulation No 1272/2008.
- K. G. Dongol, H. Koh, M. Sau and C. L. L. Chai, Adv. Synth. Catal., 2007, 349, 1015 CrossRef CAS.
- M. Guisán-Ceinos, F. Tato, E. Bruñuel, P. Calle and D. J. Cárdenas, Chem. Sci., 2013, 4, 1098 RSC.
- Y. Gartia, S. Pulla, P. Ramidi, C. C. Farris, Z. Nima, D. E. Jones, A. S. Biris and A. Ghosh, Catal. Lett., 2012, 142, 1397 CrossRef CAS.
- T. Hatakeyama, Y. Okada, Y. Yoshimoto and M. Nakamura, Angew. Chem., Int. Ed., 2011, 50, 10973 CrossRef CAS PubMed.
- C. W. Cheung, P. Ren and X. Hu, Org. Lett., 2014, 16, 2566 CrossRef CAS PubMed.
- T. Hatakeyama, N. Nakagawa and M. Nakamura, Org. Lett., 2009, 11, 4496 CrossRef CAS PubMed.
- X. Lin, F. Zheng and F.-L. Qing, Organometallics, 2012, 31, 1578 CrossRef CAS.
- R. B. Bedford, M. Huwe and M. C. Wilkinson, Chem. Commun., 2009, 600 RSC.
- R. B. Bedford, E. Carter, P. M. Cogswell, N. J. Gower, M. E. Haddow, J. N. Harvey, D. M. Murphy, E. C. Neeve and J. Nunn, Angew. Chem., Int. Ed., 2013, 52, 1285 CrossRef CAS PubMed.
- Although several of the transmetalating reagents that have been used are derived from boronic esters, all anionic, tetra-coordinated boron-containing compounds are referred to as borates (e.g. [R4B]−, [RB(OR)3]−, [R2B(OR)2]−, [R3B(OR)]−, etc.). Boronates are neutral, three-coordinated boron-containing compounds that are derived from boronic esters (e.g. B(OR)3, RB(OR)2, R2B(OR), etc.) and boranes are neutral, trialkyl or triaryl boron-containing species.
- R. B. Bedford, M. Huwe and M. C. Wilkinson, Chem. Commun., 2009, 600 RSC.
- T. Hatakeyama, T. Hashimoto, Y. Kondo, Y. Fujiwara, H. Seike, H. Takaya, Y. Tamada, T. Ono and M. Nakamura, J. Am. Chem. Soc., 2010, 132, 10674 CrossRef CAS PubMed.
- T. Hashimoto, T. Hatakeyama and M. Nakamura, J. Org. Chem., 2012, 77, 1168 CrossRef CAS PubMed.
- N. Nakagawa, T. Hatakeyama and M. Nakamura, Chem. Lett., 2015, 44, 486 CrossRef CAS.
- T. Hatakeyama, T. Hashimoto, K. K. A. D. S. Kathriarachchi, T. Zenmyo, H. Seike and M. Nakamura, Angew. Chem., Int. Ed., 2012, 51, 8834 CrossRef CAS PubMed.
- R. B. Bedford, P. B. Brenner, E. Carter, J. Clifton, P. M. Cogswell, N. J. Gower, M. F. Haddow, J. N. Harvey, J. A. Kehl, D. M. Murphy, E. C. Neeve, M. L. Neidig, J. Nunn, B. E. R. Snyder and J. Taylor, Organometallics, 2014, 33, 5767 CrossRef CAS.
- R. B. Bedford, P. B. Brenner, E. Carter, T. W. Carvell, P. M. Cogswell, T. Gallagher, J. N. Harvey, D. M. Murphy, E. C. Neeve, J. Nunn and D. R. Pye, Chem. – Eur. J., 2014, 20, 7935 CrossRef CAS PubMed.
- R. B. Bedford, T. Gallagher, D. R. Pye and W. Savage, Synthesis, 2015, 1761 CrossRef CAS.
- M. Carril, A. Correa and C. Bolm, Angew. Chem., Int. Ed., 2008, 47, 4862 CrossRef CAS PubMed.
- T. Hung, C. Huang and F. Tsai, ChemCatChem, 2012, 4, 540 CrossRef CAS.
- J. Yang, G. Shen and D. Chen, Synth. Commun., 2013, 43, 837 CrossRef CAS.
- R. Loska, C. M. R. Volla and P. Vogel, Adv. Synth. Catal., 2008, 350, 2859 CrossRef CAS.
- A. R. Hajipour and G. Azizi, Green Chem., 2013, 15, 1030 RSC.
- S. Yashuda, H. Yorimitsu and K. Oshima, Organometallics, 2010, 29, 2634 CrossRef.
- G. W. Waldhart and N. P. Mankad, J. Org. Chem., 2015, 793, 171 CrossRef CAS.
- S. Kawamura, K. Ishizuka, H. Takaya and M. Nakamura, Chem. Commun., 2010, 46, 6054 RSC.
- S. Kawamura, T. Kawabata, K. Ishizuka and M. Nakamura, Chem. Commun., 2012, 48, 9376 RSC.
- S. Kawamura, R. Agata and M. Nakamura, Org. Chem. Front., 2015, 2, 1053 RSC.
- A. D. Benischke, A. J. A. Breuillac, A. Moyeux, G. Cahiez and P. Knochel, Synlett, 2016, 27, 471 CAS.
- D. Castatgnolo and M. Botta, Eur. J. Org. Chem., 2010, 3224 CrossRef.
- K. M. Liu, J. Wei and X. F. Duan, Chem. Commun., 2015, 51, 4655 RSC.
- T. Hatakeyama, R. Imayoshi, Y. Yoshimoto, S. K. Ghorai, M. Jin, H. Takaya, K. Norisuye, Y. Sohrin and M. Nakamura, J. Am. Chem. Soc., 2012, 134, 20262 CrossRef CAS PubMed.
- Y.-Y. Lin, Y.-J. Wang, C.-H. Lin, J.-H. Cheng and C.-F. Lee, J. Org. Chem., 2012, 77, 6100 CrossRef CAS PubMed.
- W. Hu and S. Zhang, J. Org. Chem., 2015, 80, 6128 CrossRef CAS PubMed.
- R. B. Bedford, M. Nakamura, N. J. Gower, M. F. Haddow, M. A. Hall, M. Huwe, T. Hashimoto and R. A. Okopie, Tetrahedron Lett., 2009, 50, 6110 CrossRef CAS.
- S. L. Buchwald and C. Bolm, Angew. Chem., Int. Ed., 2009, 48, 5586 CrossRef CAS PubMed.
- P.-F. Larsson, A. Correa, M. Carril, P.-O. Norrby and C. Bolm, Angew. Chem., Int. Ed., 2009, 48, 5691 CrossRef CAS PubMed.
- T. Kauffmann, Angew. Chem., Int. Ed. Engl., 1996, 35, 386 CrossRef CAS.
- R. S. Smith and J. K. Kochi, J. Org. Chem., 1976, 41, 502 CrossRef CAS.
- C. L. Kwan and J. K. Kochi, J. Am. Chem. Soc., 1976, 98, 4903 CrossRef CAS.
- A. Fürstner, R. Martin, H. Krause, G. Seidel, R. Goddard and C. W. Lehmann, J. Am. Chem. Soc., 2008, 130, 8773 CrossRef PubMed.
- M. H. Al-Afyouni, K. L. Fillman, W. W. Brennessel and M. L. Neidig, J. Am. Chem. Soc., 2014, 136, 15457 CrossRef CAS PubMed.
- A. Fürstner, A. Leitner, M. Mendez and H. Krause, J. Am. Chem. Soc., 2002, 124, 13856 CrossRef.
- L. E. Aleandri, B. Bogdanovic, P. Bons, C. Dürr, A. Gaidies, T. Hartwig, S. C. Huckett, M. Lagarden and U. Wilczok, Chem. Mater., 1995, 7, 1153 CrossRef CAS.
- B. Bogdanovic and M. Schwickardi, Angew. Chem., Int. Ed., 2000, 39, 4610 CrossRef CAS.
- K. Weber, E.-M. Schnöckelborg and R. Wolf, ChemCatChem, 2011, 3, 1572 CrossRef CAS.
- R. Schoch, W. Desens, T. Werner and M. Bauer, Chem. – Eur. J., 2013, 19, 15816 CrossRef CAS PubMed.
- J. Kleimark, A. Hedström, P.-F. Larsson, C. Johansson and P.-O. Norrby, ChemCatChem, 2009, 1, 152 CrossRef CAS.
- S. L. Daifuku, J. L. Kneebone, B. E. R. Snyder and M. L. Neidig, J. Am. Chem. Soc., 2015, 137, 11432 CrossRef CAS PubMed.
- J. A. Przyojski, K. P. Veggeberg, H. D. Arman and Z. J. Tonzetich, ACS Catal., 2015, 5, 5938 CrossRef CAS.
- Y. Yang and X. Huang, Synlett, 2008, 1366 CAS.
- G. Cahiez, V. Habiak, C. Duplais and A. Moyeux, Angew. Chem., Int. Ed., 2007, 46, 4364 CrossRef CAS PubMed.
- H. Takaya, S. Nakajima, N. Nakagawa, K. Isozaki, T. Iwamoto, R. Imayoshi, N. J. Gower, L. Adak, T. Hatakeyama, T. Honma, M. Takagaki, Y. Sunada, H. Nagashima, D. Hashizume, O. Takahashi and M. Nakamura, Bull. Chem. Soc. Jpn., 2015, 88, 410 CrossRef CAS.
- C. J. Adams, R. B. Bedford, E. Carter, N. J. Gower, M. F. Haddow, J. N. Harvey, M. Huwe, M. A. Cartes, S. M. Mansell, C. Mendoza, D. M. Murphy, E. C. Neeve and J. Nunn, J. Am. Chem. Soc., 2012, 134, 10333 CrossRef CAS PubMed.
- J. J. Dunsford, I. A. Cade, K. L. Fillman, M. L. Neidig and M. J. Ingleson, Organometallics, 2014, 33, 370 CrossRef CAS.
- J. J. Dunsford, E. R. Clark and M. J. Ingleson, Dalton Trans., 2015, 44, 20577 RSC.
| This journal is © the Partner Organisations 2016 |