
Microstructure construction and composition modification of CeO2 macrospheres with superior performance†
Shuaiyu
Jiang‡
a,
Xiaodan
Yang‡
a,
Jun
Chen
a,
Xianran
Xing
a,
Lianzhou
Wang
b and
Ranbo
Yu
*ab
aDepartment of Physical Chemistry, University of Science and Technology Beijing, Beijing 100083, PR China
bNanomaterials Centre, School of Chemical Engineering and AIBN, The University of Queensland, St. Lucia, Brisbane, QLD 4072, Australia. E-mail: ranboyu@ustb.edu.cn; Fax: +86-10-62332525; Tel: +86-10-62332525
First published on 27th October 2015
Abstract
Macro/nano-composite CeO2 spheres with variable diameters of 30–300 μm assembled from nanoparticles of 5–8 nm were synthesized through the calcination of the well assembled spherical Ce(COOH)3 precursor. The controlled hydrolysis of DMF is the key factor for both the crystallization of Ce(COOH)3 and the formation of its macrospheres. These hierarchical CeO2 macrospheres possess pretty high specific surface areas of up to ∼190 m2 g−1 and show an excellent ability to remove Cr(VI) from aqueous solution. Moreover, when substituted with Bi(III), the oxygen storage capacity (OSC) of these CeO2 spheres could be improved sharply to 3243.75 μmol [O] g−1. With an easy-to-manipulate macroscale spherical morphology, these materials are easily recycled. This synthesis opens a promising route for high performance metal oxides.
Introduction
In recent years, hierarchical structures with special morphologies and novel properties have attracted more and more research interest.1–4 Because hierarchical assemblies of nanoscale building blocks may have characteristics of both the nanostructures and the microstructures. And the ordered three dimensional (3D) nanostructures may show important role for the development of a nanodevice with high performance. Moreover, the assembled nanostructures may be more environmentally friendly than the nanosized materials regarding the problem of nanosecurity. Many methods have been used to prepare hierarchical 3D structures of various functional materials, including the thermal reduction process,5 thermal oxidation process,6 oriented aggregation,7 self-assembly of building blocks through hydrophobic interactions,8 and template-assisted synthesis.9CeO2 possesses remarkable properties due to its abundant oxygen vacancy defects, high oxygen storage capacity and ability to relatively easily shuttle between III and IV oxidation states. It has been extensively studied and employed in many applications, such as an active component of three-way catalysts (TWCs),10–12 photocatalysts for water oxidation,13 oxygen ion conductors in solid oxide fuel cells,14–16 and ultraviolet (UV) blocking materials in UV shielding.17 Because of the improvement in the redox properties, the specific surface area to volume ratio, and transport properties with respect to condensed bulk materials, nanostructured and porous CeO2 or CeO2-based compounds have attracted special attention.18–28 Recently, various nano-sized CeO2 compounds have been obtained. However, improvement of the surface area was not obvious. On the other hand, synthesis of mesoporous CeO2 could give a surface area up to 200 m2 g−1,21 but the random product morphology was not desirable for further applications. To build up hierarchical 3D nanostructures would be a promising route to CeO2 with larger specific surface areas and desirable morphologies. Mesoporous CeO2 microspheres with uniform flower-like morphologies were synthesized by using glucose and acrylamide as templates, but their surface area is only 92.2 m2 g−1.27 We explored new routes to build 3D multi-shell hollow structures.13,28 Although these structures show good performance as catalysts, their lower specific surface area of ∼95 m2 g−1 is still not desirable. Therefore a facile route for the growth of CeO2 with both high surface area and uniform morphology is still in need.
In this work, an in situ structure directing formation strategy is adopted in the synthesis of hierarchically macrospherical Ce(COOH)3 precursors by controlling the hydrolysis of DMF. The macrospherical Ce(COOH)3 precursors could be easily transformed into perfect CeO2 macrospheres upon calcination. The obtained CeO2 macrospheres possessed a high surface area and showed an excellent ability to remove Cr(VI) from aqueous solution. Moreover, when doped with Bi(III), the oxygen storage capacity (OSC) of the CeO2 increased significantly with no obvious decrease of the surface area.
Experimental
All the chemicals were purchased from Beijing Chemical Reagent Company and used without further purification.Preparation of CeO2 macrospheres and hollow macrospheres
The synthesis was performed in a system of (NH4)2Ce(NO3)6–EtOH–DMF, similar to the method we have reported in previous work.29 In a typical experiment, ammonium cerium nitrate was dissolved in grain alcohol under magnetic stirring for at least 30 min, followed by addition of DMF to form a transparent orange solution, the molar ratio of (NH4)2Ce(NO3)6–EtOH–DMF was 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :86:72. The solution was aged under hydrothermal conditions at 100–150 °C for 4–6 days in a Teflon-lined stainless steel autoclave. After washing thoroughly followed by drying at 60 °C, the macrospherical Ce(COOH)3 precursor was obtained. In order to adjust the precursor formation, aqueous HAc (wt 36%) was used as the reaction media instead of EtOH, and the hollow macrospheres of the Ce(COOH)3 precursor were synthesized. These two kinds of spherical precursors were further calcined at 350 °C for 3 h using a temperature programmed muffle furnace with a heating rate of 1 °C min−1, and the porous CeO2 macrospheres were finally obtained.
:86:72. The solution was aged under hydrothermal conditions at 100–150 °C for 4–6 days in a Teflon-lined stainless steel autoclave. After washing thoroughly followed by drying at 60 °C, the macrospherical Ce(COOH)3 precursor was obtained. In order to adjust the precursor formation, aqueous HAc (wt 36%) was used as the reaction media instead of EtOH, and the hollow macrospheres of the Ce(COOH)3 precursor were synthesized. These two kinds of spherical precursors were further calcined at 350 °C for 3 h using a temperature programmed muffle furnace with a heating rate of 1 °C min−1, and the porous CeO2 macrospheres were finally obtained.
Characterization
The phases and purity of the products were examined by X-ray powder diffraction (XRD) performed on a M21XVHF22 X-ray diffractometer (Japan) with Cu Kα radiation (λ = 1.5418 Å). The morphology of the products was observed by using a field emission scanning electron microscope (LEO1530) and a high resolution transmission electron microscope (HRTEM, Philips CM-30, 3000 kV). The nitrogen adsorption–desorption isotherms at 77 K were measured using a Quantachrome Instruments AUTOSOR8–1C Powders Adsorption Analyzer.The OSC and water treatment test
For the water treatment experiment, K2Cr2O7 was used as the source of Cr(VI), the pH values of the solutions were adjusted using HCl or NaOH. Solutions containing different concentrations of Cr(VI) were prepared and adjusted to pH = 3. Then, 0.05 g of the adsorbent sample was added to 25 mL of the above solution under stirring. After a specified time, the solid and liquid were separated and Atomic Absorption Spectrometry (AA-6800) was used to measure the chromium concentration in the remaining solutions. The adsorption isotherm was obtained by varying the initial Cr(VI) concentrations and stirring for 5 h at room temperature (20 °C).The OSC measurements were carried out at 500 °C. The measurements were carried out in a flow reactor system equipped with Solenoid valves for rapid introduction of (4%)CO + (1%)Ar + He or (2%)O2 + (1%)Ar + He pluses. Typically, 30 mg powders were loaded into a 1.0 cm id. quartz tube reactor and a total gas flow rate of 300 cm3 min−1 was employed. The signals of the outlet gas were detected by using an on-line quadrupole mass spectrometer (Omnistar 200). Prior to CO step measurements, the sample was heated in (2%)O2 + (1%)Ar + He at 773 K for at least 20 min. The sample was further purged in pure He for 30 min to remove oxygen from the system and then exposed to (4%)CO + (1%)Ar + He. The accumulated amount of CO2 per gram of catalyst was monitored as a function of time.
Results and discussion
Two kinds of the macrospherical precursors were hydrothermally synthesized by adjusting the synthesis conditions. The phase purity and crystal structure of the precursors were examined by XRD. The XRD patterns of the two samples (Fig. 1a) show that they are both pure and of the same phase. All the peaks could be readily indexed as rhombohedral cerium formate (ICSD no. 069333, space group: R3m). To obtain the final CeO2 products, the precursors would undergo calcination. The calcination temperature was determined according to the thermogravimetric (TG) analysis of the precursor (see ESI Fig. S1†). A total weight loss of 33.4% was observed in the temperature range of 180–310 °C due to the pyrolysis of the organic species in the precursor. So the CeO2 could be obtained from heating the cerium formate precursor at a temperature higher than 350 °C, which could be confirmed by the XRD pattern of the calcined product (Fig. 1b). The patterns of calcined products can be indexed as a pure face-centered cubic phase of CeO2 (PDF no. 43-1002, space group: Fm3m).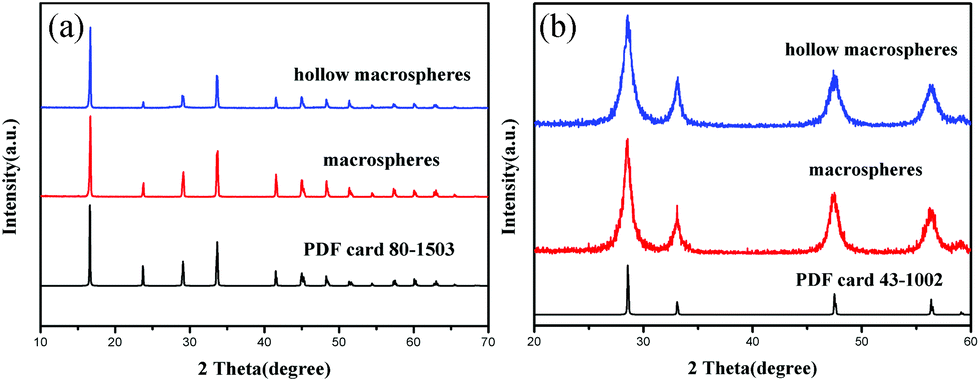 | ||
| Fig. 1 XRD patterns of (a) the macrospherical precursors and (b) calcined products. | ||
The morphology and structure of the precursors and final products were characterized by using the field emission scanning electron microscope (FE-SEM) and the high-resolution transmission electron microscope (HRTEM). It can be observed that by adjusting the hydrothermal synthesis conditions, general and hollow macrospheres of the precursor and the CeO2 could be obtained. Fig. 2a shows most of the as-synthesized Ce(COOH)3 precursor possesses a uniform spherical morphology with the average size of 300 μm, upon calcination at 350 °C these Ce(COOH)3 transformed into CeO2 spheres without any obvious shrinkage or destruction of the spherical morphologies (Fig. 2b). From the HRTEM image it can be seen that these CeO2 macrospheres are composed of many crystalline CeO2 nanoparticles with an average diameter of 5–8 nm. The hollow CeO2 macrospheres could also be obtained by calcining the as-prepared Ce(COOH)3 hollow macrospheres (Fig. 2c and d).
 | ||
| Fig. 2 FE-SEM image, (a) Ce(COOH)3 macrosphere precursor, inset is the surface of the macrospheres, (b) CeO2 macrospheres, inset is the HRTEM image of the CeO2 macrospheres, (c) Ce(COOH)3 hollow macrosphere precursor, inset is the surface of the hollow macrospheres (12 h), (d) CeO2 hollow macrospheres, cut with the blade showing the image of section. | ||
In this synthesis, DMF plays a crucial role in the formation of the microspheres. In the acid system, DMF might hydrolyze into formic acid and dimethylamine as follows:
 | (1) |
The formic acid would react with cerium sources to form Ce(COOH)3, and dimethylamine might act as the structure-directing agent to help the microsphere assembly. To determine the detailed effect of DMF, the following comparison processes have been tested. When formic acid was used instead of DMF, under the same reaction conditions, only a cluster of cerium formate just like Irish diamond could be obtained. And when formic acid and dimethylamine were used together, microrods of cerium formate with rough-and-tumble diameters could be synthesized but no macrosphere assembly could be observed (Fig. S3†). Obviously, both the hydrolysis process and products of DMF are dominant for the formation of Ce(COOH)3 macrospheres. Apparently, to realize the morphology control of the Ce(COOH)3 precursor it is necessary to control the hydrolysis rate of DMF. Because acid will enhance the hydrolysis of DMF, acetic acid and ethanol were used as the solvents, respectively. Under different hydrolysis rates of DMF, the formation of hollow and general hierarchical Ce(COOH)3 macrospheres was achieved, respectively (Fig. 2).
To gain insight into the formation process of cerium formate macrospheres, time-dependent experiments were performed. Products were collected at different stages from the reaction mixture once the precipitate had begun to appear in solution, and their morphologies were subjected to FE-SEM investigation. However, for the general Ce(COOH)3 macrospheres, it is difficult to capture the product images at different reaction stages, because the formation of the macrospheres is too fast after the Ce(COOH)3 nucleation. For the hollow Ce(COOH)3 macrospheres, the first sample was taken immediately after the formation of precipitate when reacted for 90 min, and the powder comprised broom-like microrods (Fig. S4a†). In this phase, the cerium formate formed a pyramid first, and then the top end was split into nanorods (Fig. S4a†). After 15 min, the nanorods with diameters of about 40 nm and lengths up to 6 μm began to self-assemble into microspheres. Although the nanorods grew thicker, the diameter of the microspheres remained constant. Hollow microspheres formed over the following 1.5 h (Fig. S4b†), which grew evidently bigger after 6 hours. From these four obvious morphology evolutionary stages a possible formation process is proposed as shown in Fig. 3. At first, the hydrolysis of the DMF led to the nucleation of Ce(COOH)3, which further grew into one-dimensional nanorods. With the increase of reaction time the concentration of organic amine increased, which would probably result in the increase of surface energy of the Ce(COOH)3 nanorods. To decrease the surface energy the bundles formed after treatment for 1.5 h. These bundles gradually organized into microspheres when the reaction time was extended to 3 h, which is possibly related to the structure-directing effect of the organic amine. Through the association with carboxylic acid and amino groups, hydrogen bonding patterns may direct the structure formation in the solution, generating specific supramolecular crystal architectures held together by networks of H-bonds.30,31 At the end of the reaction (12 h), a hollow macrosphere was observed. The N2 adsorption–desorption isotherms of Ce(COOH)3 and CeO2 macrospheres are given in the ESI(Fig. S5–8†), respectively. These isotherms lie between type-I and type-IV. The pore size distribution curves are shown in the inset to their own adsorption–desorption isotherm figures. The average pore diameters are shown in the ESI.† The distribution of the pore diameters is multistage. After calcination, the pore size distribution becomes wider. The BET surface areas of CeO2 macrospheres and hollow macrospheres calculated from these isotherms are 185.1 m2 g−1 and 154.7 m2 g−1, respectively.
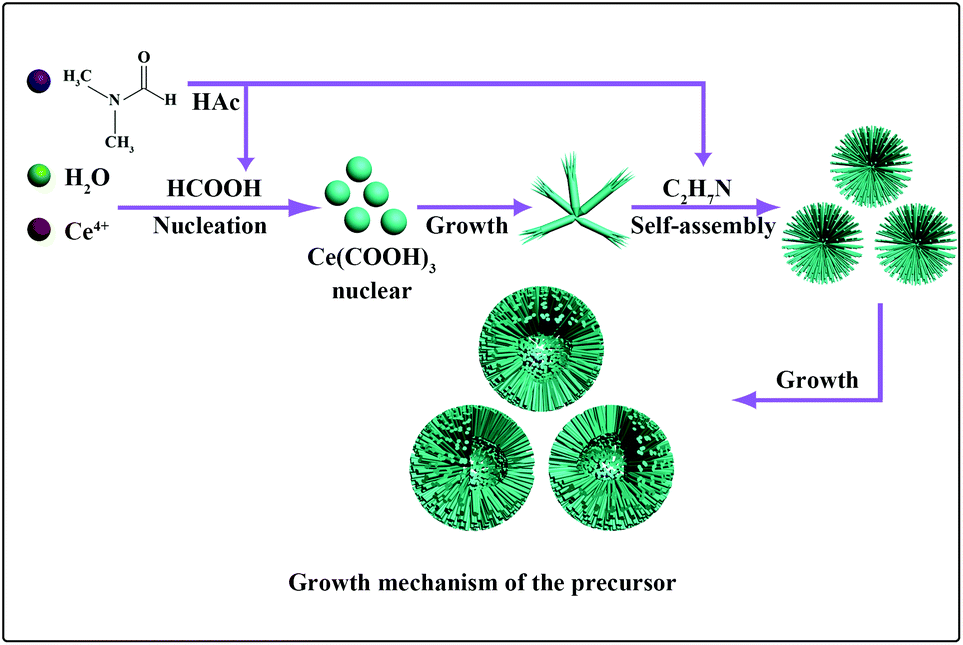 | ||
| Fig. 3 Possible formation process for the macrospheres. | ||
Clean water, free of toxic chemicals and pathogens, is vital to the world's health, and is also a critical feedstock in a variety of key industries. Some nanostructured metal oxides showed higher capacities for toxic ions and organic pollutant removal from water than bulk materials.32–34 Moreover, as the size of these oxides was several to hundred micrometers, the solid/liquid separation would be fairly easy. With the high specific surface area and hierarchical porous structure, the capacity for Cr(VI) removal from aqueous solutions of the as-synthesized CeO2 macrospheres was investigated. Chromium is considered as one of the primary highly toxic pollutants in water resources and its efficient removal from water is of great importance. Fig. S9† shows that when 0.05 g of the as-obtained CeO2 was added to 25 mL of chromium solution with an initial concentration of 21.00 mg L−1 and adjusted to pH = 3 at room temperature (20 °C), the adsorption capacity was measured as 7.9 mg Cr g−1, and the adsorption rate could reach 80%, especially, when the initial concentration of the chromium solution is 10 mg L−1, the adsorption rate could achieve 97%. The Cr removal ability of the hierarchical CeO2 macrospheres is much higher than that (adsorption rate: 65%) of the 3D flower-like CeO2 micro/nanocomposite structures.33
The total oxygen storage capacity (OSC) can express the maximum OSC and contains information on the overall reducibility of the solid. The OSC values of the as-synthesized CeO2 macrospheres and hollow macrospheres were measured as 1471.43 and 1168.75 μmol [O] g−1, respectively. For comparison, the OSC of the commercial cerium oxides with a particle size of about 50 nm and the BET specific surface area of 24.97 m2 g−1 has also been measured, and the corresponding OSC value of 611.61 μmol [O] g−1 was obtained, which is much lower than that of the as-synthesized CeO2 macrospheres. The possible reason is the higher surface area of the as-synthesized CeO2 macrospheres enhanced the oxygen storage capacity.
It is reported that when CeO2 is doped with Bi(III), the OSC can be improved, due to the increase of the surface oxygen vacancies.35 So in this work, further research studies on Bi(III) doped CeO2 macrospheres are also explored. The XRD pattern of Bi/CeO2 macrospheres (Fig. 4a) indicates the face-centered cubic phase of CeO2 has been maintained. The BET specific surface area of Bi/CeO2 could reach 192.7 m2 g−1 (Fig. S10†). EDX analysis (Fig. S11 and Table S1†) shows that the amount of Bi(III) is 4% (atom%). The FE-SEM image (Fig. 4b) shows the Bi/CeO2 macrospheres are uniform and in good distribution. There is no obvious change in the spherical morphologies after Bi(III) was doped. In the Raman spectra (Fig. 4c), a peak shift and a shoulder at around 490 cm−1 can be observed after Bi(III) was doped. These proved that the Bi(III) might be incorporated into the structure of CeO2. The OSC of the corresponding materials achieves much higher values, 3243.75 μmol [O] g−1. The dynamic OSC comparison of the CeO2 and the Bi/CeO2 macrospheres (Fig. 4d) at different temperatures in the range of 200 °C–600 °C indicated that the Bi doped sample shows much better performance than pure CeO2 macrospheres.
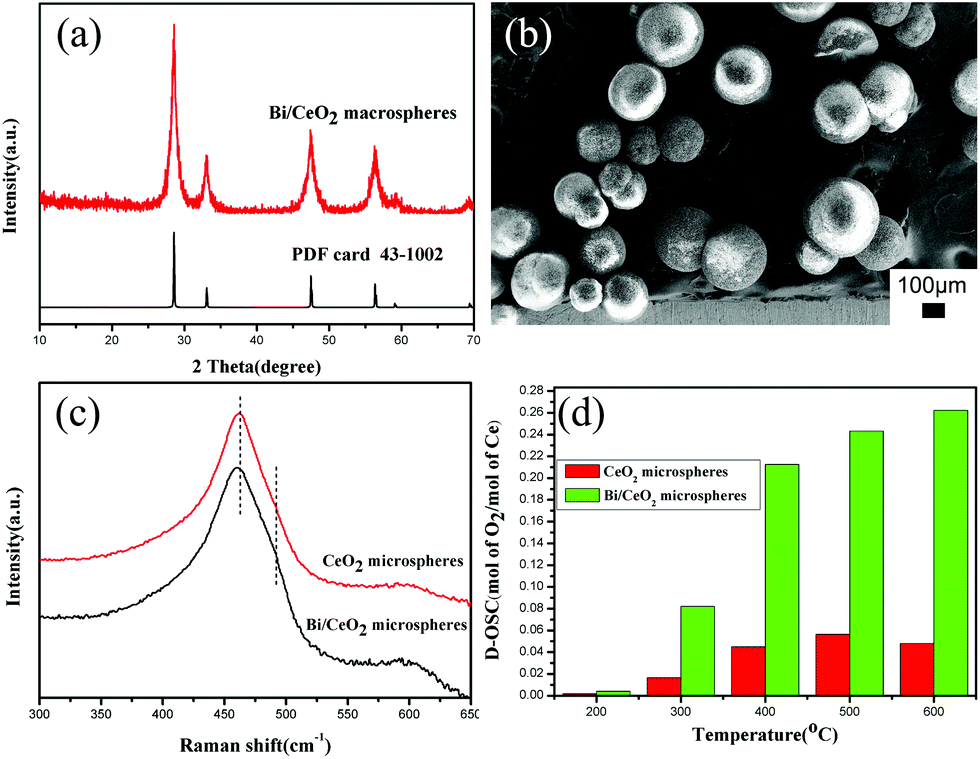 | ||
| Fig. 4 Bi doped CeO2 macrospheres: (a) XRD patterns, (b) FE-SEM image, and Raman spectra (c) and D-OSC (d) of CeO2 and Bi/CeO2 macrospheres. | ||
Conclusions
In summary, a facile route has been developed to synthesize hierarchical CeO2 macrospheres with high surface area and large pore volume. Controlled synthesis of Ce(COOH)3 precursor macrospheres with variable sizes could be realized under the low-temperature hydrothermal conditions. DMF and its hydrolysis in the reaction system contribute to both the crystallization of Ce(COOH)3 and the assembly of its macrospheres. With the high specific surface area and easy to control size, the final CeO2 macrospheres proved to be both effective sorbents for water treatment and potential catalysts with high OSC values for redox reactions. Furthermore, Bi(III) doping will further improve their OSC dramatically, which makes these materials promising candidates for techniques as catalysts, solid oxide fuel cells, and oxygen pumps. Moreover, this easily repeated synthesis route could also be used in related material preparation, and will open a new approach to functional materials with high specific surface area and performance.Acknowledgements
This work was financially supported by Grant 2014CB543401 from the National Key Program for Basic Research of 973 Program and the National Natural Science Foundation of China (No. 21271021, 51472025).Notes and references
- W. Shenton, D. Pum, U. B. Sleytr and S. Mann, Nature, 1997, 389, 585 CrossRef CAS.
- R. P. Panmand, Y. A. Sethi, S. R. Kadam, M. S. Tamboli, L. K. Nikam, J. D. Ambekar, C. J. Park and B. B. Kale, CrystEngComm, 2015, 17, 107 RSC.
- L. Passoni, L. Criante, F. Fumagalli, F. Scotognella, G. Lanzani and F. Di Fonzo, ACS Nano, 2014, 8, 12167 CrossRef CAS PubMed.
- Z. K. Zheng, W. Xie, Z. S. Lim, L. You and J. L. Wang, Sci. Rep., 2014, 4, 5721 CAS.
- Y. B. Li, Y. Bando and D. Golberg, Appl. Phys. Lett., 2003, 82, 1962 CrossRef CAS.
- A. C. Chen, X. S. Peng, K. Koczkur and B. Miller, Chem. Commun., 2004, 1964 RSC.
- B. Liu and H. C. Zeng, J. Am. Chem. Soc., 2004, 126, 8124 CrossRef CAS PubMed.
- M. R. Molla and S. Ghosh, Phys. Chem. Chem. Phys., 2014, 16, 26672 RSC.
- Z. Liu, Y. Yang, J. H. Mi, X. L. Tan and C. Lv, Int. J. Hydrogen Energy, 2013, 38, 4445 CrossRef CAS.
- E. Aneggi, C. de Leitenburg, G. Dolcetti and A. Trovarelli, Catal. Today, 2006, 114, 40 CrossRef CAS.
- L. G. Appel, J. G. Eon and M. Schmal, Phys. Status Solidi A, 1997, 163, 107 CrossRef CAS.
- A. Bruix, J. A. Rodriguez, P. J. Ramírez, S. D. Senanayake, J. Evans, J. B. Park, D. Stacchiola, P. Liu, J. Hrbek and F. Illas, J. Am. Chem. Soc., 2012, 134, 8968 CrossRef CAS PubMed.
- J. Qi, K. Zhao, G. D. Li, Y. Gao, H. J. Zhao, R. B. Yu and Z. Y. Tang, Nanoscale, 2014, 6, 4072 RSC.
- Z. L. Zhan and S. A. Bamett, Science, 2005, 308, 844 CrossRef CAS PubMed.
- H. J. Beie and A. Gnorich, Sens. Actuators, B, 1991, 4, 393 CrossRef CAS.
- P. Jasinski, T. Suzuki and H. U. Anderson, Sens. Actuators, B, 2003, 95, 73 CrossRef CAS.
- R. Si, Y. W. Zhang, L. P. You and C. H. Yan, J. Phys. Chem. B, 2006, 110, 5994 CrossRef CAS PubMed.
- A. Corma, P. Atienzar, H. Garcia and J. Y. Chane-Ching, Nat. Mater., 2004, 3, 394 CrossRef CAS PubMed.
- S. C. Laha and R. Ryoo, Chem. Commun., 2003, 2138 RSC.
- D. Terribile, A. Trovarelli, J. Llorca, C. de Leitenburg and G. Dolcetti, J. Catal., 1998, 178, 299 CrossRef CAS.
- D. M. Lyons, K. M. Ryan and M. A. Morris, J. Mater. Chem., 2002, 12, 1207 RSC.
- A. Bouchara, G. D. Soler-Illia, J. Y. Chane-Ching and C. Sanchez, Chem. Commun., 2002, 1234 RSC.
- S. Yang and L. Gao, J. Am. Chem. Soc., 2006, 128, 9330 CrossRef CAS PubMed.
- L. Yan, X. R. Xing, R. B. Yu, J. X. Deng, J. Chen and G. R. Liu, Phys. Rev. B: Condens. Matter, 2007, 390, 59 CrossRef CAS.
- L. Yan, R. B. Yu, J. Chen and X. R. Xing, Cryst. Growth Des., 2008, 8, 1474 CAS.
- R. B. Yu, L. Yan, P. Zheng, J. Chen and X. R. Xing, J. Phys. Chem. C, 2008, 112, 19896 CAS.
- C. W. Sun, J. Sun, G. L. Xiao, H. R. Zhang, X. P. Qiu, H. Li and L. Q. Chen, J. Phys. Chem. B, 2006, 110, 13445 CrossRef CAS PubMed.
- P. F. Xu, R. B. Yu, H. Ren, L. B. Zong, J. Chen and X. R. Xing, Chem. Sci., 2014, 5, 4221 RSC.
- X. Yao, X. D. Yang, R. B. Yu, P. F. Xu, J. Chen and X. R. Xing, Mater. Res. Bull., 2015, 61, 22 CrossRef CAS.
- J. Zhang, S. J. Liu, J. Lin, H. S. Song, J. J. Luo, E. M. Elssfah, E. Ammar, Y. Huang, X. X. Ding, J. M. Gao, S. R. Qi and C. G. Tang, J. Phys. Chem. B, 2006, 110, 14249 CrossRef CAS PubMed.
- J. M. Lehn, Supramolecular chemistry concepts and perspectives, Angew. Chem., Int. Ed. Engl., 1995, 34, 2563 Search PubMed.
- P. Li, D. E. Miser, S. Rabiei, R. T. Yadav and M. R. Hajaligol, Appl. Catal., B, 2003, 43, 151 CrossRef CAS.
- R. C. Wu, J. H. Qu and Y. S. Chen, Water Res., 2005, 39, 630 CrossRef CAS PubMed.
- L. S. Zhong, J. S. Hu, A. M. Cao, Q. Liu, W. G. Song and L. J. Wan, Chem. Mater., 2007, 19, 1648 CrossRef CAS.
- N. Imanaka, T. Masui, K. Koyabu, K. Minami and T. Egawa, Adv. Mater., 2007, 19, 1608 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: The TG-DSC isotherm, crystal structure, the N2 adsorption–desorption isotherms, FE-SEM images, SEM-EDAX. See DOI: 10.1039/c5qi00140d |
| ‡ These authors contributed equally to this work. |
| This journal is © the Partner Organisations 2016 |