
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Facile synthesis of multiarmed and multicomponent star polymers by a new iterative methodology using (formyl-protected 1,3-dioxolane)-end-functionalized polymer anions†
Raita
Goseki
 *ab,
Shotaro
Ito
a,
Emi
Akemine
a and
Akira
Hirao
cd
*ab,
Shotaro
Ito
a,
Emi
Akemine
a and
Akira
Hirao
cd
aDepartment of Organic and Polymeric Materials, Tokyo Institute of Technology, 2-12-1, Ookayama, Meguro-ku, Tokyo 152-8550, Japan. E-mail: rgoseki@polymer.titech.ac.jp; Fax: +81 3 5734 2131; Tel: +81 3 5734 2131
bDepartment of Chemical Science and Engineering, Tokyo Institute of Technology, 2-12-1, Ookayama, Meguro-ku, Tokyo 152-8552, Japan
cDepartment of Chemical Engineering, National Taiwan University, No. 1, Sec. 4, Roosevelt Road, Taiwan
dDepartment of Applied Chemistry, National Chiao Tung University, 1001 University Road, Hsinchu 30010, Taiwan
First published on 2nd September 2016
Abstract
A new and efficient iterative methodology using (formyl-protected 1,3-dioxolane (DOL))-end-functionalized polymer anions was carried out for the facile synthesis of multiarmed and multicomponent μ-star polymers. Using this methodology, both the arm introduction and DOL reintroduction steps are simultaneously performed by the linking reaction of the DOL-end-functionalized polymer anion with the formyl group regenerated from the DOL reintroduced in advance and repeated several times. By combining the DOL-end-functionalized polymer anions with non-DOL-functionalized polymer anions in the linking reaction, synthetically-difficult and well-defined complex μ-star polymers ranging from the ABC, ABCD, ABCDE, and ABCD2, to the ABCD2E2 types were successfully obtained. Moreover, synthesis of A3B2 and A3B2C4 star polymers was also achieved with the same methodology using (DOL)2-end-functionalized polymer anions. The arm segments were polystyrene, polystyrene derivatives, polyisoprene, poly(2-vinylpyridine), and poly(alkyl methacrylate(s)), which were introduced into the μ-star polymers via the formyl or α-phenylacrylate reaction site(s).
Introduction
Asymmetric star-branched polymers with chemical structures that emanate a number of different arm segments from a core, often named μ-star polymers, have attracted a great deal of attention in the field of nanoscience as multiphase materials creating characteristic nanostructures which are self-assembled in the bulk, as thin films, and in selected solvents.1–21 These nanostructures can be tailored by varying the star-branched architecture, and the composition, and chemical structure of the arm. Based on this, μ-star polymers are expected to be promising next-generation multiphase materials with future applications.μ-Star polymers composed of more than three different arm segments are quite limited in synthesis, even now, because the same number of selective reactions corresponding to the arm numbers is required. In order to solve this synthetic limitation, we have been developing several new methodologies on the basis of an iterative concept since 2001.22–47 A typical iterative methodology is shown in Scheme 1. An end-functionalized polymer (A) with 1,1-diphenylethylene (DPE) was prepared by the reaction of a 4-bromobutyl-substituted DPE, 1, with a DPE-end-capped living anionic polymer (A). A living polymer (B) reacted with the DPE terminus to link the polymer chain with the “A” chain. A new DPE anion was generated at the linking point, followed by a reaction with 1 to reintroduce the reaction site (DPE). A block copolymer of A-b-B with DPE between the two blocks was thus obtained. Since the resulting polymer possessed a DPE reaction site, the same reaction process using a living polymer (C) was iterated to synthesize an ABC core-DPE-functionalized star polymer. By iterating the process, both ABCD and ABCDE star polymers were successively obtained.
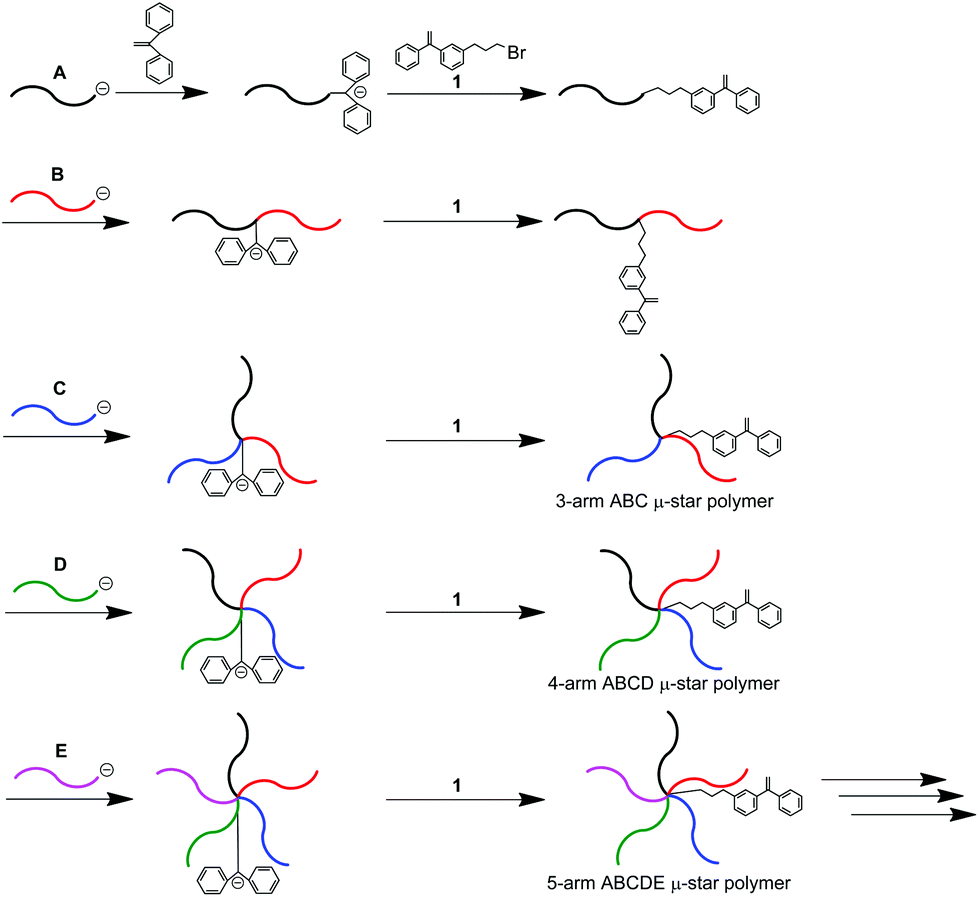 | ||
| Scheme 1 μ-Star polymer synthesis by an iterative methodology using 1. | ||
Very importantly, the need for the same number of selective reactions corresponding to arm numbers could be completely avoided in this methodology, since each arm segment was always introduced by a linking reaction in each process. With the above-mentioned and modified methodologies, we successfully synthesized new complex μ-star polymers, such as ABCDEF, ABCDEFG, A3B3C3, AB2C4, AB2C4D8, and even AB2C4D8E16 types.6,31,35–37,48–54
The living polymers of styrene, 1,3-butadiene, and isoprene quantitatively reacted with the DPE reaction site(s) to introduce their polymers as arms. Contrastingly, poly(2-vinylpyridine) (P2VP) and poly(alkyl methacrylate)s (PRMA)s with useful functionalities cannot be introduced because their living chain-end anions are not nucleophilic enough to react with the DPE reaction site(s). To realize a better generality and versatility of the iterative methodology, we explored a more reactive site capable of reacting with the less nucleophilic living P2VP and PRMAs and found that an α-phenylacrylate (PA) function satisfied our purpose,47,48,55–59 as shown in Scheme 2.
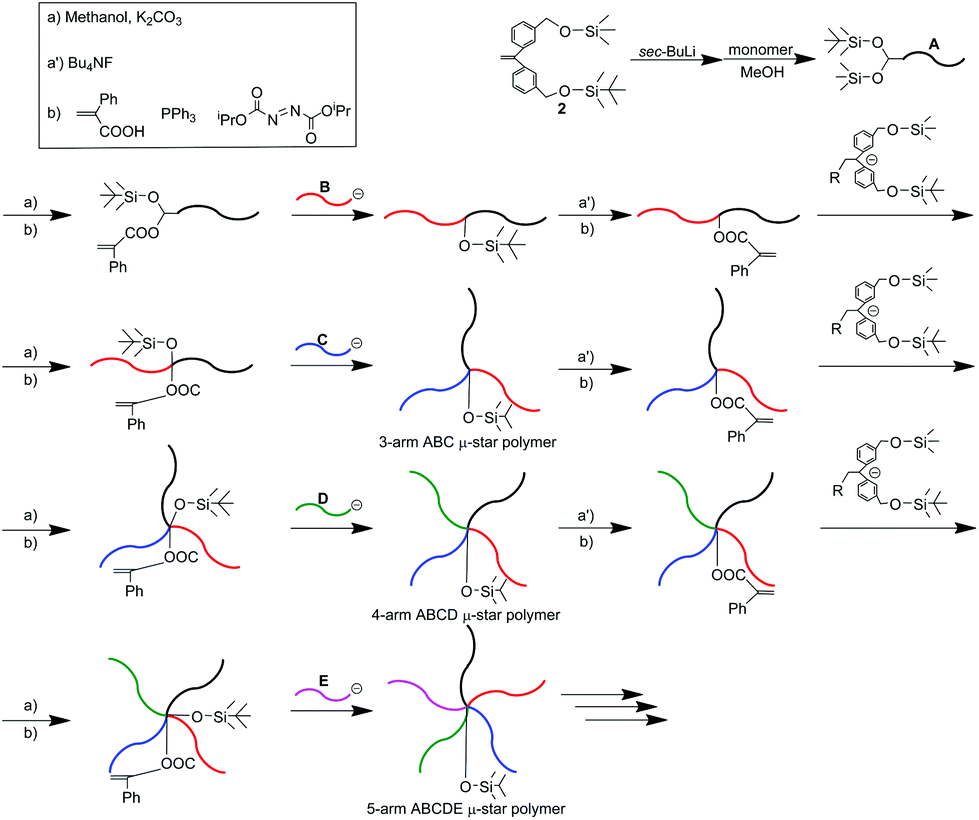 | ||
| Scheme 2 μ-Star polymer synthesis by an iterative methodology using a functional DPE anion substituted with different two silyl ethers. | ||
In this methodology, two different hydroxyl-protected functionalities, trimethylsilyl (TMS) and tert-butyldimethylsilyl (TBS) ethers, were adopted. The two silyl ethers, convertible to the PA reaction sites, were first introduced into the initiating chain-end of the methacrylate-based polymer (A) by the living anionic polymerization of a methacrylate monomer with a functional DPE anion, prepared from sec-BuLi, and DPE substituted with the two silyl ethers, 2. The TMS ether was selectively deprotected, followed by subsequent esterification to convert it to a PA reaction site. The resulting (A) polymer, functionalized with the PA and TBS termini, was reacted with a living PRMA (B) to afford an A-b-B molecule with the TBS group between A and B. The TBS ether was then deprotected with (C4H9)4NF, followed by esterification to convert it to a PA reaction site. Both of the two silyl ethers were again reintroduced by the reaction of the PA reaction site with the same DPE anion substituted with the same two silyl ethers as those used above. As seen in Scheme 2, the same process was iterated, resulting in quantitative formation of the ABC, ABCD, and ABCDE star polymers. Throughout the synthesis, the less nucleophilic living anionic P2VP and (PRMA)s were found to undergo the quantitative linking reaction with the PA reaction site to introduce their polymer segments as arms.55
We subsequently reported a second successful methodology, in which a formyl-protected 1,3-dioxolane (DOL) group was employed to repeat the reaction process. A DOL-substituted DPE derivative, 3, was prepared and changed to a functional DPE anion by treatment with sec-BuLi. MMA was then polymerized with this anion. DOL introduced at the initiating end was deprotected to regenerate the formyl group. The same DPE anion substituted with DOL reacted with the formyl terminus to reintroduce DOL together with the production of a hydroxyl group. The hydroxyl group was converted to a PA reaction site by esterification with α-phenylacryloyl chloride, followed by the reaction with living poly(benzyl methacrylate) (PBnMA) to quantitatively link PBnMA to the PMMA chain. During the esterification and linking reactions, DOL remained unchanged.
After deprotection of DOL, the same process with living poly(allyl methacrylate) (C) was repeated to afford an ABC star polymer, in which DOL was at the core. Repetition of the process twice enabled successive synthesis of the ABCD and ABCDE star polymers.57 Thus, the second methodology also satisfactorily worked for multiarmed and multicomponent star polymer synthesis. The difference between the first and second iterative methodologies is that two different functionalities sequentially converting to PA reaction sites are needed to repeat the reaction process in the first methodology, while the use of only a formyl-protected DOL group is sufficient to perform the same role in the latter methodology.
We now report a more efficient iterative methodology using DOL-end-functionalized polymer anions as an extension of the above-mentioned second methodology for the facile synthesis of multiarmed and multicomponent μ-star polymers.
Experimental section
Materials and characterization
Purification and preparation of the solvents and monomers used in this study were described in our previous papers.41,52,53,57,58 The general synthetic procedure for μ-star polymers and their characterization is described in the ESI.†Results and discussion
μ-Star polymer synthesis by a new iterative methodology using DOL-end-functionalized polymer anions (1)
As already mentioned, we reported μ-star polymer synthesis by an iterative methodology using a DOL-substituted DPE anion (see Scheme 3). Herein, we propose a new iterative methodology using DOL-end-functionalized polymer anions, which is more efficient than the above-mentioned original methodology. In this methodology, both the arm introduction and DOL reintroduction steps are simultaneously performed in one linking reaction. Accordingly, one of the two reaction steps is saved in each process. This has the great advantage of reducing the number of reaction steps in multiarmed and multicomponent star polymer synthesis which requires several reaction processes.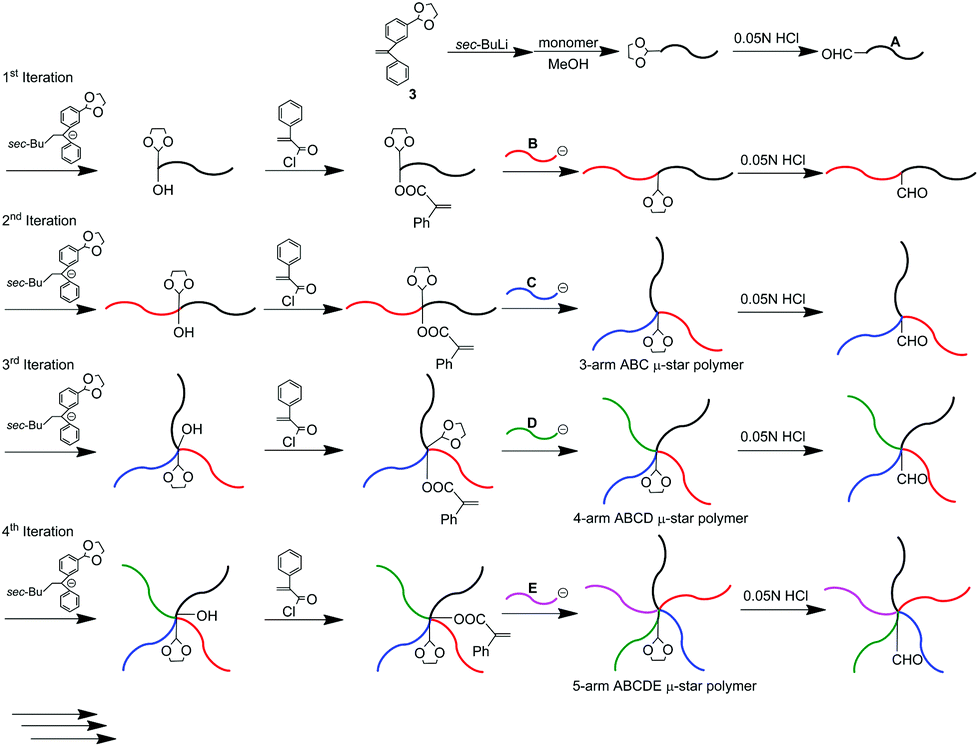 | ||
| Scheme 3 μ-Star polymer synthesis by an iterative methodology using a DOL-substituted DPE anion. | ||
As seen in Scheme 4, the living polymerization of MMA with the DOL-substituted DPE anion prepared from 3 and sec-BuLi is carried out to prepare a DOL-end-functionalized PMMA (A). The formyl terminus was regenerated by deprotection of DOL with a catalytic amount of p-toluene sulfonic acid. In the 1H NMR spectrum, quantitative formyl regeneration was confirmed by a new signal at 9.86 ppm assigned to the formyl proton. The resulting formyl-end-functionalized PMMA was reacted with the DOL-end-functionalized polystyrene (PS) anion (in a 1.2-fold excess) prepared from living PS and 3 to link the PS. The SEC profile showed two peaks for the linked product and the excess DOL-end-functionalized PS anion (Fig. 1(a)). The linking efficiency was estimated to be quantitative by comparing these two peak areas. The linked product was isolated by fractional precipitation and found to possess a sharp SEC peak (Fig. 1(b)) and a predictable molecular weight.
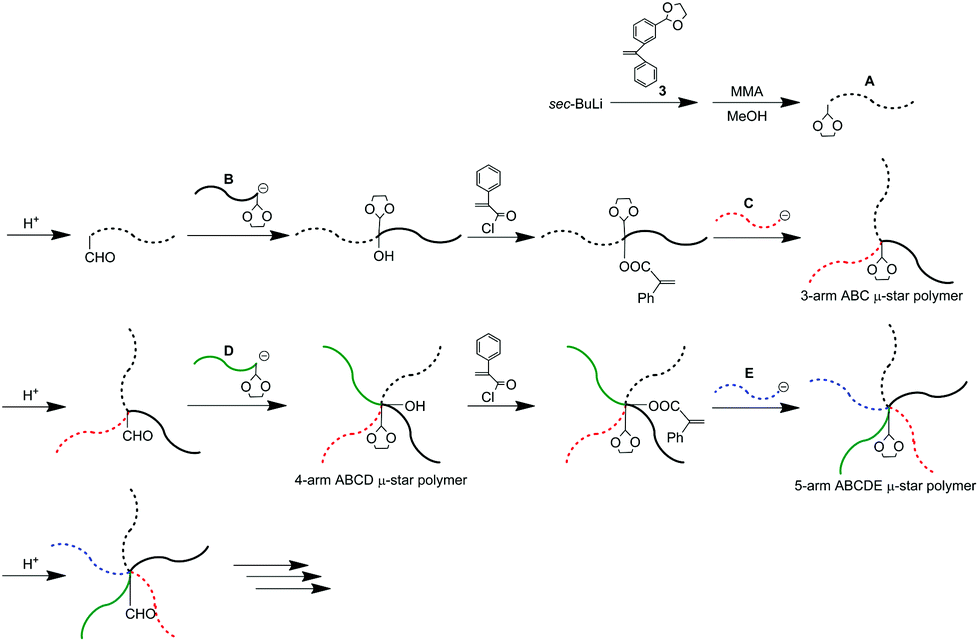 | ||
| Scheme 4 μ-Star polymer synthesis by a new methodology using DOL-end-functionalized polymer anions. | ||
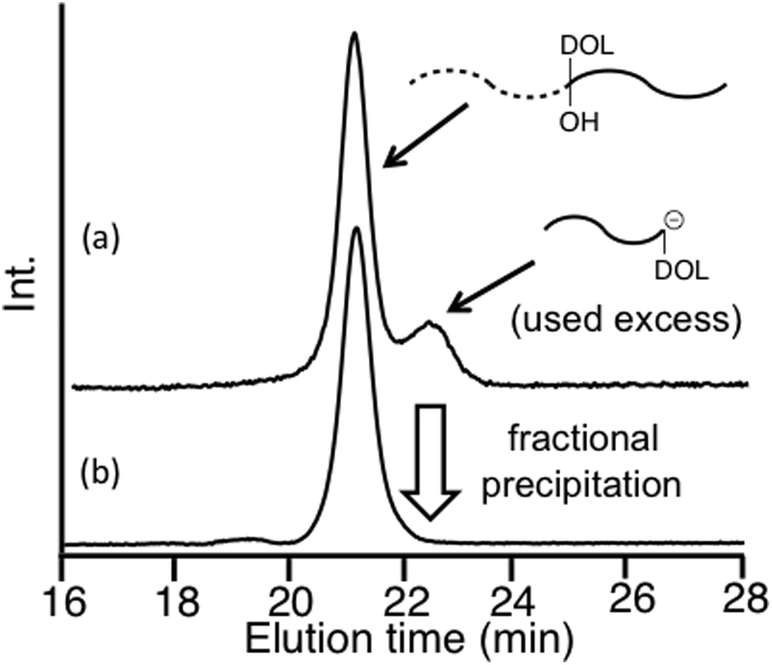 | ||
| Fig. 1 SEC profiles of the reaction mixture (a) and the isolated PMMA-b-PS with DOL and OH groups between the two blocks (b). | ||
Resonance peaks for reintroduced DOL were observed in the 1H NMR spectrum, while a signal for the hydroxyl group produced by the linking reaction was not clearly found in the same spectrum. After conversion of the hydroxyl group into the PA reaction site,57 NMR analysis based on the integrated ratio of the PA group indicated that the hydroxyl group was quantitatively produced.
The thus-prepared PMMA-b-PS with PA and DOL groups between the two blocks was reacted with the living anionic PBnMA (C). The quantitative linking efficiency was also estimated from the SEC profile of the reaction mixture. The isolated polymer was observed to be the expected ABC star polymer with a well-controlled structure (see Table 1).
| Type | M n (kg mol−1) | M w/Mn | A/B/C/D/E (E′) | |||
|---|---|---|---|---|---|---|
| Calcd | SEC | RALLSa | SEC | Calcd | Obsdb | |
a SEC–RALLS equipped with triple detectors.
b ![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 1H NMR; A/B/C/D/E (E′) = PMMA/PS/PBnMA/PαMS/PCPPHM (P2VP). 1H NMR; A/B/C/D/E (E′) = PMMA/PS/PBnMA/PαMS/PCPPHM (P2VP).
|
||||||
| A | 11.5 | 13.6 | — | 1.03 | 100/0/0/0/0 | 100/0/0/0/0 |
| AB | 27.2 | 26.8 | — | 1.03 | 51/49/0/0/0 | 53/47/0/0/0 |
| ABC | 37.5 | 33.7 | 37.8 | 1.04 | 41/39/20/0/0 | 41/37/22/0/0 |
| ABCD | 48.4 | 37.1 | 47.7 | 1.03 | 32/31/16/21/0 | 34/29/18/19/0 |
| ABCDE | 57.4 | 40.1 | 56.2 | 1.04 | 30/29/15/20/6 | 32/27/16/18/7 |
| ABCDE′ | 59.1 | 43.5 | 57.2 | 1.04 | 26/24/12/16/23 | 27/20/13/14/26 |
After deprotection of the DOL group, the same process was twice iterated. A DOL-end-functionalized PαMS anion (D), prepared from the living PαMS and 3, and the living anionic poly(6-(4-(4-cyanophenyl)phenoxy)hexyl methacrylate) (PCPPHMA) (E) were used, respectively. Both the linking reactions proceeded with almost quantitative efficiencies to afford the ABCD and ABCDE star polymers. SEC profiles of all of the polymers, shown in Fig. 2, exhibit that each of the peaks is sharp in distribution and moves toward the higher molecular weight region by iterating the process. The final ABCDE μ-star polymer, composed of PMMA, PS, PBnMA, PαMS, and PCPPHMA arms, possessed an expected molecular weight determined by SEC–RALLS (Mn = 56.2 kg mol−1) and a narrow polydispersity index of 1.04. The introduction of the E arm segment into the μ-star seems interesting because it shows liquid crystalline behaviour.
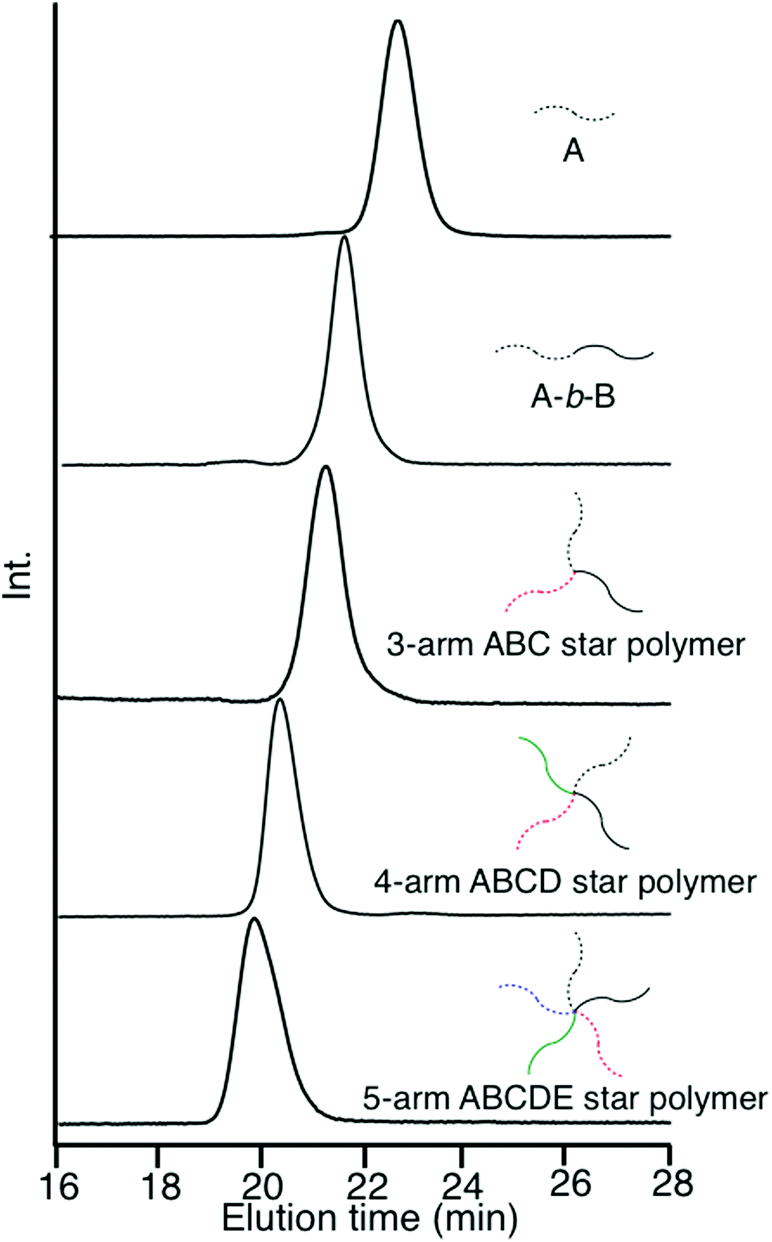 | ||
| Fig. 2 SEC profiles from the A, AB, 3-arm ABC, and 4-arm ABCD, to the 5-arm ABCDE μ-star polymers. | ||
To evaluate the possible introduction of the P2VP (E′) arm into the μ-star polymer, the living P2VP was reacted in situ with the same ABCD star core-functionalized with PA and DOL groups as that used in the above star polymer synthesis. Since the resulting polymer was the expected ABCDE′ star polymer, the living anionic P2VP was found to readily and quantitatively react with the PA reaction site (see Table 1).
One more ABCDE type star polymer was similarly synthesized using polymer anions prepared from all of the functional styrene derivatives. For this synthesis, the DOL-end-functionalized poly(4-methoxystyrene) (PMOS) anion, poly(4-methylstyrene) (PMS) anion, DOL-end-functionalized PS anion, PαMS anion, and DOL-end-functionalized poly(4-tert-butyldimethylsilyloxystyrene) (PTBSOS) anion were used in each reaction process. The PMOS (A), PS (C), and PTBSOS (E) anions were prepared by the reaction of the corresponding living anionic polymers with 3, while both the non-DOL-end-functionalized PMS (B) and PαMS (D) anions corresponded to DPE-end-capped polymer anions. All the linking reactions were observed to proceed almost quantitatively, resulting in the successful synthesis of the ABC, ABCD, and ABCDE star polymers (see Table 2). The PTBSOS arm was quantitatively converted to a hydrophilic poly(4-vinylphenol) segment, which showed an ionic character and water-solubility under basic conditions.
| Type | M n (kg mol−1) | M w/Mn | A/B/C/D/E | |||
|---|---|---|---|---|---|---|
| Calcd | SEC | RALLSa | SEC | Calcd | Obsdb | |
|
a SEC–RALLS equipped with triple detectors.
b 1H NMR; A/B/C/D/E = PMOS/PMS/PS/PαMS/PTBSOS.
|
||||||
| A | 11.8 | 11.2 | 11.6 | 1.03 | 100/0/0/0/0 | 100/0/0/0/0 |
| AB | 21.7 | 21.8 | 22.4 | 1.03 | 49/51/0/0/0 | 48/52/0/0/0 |
| ABC | 34.1 | 29.7 | 32.3 | 1.03 | 30/31/39/0/0 | 29/30/41/0/0 |
| ABCD | 43.0 | 36.6 | 43.4 | 1.04 | 23/24/30/23/0 | 21/25/32/22/0 |
| ABCDE | 54.4 | 44.5 | 54.7 | 1.04 | 20/21/27/21/11 | 19/22/29/20/10 |
The use of the DOL-end-functionalized polymer anions significantly reduced the total number of reaction steps. For example, the number of reaction steps was reduced from sixteen to eight in the ABCDE star synthesis counting from the formyl-end-functionalized polymer, as seen in Schemes 3 and 4. Thus, the new methodology obviously works more efficiently than the previous one using the DOL-substituted DPE anion. Since the final ABCDE star polymers are still DOL-core-functionalized, the same iterative process may possibly be continued to further synthesize more armed and component stars.
As can be seen in Scheme 4, the DOL-end-functionalized polymer anion is used alternately with the non-DOL-end-functionalized polymer anion in each linking reaction step. Both the formyl group regenerated by deprotection of DOL reintroduced in advance and the PA function prepared from the produced hydroxyl group worked as the reaction sites for the polymer anions. Very importantly, whenever the DOL-end-functionalized polymer anion is used in the linking reaction, both steps, i.e.; the arm introduction and DOL reintroduction, are simultaneously performed. The use of the formyl group is advantageous in this regard such that the hydroxyl group’s conversion to the PA reaction site is always produced by the linking reaction of the polymer anion with the formyl group, and therefore, the two functionalities used in the methodology shown in Scheme 2 are not necessary to continue the iterative process.
μ-Star polymer synthesis by a new iterative methodology using DOL-end-functionalized polymer anions (2)
As often mentioned, the DOL-end-functionalized polymer anion plays a key role to simultaneously introduce an arm segment and reintroduce the DOL group in one linking reaction. By alternately using the DOL-functionalized polymer anion with the non-DOL-end-functionalized polymer anion, the process was iterated to smoothly synthesize the μ-star polymers. A question now arises about what kind of μ-star polymers can be synthesized by the same methodology, in which the DOL-end-functionalized polymer anion is always used in the linking reaction.As illustrated in Scheme 5, the starting DOL-end-functionalized polymer anion (A) is prepared and quantitatively converted to formyl-end-functionalized (A) simply by deprotection of the DOL terminus. A DOL-end-functionalized polymer anion (B) then reacted with the resulting formyl-functionalized (A), resulting in A-b-B, in which the OH and DOL were functionalized between the A and B chains. After conversion of the produced hydroxyl group to the PA reaction site, a DOL-end-functionalized polymer anion (C) reacted with the resulting PA-functionalized A-b-B to give an ABC star polymer with two DOL groups at the core. Obviously, the resulting star polymer obtained at this stage is different in DOL number from the ABC star polymer with one DOL at the core by the methodology shown in Scheme 4.
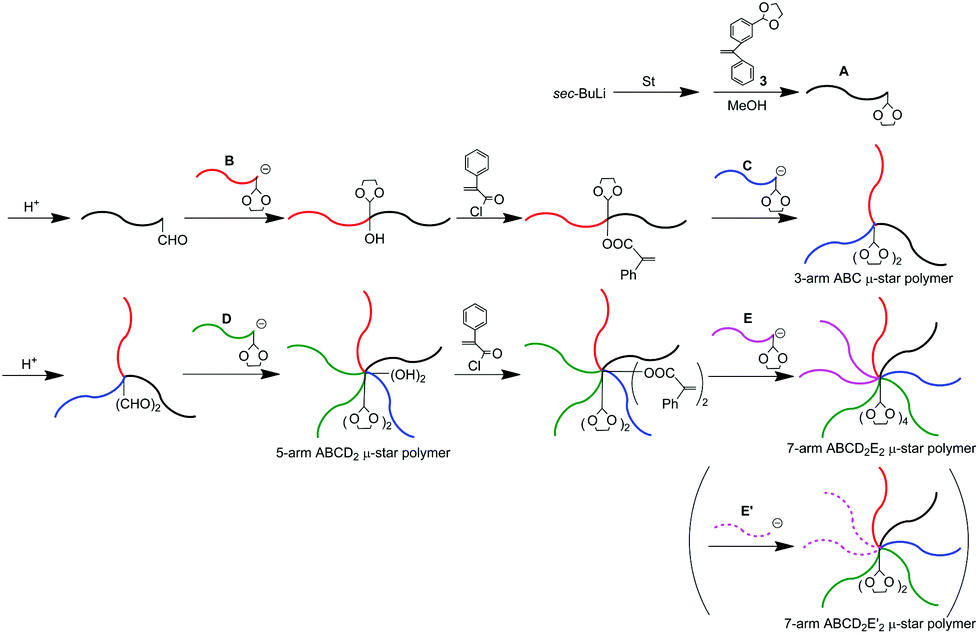 | ||
| Scheme 5 μ-Star polymer synthesis by an iterative methodology using DOL-end-functionalized polymer anions in all linking reactions. | ||
Two formyl groups were regenerated by deprotection of the two DOL groups, followed by subsequent linking with a DOL-end-functionalized anion (D), affording a 5-arm ABCD2 star polymer. Two hydroxyl groups were produced and two DOL groups were reintroduced at the core at the same time by the linking reaction. After converting the two hydroxyl groups to two PA reaction sites, the same process using a DOL-end-functionalized anion (E) was iterated to synthesize an ABCD2E2 star polymer. The number of DOL groups at the core increases from two to four. For these μ-star polymer syntheses, five different DOL-end-functionalized polymer anions were prepared by reacting 3 with the living anionic polymers of four styrene derivatives and isoprene (Ip) and sequentially reacted in situ with the regenerated formyl and the PA reaction sites. All of the linking reactions were estimated from the SEC profiles to be quantitative. The SEC profiles of all the linked products exhibit sharp monomodal distributions (see Fig. 3). The well-defined structures of the synthesized ABC, ABCD2, and ABCD2E2 star polymers were all confirmed based on the characterization results summarized in Table 3.
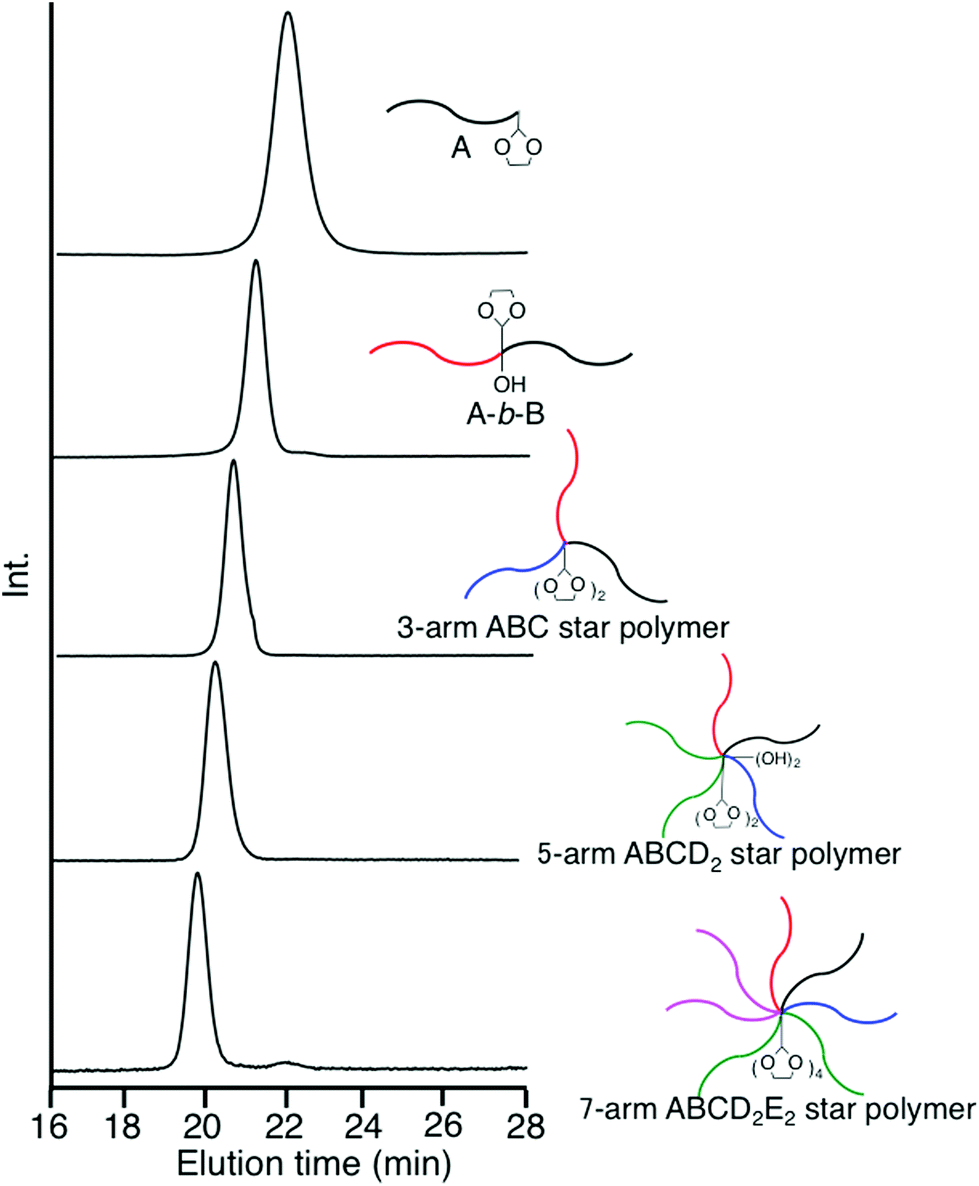 | ||
| Fig. 3 SEC profiles from A, AB, 3-arm ABC, and 5-arm ABCD2, to 7-arm ABCD2E2. | ||
| Type | M n (kg mol−1) | M w/Mn | A/B/C/D/E (or E′) | |||
|---|---|---|---|---|---|---|
| Calcd | SEC | RALLSa | SEC | Calcd | Obsdb | |
|
a SEC–RALLS equipped with triple detectors.
b 1H NMR; A/B/C/D/E (E′) = PS/PMS/PαMS/PMOS/PIp (PMMA).
|
||||||
| A | 10.4 | 10.1 | — | 1.05 | 100/0/0/0/0 | 100/0/0/0/0 |
| AB | 20.8 | 20.3 | 20.6 | 1.03 | 52/48/0/0/0 | 53/47/0/0/0 |
| ABC | 30.5 | 24.9 | 30.1 | 1.03 | 36/34/30/0/0 | 36/36/28/0/0 |
| ABCD2 | 45.4 | 33.4 | 43.0 | 1.04 | 25/23/21/31/0 | 27/24/18/31/0 |
| ABCD2E2 | 64.6 | 42.6 | 63.2 | 1.04 | 16/14/12/17/41 | 16/14/10/20/40 |
| ABCD2E′2 | 60.8 | 40.9 | 62.3 | 1.04 | 17/16/15/20/32 | 18/16/12/20/34 |
Since the final μ-star polymer has four DOL groups, the same process can be iterated to synthesize more armed μ-star polymers, such as the 11-arm ABCD2E2F4 and 15-arm ABCD2E2F4G4 stars (see Scheme S1†).
By reacting the living PMMA (E′), instead of the DOL-end-functionalized PIp anion, with the 5-arm core-((PA)2 and (DOL)2)-functionalized ABCD2 μ-star polymer at the final stage shown in Scheme 5, the same architectural 7-arm star-branched ABCD2E′2 polymer was synthesized (see Table 3). In this star polymer, however, the number of DOL groups at the core remained unchanged at two. Therefore, the next possible μ-star would be a 9-arm ABCD2E′2F2 star polymer with two hydroxyl and two DOL groups at the core (see also Scheme S1†). Thus, the occasional use of the non-DOL-functionalized polymer anion instead of the DOL-end-functionalized polymer anion may advantageously extend the synthetic possibility to obtain a greater variety of μ-star polymers.
μ-Star polymer synthesis by an iterative methodology using (DOL)2-end-functionalized polymer anions
The utility of a (DOL)2-end-functionalized polymer anion in the iterative methodology was examined for significantly increasing the introduced arm number (Scheme 6). The living PS reacted with a (DOL)2-substituted DPE derivative, 4, and the introduced two DOL termini were quantitatively deprotected by a catalytic amount of p-toluene sulfonic acid in a similar way to that already mentioned. The subsequent linking reaction was carried out between the resulting (formyl)2-end-functionalized PS and the above-used (DOL)2-end-functionalized PS anion. The quantitative reaction efficiency was estimated by SEC analysis of the reaction mixture. The introduction of four DOL groups into the resulting polymer was ascertained by 1H NMR. Thus, a 3-arm PS star (A3) with two hydroxyl and four DOL functions at the core was synthesized.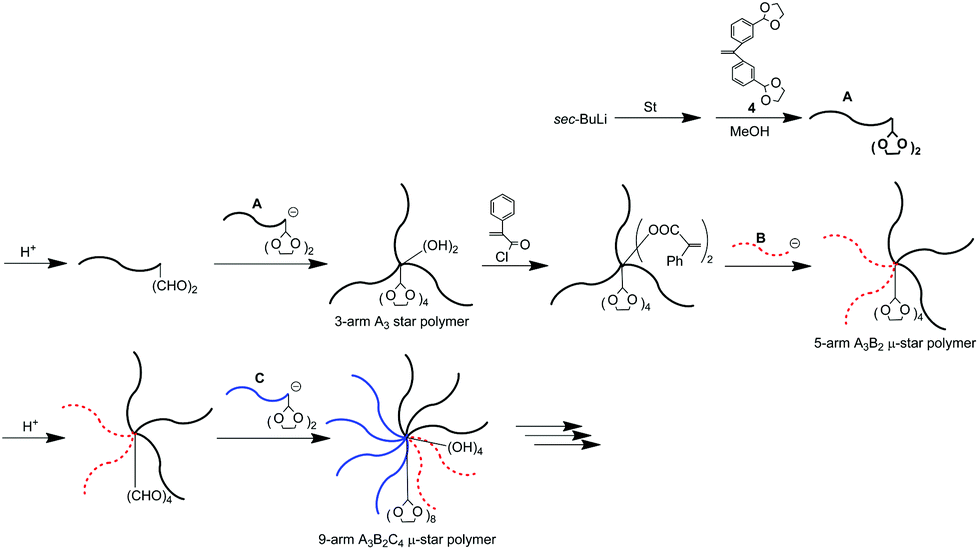 | ||
| Scheme 6 μ-Star polymer synthesis by an iterative methodology using (DOL)2-end-functionalized polymer anions. | ||
The two hydroxyl groups were converted to two PA reaction sites and the living anionic PMMA (B) reacted with PA-functionalized A3. An A3B2 star with four DOL groups was obtained (Table 4). One more A3B′2 star core-functionalized with four DOL groups was similarly synthesized by the quantitative reaction of the living PCPPHMA (B′) with the above-mentioned core-((PA)2 and (DOL)4)-functionalized A3. Thus, a liquid crystalline polymer segment could also be introduced into the star.
| Type | M n (kg mol−1) | M w/Mn | A/B/C | |||
|---|---|---|---|---|---|---|
| Calcd | SEC | RALLSa | SEC | Calcd | Obsdb | |
|
a SEC–RALLS equipped with triple detectors.
b 1H NMR; A/B (B′)/C = PS/PMMA (PCPPHM)/PαMS.
|
||||||
| A | 4.94 | 4.89 | — | 1.05 | 100/0/0 | 100/0/0 |
| A3 | 15.3 | 14.3 | 14.9 | 1.04 | 100/0/0 | 100/0/0 |
| A3B2 | 26.7 | 21.6 | 27.5 | 1.04 | 63/37/0 | 68/32/0 |
| A3B′2 | 29.8 | 22.4 | 29.4 | 1.03 | 57/43/0 | 60/40/0 |
| A3B2C4 | 47.3 | 28.5 | 46.5 | 1.03 | 33/24/43 | 32/22/46 |
Next, the four DOL groups at the core were deprotected to convert to an A3B2 μ-star polymer with four formyl groups. A (DOL)2-end-functionalized PαMS anion (C) was newly prepared from the living PαMS and 4 and reacted with the resulting formyl-functionalized A3B2 star. The quantitative reaction resulted in the formation of a well-defined A3B2C4 star polymer, whose core was functionalized with four hydroxyl and eight DOL groups (Table 4). Thus, the numbers of the DOL and hydroxyl groups were drastically increased in fewer reaction steps. This is therefore expected to allow for the ready synthesis of more armed μ-stars by developing the methodology using (DOL)2-end-functionalized polymer anions.
Conclusions
We have developed a new and efficient iterative methodology using DOL-end-functionalized polymer anions for the facile synthesis of multiarmed and multicomponent stars. With this methodology, various well-defined multiarmed and multicomponent star polymers were successfully synthesized as follows: the ABC, ABCD, ABCDE, ABCD2, and ABCD2E2 star polymers. Since both the arm introduction and DOL regeneration steps are simultaneously performed by one linking reaction of the DOL-end-functionalized polymer anion with the regenerated formyl group, the number of reaction steps could be significantly reduced for complex μ-star synthesis. Furthermore, the occasional use of the DOL-end-functionalized polymer anion with the non-DOL-functionalized polymer anion allows for the possible synthesis of more varied μ-star polymers. Thus, this newly developed methodology works more efficiently and versatilely than the original methodology using the DOL-substituted DPE anion.The synthetic possibility for more armed μ-star polymers was examined by developing the same iterative methodology using (DOL)2-end-functionalized polymer anions. With this methodology, an A3B2C4 star polymer could be readily synthesized in a few reaction steps. Further synthesis of more complex star polymers is currently under investigation.
Acknowledgements
This work was partly funded by JSPS KAKENHI, Grant Number JP15K17907, for Scientific Research from the Japan Society for the Promotion of Science (JSPS).References
- J. M. Ren, T. G. McKenzie, Q. Fu, E. H. H. Wong, J. Xu, Z. An, S. Shanmugam, T. P. Davis, C. Boyer and G. G. Qiao, Chem. Rev., 2016, 116, 6743–6836 CrossRef CAS PubMed.
- D. L. Lohse and N. Hadjichristidis, Curr. Opin. Colloid Interface Sci., 1997, 2, 171–176 CrossRef CAS.
- H. Hückstuädt, A. Göpfert and V. Abetz, Macromol. Chem. Phys., 2000, 201, 296–307 CrossRef.
- K. Yamauchi, H. Takahashi, H. Hasegawa, H. Iatrou, N. Hadjichristidis, T. Naneko, Y. Nishikawa, H. Jonnai, T. Matsui, H. Nishioka, M. Shimizu and H. Furukawa, Macromolecules, 2003, 36, 6962–6966 CrossRef CAS.
- T. M. Birshtein, A. A. Polotsky and V. Abetz, Macromol. Theory Simul., 2004, 13, 512–519 CrossRef CAS.
- A. Takano, S. Wada, S. Sato, T. Araki, K. Hirahara, T. Kazama, S. Kawahara, Y. Isono, A. Ohno, N. Tanaka and Y. Matsushita, Macromolecules, 2004, 37, 9941–9946 CrossRef CAS.
- K. Hayashida, A. Takano, S. Arai, Y. Shiohara, Y. Amemiya and Y. Matsushita, Macromolecules, 2006, 39, 9402–9408 CrossRef CAS.
- Y. Matsushita, Polym. J., 2008, 40, 177–183 CrossRef CAS.
- Z. Li, M. A. Hillmyer and T. P. Lodge, Langmuir, 2006, 22, 9409–9417 CrossRef CAS PubMed.
- Z. Li, M. A. Hillmyer and T. P. Lodge, Nano Lett., 2006, 6, 1245–1249 CrossRef CAS PubMed.
- Z. Li, M. A. Hillmyer and T. P. Lodge, Macromolecules, 2006, 39, 765–771 CrossRef CAS.
- A. Walther and A. H. E. Müller, Chem. Commun., 2009, 1127–1129 RSC.
- A. Gitsas, G. Floudas, M. Mondeshki, I. Lieberwirth, H. W. Spiess, H. Iatrou, N. Hadjichristidis and A. Hirao, Macromolecules, 2010, 43, 1874–1881 CrossRef CAS.
- S. Junnila, N. Houbenov, S. Hanski, H. Iatrou, A. Hirao, N. Hadjichristidis and O. Ikkala, Macromolecules, 2010, 43, 9071–9076 CrossRef CAS.
- Y. Matsushita, K. Hayashida and A. Takano, Macromol. Rapid Commun., 2010, 31, 1579–1587 CrossRef CAS PubMed.
- A. O. Moughton, M. A. Hillmyer and T. P. Lodge, Macromolecules, 2012, 45, 2–19 CrossRef CAS.
- K. Aissou, H. K. Choi, A. Nunns, I. Manners and C. A. Ross, Nano Lett., 2013, 13, 835–839 CrossRef CAS PubMed.
- T. Isono, I. Otsuka, Y. Kondo, S. Halila, S. Fort, C. Rochas, T. Satoh, R. Borsali and T. Kakuchi, Macromolecules, 2013, 46, 1461–1469 CrossRef CAS.
- T. Isono, Y. Kondo, I. Otsuka, Y. Nishiyama, R. Borsali, T. Kakuchi and T. Satoh, Macromolecules, 2013, 46, 8509–8518 CrossRef CAS.
- Y. Rho, C. Kim, T. Higashihara, S. Jin, J. Jung, T. J. Shim, A. Hirao and M. Ree, ACS Macro Lett., 2013, 2, 849–855 CrossRef CAS.
- R. Goseki, A. Hirao, M. Kakimoto and T. Hayakawa, ACS Macro Lett., 2013, 2, 625–629 CrossRef CAS.
- H. Iatrou and N. Hadjichristidis, Macromolecules, 1992, 25, 4649–4651 CrossRef CAS.
- T. Fujimoto, H. Zang, T. Kazama, Y. Isono, H. Hasegawa and T. Hashimoto, Polymer, 1992, 33, 2208–2213 CrossRef CAS.
- S. Sioula, N. Hadjichristidis and E. L. Thomas, Macromolecules, 1998, 31, 5172–5277 Search PubMed.
- A. Hirao, T. Higashihara and M. Hayashi, Macromol. Chem. Phys., 2001, 202, 3165–3173 CrossRef CAS.
- T. Higashihara, M. Hayashi and A. Hirao, Macromol. Chem. Phys., 2002, 203, 166–175 CrossRef CAS.
- T. Higashihara and A. Hirao, Macromolecules, 2002, 35, 7238–7245 CrossRef.
- T. Higashihara and A. Hirao, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 4535–4547 CrossRef CAS.
- T. Higashihara, M. Nagura, K. Inoue, M. Haraguchi and A. Hirao, Macromolecules, 2005, 38, 4577–4587 CrossRef CAS.
- Y. Zhao, T. Higashihara, K. Sugiyama and A. Hirao, J. Am. Chem. Soc., 2005, 127, 14158–14159 CrossRef CAS PubMed.
- A. Hirao, T. Higashihara, M. Nagura and T. Sakurai, Macromolecules, 2006, 39, 6081–6091 CrossRef CAS.
- P. Fragouli, H. Iatrou, N. Hadjichristidis, T. Sakurai, Y. Matsunaga and A. Hirao, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 6587–6599 CrossRef CAS.
- A. Mavroudis and N. Hadjichristidis, Macromolecules, 2006, 39, 535–540 CrossRef CAS.
- X. Wang, J. He and Y. Yang, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 4818–4828 CrossRef CAS.
- Y. Zhao, T. Higashihara, K. Sugiyama and A. Hirao, Macromolecules, 2007, 40, 228–238 CrossRef CAS.
- A. Hirao, T. Higashihara and K. Inoue, Macromolecules, 2008, 41, 3579–3587 CrossRef CAS.
- T. Higashihara, T. Sakurai and A. Hirao, Macromolecules, 2009, 42, 6009–6014 Search PubMed.
- T. Hirano, H.-S. Yoo, Y. Ozama, A. A. El-Magd, K. Sugiyama and A. Hirao, J. Inorg. Organomet. Polym., 2010, 20, 445–456 CrossRef CAS.
- A. A. El-Magd, K. Sugiyama and A. Hirao, Macromolecules, 2011, 44, 826–834 CrossRef.
- N. Hadjichristidis, M. Pitsikalis, S. Pispas and H. Iatrou, Chem. Rev., 2001, 101, 3747–3792 CrossRef CAS PubMed.
- T. Higashihara, M. Nagura, K. Inoue, M. Haraguchi and A. Hirao, Macromolecules, 2005, 38, 4577–4587 CrossRef CAS.
- N. Hadjichristidis, H. Iatrou, M. Pitsikalis, S. Pispas and A. Avgeropoulos, Prog. Polym. Sci., 2005, 30, 725–782 CrossRef CAS.
- A. Hirao, T. Higashihara and M. Hayashi, Polym. J., 2008, 40, 923–941 CrossRef CAS.
- T. Higashihara, M. Hayashi and A. Hirao, Prog. Polym. Sci., 2011, 36, 323–375 CrossRef CAS.
- A. Hirao, K. Murano, T. Oie, M. Uematsu, R. Goseki and Y. Matsuo, Polym. Chem., 2011, 2, 1219–1233 RSC.
- A. Hirao, M. Hayashi, T. Higashihara and N. Hadjichristidis, Recent Developments in Miktoarm Star Polymers with More than Three different Arms, in Complex Macromolecular Architectures: Synthesis, Characterization, and Self-Assembly, ed. N. Hadjichristidis, A. Hirao, Y. Tezuka and F. Du Prez, John Wiley & Sons, Singapore, 2011, pp. 197–132 Search PubMed.
- A. Hirao, R. Goseki and T. Ishizone, Macromolecules, 2014, 47, 1883–1905 CrossRef CAS.
- S. Ito, R. Goseki, T. Ishizone and A. Hirao, Eur. Polym. J., 2013, 49, 2545–2566 CrossRef CAS.
- T. Higashihara, K. Inoue, M. Nagura and A. Hirao, Macromol. Res., 2006, 14, 287–299 CrossRef CAS.
- A. Hirao, T. Higashihara and M. Hayashi, Macromol. Chem. Phys., 2001, 202, 3165–3173 CrossRef CAS.
- T. Higashihara, M. Hayashi and A. Hirao, Macromol. Chem. Phys., 2002, 203, 166–175 CrossRef CAS.
- A. Hirao and T. Higashihara, Macromolecules, 2002, 35, 7238–7245 CrossRef CAS.
- T. Higashihara, M. Nagura, K. Inoue, N. Haraguchi and A. Hirao, Macromolecules, 2005, 38, 4577–4587 CrossRef CAS.
- T. Higashigara, K. Inoue and A. Hirao, Macromol. Symp., 2010, 296, 53–62 CrossRef.
- S. Ito, R. Goseki, S. Senda and A. Hirao, Macromolecules, 2012, 45, 4997–5011 CrossRef CAS.
- S. Ito, R. Goseki, T. Ishizone, S. Senda and A. Hirao, Macromolecules, 2013, 46, 819–827 CrossRef CAS.
- R. Goseki, Y. Ozama, E. Akemine, S. Ito, S. Ehara and A. Hirao, Polymer, 2013, 54, 2049–2057 CrossRef CAS.
- S. Ito, R. Goseki, I. Manners, T. Ishizone and A. Hirao, Macromol. Chem. Phys., 2015, 216, 1523–1533 CrossRef CAS.
- R. Goseki, K. Togii, S. Tanaka, S. Ito, T. Ishizone and A. Hirao, Macromolecules, 2015, 48, 2370–2377 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, 1H NMR spectra, SEC profiles (Fig. S1–S5), and scheme of synthetic strategy for the complex μ-star polymer. See DOI: 10.1039/c6py01341d |
| This journal is © The Royal Society of Chemistry 2016 |