
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhoto-polymerizable, low shrinking modular construction kit with high efficiency based on vinylcyclopropanes†
Paul
Pineda Contreras
and
Seema
Agarwal
*
Macromolecular Chemistry II and Bayreuth Center for Colloids and Interfaces, Universität Bayreuth, Universitätsstrasse 30, 95440 Bayreuth, Germany. E-mail: agarwal@uni-bayreuth.de; Tel: +49-921-553397
First published on 11th April 2016
Abstract
The successful development of a universal hydrogen bond (H-bond) concept, specific for bi-functional vinylcyclopropane (VCP) ester–amide derivatives is reported here. Thereby, uniformity within the intermolecular hydrogen bond strength as investigated by variable temperature NMR and FT-IR measurements provides an excellent control of high reactivity, nearly regardless of the chosen spacer-unit, offering the system a possible application as a modular construction kit for cross-linked networks with varied properties. Two entirely diversified VCP ester–amide systems were prepared. The performance benchmark of a flexible, high molar mass macro-monomer VCPPPG2000, implementing a polypropyleneglycol Genamine D01/2000 macro-spacer, and two rigid, low molar mass monomers VCP-m-phenyl and VCP-p-phenyl outlined significantly the strength of the system. Extremely low volume shrinkages between 1.4–4.5% and a wide range of E-moduli could be achieved. An easy control of adjusting final characteristics by varying the co-monomer content in co-networks was shown, especially without obtaining any significant disadvantage in the curing behavior. The VCP ester–amides offer the possibility to replace some currently used dimethacrylate resins within several high-end applications, especially where low volume shrinkage and high polymerization efficiency are required.
Introduction
Bi-functional, cross-linkable resins featuring a variety of precise specifications are of great interest within a wide spectrum of applications, such as electronics,1 coatings,2,3 lithography,4,5 dental applications,6 3D microstructures7 and post-polymerization modifications.8 However, since the final properties of resins have to match precisely with the complex specifications, there is a demand for monomer-systems providing universality and high precision.1,9 A suitable method could be to apply low shrinking modular construction kits, merging rather specific techniques within one certain system.Nevertheless, monomer-systems based on methacrylates pose serious constraints, due to a high discrepancy in photo-curing behaviour, using cross-linkable resins with varied spacer-elements.10,11 Likewise the observed polymerization shrinkage during curing of methacrylate systems is too high, which can result in high internal compressive stress, leading to microcracks, incomplete fillings, poor adhesion and a short life span of cured resin.12 Based on these two issues methacrylate systems are not appropriate for low shrinking modular construction kits. An alternative concept, partly developed from the knowledge of low-molecular-weight gelators and supramolecular structures, establishes a further promise on weak forces, such as hydrogen bonds (H-bonds), presuming a partial self-assembly of the molecules. In particular, if a partial self-assembly is taking place selectively at a polymerizable group, a fast and selective curing, as well as targeted control can be assumed, likewise it is required for a modular kit.
Thereby, vinylcyclopropane (VCP) ester–amide derivatives can play a decisive role in the future. Recently we have shown in a special case, that intermolecular amide H-bonds could induce a very effective partial preorganization of bi-functional VCPs molecules, which strongly increased the polymerization behavior.13 Further, the radical ring-opening polymerization (RROP) of VCP units can significantly reduce the volume shrinkage on polymerization. Therefore, the shrinkage is lower than those recorded for e.g. for methacrylate resins, making VCPs attractive as low-shrinking monomers.14,15
However, so far the advances within VCP resins have been mainly focused on particular characteristics, e.g. reducing the volume shrinkage and improving the reactivity, the ring-opening efficiency and mechanical properties, especially among researchers in the field of dental fillings.16–18
Herein, we report the design of the first highly efficient modular construction kit based on bi-functional VCP ester–amides, extending the general concept of hydrogen-bonding to a universal tool for an extensive application of VCPs as low shrinking resin. The H-bond strength of two diverse VCP ester–amide derivatives was studied in detail, first for a flexible, high molar mass macro-monomer (VCPPPG2000, Mw ∼ 2230 g mol−1), and secondly for a rigid, low molar mass monomer (VCP-m-phenyl, Mw = 468.5 g mol−1) by temperature dependent NMR- and FT-IR-measurements. Further, a detailed overview of mechanical strength by three point bending experiments, and thermal properties of the cured networks and co-networks by differential scanning calorimetry (DSC) and thermogravimetric (TGA) analysis is provided. In addition to the VCP-m-phenyl its constitution isomer of a para-phenyl substituted VCP ester–amide resulted in a partial-crystalline system. Thus, the cured and un-cured resins have been studied in detail by nuclear magnetic resonance (NMR) measurements in liquid and solid-state, by temperature dependent polarized microscopy as well as by temperature dependent X-ray diffraction (XRD).
Experimental section
Materials
m-Xylylenediamine (99%), p-xylylenediamine (99%), trans-1,4-dibromo-2-butene (99%), diethyl malonate (99%), potassium hydroxide (KOH, >85%), N,N′-dicyclohexylcarbodiimide (DCC, 99%), ethyl 4-(dimethylamino)benzoate (EDMAB, 99%), triethylamine (99%), camphorquinone (CQ, 97%), 1,3,5-trimethylbenzen (mesitylene, >99.8%) and dichloromethane (>99.5%) were supplied by Sigma Aldrich and used without further purification. 1-Hydroxybenzotriazole hydrate (HOBt, >97%, 12% water, Sigma Aldrich) was dried in vacuum prior use. Sodium (>99%, Sigma Aldrich) was purified by melting in dry xylole prior use. Genamine D01/2000 (99%) was provided by Clariant and used as received (determined by (MALDI-TOF-MS): Mn = 1980 g mol−1, Mw = 2027 g mol−1, Đ = 1.02). Urethane-dimethacrylate (UDMA), dodecanediol-dimethacrylate (1,12-DMA) and a 6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 mixture of bisphenol-A-glycidyl methacrylate (BisGMA) and triethylene-glycol-dimethacrylate (TEGDMA) have been provided by the Kettenbach GmbH (Eschenburg, Germany) and were used as received.
:4 mixture of bisphenol-A-glycidyl methacrylate (BisGMA) and triethylene-glycol-dimethacrylate (TEGDMA) have been provided by the Kettenbach GmbH (Eschenburg, Germany) and were used as received.
Analytical methods
1H- (300 MHz) and 13C-NMR spectra (75 MHz) were recorded on a Bruker Ultrashied-300 spectrometer at room temperature in CDCl3. The spectra were calibrated on the solvent signal (δ(1H) = 7.26 ppm; δ(13C) = 77.16 ppm). Variable 1H temperature experiments at a concentration of 5 mM in CDl3 have been performed in a range from 20 °C to 60 °C. High-resolution solid state 13C-CP/MAS (cross polarization/magic angle spinning) NMR Spectra were measured by a Bruker spectrometer operating at 100 MHz, using a pulse experiment with a 30 s pulse delay for 4096 scans. High resolution mass spectra, coupled online to a HPLC system, were recorded on a Q-Exactive Orbitrap (Thermo Scientific). MALDI-TOF MS analysis were performed on a Bruker Reflex III instrument equipped with a N2 laser (λ = 337 nm) in linear and reflectron mode. The acceleration voltage was 20 kV. Dithranol (Sigma Aldrich, >98%) was used as matrix material and sodium trifluoroacetate (Sigma Aldrich, 98%) as ionization salt. The samples were prepared with the dried droplet method from THF solution by mixing matrix, polymer and salt in a ratio of 20:5:1 and applying approximately 1 μL to the target spot. Temperature dependent FT-IR spectra were recorded by a Nicolet Nexus 470 spectrometer with a heated attenuated total reflectance (ATR) unit. Chromatographic purifications were performed using a GRACE Reveleris X2 apparatus. Technical grade solvents, which have been distilled prior use, have been utilized. A dynamic-mechanical analyzer (DMTA IV, Rheometric Scientific) has been used to determine the E-moduli by three-point bending investigations, respectively at 25 °C. A Mettler thermal DSC analyzer (821c) was used for the thermal characterization of the monomer resins and cross-linked polymers. The DSC scans were recorded in nitrogen atmosphere and heating rates of 10 K min−1. Thermogravimetric analysis were done with a TG 209 F1 (Netzsch) by heating the samples up to 800 °C, respectively under nitrogen atmosphere as well as under synthetic air. 85 μm corundum crucibles, heating rates of 10 K min−1 and about 10–15 mg of each sample were applied. An AccuPyc 1330 gas pycnometer (Micromeritics) operating with helium gas was used to determine the density of monomers and cross-linked polymers. Temperature dependent XRD patterns were carried out on an X-ray powder diffractometer of the type PANalytical XPert Pro, applying a reaction chamber to measure from 25 to 90 °C in a 2 Theta range from 7 to 35, respectively.
Synthesis of polymer networks
To perform the photo initiation a blue light LED reactor with a light power of ∼2.0 mW cm−2 at 465 nm was used as radiation source. 1 mol% of the photo-initiator mixture CQ:EDMAB in a molar ratio of 1:2 was used. The initiator was dissolved in little DCM and added to the resin. The DCM was evaporated in vaccum prior to curing. For DMTA measurements specimens were produced by filling a stainless steel profile with a dimension of 25 × 3 × 1 mm with the monomer/initiator mixture. The profile was enclosed and fixed both on top and on the bottom with a 50 μm thick hostophan sheet and irradiated for 2 h. The cured specimens were pressed out carefully and stored for 24 h at room temperature prior to further testing. For density measurements the monomer initiator mixture was inserted into a 2 mL standard test tube, flushing the tubes with an argon stream for 30 s and irradiated for 2 h. Afterwards the samples were taken out of the glass tubes and stored for 24 h at 100 °C prior measuring the densities. The partial-crystalline VCP-p-phenyl was polymerized in 50 weight% solution of chloroform/toluene (1:1). The reaction mixture was degassed by three freeze–pump–thaw cycles and polymerized within the LED reactor under stirring over 12 h. The polymerization was stopped by putting the test tube into liquid nitrogen; the samples were taken out of the glass tube, transferred into centrifuge tubes and extracted three times by over 25-fold excess of acetone. Afterwards, the resulting polymer was dried in vacuum at 100 °C for 24 h.
Determination of residual monomer amount
The quantification of the residual amount of uncured monomer was performed by 1H-NMR measurements. Therefore the weighed and cured samples have been immediately immersed after radiation in 2.0 mL of CDCl3 (as extraction fluid) in 5 mL bottles for 24 h. The residual monomer amounts have been calculated by comparing the integration areas of the respective 1H-NMR proton signals to the proton signals of mesitylene, which has been added to the CDCl3 as internal standard in 2.8444 mg mL−1 amount. To achieve a high signal to noise resolution for each 1H-NMR experiment a number of scans of 2048 has been chosen.Synthesis of diethyl 2-vinylcyclopropane-1,1-dicarboxylate and 1-(ethoxycarbonyl)-2-vinylcyclopropanecarboxylic acid
Diethyl 2-vinylcyclopropane-1,1-dicarboxylate and the hydrolyzed diethyl 2-vinylcyclopropane-1,1-dicarboxylate were obtained as per published procedure.14,19General procedure to obtain bi-functional VCP ester–amide derivatives
In a round-bottom flask purged with argon 0.5 eq. (0.051 mol) of the diamine compound were dissolved/diluted in 200 mL of anhydrous DCM. The solution was transferred to a funnel, which was connected to 1000 mL three-necked reaction flask, charged with 200 mL of anhydrous DCM, 1.1 eq. (0.112 mol) of 1-(ethoxycarbonyl)-2-vinylcyclopropane-carboxylic acid, 1.1 eq. (0.112 mol) of DCC and 1.15 eq. (0.117 mol) of HOBt. The three-necked reaction flask was stirred for 15 min at room temperature, cooled to 0 °C and then 2.1 eq. (0.214 mol) of triethylamine were added. Afterwards the diamine solution with a drip rate of about 40 mL h−1 was added slowly. The reaction solution was stirred for 12 h at room temperature. Subsequently the reaction mixture was filtered, the pale yellow solution was washed two times with water (100 mL), once with 1 molar HCl solution (100 mL) and three times with neutral water (50 mL). After drying over magnesium sulfate, the solution was deposed on silica gel and purified by flash chromatography (general eluents: hexane and ethyl acetate). To the purified organic phase 1500–2500 ppm of butylated hydroxytoluene were added. The organic phase was evaporated in vacuum and the bi-functional VCP–amide derivative dried in vacuo. The average yield after purification was 87%. A detailed monomer characterization of the synthesized VCPPPG2000, VCP-m-phenyl and VCP-p-phenyl monomers is provided in the ESI in Fig. S1–S8.†Results and discussion
The availability of targeted amendments to control selective characteristics by varying the spacer unit, without major transformation e.g. in reactivity is crucial for designing a modular construction kit for photo-polymerization purposes. Due to the existence of selective intermolecular H-bonds exactly at the VCP-unit, VCP ester–amide derivatives provide a clear advantage as the curing behaviour is actually independent of the chosen spacer unit. Thereby, Fig. 1A illustrates the universal concept of partial self-assembly for any VCP ester–amide derivative. Moreover, the proposed RROP mechanism of VCP ester–amides (Fig. 1B) clarifies the general efficiency of the intermolecular H-bonds, which are preferred because of N–H⋯O donor–acceptor interactions and due to chelating effect.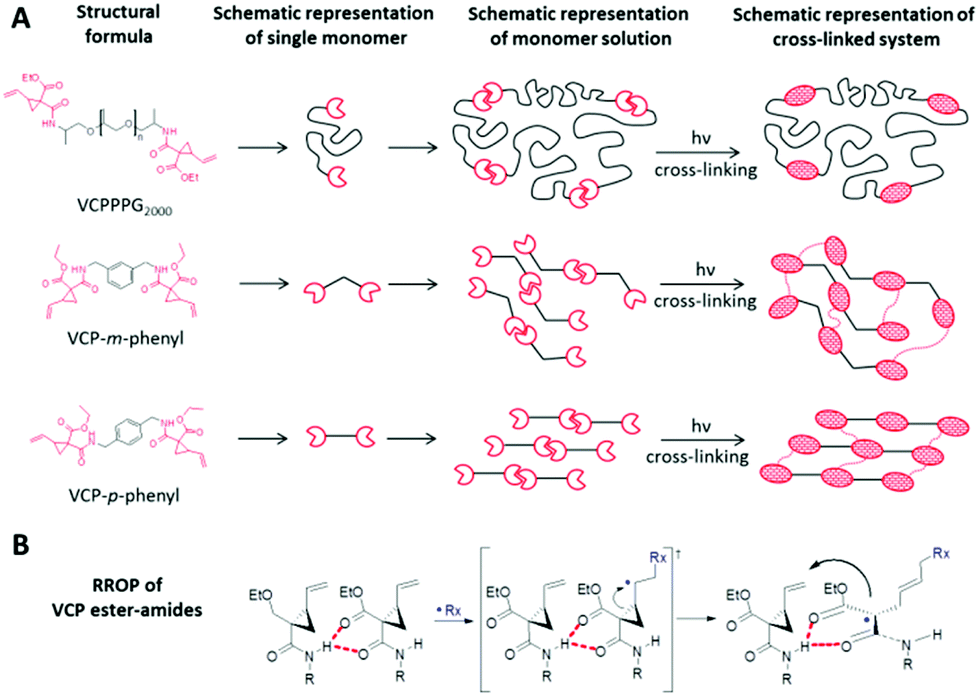 | ||
| Fig. 1 (A) Schematic aspect of the H-bond mediated partial self-assembly of VCP ester–amide units. The alignment is primary taking place at the functional group. (B) Proposed RROP mechanism of amide-based VCPs. The partial self-assembly and the enhanced orbital arrangements have to be taken into consideration. | ||
To provide a reasonable illustration, two entirely diversified VCP ester–amide systems were prepared. Thus, first a flexible, high molar mass macro-monomer VCPPPG2000 and secondly two rigid, low molar mass monomers VCP-m-phenyl and VCP-p-phenyl have been synthesized and compared subsequently.
To study the H-bond relationship, both variable-temperature NMR- (Fig. 2) as well as FT-IR-measurements (see Fig. S9 and S10 in the ESI†) have been carried out, for VCPPPG2000 and VCP-m-phenyl. Thereby the evaluation of the temperature dependency of the amide proton signal revealed the presence of H-bond. The corresponding signal was shifted to higher magnetic field, as with increased temperature a decreased deshielding effect is anticipated.20,21 The partial regression line of the chemical shift confirmed independently for both VCP ester–amides (R2 = 0.999), that within the investigated temperature range of 20–60 °C the H-bonds were weaken, but not disrupted. Further, the corresponding slopes of the linear regressions were more or less identical, −4.32 × 10−3 ppm K−1 for VCPPPG2000 and −4.85 × 10−3 ppm K−1 for VCP-m-phenyl, which confirmed the uniformity of the VCP ester–amide H-bond strength.
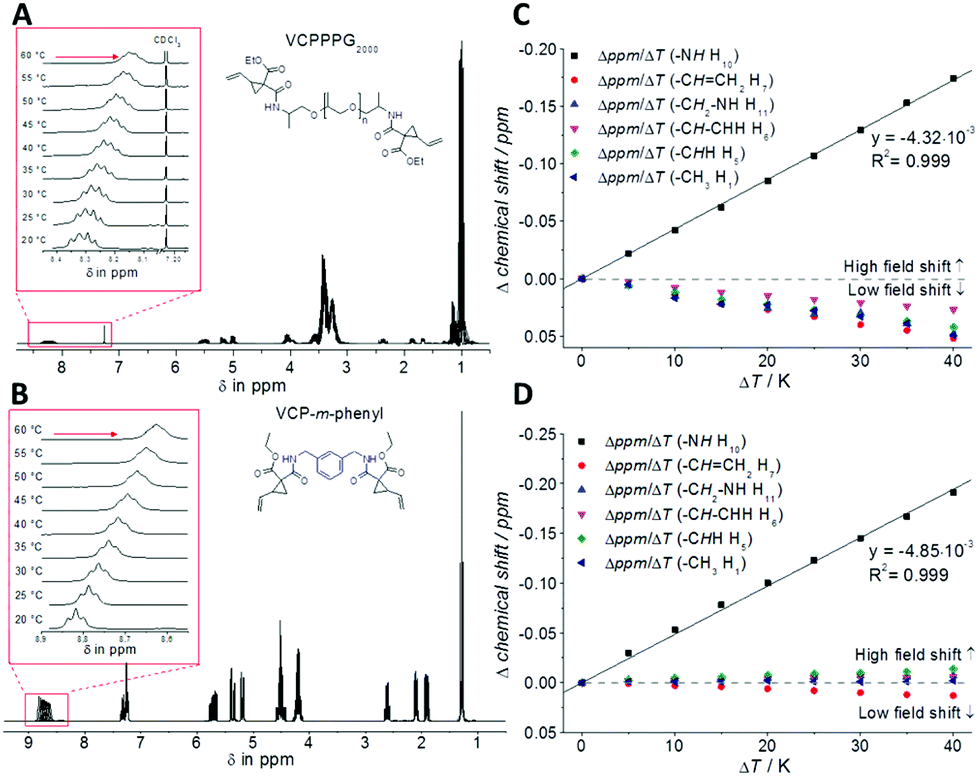 | ||
| Fig. 2 (A + B) Variable-temperature 1H-NMR experiments of VCPPPG2000 and VCPP-m-phenyl (0.5 mM in CDCl3) between 20–60 °C. (C + D) Graphical plot of chemical shift within 1H-NMR experiments. With increased temperature a decreased deshielding effect is anticipated, as a partial rupture of the H-bonds occurs. | ||
In addition to the 1H-NMR experiments, temperature dependent FTIR measurements for the bulk resins underlined this uniformity within the H-bonds. In analogy to the NMR-experiments, an increase in temperature weakened the H-bonds, thus the characteristic IR bands showed an alteration in shape and shift, while non-H-bonded bands remained unchanged. Thus, especially the N–H (3355 cm−1) and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration (1652 cm−1) shifted to higher frequencies with increased temperature (+4 cm−1 and +5 cm−1 for VCPPPG2000 and VCP-m-phenyl, respectively). However, the evidence of the H-bond effect appeared not as pronounced as e.g. compared to the well-known urethane-dimethacrylate (UDMA) (see Fig. S11 in the ESI†), since the urethane group contributes to higher coplanarity and higher number of available acceptor atoms compared to VCP ester–amides.22 Hence for UDMA the N–H (3358 cm−1) and CO stretching vibration (1702 cm−1) shifted by +20 cm−1 and +12 cm−1 to higher frequencies. Nevertheless, this did not affect the general curing efficiency of VCP ester–amides, as the H-bonds were sufficiently strong to provide partial preorganization of monomer molecules.
O stretching vibration (1652 cm−1) shifted to higher frequencies with increased temperature (+4 cm−1 and +5 cm−1 for VCPPPG2000 and VCP-m-phenyl, respectively). However, the evidence of the H-bond effect appeared not as pronounced as e.g. compared to the well-known urethane-dimethacrylate (UDMA) (see Fig. S11 in the ESI†), since the urethane group contributes to higher coplanarity and higher number of available acceptor atoms compared to VCP ester–amides.22 Hence for UDMA the N–H (3358 cm−1) and CO stretching vibration (1702 cm−1) shifted by +20 cm−1 and +12 cm−1 to higher frequencies. Nevertheless, this did not affect the general curing efficiency of VCP ester–amides, as the H-bonds were sufficiently strong to provide partial preorganization of monomer molecules.
In compliance to the uniform strength of H-bond interaction, for the entirely diversified VCP ester–amides resins VCPPPG2000, VCP-m-phenyl and VCPMe3hexyl (extended monomer from recently published literature,13 mentioned for clarification purposes) almost identical curing behaviors were observed (Fig. 3). Hereby curing experiments were carried out by photo-polymerizations using a mixture of camphorquinone (CQ) and ethyl 4-(dimethylamino)-benzoate (EDMAB) in a molar ratio of 1:2 as initiator. The exposure with a commercial blue light-emitting diode (LED) could confirm very fast kinetic and high overall conversions. For all investigated VCP ester–amides an overall conversion of >96% could be observed. In comparison, VCP-resins without H-bonded interactions cannot reach high overall conversion even after prolonged curing times.14 Thus VCP ester–amides like VCPPPG2000, VCP-m-phenyl as well as the previous studied VCPMe3hexyl have to be seen as pioneers defining a new, universal class of highly efficient and low shrinking resins. However, it is worth noting, that VCPPPG2000 reached a conversion of 92% after 30 s of exposure. In this particular case, the slightly enhanced conversion of VCPPPG2000 was explained by its low Tg of −55 °C (Table 1). Therefore, a high mobility and continuous diffusion to further radical centers is allowed.
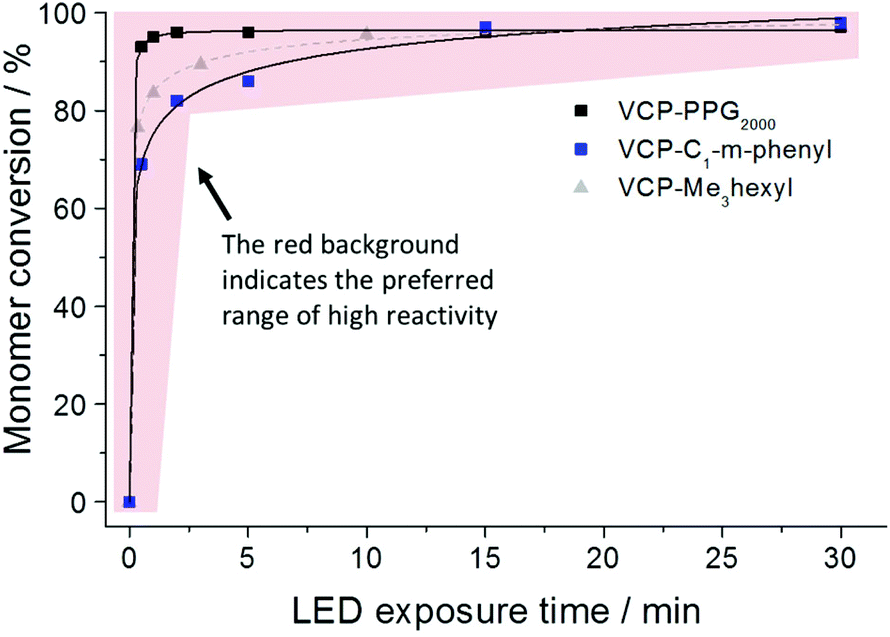 | ||
| Fig. 3 Determined monomer conversion for the photo-polymerization of VCPPPG2000, VCP-m-phenyl and VCPMe3hexyl (1 mol% initiator CQ:EDMAB in a molar ratio of 1:2). The curing was performed under a controlled atmosphere of nitrogen using a commercial LED source (2.013 mW cm−2 for 465 nm). | ||
| Entry | Polymer |
T
ga/°C |
T
5%b (N2)/°C |
T
5%c (air)/°C |
Water uptaked/% |
E
mode/MPa |
|---|---|---|---|---|---|---|
| a Determined by DSC. b Determined by TGA (50 mL min−1 N2 as purge gas). c Determined by TGA (50 mL min−1 synthetic air as purge gas). d Determined by TGA as mass loss between 30–200 °C, after storage for 48 h at 50 °C in water, respectively. e After curing specimens of the dimension 25 × 3 × 1 mm3 by photo-polymerization and additional annealing for 12 h at 100 °C, respectively. | ||||||
| 1 | VCPPPG2000 | −55 | 340 | 239 | 1.3 | 0.15 |
| 2 | VCP-m-phenyl | 147 | 329 | 327 | 2.9 | 232.3 |
| 3 | VCP-p-phenyl | 189 | 339 | 321 | 2.3 | — |
| 4 | UDMA | 119 | 316 | 291 | 2.8 | 224.1 |
Hence, compared to methacrylate systems, VCP ester–amides show fast curing kinetics irrespective of the spacer unit, but is superior defining the final network characteristics as discussed in Fig. 4.
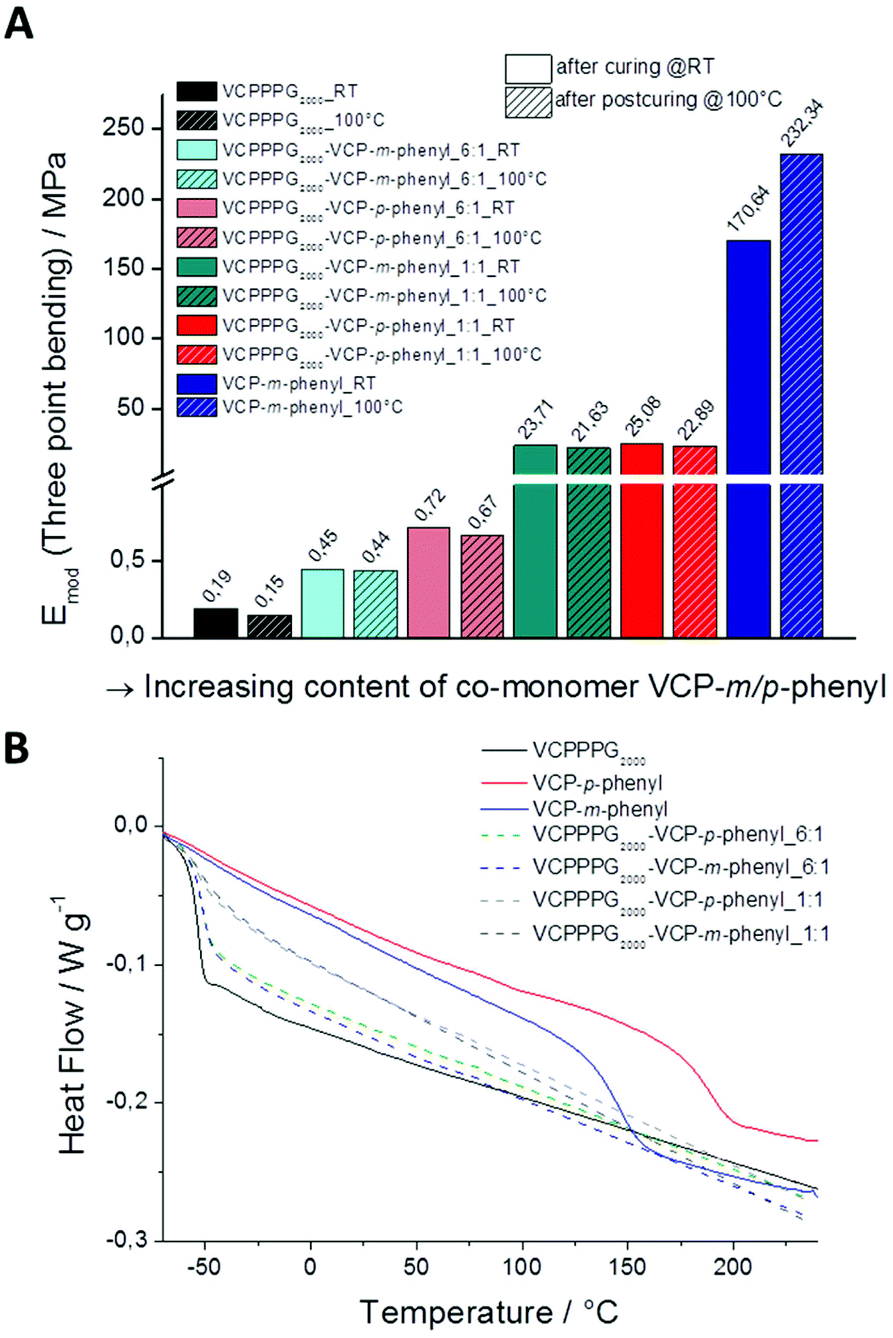 | ||
| Fig. 4 (A) Determined E-moduli of cured VCPPPG2000 and VCPP-m-phenyl specimens, as well as by co-networks in ratios of 6:1 and 1:1 of VCPPPG2000, VCPP-m-phenyl and VCPP-p-phenyl. With increasing content of the co-monomer VCP-m/p-phenyl, the moduli could be raised up to 1200 times to higher values. (B) Second DSC heating trace of cured VCP specimens, illustrating the glass transition Tg of the cured samples. | ||
Thereby, we investigated the mechanical properties of cured VCP ester-amid specimens by three point bending experiments, for VCPPPG2000 and VCPP-m-phenyl, as well as for the co-networks of VCPPPG2000, VCPP-m-phenyl and VCPP-p-phenyl in different ratios (Fig. 4A). The mechanical properties of the cured specimens have been investigated once directly after curing, and secondly after annealing those 12 h at 100 °C. Thus the additional potential modulus, after partial relaxation of internal network tensions (sub-Tg relaxation) could be determined.23,24 Subsequent extractions with CDCl3 confirmed conversions higher than 98% for all specimens. Cured VCPPPG2000 specimens showed E-moduli of 0.19 ± 0.001 MPa, reflecting predominantly the characteristic of a highly cross-linked organogel. Anealing at 100 °C did not affect significantly the modulus (0.15 ± 0.001 MPa), as the Tg for these cured specimens was much lower as the curing temperature (25 °C). In contrast, for cured VCP-m-phenyl specimens the E-modulus could be increased by +36%, since the softening temperature for these specimens was much higher (147 °C).
Further, we could easily control the E-moduli by varying the co-monomer content in the co-networks. With increasing content of the co-monomer VCP-m/p-phenyl, the moduli could be raised continuously from 0.19 ± 0.001 MPa for VCPPPG2000, up to a value of 232.3 ± 0.5 MPa for the cured VCP-m-phenyl. Thus, the moduli could be raised up to 1200 times to higher values. Thereby cured VCP-m-phenyl resins can compete clearly with commercially available UDMA resins, which provide moduli of 168.8 ± 0.5 MPa and 224.1 ± 0.4 MPa, respectively. In our previous study, VCPMe3hexyl showed E-moduli of 130.9 ± 0.7 MPa and 181.9 ± 0.5 MPa, respectively just after curing and post-curing.13 This supports the above hypothesis by a third example, that primarily the spacer unit is defining the network properties, whereas the curing behaviour remains constant. Furthermore, according to the high Tg of 147 and 189 °C for the cured VCP-m-phenyl and VCP-p-phenyl resins (Table 1) the mechanical properties of these networks can be utilized over a wide temperature range.
Moreover, the corresponding thermal characteristics of these cured resins exhibited high potential as well. High overall thermal stabilities could be observed (T5% > 320 °C) under nitrogen atmosphere as well under air. In contrast, several di-methacrylate networks such as UDMA showed a significant lower stability under identical conditions. In particular, under an atmosphere of air a fast oxidation of the di-methacrylate networks occurred at temperatures of already 290 °C (Table 1 and Fig. S12A in the ESI†). Further, the water absorption of the cured VCP ester–amide networks was less than 3%, as determined by TGA (see Fig. S12B in the ESI†). Especially with regard to the applicability for coatings and dental materials a low water-uptake is required in order to provide durability.25,26
Subsequently, the cured VCP ester–amide networks were analyzed by 13C-CP/MAS solid-state spectroscopy. Due to the absence of the carbon atom of the terminal vinyl bond at 119.7 ppm an extraordinary high cross-linking density of the cured networks could be confirmed, respectively for VCP-m-phenyl (Fig. 5) and VCP-p-phenyl (S13 in the ESI†). Only a very small shoulder remained, hardly perceived, which indicated a negligible amount of remaining vinyl double bonds.
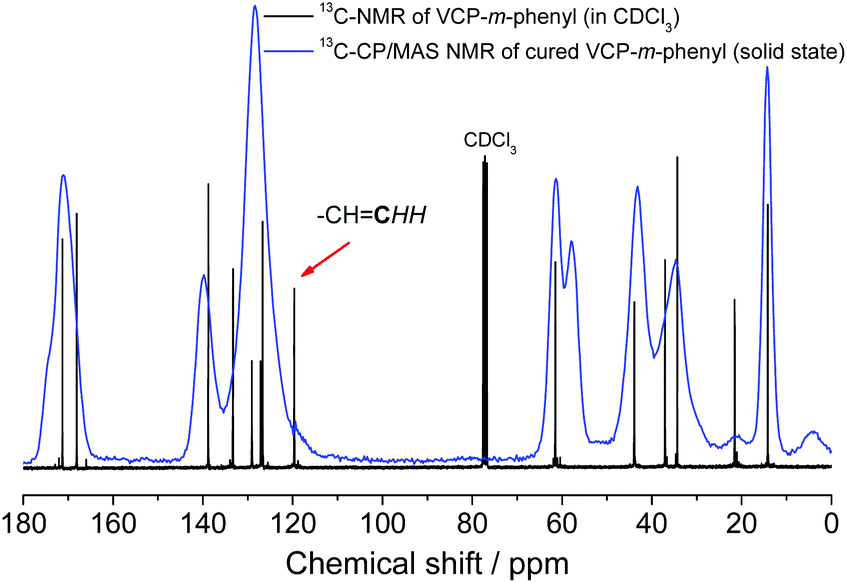 | ||
| Fig. 5 13C-NMR comparison: 13C NMR (CDCl3, 75 MHz) of un-cured VCP-m-phenyl and 13C-CP/MAS NMR (100 MHz) of cured VCP-m-phenyl. The signal at 119.7 ppm defines the carbon atom of the terminal vinyl bond. After curing within the 13C-CP/MAS NMR spectra only a shoulder, hardly perceived at 119.7 ppm, is observed, which indicates a high cross-linking density of the cured VCP-m-phenyl. | ||
Further to depict the general potential of reducing the volume-shrinkage during polymerization by applying RROP of VCP ester–amide resins, the corresponding state variables volume shrinkage, monomer- and polymer density for cured and uncured states were provided within Fig. 6. Very low volume changes between −4.5% to −1.4% have been determined for the VCP ester–amide resins, as well as for their co-networks in different ratios. It is remarkable, that the cured VCP-m-phenyl showed a reduction by 50% in volume shrinkage compared e.g. to UDMA, without compromising mechanical performance. VCPPPG2000 showed an extremely low volume shrinkage (−1.4%), represented by its increased molecular weight as macro-monomer.9,27
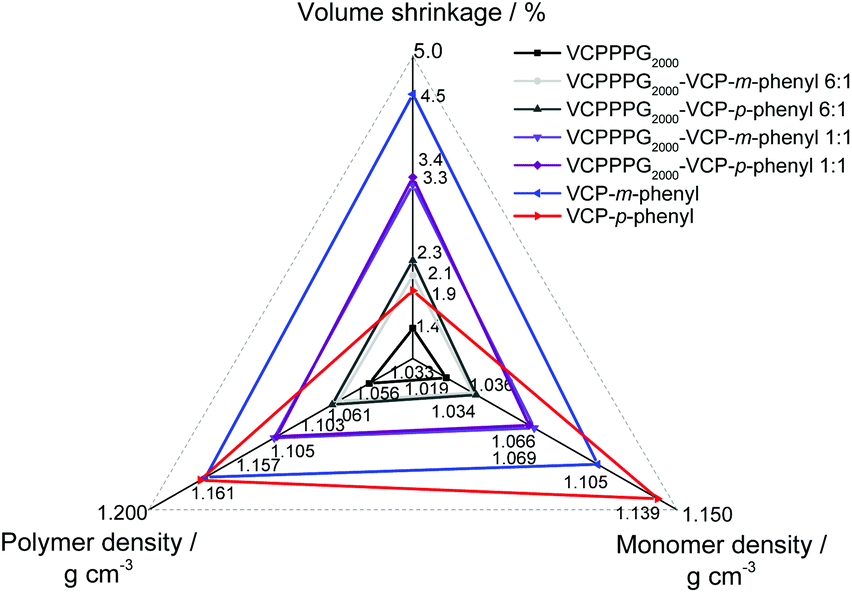 | ||
| Fig. 6 Spider chart showing the dependency between the monomer- and polymer density to the volume shrinkage. Only the partial-crystalline VCP-p-phenyl shows a disproportion within the correlation, based due to the denser partial-crystalline monomer structure of VCP-p-phenyl (1.139 g cm−3). | ||
In this context we observed a significant difference between the cured isomers VCP-m-phenyl and VCP-p-phenyl. While VCP-m-phenyl showed a volume change of −4.5%, for VCP-p-phenyl a volume change of −1.9% was observed. In comparison to the amorphous VCP-m-phenyl, its isomer VCP-p-phenyl occurs as a heterogeneous system, combining a liquid resin with partial crystallinity. Thereby the DSC curve of VCP-p-phenyl showed a melting at Tm = 67 °C, whereas VCP-m-phenyl remained completely amorphous (Fig. 7A). In agreement with the DSC measurement, temperature variable polarizing microscopy showed at the same temperature range a transition to an isotropic melt, respectively for VCP-p-phenyl (Fig. 7B).
 | ||
| Fig. 7 (A) DSC curves of first heating cycles of un-cured VCP-m/p-phenyl resins. First the transition from a molecular glass to a resin (Tg − 15 °C) is passed, respectively for both VCPs. In the further heating VCP-m-phenyl shows a complete amorphous behavior, whereas for VCP-p-phenyl a melting (onset at 67 °C) is observed. At a temperature range of ∼99 °C an auto-polymerization of both resins is observed. (B) Polarized microscopy images of the partial-crystalline VCP-p-phenyl resin. Beneath the melting transition both amorphous as well as crystalline areas can be observed. When the melting range is passed the VCP-p-phenyl resin becomes fully isotropic. | ||
Thus an increased density of VCP-p-phenyl (1.139 g cm−3) was observed in comparison to VCP-m-phenyl (1.105 g cm−3). A complete crystallization of VCP-p-phenyl is hindered, due to the general isomeric structure of VCP ester–amides (see Scheme S1 in the ESI†). Furthermore, once the crystalline phase is melted the reverse crystallization occurs within a longer time lag over weeks, thus within the XRD diffractogram no reflexes returned after cooling the resin back to room temperature (see Fig. S14 within the ESI†).
Generally, the volume change is calculated by the densities of the cured and uncured resins. As in most cases the density of the cured resin is higher, negative volume changes are observed. However, volume expandable resins are known within the literature as well.28,29 Yet, their applicability is restricted, as the volume expansion is mainly based to the transition of a denser crystalline monomer structure compared to a less compressed, amorphous polymer structure. In fact, this attitude is lost, if the crystalline phase is melted or diluted by any further phase, like it can be observed also for the co-networks of VCP-p-phenyl (Fig. 5). In this sense heterogeneous systems, like VCP-p-phenyl, have to be seen as an intermediate stage between both physical states, offering a further alternative to reduce the volume shrinkage. Especially the connection of an easy processable wax-like structure and the low volume shrinkage exhibit further potential for composite formulations, and will be investigated in a following work.
Conclusions
A uniformity of the VCP ester–amide H-bond strength was shown, providing a universal concept of applying VCP ester–amides as suitable low-shrinking resins. With an excellent control of high reactivity, nearly regardless of the spacer-unit chosen, VCP ester–amides offer a clear advantage in comparison to methacrylate based systems. Thus with an appropriate variation of spacer-unit, selective characteristics, like e.g. optical properties or mechanical strength could be adjusted in a simple way, without obtaining any significant disadvantage within the curing behaviour. Based on this concept, new VCP ester–amide resins were presented, showing very low volume changes between −1.4 to −4.5%, a broad variety in mechanical strength between 0.19 to 232 MPa and a high overall thermal stability even under an atmosphere of air. Thereby the network properties could be continuously adjusted by varying the co-monomer content in co-networks. Moreover, due to the inevitable isomeric mixtures of VCP ester–amides, a heterogeneous system could be achieved, combining liquid phase with partial crystallinity, offering an intermediate stage between both physical states. The presented concept of applying VCP ester–amides as low shrinking resins for a modular construction kit is facile and variable. Thus VCP ester–amides offer vast potential for commercial use; especially as the number of combinations possibilities respectively for specified spacer units are yet not exploited.Acknowledgements
We sincerely thank J. Schöbel for MALDI-TOF analysis, and Dr R. Giesa (MCI), Prof. J. Senker and Dr R. Siegel (both ACIII), Prof. J. Breu and Dr W. Milius (both ACI), Prof. M. Willert-Porada and I. Otto (both Chair of Materials Processing) all of University of Bayreuth for providing diverse access to DMTA, solid-state NMR technique, XRD measurements and a gas-pycnometer.Notes and references
- Y. Fuchs, O. Soppera and K. Haupt, Anal. Chim. Acta, 2012, 717, 7 CrossRef CAS PubMed.
- Y. Jian, Y. He, L. Zhao, A. Kowalczyk, W. Yang and J. Nie, Adv. Polym. Technol., 2013, 32, 21331 CrossRef.
- E. Andrzejewska, Prog. Polym. Sci., 2001, 26, 605 CrossRef CAS.
- C. Decker, Macromol. Rapid Commun., 2002, 23, 1067 CrossRef CAS.
- C. Gorsche, K. Seidler, P. Knaack, P. Dorfinger, T. Koch, J. Stampfl, N. Moszner and R. Liska, Polym. Chem., 2016, 7, 257 RSC.
- K. L. van Landuyt and J. Snauwaert, et al. , Biomaterials, 2007, 28, 3757 CrossRef CAS PubMed.
- A. S. Quick, A. de los S. Pereira, M. Bruns, T. Bückmann, C. Rodriguez-Emmenegger, M. Wegener and C. Barner-Kowollik, Adv. Funct. Mater., 2015, 25, 3735 CrossRef CAS.
- D. H. Seuyep Ntoukam, G. A. Luinstra and P. Theato, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 2841 CrossRef CAS.
- K. S. Anseth, C. N. Bowman and N. A. Peppas, J. Polym. Sci., Part A: Polym. Chem., 1994, 32, 139 CrossRef CAS.
- I. Sideridou, V. Tserki and G. Papanastasiou, Biomaterials, 2002, 23, 1819 CrossRef CAS PubMed.
- S. H. Dickens, J. W. Stansbury, K. M. Choi and C. J. E. Floyd, Macromolecules, 2003, 36, 6043 CrossRef CAS.
- J. W. Stansbury, M. Trujillo-Lemon, H. Lu, X. Ding, Y. Lin and J. Ge, Dent. Mater., 2005, 21, 56 CrossRef CAS PubMed.
- P. Pineda Contreras, C. Kuttner, A. Fery, U. Stahlschmidt, V. Jerome, R. Freitag and S. Agarwal, Chem. Commun., 2015, 51, 11899 RSC.
- P. Pineda Contreras, P. Tyagi and S. Agarwal, Polym. Chem., 2015, 6, 2297 RSC.
- N. Moszner and U. Salz, Macromol. Mater. Eng., 2007, 292, 245 CrossRef CAS.
- N. Moszner, F. Zeuner, T. Völkel and V. Rheinberger, Macromol. Chem. Phys., 1999, 200, 2173 CrossRef CAS.
- F. Sanda and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 265 CrossRef CAS.
- V. Alupei and H. Ritter, e-Polym., 2002, 2, 724 Search PubMed.
- V. Alupei and H. Ritter, Macromol. Rapid Commun., 2001, 22, 1349 CrossRef CAS.
- K. D. Kopple, M. Ohnishi and A. Go, J. Am. Chem. Soc., 1969, 91, 4264 CrossRef CAS.
- V. Berl, M. Schmutz, M. J. Krische, R. G. Khoury and J.-M. Lehn, Chem. – Eur. J., 2002, 8, 1227 CrossRef CAS.
- M. T. Lemon, M. S. Jones and J. W. Stansbury, J. Biomed. Mater. Res., Part A, 2007, 83, 734 CrossRef PubMed.
- J. Lange, S. Toll, J.-A. E. Månson and A. Hult, Polymer, 1997, 38, 809 CrossRef CAS.
- J. Tu, S. J. Tucker, S. Christensen, A. R. Sayed, W. L. Jarrett and J. S. Wiggins, Macromolecules, 2015, 48, 1748 CrossRef CAS.
- S. G. Pereira, T. G. Nunes and S. Kalachandra, Biomaterials, 2002, 23, 3799 CrossRef CAS PubMed.
- J. Park, G. Lee, H. Ooshige, A. Nishikata and T. Tsuru, Corros. Sci., 2003, 45, 1881 CrossRef CAS.
- J. E. Klee and U. Lehmann, J. Org. Chem., 2009, 5, 72 Search PubMed.
- H. Chiba, K. Kitazume, S. Yamada and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 2015, 54, 39 CrossRef.
- N. Moszner and U. Salz, Prog. Polym. Sci., 2001, 26, 535 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Monomer characterization, TGA curves, 13C solid state NMR, temperature variable IR spectra and X-ray scattering diffractograms. See DOI: 10.1039/c6py00411c |
| This journal is © The Royal Society of Chemistry 2016 |