
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tandem vinyl insertion-/ring-opening metathesis copolymerization with ansa-6-[2-(dimesitylboryl)-phenyl]pyrid-2-ylamido zirconium complexes: role of trialkylaluminum and MAO†
Min
Wang
a,
Dongren
Wang
a,
Laura
Widmann
a,
Wolfgang
Frey
b and
Michael R.
Buchmeiser
*ac
aLehrstuhl für Makromolekulare Stoffe und Faserchemie, Institut für Polymerchemie, Universität Stuttgart, Pfaffenwaldring 55, D-70569 Stuttgart, Germany. E-mail: michael.buchmeiser@ipoc.uni-stuttgart.de; Fax: +49(0)-711-685-64050
bInstitut für Organische Chemie, Universität Stuttgart, Pfaffenwaldring 55, D-70569 Stuttgart, Germany
cInstitut für Textilchemie und Chemiefasern (ITCF) Denkendorf, Körschtalstr. 26, D-73770 Denkendorf, Germany
First published on 15th February 2016
Abstract
The novel dialkylzirconium complexes L′ZrR2, (R = CH3, Bn = benzyl, CH2SiMe3, L′ = Me2Si{η5-tetramethylcyclopentadienyl}{6-[2-(dimesitylboryl)phenyl]pyrid-2-ylamido}) were synthesized. Upon activation with 1 equiv. of [Ph3C]+[B(C6F5)4]−, both L′Zr(CH3)2 and L′Zr(Bn)2 are quantitatively converted in situ into [L′Zr(CH3)]+[B(C6F5)4]− and [L′Zr(Bn)]+[B(C6F5)4]− while only 28 mol% conversion is observed with L′Zr(CH2SiMe3)2. The aluminum-free cationic catalysts [L′Zr(CH3)]+[B(C6F5)4]−, [L′Zr(Bn)]+[B(C6F5)4]− and [L′Zr(CH2SiMe3)]+[B(C6F5)4]− initiate ring-opening metathesis polymerization (ROMP) of NBE to form predominantly cis-poly(NBE)ROMP. Upon activation with [Ph3C]+[B(C6F5)4]− and AliBu3, L′Zr(CH3)2, L′Zr(Bn)2 and L′Zr(CH2SiMe3)2 also exhibit moderate catalytic activity in the copolymerization of ethylene (E) with NBE. The resulting copolymers do not contain any ROMP-derived poly(NBE), which is in stark contrast to the complexes’ homopolymerization propensity for NBE. Upon activation with methylalumoxane (MAO), L′Zr(CH3)2 and L′Zr(Bn)2 produce pure vinyl-insertion polymerization-derived poly(NBE)-co-poly(E) while L′Zr(CH2SiMe3)2 allows for the synthesis of predominantly trans-poly(NBE)ROMP-co-poly(NBE)VIP-co-poly(E)-based copolymers via an α-H+ elimination/addition process. Our findings are discussed on the basis of the instability of the alkylidenes in the presence of E and the blocking effect of aluminum alkyls on ROMP via coordination to the pyridyl-moiety in the cationic complexes.
Introduction
Group 4 transition-metal monocyclopentadienyl-amido complexes are of considerable interest due to their remarkable activity in α-olefin polymerization.1–3 The ability to additionally incorporate bulkier olefins, e.g., norborn-2-ene (NBE), has attracted significant attention.4 Introduction of alkyl substituents instead of halides has a significant impact on the reactivity of group 4 initiators and affects both catalytic activity and polymer properties such as molecular weight and comonomer incorporation.5,6 Generally, metal alkyl cations, formed from the neutral dialkyl progenitor complexes upon activation with co-catalysts, e.g., [HNMe2Ph]+[B(C6F5)4]−, B(C6F5)3 or trityl borate [Ph3C]+[B(C6F5)4]−,7 are assumed to represent the active species in olefin polymerization.Our previous studies8 revealed that group 4 metal dichloro complexes of the general formula L′MCl2 (M = Ti, Zr; L′ = Me2Si{η5-tetramethylcyclopentadienyl}{6-[2-(R′2-boryl)phenyl]pyrid-2-ylamido}, R′ = e.g., mesityl, ethyl, Scheme 1) activated by methylalumoxane (MAO) allow the synthesis of ethylene-norborn-2-ene (E-NBE) copolymers with both ring-opening metathesis polymerization (ROMP)- and vinyl-insertion polymerization (VIP)-derived poly(NBE) sequences within one single polymer chain, that is poly(NBE)ROMP-co-poly(NBE)VIP-co-poly(E) (Fig. 1). This was explained by a switch from VIP to ROMP via α-H+ elimination from the growing polymer chain by the pyridyl nitrogen.
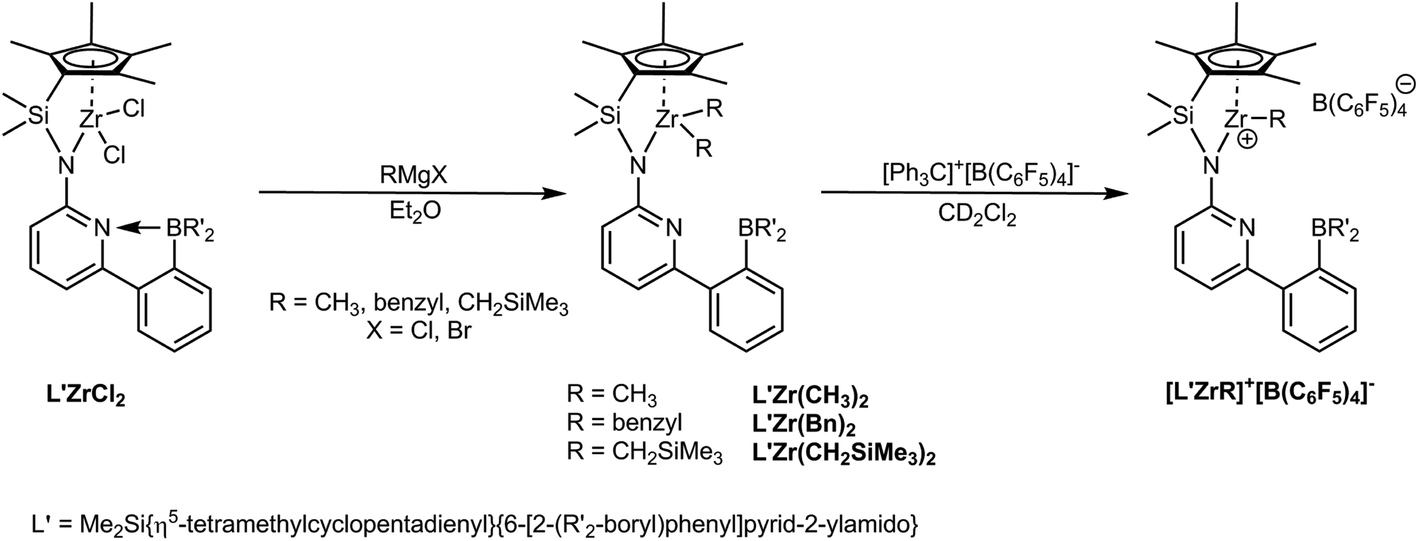 | ||
| Scheme 1 Synthesis of L′Zr(CH3)2, L′Zr(Bn)2, L′Zr(CH2SiMe3)2 and the corresponding cationic complexes. Here, R′ = mesityl. | ||
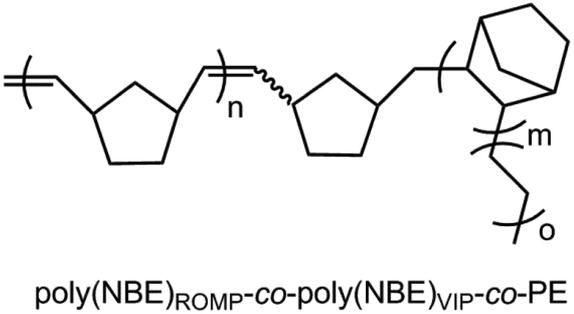 | ||
| Fig. 1 Proposed structure of ROMP/VIP-derived poly(NBE)-co-poly(E) produced by L′MCl2/MAO.9 | ||
This switch can be tuned via the temperature-dependent dissociation of the N–B bond9 for which detailed variable-temperature 1H and 11B NMR measurements have been carried out. With the aid of a variety of different pre-catalysts, the following key features have already been identified:8–14 (i) a crowded ligand sphere around the metal favors the α-H+ elimination process, i.e. the switch from VIP to ROMP, (ii) high NBE concentrations are required to support this switch from VIP to ROMP, and (iii) the propensity of the pyridine group to coordinate to boron can be governed by the sterics of the substituents at boron.
Unfortunately, the catalytic system L′ZrCl2/MAO displays a low activity in E-NBE copolymerization (≤6 kg of polymer mol−1catalyst h−1 bar−1), which has been attributed to the bulky ligand sphere and the propensity of the alkylidene to undergo cross metathesis with ethylene.12 Generally, cationic M-alkyl catalysts derived from L′MR2via borane- or borate-activation have been demonstrated to display higher polymerization activity than the corresponding cationic M-alkyl catalysts derived from L′MCl2/MAO. This can be explained by the fact that catalysts of the type [L′MR]+[BArF]− are truly catalytic species while the systems L′MR+/MAO in fact exist in the form of ion pairs or even adducts of the general formula [L′M(μ-R)2AlR2]+[RMAO]−.7,15–17
Here, we report on structural modifications on the half-sandwich zirconium dichlorides (L′ZrCl2) by replacement of both chloro ligands by dialkyl groups (alkyl = CH3, benzyl, CH2SiMe3). The desired cationic complexes were then prepared via treatment of the dialkyl complexes with a stoichiometric amount of [Ph3C]+[B(C6F5)4]−. The catalytic performance of the cationic complexes in the homopolymerization of both NBE and E as well as in the copolymerization of E with NBE was explored in comparison to the parent dichloro-complex L′ZrCl2, activated by MAO.
Results and discussion
L′ZrCl2 was prepared as reported.12 Treatment of L′ZrCl2 with the corresponding Grignard reagents in diethyl ether18–24 resulted in the formation of the dialkylzirconium complexes L′Zr(CH3)2, L′Zr(Bn)2 (Bn = benzyl) and L′Zr(CH2SiMe3)2. The synthetic routes are outlined in Scheme 1. Crystals suitable for single-crystal X-ray diffraction were grown from saturated diethyl ether solution. L′Zr(CH3)2 crystalizes in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , a = 868.85(10), b = 1218.49(12), c = 1810.5(2) pm, α = 79.834°, β = 85.356°, γ = 81.823°, Z = 2 (Fig. 2). The Zr(1)–Namide distance is 216.0(2) pm, which is longer than in L′ZrCl2 (210.85 pm). The pyridyl nitrogen is coordinated to zirconium (Zr(1)–N(2) = 253.9 pm) and not to the boron atom. Both the Zr–Npyridine and the Zr–Namide distances are longer than in L′ZrCl2 (Table 1), which is attributed to the electron-donating property of the dimethyl groups.
, a = 868.85(10), b = 1218.49(12), c = 1810.5(2) pm, α = 79.834°, β = 85.356°, γ = 81.823°, Z = 2 (Fig. 2). The Zr(1)–Namide distance is 216.0(2) pm, which is longer than in L′ZrCl2 (210.85 pm). The pyridyl nitrogen is coordinated to zirconium (Zr(1)–N(2) = 253.9 pm) and not to the boron atom. Both the Zr–Npyridine and the Zr–Namide distances are longer than in L′ZrCl2 (Table 1), which is attributed to the electron-donating property of the dimethyl groups.
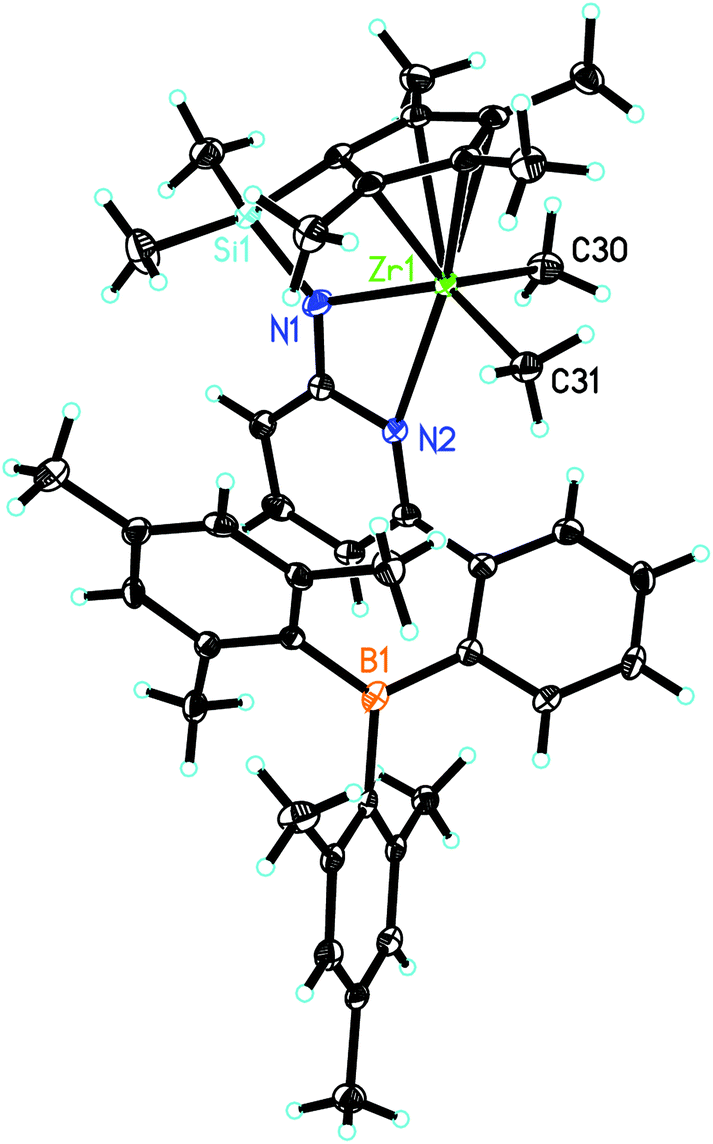 | ||
| Fig. 2 Single-crystal X-ray structure of L′Zr(CH3)2. Selected bond lengths [pm] and angles [°]: Zr(1)–N(1) 216.0(2), Zr(1)–C(30) 224.4(2), Zr(1)–C(31) 226.9(3), Zr(1)–C(34) 247.3(2), Zr(1)–C(35) 248.4(3), Zr(1)–C(38) 251.4(2), Zr(1)–N(2) 253.9(2), Zr(1)–C(36) 258.1(3), Zr(1)–C(37) 258.1(2), Zr(1)–C(1) 283.7(3); N(1)–Zr(1)–C(30) 107.52(10), N(1)–Zr(1)–C(31) 132.95(9), C(30)–Zr(1)–C(31) 102.70(10), N(1)–Zr(1)–N(2) 56.29(7), C(30)–Zr(1)–N(2) 90.31(8), C(31)–Zr(1)–N(2) 88.90(8). | ||
| # | L′ZrCl2 | L′Zr(CH3)2 | L′Zr(Bn)2 | L′Zr(CH2SiMe3)2 |
|---|---|---|---|---|
| Zr–Namide | 210.85 | 216.0 | 208.9 | 213.23 |
| Zr–Npyridine | 249.61 | 253.9 | — | — |
| Zr–Calkyl | — | 224.4/226.9 | 226.2/227.9 | 224.3/225.5 |
L′Zr(Bn)2 crystalizes in the monoclinic space group P21/c, a = 2791.4(2), b = 1601.47(9), c = 1021.51(7) pm, α = 90°, β = 95.322°, γ = 90°, Z = 4 (Fig. 3). The pyridyl nitrogen in L′Zr(Bn)2 is neither coordinated to zirconium nor to the boron atom, which is in stark contrast to the situation in L′ZrCl2 and L′Zr(CH3)2. We tentatively attribute this finding to the increased steric demand of the benzyl groups. The Zr(1)–Namide distance is 208.9(6) pm, which is significantly shorter than in L′Zr(CH3)2 (216.0 pm), attributable to the fact that the pyridine is uncoordinated. The angles C(42)–C(41)–Zr(1) and C(49)–C(48)–Zr(1) are 123.6(3)° and 91.1(3)°, respectively, which reveals that two benzyl ligands are inequivalent in the solid state, one adopting an η1-bonding mode (123.6°) and the other η2-bonding25–30 (91.1°). In contrast to the solid state, the two benzyl groups in L′Zr(Bn)2 are magnetically equivalent in solution (CD2Cl2), with two diastereotopic benzylic protons at δ = 2.06 and 1.93 ppm (J = 52.0 Hz).
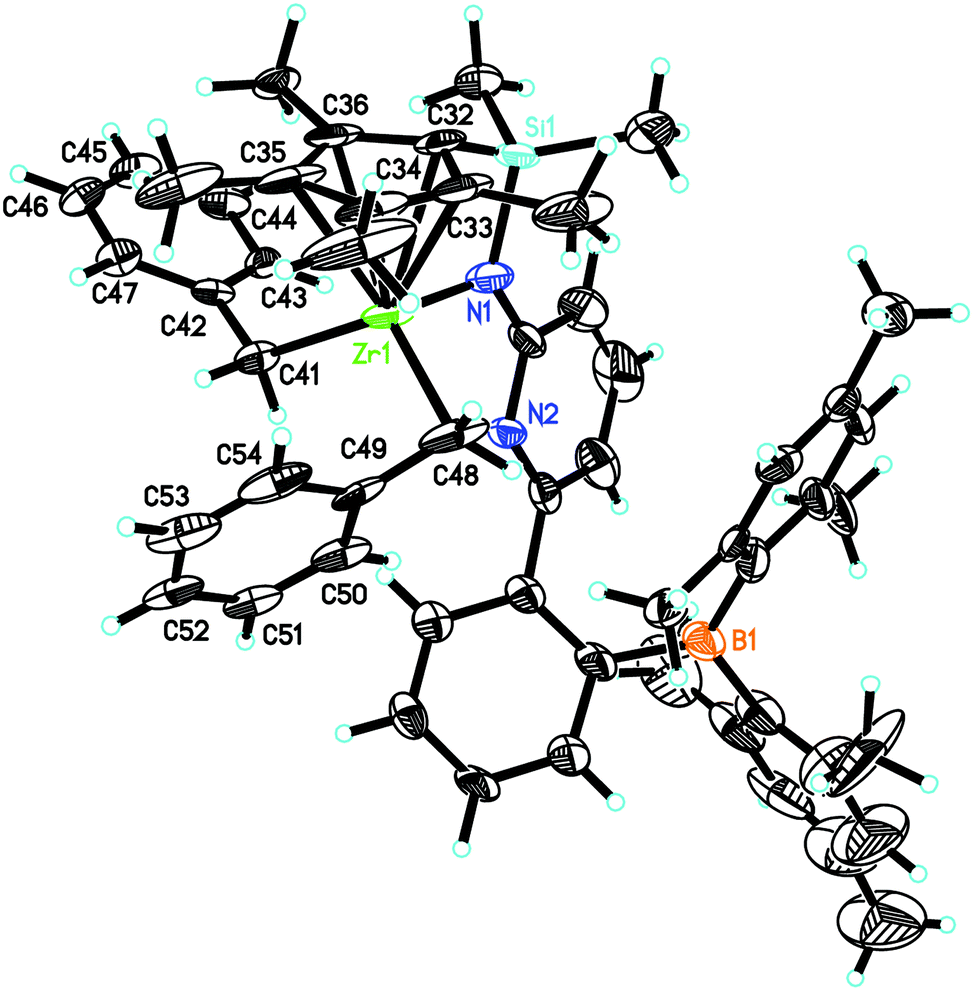 | ||
| Fig. 3 Single-crystal X-ray structure of L′Zr(Bn)2. Selected bond lengths [pm] and angles [°]: Zr(1)–N(1) 208.9(6), Zr(1)–C(48) 226.2(5), Zr(1)–C(41) 227.9(5), Zr(1)–C(32) 243.3(5), Zr(1)–C(33) 247.4(5), Zr(1)–C(34) 260.0(6), Zr(1)–C(35) 257.7(7), Zr(1)–C(36) 251.4(5); N(1)–Zr(1)–C(48) 109.8(2), N(1)–Zr(1)–C(41) 102.59(19), C(48)–Zr(1)–C(41) 117.38(19), C(42)–C(41)–Zr(1) 123.6(3), C(49)–C(48)–Zr(1) 91.1(3). | ||
L′Zr(CH2SiMe3)2 (Fig. 4) crystalizes in the triclinic space group P, a = 1114.92(5), b = 1131.88(5), c = 2027.72(9) pm, α = 89.308°, β = 82.967°, γ = 77.549°, Z = 2. The pyridyl nitrogen is neither coordinated to Zr nor to B, similar to L′Zr(Bn)2. The Zr(1)–Namide distance is 213.23(11) pm, which is shorter than in L′Zr(CH3)2 (216.0 pm) but longer than in L′Zr(Bn)2 (208.9 pm).
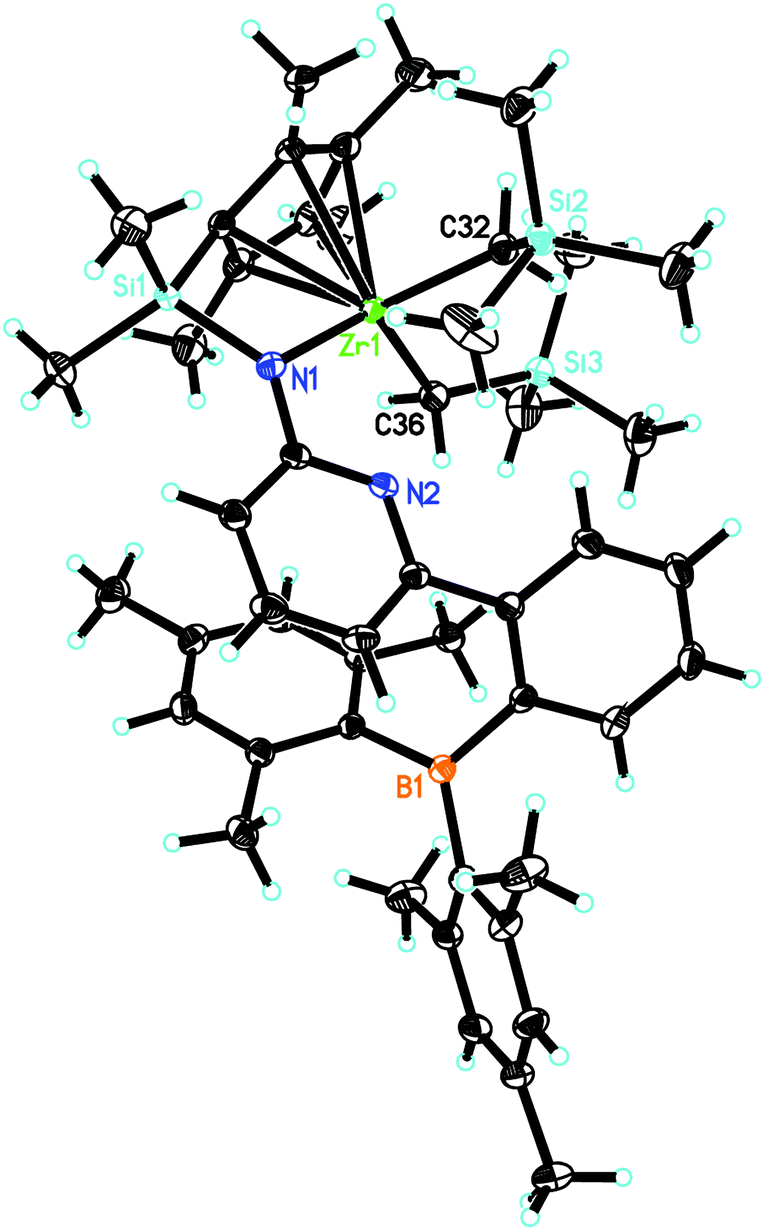 | ||
| Fig. 4 Single-crystal X-ray structure of L′Zr(CH2SiMe3)2. Selected bond lengths [pm] and angles [°]: Zr(1)–N(1) 213.23(11), Zr(1)–C(32) 224.30(13), Zr(1)–C(36) 225.50(14), Zr(1)–C(40) 246.68(13), Zr(1)–C(44) 248.05(13), Zr(1)–C(41) 252.47(13), Zr(1)–C(43) 259.96(13), Zr(1)–C(42) 261.18(13); N(1)–Zr(1)–C(32) 111.58(5), N(1)–Zr(1)–C(36) 103.35(5), C(32)–Zr(1)–C(36) 103.35(5), Si(2)–C(32)–Zr(1) 130.85(7), Si(3)–C(36)–Zr(1) 126.57(7). | ||
The corresponding cationic complexes were created in situ by abstraction of one alkyl group from the corresponding dialkyl complexes using 1 equiv. of [Ph3C]+[B(C6F5)4]− in CD2Cl2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 31,32 (Scheme 1). For L′Zr(CH3)2 and L′Zr(Bn)2, 1H NMR spectroscopy was indicative for the quantitative conversion of the dialkyl complexes to the corresponding cationic complexes. However, addition of [Ph3C]+[B(C6F5)4]− to a CD2Cl2 solution of L′Zr(CH2SiMe3)2 resulted only in ∼28 mol% formation of the cationic complex, probably due to the sterics of two CH2SiMe3 groups at the metal and their low reactivity with [Ph3C]+[B(C6F5)4]−. Among these monoalkyl cationic complexes, isolation of the methyl cation with one diethyl ether molecule coordinated to Zr as indicated by NMR and elemental analysis was successful.
31,32 (Scheme 1). For L′Zr(CH3)2 and L′Zr(Bn)2, 1H NMR spectroscopy was indicative for the quantitative conversion of the dialkyl complexes to the corresponding cationic complexes. However, addition of [Ph3C]+[B(C6F5)4]− to a CD2Cl2 solution of L′Zr(CH2SiMe3)2 resulted only in ∼28 mol% formation of the cationic complex, probably due to the sterics of two CH2SiMe3 groups at the metal and their low reactivity with [Ph3C]+[B(C6F5)4]−. Among these monoalkyl cationic complexes, isolation of the methyl cation with one diethyl ether molecule coordinated to Zr as indicated by NMR and elemental analysis was successful.
Dialkyl complexes are also valuable progenitors for the formation of metal alkylidenes through thermally induced α-H elimination.33 Alternatively, α-hydrogen abstraction can be induced by the addition of phosphines (PMe3, PPhMe2, PPh2Me, etc.).34–39 The resulting alkylidenes are the key species in metathesis polymerization. Unfortunately, all attempts to generate metal alkylidenes through thermolysis or by the addition of phosphines to the neutral alkyl compounds were unsuccessful. Thus, in toluene-d8, L′Zr(CH3)2, L′Zr(Bn)2, L′Zr(CH2SiMe3)2 were found to be thermally stable at least up to 80 °C. The same accounts for [L′Zr(CH3)]+, [L′Zr(Bn)]+ and [L′Zr(CH2SiMe3)]+[B(C6F5)4]− in o-C6D4Cl2. Even in the presence of 3–7 equiv. of PMe3 or PPhMe2 with respect to Zr, no transformation of the Zr-dialkyls to the corresponding Zr-alkylidenes was observed by 1H NMR.
NBE homopolymerization and E-NBE copolymerization
Ring-opening metathesis homopolymerization of NBE by the action of in situ generated [L′Zr(CH3)]+, [L′Zr(Bn)]+ and [L′Zr(CH2SiMe3)]+[B(C6F5)4]− was carried out in toluene at different temperatures (Table 2). Notably, productivity of [L′Zr(Bn)]+[B(C6F5)4]− at 80 °C was remarkably higher (140 kg of polymer mol−1catalyst h−1) than that of [L′Zr(CH3)]+ and [L′Zr(CH2SiMe3)]+[B(C6F5)4]−, which is attributable to a higher propensity of [L′Zr(Bn)]+ to form L′Zr![[double bond, length as m-dash]](https://www.rsc.org/images/entities/b_char_e001.gif) CHPh. Interestingly, all polymers were purely ROMP-derived poly(NBE) and did not contain any VIP-type poly(NBE) as evidenced by 13C NMR (Fig. S1b–S4b, ESI†). The cis-content of the polymers increased with decreasing polymerization temperature (52 < σcis < 85%, Fig. S1a–S4a, ESI†) and correlated with Tg;40 however, it was lower (Fig. S1a–S4a, ESI†) than that in poly(NBE)s prepared by the action of L′ZrCl2/MAO (86 < σcis < 95%).12 This points towards stronger ion pairing of the cations with MAO than with the [B(C6F5)4]− anion and consequently a significant influence of the large MAO on the geometry of the active centers. In terms of mechanism, the formation of poly(NBE)ROMP must proceed by the formation of zirconium alkylidenes,34–39,41–48 promoted by the approach of NBE molecules or by the NBE insertion into the metal–carbon bond of the parent cationic system followed by α-hydrogen abstraction (Scheme 2, vide infra). However, no alkylidene species were spectroscopically detectable. Obviously, only minor amounts of the corresponding cations convert into alkylidenes. The comparably high polydispersity indexes are tentatively attributed to secondary metathesis, i.e. to back-biting.
CHPh. Interestingly, all polymers were purely ROMP-derived poly(NBE) and did not contain any VIP-type poly(NBE) as evidenced by 13C NMR (Fig. S1b–S4b, ESI†). The cis-content of the polymers increased with decreasing polymerization temperature (52 < σcis < 85%, Fig. S1a–S4a, ESI†) and correlated with Tg;40 however, it was lower (Fig. S1a–S4a, ESI†) than that in poly(NBE)s prepared by the action of L′ZrCl2/MAO (86 < σcis < 95%).12 This points towards stronger ion pairing of the cations with MAO than with the [B(C6F5)4]− anion and consequently a significant influence of the large MAO on the geometry of the active centers. In terms of mechanism, the formation of poly(NBE)ROMP must proceed by the formation of zirconium alkylidenes,34–39,41–48 promoted by the approach of NBE molecules or by the NBE insertion into the metal–carbon bond of the parent cationic system followed by α-hydrogen abstraction (Scheme 2, vide infra). However, no alkylidene species were spectroscopically detectable. Obviously, only minor amounts of the corresponding cations convert into alkylidenes. The comparably high polydispersity indexes are tentatively attributed to secondary metathesis, i.e. to back-biting.
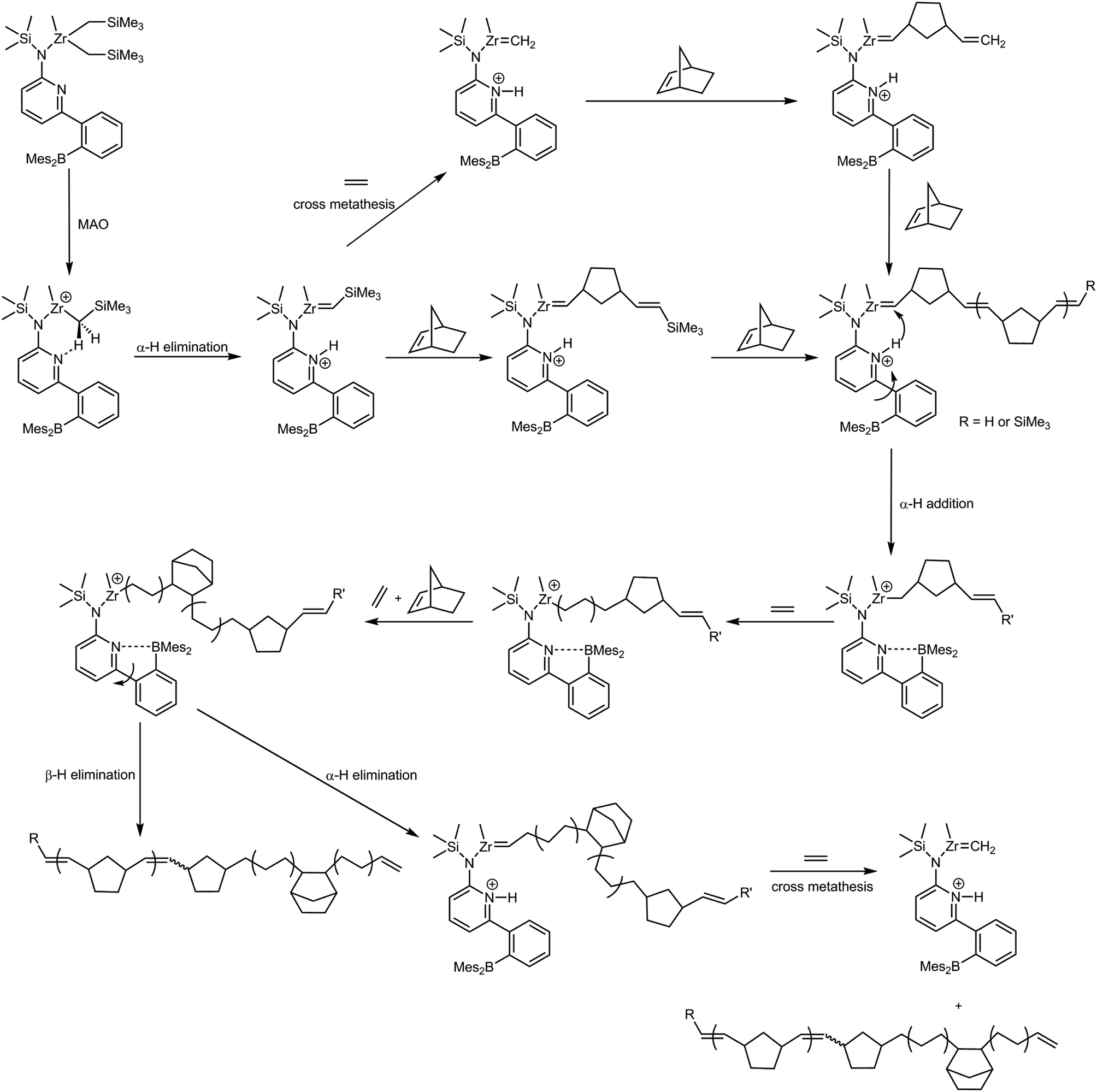 | ||
| Scheme 2 Proposed mechanism for the formation of poly(NBE)ROMP-co-poly(NBE)VIP-co-poly(E) by the action of L′Zr(CH2SiMe3)2/MAO. | ||
a
| # | Catalyst | T (°C) | A |
M
nc (g mol−1) |
Đ |
T
gd (°C) |
cis (%) |
|---|---|---|---|---|---|---|---|
|
a 100 mL Schlenk flask, total volume of reaction mixture: 50 mL of toluene, [catalyst] = 1 × 10−4 mol L−1, catalyst:[Ph3C]+[B(C6F5)4]−:NBE = 1:1:10000, t = 1 h.
b Activity in kg of polymer mol−1catalyst h−1.
c Molecular weights (Mn) and polydispersity indexes (Đ) determined by HT-GPC in 1,2,4-trichlorobenzene vs. PS.
d Measured by DSC.
e Determined by 1H NMR analysis in 1,1,2,2-tetrachloroentane-d2.
|
|||||||
| 1 | L′Zr(CH3)2 | 50 | 10 | 170000 |
2.7 | 58 | 85 |
| 2 | L′Zr(CH3)2 | 65 | 12 | 92000 |
2.5 | 56 | 71 |
| 3 | L′Zr(CH3)2 | 80 | 45 | 10000 |
1.6 | 47 | 52 |
| 4 | L′Zr(Bn)2 | 50 | 2 | — | — | 48 | 69 |
| 5 | L′Zr(Bn)2 | 65 | 10 | 100000 |
3.1 | 53 | 70 |
| 6 | L′Zr(Bn)2 | 80 | 140 | 14000 |
2.2 | 39 | 72 |
| 7 | L′Zr(CH2SiMe3)2 | 50 | 5 | 200000 |
2.1 | 45 | 72 |
| 8 | L′Zr(CH2SiMe3)2 | 65 | 10 | 43000 |
1.1 | 50 | 71 |
| 9 | L′Zr(CH2SiMe3)2 | 80 | 9 | 21000 |
1.9 | 53 | 61 |
Cationic group 4 transition-metal alkyl complexes have been reported to exhibit high catalytic activity and efficient incorporation of bulky comonomers in the absence of aluminum co-catalysts.5,6,49,50 It was therefore of interest to explore the copolymerization of E with NBE with [L′Zr(CH3)]+, [L′Zr(Bn)]+ and [L′Zr(CH2SiMe3)]+[B(C6F5)4]−. Unfortunately, none of these catalysts was capable of homopolymerizing E or copolymerizing E with NBE to yield high molecular weight (co-) polymers without the involvement of aluminum alkyl activators. For reasons, vide infra.
Upon in situ activation with [Ph3C]+[B(C6F5)4]− and triisobutylaluminum (AliBu3),51–60L′Zr(CH3)2, L′Zr(Bn)2 and L′Zr(CH2SiMe3)2 showed moderate catalytic activity in E-NBE copolymerization ranging from 15 to 80 kg of polymer mol−1catalyst h−1 bar−1, which is significantly higher than the activity of L′ZrCl2/MAO (≤4 kg of polymer mol−1catalyst h−1 bar−1)12 under similar conditions (Table 3, entries 2–6). It is worth noting that L′Zr(Bn)2 exhibited the highest productivity (80 kg of polymer mol−1catalyst h−1 bar−1) in E-NBE copolymerization, in line with its highest activity in NBE homopolymerization (vide supra). This high activity of [L′Zr(Bn)]+[B(C6F5)4]− might be attributed to a reversible η2–η1 rearrangement of the benzylic group, which stabilizes the active sites and reduces deactivation.15 Generally, NBE incorporation into the copolymers was low (≤2 mol%) compared to the one obtained with L′ZrCl2/MAO (28 mol%) under similar conditions.12 Solely VIP-derived poly(NBE) moieties were observed in poly(E)-co-poly(NBE) (Fig. 5). The characteristic resonances at δ = 47.0 (C2/C3), 41.5 (C1/C4) and 32.9 (C7) ppm are assignable to alternating syndiotactic (alt-st, E-NBE-E-NBE)/isolated (E-NBE-E-E) VIP-derived sequences while the signal at δ = 29.5 ppm corresponds to PE sequences.61,62 Also, no signals for alternating isotactic (alt-it, E-NBE-E-NBE) units, NBE diads (E-NBE-NBE-E) or triads (E-NBE-NBE-NBE-E) were observed. Although the alt-st and isolated NBE sequences cannot be distinguished as a result of overlapping signals, it is with respect to the low NBE incorporation and the bulky ligand sphere reasonable to propose isolated NBE sequences. 13C NMR analysis proved that PE sequences in the copolymers were mostly mainly linear with low degrees of branching (∼1 branch per 1000 carbons), indicative for a low β-hydride elimination and chain-walking propensity.
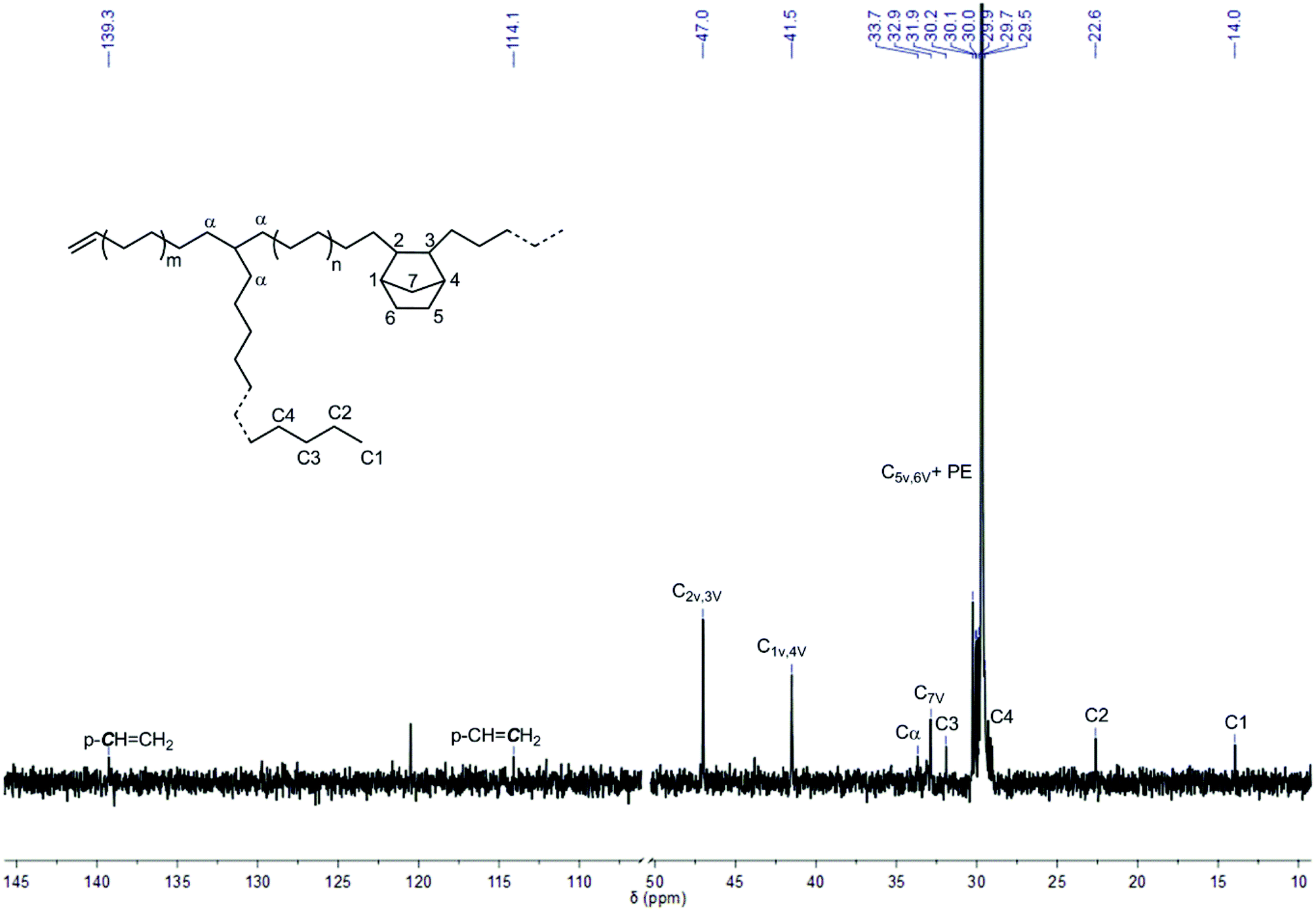 | ||
| Fig. 5 13C NMR spectrum of poly(E)-co-poly(NBE)VIP produced by L′Zr(CH3)2/[Ph3C]+[B(C6F5)4]−/AliBu3 (Table 3, entry 4). The signal at δ = 120.5 ppm stems from the impurity in 1,1,2,2-tetrachloroethane-d2. | ||
a and E-NBE copolymerization by the action of L′Zr(CH3)2, L′Zr(Bn)2 and L′Zr(CH2SiMe3)2 activated by [Ph3C]+[B(C6F5)4]−/AliBu3a and MAOb
| # | Catalyst | Monomers | T (°C) | A | C NBE (mol%) |
M
ne (g mol−1) |
Đ |
T
mf (°C) |
|---|---|---|---|---|---|---|---|---|
|
a 250 mL of toluene (including the volume of monomer), t = 1 h, [catalyst] = 2 × 10−5 mol L−1, catalyst:[Ph3C]+[B(C6F5)4]−:AliBu3 = 1:1:350 or catalyst:[Ph3C]+[B(C6F5)4]−:AliBu3:NBE = 1:1:350:20000, pethylene = 4 bar.
b Catalyst:MAO:NBE = 1:2000:20000, pethylene = 4 bar.
c Activity in kg of polymer mol−1catalyst h−1 bar−1.
d NBE content in the copolymer [mol%] as estimated by 13C NMR spectroscopy.
e Molecular weights (Mn) and polydispersity indexes (Đ) determined by HT-GPC in 1,2,4-trichlorobenzene vs. PS.
f Measured by DSC.
|
||||||||
| 1a | L′Zr(CH3)2 | E | 50 | 30 | — | 1000000 |
2.9 | 129 |
| 2a | L′Zr(CH3)2 | E/NBE | 30 | 15 | 1.7 | 570000 |
2.6 | 131 |
| 3a | L′Zr(CH3)2 | E/NBE | 50 | 60 | 1.5 | 300000 |
4.3 | 129 |
| 4a | L′Zr(CH3)2 | E/NBE | 80 | 42 | 1.7 | 32000 |
7.6 | 129 |
| 5a | L′Zr(Bn)2 | E/NBE | 50 | 80 | 0.5 | 340000 |
6.3 | 131 |
| 6a | L′Zr(CH2SiMe3)2 | E/NBE | 50 | 45 | 2.0 | >6000000 |
— | 130 |
| 7b | L′Zr(CH3)2 | E/NBE | 50 | 4 | 1.1 | >6000000 |
— | 125 |
| 8b | L′Zr(Bn)2 | E/NBE | 50 | 4 | 2.0 | 640000 |
2.7 | 128 |
| 9b | L′Zr(CH2SiMe3)2 | E/NBE | 30 | 5 | 3 | 490000 |
2.5 | 123 |
| 10b | L′Zr(CH2SiMe3)2 | E/NBE | 50 | 7 | 25 | 220000 |
2.4 | 121 |
| 11b | L′Zr(CH2SiMe3)2 | E/NBE | 80 | 3 | 19 | 660000 |
4.6 | 126 |
In addition, a terminal vinyl group was observed in all PE sequences of copolymers as clearly demonstrated by multiplets at δ = 5.9 and 5.0 ppm in the 1H NMR spectrum (Fig. S5, ESI†) and at δ = 139.3 and 114.1 ppm in the 13C NMR spectrum14 (Fig. 5). Similarly, the PE prepared from L′Zr(CH3)2/[Ph3C]+[B(C6F5)4]−/AliBu3 showed vinyl terminals (Fig. S6, ESI†); the PE itself was mainly linear with 2 branches per 1000 carbons as estimated by 13C NMR (Fig. S7, ESI†). In combination with the melting point (Tm) of poly(E)-co-poly(NBE), which ranged from 129 to 131 °C and a single peak in the GPC, the structure of the copolymers produced by L′Zr(CH3)2, L′Zr(Bn)2 and L′Zr(CH2SiMe3)2 activated with [Ph3C]+[B(C6F5)4]− and AliBu3 was VIP-type poly(E)-co-poly(NBE) with few branches (Fig. 5 and Fig. S8–S9, ESI†). With L′Zr(CH3)+/AliBu3, an increase in polymerization temperature from 30 to 80 °C resulted in a dramatic decrease in molecular weight (Mn) from 570000 to 32000 g mol−1 and a significant increase in the polydispersity index (Đ) from 2.6 to 7.6, indicative for substantial β-hydride elimination/chain transfer to monomer.
Generally, AlR3 plays an essential role in the polymerization process. In VIP, heterobimetallic complexes termed as [L′M(μ-R)2AlR2]+ are considered to be the dormant species.15,63–65 Consequently, subsequent dissociation of AlR3 to form metal alkyl cations is the key step in olefin polymerization.15,66 The reversible coordination/decoordination of the pyridyl group in L′ZrCl2 has been shown to play a crucial role in the α-hydrogen abstraction process.12 Unlike L′ZrCl2, L′Zr(CH3)2 displays only the “open” structure with no pyridyl coordination to boron as evidenced by 11B NMR67–69 (Fig. S10, ESI†), which is attributed to the electron richer nature of the complex. The boron signals in the 11B NMR of L′Zr(Bn)2 and L′Zr(CH2SiMe3)2 are too weak and do not allow any conclusive structure assignment (i.e. coordinated vs. non-coordinated). However, in view of the similar electronic nature of the benzyl and CH2SiMe3 groups and the solid state structures it is reasonable to assume that the pyridine group is not coordinated to boron in the temperature range of polymerization. The finding that [L′ZrR]+[B(C6F5)4]− (R = CH3, Bn, CH2SiMe3) forms ROMP-derived poly(NBE) in the absence but not in the presence of AliBu3 and that [L′ZrR]+[B(C6F5)4]− (R = CH3, Bn, CH2SiMe3) does form VIP-derived poly(NBE)-co-poly(E) in the presence but not in the absence of AliBu3 strongly suggests that AliBu3 binds to the pyridyl-moiety in [L′ZrR]+, thereby terminating its capability to induce α-hydrogen abstraction, a process that occurs in the absence of AliBu3. Accordingly, the cationic complexes are capable of forming VIP-derived poly(NBE)-co-poly(E) in the presence of AliBu3, but not in its absence. In the absence of AliBu3, α-H+ elimination dominates and poly(NBE)ROMP forms. Notably, the isolated diethyl ether adduct [L′Zr(CH3)Et2O]+[B(C6F5)4]− is inactive in NBE homopolymerization in the absence of AliBu3 and inactive in E-NBE copolymerization in the presence of AliBu3, which clearly reveals the irreversible blocking effect of Et2O on the coordination of olefin. It is also worth pointing out that any interaction between the metal and the [B(C6F5)4]− anion, whether via the fluorines or via the phenyl-group itself, can be excluded to the greatest possible extent by the fact that the 19F NMR spectra of [L′ZrR]+[B(C6F5)4]− (R = CH3, Bn) do not show any differences to the one of [Ph3C]+[B(C6F5)]− (Fig. S11 and S33, S36, S39, ESI†). All in all, the inability of L′ZrR+ (R = CH3, Bn, CH2SiMe3) to promote ROMP in the presence of AliBu3 and to promote vinyl insertion copolymerization of E with NBE in the absence of AliBu3 is attributed to the following three key features: (i) the instability of the alkylidenes in the presence of E, (ii) the blocking of the pyridyl ligand in [L′ZrR]+ by AliBu3 and (iii) the competition of a coordinating solvent for the coordination site.
Next, the performance of L′ZrR2/MAO (R = CH3, Bn, CH2SiMe3) in E-NBE copolymerization was explored in comparison to the one of L′ZrCl2/MAO and [L′ZrR]+[B(C6F5)]−/AliBu3 (R = CH3, Bn, CH2SiMe3) (Table 3, entries 7–11). Catalytic activities were lower for the systems L′ZrR2/MAO (3–7 kg of polymer mol−1catalyst h−1 bar−1) and L′ZrCl2/MAO (1–4 kg of polymer mol−1catalyst h−1 bar−1) compared to [L′ZrR]+[B(C6F5)]−/AliBu3 (15–80 kg of polymer mol−1catalyst h−1 bar−1), which is in line with literature reports7,15–17 (vide supra).
For the copolymers produced by L′Zr(CH3)2/MAO and L′Zr(Bn)2/MAO, only VIP-derived poly(NBE)-co-poly(E) was obtained. Low NBE incorporation, i.e. 1.1 and 2.0 mol%, respectively, was observed along with vinyl terminals and few long chain branches (Fig. S12–S13, ESI†). By contrast, L′Zr(CH2SiMe3)2/MAO produced copolymers with both ROMP- and VIP-derived NBE units in the same polymer chain, that is poly(NBE)ROMP-co-poly(NBE)VIP-co-PE. At 30 °C, the ratio of poly(NBE)ROMP:poly(NBE)VIP:PE was 1:2:97 (Fig. S14–S15, ESI†) and increased to 22:3:75 with increasing temperature (50 °C) (Fig. 6a and Fig. S16–S18, ESI†). Again, terminal vinyl groups and few long chain branches were observed. A further increase in the temperature (80 °C) resulted in a decrease in the proportion of ROMP-type poly(NBE) units in the copolymers (14:5:81) (Fig. S19–S20, ESI†).
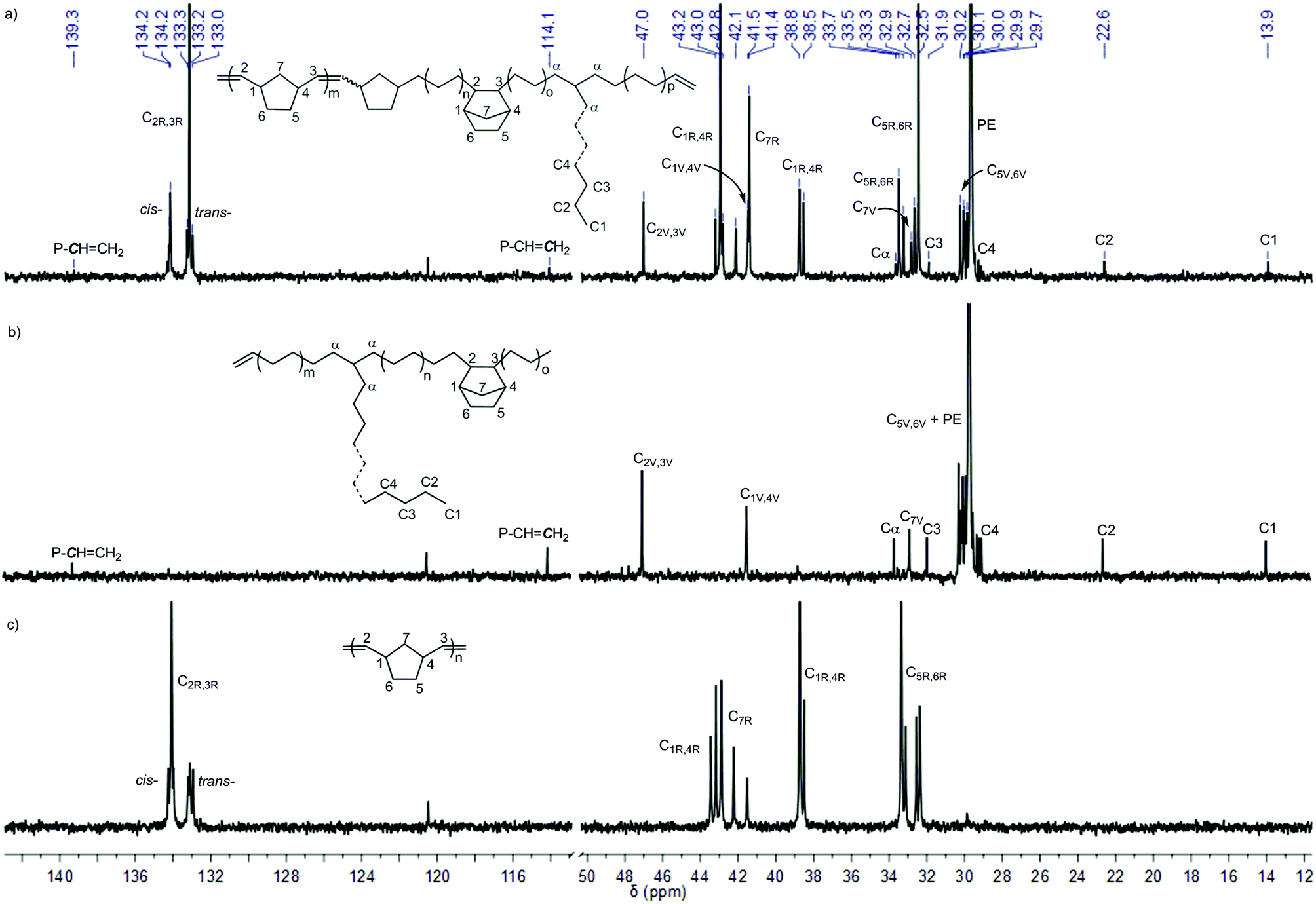 | ||
| Fig. 6 13C NMR spectra of (a) poly(NBE)ROMP-co-poly(NBE)VIP-co-poly(E) produced by L′Zr(CH2SiMe3)2/MAO (Table 3, entry 10), (b) poly(NBE)VIP-co-poly(E) produced by L′Zr(Bn)2/MAO (Table 3, entry 8) and (c) poly(NBE)ROMP produced by L′Zr(CH2SiMe3)2/[Ph3C]+[B(C6F5)4]− (Table 2, entry 8). The signal at δ = 120.5 ppm stems from the impurity in 1,1,2,2-tetrachloroethane-d2. | ||
Signals at δ = 47.0 (C2,3), 41.5 (C1,4), 32.9 (C7) and 30.2–29.9 (C5,6) ppm can be attributed to alt-st/isolated VIP-derived poly(NBE) units; those at δ = 29.7 ppm are assignable to PE. As outlined above, isolated VIP-type NBE sequences interrupted by PE sequences are reasonable. This incapability to form consecutive VIP-derived NBE–NBE sequences also accounts for the finding that no VIP-derived sequences are visible in poly(NBE)ROMP. The signals at δ = 134.2, 133.2 (C2,3), 43.0, 38.8 (C1,4), 42.8, 42.1, 41.4 (C7) and 33.5, 32.5 (C5,6) are unambiguously assignable to ROMP-derived NBE sequences. In contrast to poly(NBE)ROMP-co-poly(NBE)VIP-co-PE prepared by a structurally similar Ti-catalyst,8 copolymers had a substantially higher trans-content with more tttt and cccc sequences (t = trans, c = cis). Notably, these signals prevail even after extensive hot extraction with THF. This together with the finding that poly(NBE)ROMP is a high cis polymer but poly(NBE)ROMP-co-poly(NBE)VIP-co-PE is predominantly trans and the unimodal GPC traces (Fig. S21†) strongly suggest that the ROMP-derived sequences are part of the entire polymer chain. Further evidence for the proposed copolymer structure (Fig. 6a) comes from the absence of any glass transition (Fig. S22†) attributable to pure poly(NBE)ROMP. This absence of any poly(NBE)ROMP-derived Tg in combination with the high incorporation of ROMP-derived poly(NBE) sequences (vide supra) also points towards a multi-block structure, however, without real proof.
Our plausible explanation for the formation of this unique polymer structure is an α-H+ elimination/addition process as outlined in Scheme 2. According to our proposed scheme, L′Zr(CH2SiMe3)2 is activated by MAO to form the cationic species L′Zr(CH2SiMe3)+, which through α-H+ elimination yields a Zr-alkylidene (L′ZrCHSiMe3) that promotes ROMP of NBE. As outlined earlier,8,12,13 substantial steric congestion is required to induce the switch from VIP to ROMP. In that regards, the CH2SiMe3 group acts similar, though less effective, than a NBE group between the metal and the polymer chain. In the presence of ethylene, L′ZrCHSiMe3 yields a Zr-methylidene (L′ZrCH2) via cross metathesis, which is also ROMP-active, too. The previous observation that with catalysts containing the 6-(2-BR′2-phenyl)pyrid-2-ylamido motif (R′ = ethyl, mesityl) high NBE concentrations are needed to form any ROMP-derived sequences8,10–13 strongly suggests that cross metathesis of any Zr-alkylidene with E to form Zr-methylidene is the predominant reaction in these systems. This is in line with the high diffusivity of E. The transient Zr-methylidene is presumably exhausted to most extent as a result of its low stability, e.g. via bimolecular decomposition,12 which accounts for the low ROMP propensity and the low productivity of these pre-catalysts upon activation with MAO in E-NBE copolymerization. As already outlined, this accounts for the finding that no polymer is obtained in the copolymerization of E with NBE by the action of [L′Zr(CH3)]+, [L′Zr(Bn)]+ or [L′Zr(CH2SiMe3)]+[B(C6F5)4]− in the absence of AliBu3. In case polymerizations are activated with MAO, an α-hydrogen addition to the ROMP-active species can re-establish the VIP-active species, which now incorporates E and NBE. During E-NBE insertion copolymerization, E incorporation is favored as evidenced by the high E content in the resulting E-NBE copolymers.
The proposed switch from ROMP to VIP, i.e. α-H+ addition, is remarkable, since for such a step the pyridinium (Py-H+) moiety formed in course of α-H+ abstraction must be stable in the presence of MAO at least for a short time. In contrast to AliBu3 and most probably because of its size, MAO neither effectively blocks the pyridine moiety nor instantaneously deprotonates the Py-H+ moiety, which explains both for the ROMP propensity in the presence of MAO and the ROMP-inactivity in the presence of AliBu3. In fact, as outlined earlier,8 higher MAO concentrations result in larger fractions of ROMP-derived units in E-NBE copolymers. Any additional α-H+ elimination in course of the copolymerization would regenerate the ROMP-active species, however, in view of the high propensity of the system to undergo cross metathesis with E (vide supra), only very few additional ROMP-derived poly(NBE) sequences can be expected to form (presumably <1% with respect to the initial amount of pre-catalyst). Instead, because of the instability of the Zr-methylidenes at elevated temperature, polymerization quickly comes to an end, which is indeed observed. What would be needed to boost productivity is an internal olefin that forms a more stable alkylidene in course of the cross metathesis with the Zr-alkylidene. Unfortunately, internal olefins are unable to undergo VIP.
Conclusions
Three new complexes, L′Zr(CH3)2, L′Zr(Bn)2 and L′Zr(CH2SiMe3)2, based on modifications of L′ZrCl2 have been synthesized. These compounds are thermally stable and do not allow for the generation of alkylidenes, neither via thermolysis nor via the addition of phosphines. The corresponding cations were prepared in situ upon activation with [Ph3C]+[B(C6F5)4]−. The aluminum-free monoalkyl cations are able to produce predominantly cis, ROMP-derived poly(NBE) instead of VIP-derived poly(NBE) in NBE homopolymerization. The formation of alkylidenes in the absence of any aluminum reagent is proposed. Dialkyl complexes activated with [Ph3C]+[B(C6F5)4]− and AliBu3 exhibit moderate catalytic activity in E-NBE copolymerization, producing only VIP-type poly(NBE) and branched PE with vinyl terminals. Polymer structures are considered to be a result of the high propensity of AliBu3 to react with the pyridyl-moiety in [L′ZrR]+ (R = CH3, Bn, CH2SiMe3). Similar to L′ZrCl2/MAO, which affords E-NBE copolymers containing both ROMP- and VIP-derived poly(NBE) in the PE chain with low productivity, L′Zr(CH2SiMe3)2/MAO allows for the synthesis of copolymers containing both (mostly trans) ROMP- and VIP-derived NBE sequences within the same polymer chain, i.e. poly(NBE)ROMP-co-poly(NBE)VIP-co-poly(E), through an α-H+ elimination/addition process. By contrast, L′Zr(CH3)2/MAO and L′Zr(Bn)2/MAO produce poly(NBE)VIP-co-poly(E) without any ROMP-derived poly(NBE) sequences. We attribute the incapability of L′Zr(CH3)2/MAO and L′Zr(Bn)2/MAO to promote the ROMP of NBE to the low stability of L′ZrCHR (R = H, Ph) and their high propensity to undergo cross-metathesis with E. Finally, implications on the copolymerization of α-olefins (E) with NBE using “standard” metallocenes are clear. Apart from high NBE concentrations (catalyst:NBE > 1:5000), copolymerizations must be MAO-co-catalyzed and the catalyst, which ever, must allow for α-H+ elimination in order to observe ROMP-derived structures. This is in most copolymerization systems not the case. In fact, particularly industrial large-volume systems sometimes contain substantial amounts of aluminum alkyls, which not only promote chain transfer and increase productivity70–72 but also effectively prevent the formation of any ROMP-active sites by blocking any Lewis-basic groups.
Experimental
All manipulations were carried out using standard Schlenk or dry box techniques under an argon or nitrogen atmosphere unless specified otherwise. Deuterated solvents for NMR measurements were degassed by several freeze–pump–thaw cycles and stored inside a glove box. Benzene-d6 and toluene-d8 were dried and distilled from sodium/benzophenone; CD2Cl2 was distilled from P2O5; C2D2Cl4 and o-C6D4Cl2 were distilled from CaH2. THF, diethyl ether, toluene, n-pentane and CH2Cl2 were deoxygenated by sparging with N2 and passed through a triple-column solvent purification system (MBraun, Garching, Germany). Commercially available reagents for synthesis, i.e. methylmagnesium bromide solution (3.0 M in diethyl ether), benzylmagnesium chloride solution (1.0 M in diethyl ether), (trimethylsilyl)methylmagnesium chloride solution (1.0 M in diethyl ether), triisobutylaluminum solution (1.1 M in toluene) and [Ph3C]+[B(C6F5)4]− were used without further purification. Methylalumoxane (MAO, 10 wt% solution in toluene) was purchased from Sigma-Aldrich, Germany. The toluene was removed in vacuo and the remaining white powder was dried in vacuo at 80 °C overnight to remove any free AlMe3. Celite was dried in vacuo at 180 °C for two days prior to use. Before charging the autoclave, ethylene (E) gas was purified by passing it through columns filled with Cu-catalyst (BASF R3-11G) and 3 Å molecular sieves. All homopolymerization reactions of NBE were performed in Schlenk tubes under N2 atmosphere. The homopolymerization of E and all copolymerizations of E with NBE were carried out in a Büchi-Uster pressure reactor equipped with a Huber thermostat. The ethylene pressure was kept constant and E-consumption was monitored with the aid of a Büchi pressflow bpc 6010 flow controller.
1H and 13C NMR spectra were recorded at 400 and 100 MHz, respectively, on a Bruker Avance III 400 spectrometer at 25 °C unless noted otherwise. Chemical shifts are reported in ppm and referenced to tetramethylsilane (TMS). All 1H and 13C NMR data of the ethylene homo- and E-NBE copolymers were measured at 100 °C except where noted. Molecular weights and molecular weight distributions were obtained by high-temperature gel permeation chromatography (HT-GPC) on an Agilent PL-GPC 220 system equipped with three consecutive PL gel 5 μm MIXED-C 300 × 7.5 mm columns with 1,2,4-trichlorobenzene as the solvent at 160 °C. The flow rate was set to 1 mL min−1. The GPC system was calibrated with narrow polystyrene (PS) standards purchased from Polymer Labs with the molecular weights in the range of 162–6035000 g mol−1 (Easi Vial-red, yellow and green, Fig. S23†). Melting points and glass transition temperature were measured by differential scanning calorimetry (DSC) under N2 atmosphere on a Perkin-Elmer DSC 4000 at a heat rate of 10 °C min−1.
L′Zr(CH3)2
To a solution of L′ZrCl2 (300 mg, 0.396 mmol) in diethyl ether (20 mL) at −35 °C was added MeMgBr (3.0 M in diethyl ether, 0.30 mL, 0.911 mmol). The solution became a suspension within 10 min and was stirred overnight at room temperature in the dark. After the removal of the solvent, the yellow residue was dissolved in toluene (10 mL) and the insoluble solid was filtered off through a pad of celite. The solvent was removed in vacuo to give a brown solid. Yield: 270 mg (80.5%). The solid was recrystallized from diethyl ether at −35 °C to give light-yellow crystals. 1H NMR (C6D6): δ = 8.56 (dd, J = 7.7, 0.7 Hz, 1H), 7.62 (dd, J = 7.6, 1.0 Hz, 1H), 7.40 (td, J = 7.5, 1.4 Hz, 1H), 7.12 (td, J = 7.5, 1.2 Hz, 1H), 6.80 (dd, J = 7.9, 7.5 Hz, 1H), 6.72 (dd, J = 7.4, 1.0 Hz, 1H), 6.65 (s, 4H), 5.46 (dd, J = 8.0, 1.0 Hz, 1H), 2.14 (d, J = 10.9 Hz, 18H), 2.02 (d, J = 8.9 Hz, 12H), 0.46–0.41 (m, 6H), 0.12 (s, 6H, Zr–CH3). 1H NMR (CD2Cl2): δ = 8.09 (d, J = 7.7 Hz, 1H), 7.50 (td, J = 7.4, 1.7 Hz, 1H), 7.43–7.33 (m, 2H), 7.08 (d, J = 7.5 Hz, 1H), 6.62 (s, 4H), 6.53 (d, J = 7.3 Hz, 1H), 5.64 (d, J = 8.1 Hz, 1H), 2.18 (s, 6H), 2.04 (s, 6H), 1.96 (d, J = 11.7 Hz, 18H), 0.51–0.41 (m, 6H), −0.38 (s, 6H, Zr–CH3). 13C NMR (C6D6): δ = 164.8, 157.9, 148.8, 145.0, 143.4, 141.1, 139.1, 138.8, 136.2, 130.8, 130.2, 128.9, 128.7, 127.1, 123.8, 117.4, 109.0, 97.3, 41.4 (Zr–CH3), 24.2, 21.3, 14.0, 11.5, 3.3. Elemental analysis calcd (%) for C42H53BN2SiZr(CH2Cl2)0.5: C 67.30, H 7.18, N 3.69; found: C 67.35, H 7.45, N 3.62.L′Zr(Bn)2
To a solution of L′ZrCl2 (100 mg, 0.132 mmol) in diethyl ether (8 mL) at −35 °C was added benzylMgCl (1.0 M in diethyl ether, 0.29 mL, 0.29 mmol). The solution became a suspension within 5 min and was stirred overnight at room temperature in the dark. After the removal of the insoluble solid through celite, the solution was concentrated to ∼1 mL and cooled to −35 °C overnight to afford a yellow solid. Yield: 68 mg (48.7%). The solid was recrystallized from diethyl ether at −35 °C to give yellow crystals. 1H NMR (CD2Cl2): δ = 7.50 (dd, J = 7.6, 0.7 Hz, 1H), 7.45–7.33 (m, 2H), 7.29 (td, J = 7.4, 1.2 Hz, 1H), 7.21 (dd, J = 8.0, 7.4 Hz, 1H), 6.98 (t, J = 7.7 Hz, 4H), 6.77 (t, J = 7.3 Hz, 2H), 6.69–6.59 (m, 9H), 5.77 (dd, J = 8.1, 0.8 Hz, 1H), 2.20 (s, 6H), 2.06, 1.93 (d, J = 52 Hz, 2H, Zr–CH2Ph), 2.03, 1.90 (d, J = 52 Hz, 2H, Zr–CH2Ph), 1.95 (s, 12H), 1.86 (s, 6H), 1.76 (s, 6H), 0.49 (s, 6H). 13C NMR (CD2Cl2): δ = 164.7, 158.8, 148.3, 147.4, 145.7, 143.3, 141.02, 138.9, 138.1, 136.0, 130.7, 130.4, 129.2, 128.6, 128.4, 128.2, 127.0, 125.4, 121.6, 118.3, 108.9, 99.0, 68.3 (Zr–CH2Ph), 23.9, 21.3, 13.8, 11.3, 2.7. Elemental analysis calcd (%) for C54H61BN2SiZr(CH2Cl2)0.5: C 71.88, H 6.86, N 3.08; found: C 71.98, H 7.08, N 3.13.L′Zr(CH2SiMe3)2
To a solution of L′ZrCl2 (100 mg, 0.132 mmol) in diethyl ether (8 mL) at −35 °C was added Me3SiCH2MgCl (1.0 M in diethyl ether, 0.29 mL, 0.29 mmol). The solution became a suspension within 10 min and was stirred overnight at room temperature in the dark. After the removal of the insoluble solid through celite, the solution was concentrated to ∼0.5 mL and cooled to −35 °C overnight to give yellow crystals. Yield: 76 mg (61.3%). 1H NMR (CD2Cl2): δ = 8.07 (d, J = 7.4 Hz, 1H), 7.56 (td, J = 7.4, 1.5 Hz, 1H), 7.42 (dd, J = 7.5, 1.4 Hz, 1H), 7.36 (td, J = 7.4, 1.0 Hz, 1H), 7.07 (t, J = 7.7 Hz, 1H), 6.62 (s, 4H), 6.56 (d, J = 7.3 Hz, 1H), 5.68 (d, J = 8.0 Hz, 1H), 2.19 (d, J = 2.7 Hz, 12H), 2.10 (s, 6H), 1.97 (s, 12H), 0.58, 0.10 (d, J = 192 Hz, 2H, Zr–CH2SiMe3), 0.54 (m, 6H), 0.56, 0.07 (d, J = 196 Hz, 2H, Zr–CH2SiMe3), −0.20 (s, 18H). 13C NMR (CD2Cl2): δ = 164.2, 158.1, 147.6, 146.9, 143.3, 141.0, 138.8, 137.4, 136.4, 131.7, 130.1, 128.5, 128.4, 128.2, 124.8, 118.4, 108.6, 98.7, 58.2 (Zr–CH2SiMe3), 24.0, 21.3, 15.5, 14.9, 12.6, 3.6, 2.9. Elemental analysis calcd (%) for C48H69BN2Si3Zr(Et2O): C 66.83, H 8.52, N 3.00; found: C 66.36, H 8.49, N 3.26.In situ preparation of [L′Zr(CH3)]+[B(C6F5)4]−
To a solution of L′Zr(CH3)2 (10.0 mg, 11.8 μmol) in CD2Cl2 (0.3 mL) was added a solution of [Ph3C]+[B(C6F5)4]− (10.9 mg, 11.8 μmol) in CD2Cl2 (0.3 mL) at room temperature. 1H and 13C NMR spectra were measured immediately and were indicative for the complete formation of [L′Zr(CH3)]+[B(C6F5)4]−. 1H NMR (CD2Cl2): δ = 7.87 (dd, J = 8.2, 7.7 Hz, 1H), 7.84–7.77 (m, 2H), 7.57 (d, J = 6.6 Hz, 1H), 7.35 (s, 1H), 7.31–7.24 (m, 8H), 7.24–7.18 (m, 3H), 7.11 (ddd, J = 2.9, 2.2, 1.1 Hz, 6H), 6.95 (d, J = 1.8 Hz, 1H), 6.70 (d, J = 1.4 Hz, 1H), 6.56 (dd, J = 8.3, 1.0 Hz, 1H), 6.45 (s, 1H), 2.45–2.37 (m, 9H), 2.27 (s, 3H), 2.24 (s, 3H), 2.18 (s, 3H, Ph3C–CH3), 2.09 (d, J = 4.6 Hz, 6H), 1.92 (s, 3H), 1.74 (s, 3H), 1.49 (s, 3H), 0.95 (s, 3H), 0.56 (s, 3H), −0.05 (s, 3H, Zr–CH3). 13C NMR (CD2Cl2): δ = 164.2, 160.2, 150.2, 150.2, 149.5, 144.2, 142.3, 139.6, 138.3, 137.9, 136.5, 135.5, 135.1, 132.5, 131.4, 131.0, 130.8, 130.3, 129.6, 129.1, 128.5, 128.2, 127.5, 126.7, 126.3, 123.7, 115.2, 111.3, 104.6, 52.9 (Zr–CH3), 37.9, 30.6, 28.4, 26.3, 25.9, 22.6, 20.5, 17.0, 15.8, 12.8, 11.7, 7.1, 3.8. 19F NMR (CD2Cl2): δ = −133.0 (d, J = 13.0 Hz), −163.7 (t, J = 21.1 Hz), −165.1 to −170.3 (m).[L′Zr(CH3)Et2O]+[B(C6F5)4]−
A solution of [Ph3C]+[B(C6F5)4]− (54.5 mg, 59.0 μmol) in CH2Cl2 (1.5 mL) was added to a solution of L′Zr(CH3)2 (50.0 mg, 59.0 μmol) in CH2Cl2 (1.5 mL) at room temperature. The mixture was stirred for 10 min; then the solvent was removed in vacuo. The resulting residue was recrystallized from diethyl ether and n-pentane at room temperature to obtain a yellow solid. Yield: 65 mg (73.6%). 1H NMR (CD2Cl2): δ = 7.91–7.85 (m, 1H), 7.85–7.77 (m, 2H), 7.57 (dd, J = 7.6, 0.9 Hz, 1H), 7.35 (s, 1H), 7.30–7.23 (m, 2H), 6.95 (d, J = 1.6 Hz, 1H), 6.70 (s, 1H), 6.56 (dd, J = 8.3, 0.9 Hz, 1H), 6.44 (s, 1H), 3.45 (q, J = 7.0 Hz, 4H), 2.46–2.36 (m, 9H), 2.26 (d, J = 11.2 Hz, 6H), 2.09 (d, J = 5.4 Hz, 6H), 1.92 (s, 3H), 1.74 (s, 3H), 1.49 (s, 3H), 1.16 (t, J = 7.0 Hz, 6H), 0.95 (s, 3H), 0.56 (s, 3H), −0.06 (s, 3H, Zr–CH3). 13C NMR (CD2Cl2): δ = 164.2, 160.2, 150.3, 150.2, 149.8, 144.2, 142.3, 139.6, 138.3, 137.9, 136.5, 135.5, 135.1, 132.5, 131.4, 131.0, 130.8, 130.3, 129.6, 128.5, 127.5, 126.7, 123.7, 115.2, 111.3, 104.6, 37.9, 28.4, 26.3, 25.9, 22.6, 20.5, 17.0, 15.8, 12.8, 11.7, 7.1, 3.8. The signal of Zr–CH3 in the 13C NMR spectrum was invisible due to an overlap with the signal of CD2Cl2. 19F NMR (CD2Cl2): δ = −133.1 (d, J = 13.1 Hz), −163.7 (t, J = 20.9 Hz), −166.4 to −168.9 (m). Elemental analysis calcd (%) for C69H60B2F20N2OSiZr(CH2Cl2)0.5: C 55.78, H 4.11, N 1.87; found: C 55.60, H 4.20, N 1.96.In situ preparation of [L′Zr(Bn)]+[B(C6F5)4]−
To a solution of L′Zr(Bn)2 (13.0 mg, 12.3 μmol) in CD2Cl2 (0.3 mL) was added a solution of [Ph3C]+[B(C6F5)4]− (11.3 mg, 12.3 μmol) in CD2Cl2 (0.3 mL) at room temperature. 1H and 13C NMR spectra were measured immediately and were indicative for the complete conversion of [L′Zr(Bn)]+[B(C6F5)4]−. 1H NMR (CD2Cl2): δ = 8.01 (t, J = 7.9 Hz, 1H), 7.80–7.75 (m, 1H), 7.64 (dd, J = 13.9, 7.2 Hz, 2H), 7.29 (t, J = 7.3 Hz, 2H), 7.26–7.12 (m, 16H), 7.04 (t, J = 7.4 Hz, 1H), 6.94 (dt, J = 10.5, 7.8 Hz, 4H), 6.81 (dd, J = 16.3, 8.1 Hz, 3H), 6.63 (d, J = 7.9 Hz, 3H), 6.47–6.38 (m, 3H), 3.95 (s, 2H, Ph3C–CH2Ph), 2.41 (s, 3H), 2.40 (s, 3H), 2.38 (s, 3H), 2.37, 1.57 (d, J = 320 Hz, 1H, Zr–CH2Ph), 2.34, 1.54 (d, J = 320 Hz, 1H, Zr–CH2Ph), 2.29 (s, 3H), 2.24 (s, 3H), 2.22 (s, 3H), 2.07 (s, 3H), 1.93 (s, 3H), 1.51 (s, 3H), 1.08 (s, 3H), 0.81 (s, 3H), 0.68 (s, 3H). 13C NMR (CD2Cl2): δ = 164.1, 160.6, 151.3, 149.2, 149.1, 147.2, 143.9, 142.9, 141.2, 141.0, 139.0, 136.5, 135.4, 135.1, 131.7, 131.5, 131.4, 130.7, 130.2, 130.1, 130.1, 129.8, 129.1, 128.7, 128.7, 128.0, 127.6, 126.7, 126.5, 126.3, 126.3, 126.3, 124.3, 123.9, 114.8, 111.8, 105.1, 64.0 (Zr–CH2Ph), 59.0 (Ph3C–CH2Ph), 46.4 (Ph3C–CH2Ph), 28.0, 26.7, 23.5, 23.2, 20.6, 20.5, 16.7, 16.6, 13.4, 11.8, 7.0, 3.2. 19F NMR (CD2Cl2): δ = −133.0 (d, J = 13.5 Hz), −163.7 (t, J = 21.1 Hz), −166.4 to −169.3 (m).In situ preparation of [L′Zr(CH2SiMe3)]+[B(C6F5)4]− (partial conversion)
To a solution of L′Zr(CH2SiMe3)2 (4.7 mg, 5.0 μmol) in CD2Cl2 (0.3 mL) was added a solution of [Ph3C]+[B(C6F5)4]− (4.6 mg, 5.0 μmol) in CD2Cl2 (0.3 mL) at room temperature and the mixture was stirred overnight. The 1H NMR spectrum revealed ∼28 mol% conversion of L′Zr(CH2SiMe3)2 to [L′Zr(CH2SiMe3)]+[B(C6F5)4]− as evidenced by the ratio of intensities of (converted aromatic protons)/(converted + non-converted aromatic protons) (Fig. S40, ESI†).General procedure for NBE homopolymerization
All preparations were carried out inside a glove box. Defined amounts of NBE were dissolved in toluene (40 mL) inside a Schlenk tube. Then a toluene solution (5 mL) of a defined amount of the pre-catalyst was added. The mixture was stirred for 5 min before the addition of a defined amount of [Ph3C]+[B(C6F5)4]− in toluene (5 mL) and heated to the desired temperature. Polymerizations were quenched after 1 h by the addition of methanol (10 mL). The mixture was then poured into methanol (500 mL) containing concentrated HCl (10 mL). The polymer was collected by filtration and adequately washed with methanol and then dried in vacuo at 50 °C for 2 days.General procedure for E homo- and E-NBE copolymerization
Samples were prepared inside a glove box. Polymerization reactions were conducted by using a Büchi glass reactor (500 mL), which was dried at 120 °C in vacuo for 2 h, cooled to 30 °C and purged with Ar gas before use. (a) [Ph3C]+[B(C6F5)4]− and AliBu3-activation: a solution of NBE in toluene (ca. 240 mL), a solution of the pre-catalyst in toluene (5 mL) and a solution of a defined amount of AliBu3 in toluene were quickly introduced into the reactor and stirred (300 rpm) for 5 min at 30 °C before the addition of a solution of [Ph3C]+[B(C6F5)4]− in toluene (5 mL). (b) MAO-activation: a solution of NBE in toluene (ca. 240 mL) and a solution of a defined amount of MAO in toluene (5 mL) were quickly introduced into the reactor and stirred (300 rpm) for 5 min at 30 °C before the addition of a solution of pre-catalysts in toluene (5 mL). The reactor was pressurized with ethylene gas once the mixture had reached the desired temperature. The polymerization reaction was quenched after 1 h by the addition of methanol (10 mL). The resulting mixture was then poured into methanol (500 mL) containing concentrated HCl (10 mL). The polymer was collected by filtration and washed with methanol. All the E-NBE copolymers were extracted extensively with THF at 50 °C overnight before filtration. The resulting polymers were adequately washed with methanol and then dried in vacuo at 50 °C for 2 days.X-ray measurements and structure determination
Data were collected on a Bruker Kappa Apex 2 duo diffractometer at 100 K. Structures were solved using direct methods with refinement by full matrix least-squares of F2, with the program system SHELXL 97 in connection with a multi-scan absorption correction.73 All non-hydrogen atoms were refined anisotropically.Acknowledgements
Financial support by the Deutsche Forschungsgemeinschaft (DFG, grant-nr: BU2174/14-1) is gratefully acknowledged.References
- A. L. McKnight and R. M. Waymouth, Chem. Rev., 1998, 98, 2587–2598 CrossRef CAS PubMed.
- G. J. P. Britovsek, V. C. Gibson and D. F. Wass, Angew. Chem., Int. Ed., 1999, 38, 428–447 CrossRef CAS.
- W. Kaminsky, J. Chem. Soc., Dalton Trans., 1998, 1413–1418 RSC.
- X. Li and Z. Hou, Coord. Chem. Rev., 2008, 252, 1842–1869 CrossRef CAS.
- J. Klosin, P. P. Fontaine and R. Figueroa, Acc. Chem. Res., 2015, 48, 2004–2016 CrossRef CAS PubMed.
- Y.-X. Chen and T. J. Marks, Organometallics, 1997, 16, 3649–3657 CrossRef CAS.
- E. Y.-X. Chen and T. J. Marks, Chem. Rev., 2000, 100, 1391–1434 CrossRef CAS PubMed.
- M. R. Buchmeiser, S. Camadanli, D. Wang, Y. Zou, U. Decker, C. Kühnel and I. Reinhardt, Angew. Chem., 2011, 123, 3628–3633 ( Angew. Chem., Int. Ed. , 2011 , 50 , 3566–3571 ) CrossRef.
- M. R. Buchmeiser, Curr. Org. Chem., 2013, 17, 2764–2775 CrossRef CAS.
- G. V. Narayana, G. Xu, D. Wang, W. Frey and M. R. Buchmeiser, ChemPlusChem, 2014, 79, 151–162 CrossRef CAS.
- G. V. Narayana, G. Xu, D. Wang, M. Speiser, W. Frey and M. R. Buchmeiser, Macromol. Chem. Phys., 2014, 215, 2007–2013 CrossRef CAS.
- M. Wang, G. Xu, D. Wang, Y. Zou, W. Frey and M. R. Buchmeiser, Polym. Chem., 2015, 6, 3290–3304 RSC.
- G. Xu, G. V. Narayana, M. Speiser, D. Wang, Y. Zou and M. R. Buchmeiser, Macromol. Chem. Phys., 2014, 215, 893–899 CrossRef CAS.
- Y. Zou, D. Wang, K. Wurst, C. Kühnel, I. Reinhardt, U. Decker, V. Gurram, S. Camadanli and M. R. Buchmeiser, Chem. – Eur. J., 2011, 17, 13832–13846 CrossRef CAS PubMed.
- M. Bochmann, J. Chem. Soc., Dalton Trans., 1996, 255–270 RSC.
- R. Collins, A. Russell and P. Mountford, Appl. Petrochem. Res., 2015, 1–19 Search PubMed.
- M. Bochmann, J. Organomet. Chem., 2004, 689, 3982–3998 CrossRef CAS.
- L. Resconi, R. L. Jones, A. L. Rheingold and G. P. A. Yap, Organometallics, 1996, 15, 998–1005 CrossRef CAS.
- J. Pinkas, I. Cisarova, J. Kubista, M. Horacek and M. Lamac, Dalton Trans., 2013, 42, 7101–7110 RSC.
- W.-Y. Lee and L.-C. Liang, Inorg. Chem., 2008, 47, 3298–3306 CrossRef CAS PubMed.
- K. C. Jantunen, B. L. Scott and J. L. Kiplinger, J. Alloys Compd., 2007, 444–445, 363–368 CrossRef CAS.
- P. D. Bolton, N. Adams, E. Clot, A. R. Cowley, P. J. Wilson, M. Schröder and P. Mountford, Organometallics, 2006, 25, 5549–5565 CrossRef CAS.
- L.-C. Liang, P.-S. Chien, Y.-C. Hsiao, C.-W. Li and C.-H. Chang, J. Organomet. Chem., 2011, 696, 3961–3965 CrossRef CAS.
- S. Gentil, N. Pirio, P. Meunier, J. C. Gallucci, J. D. Schloss and L. A. Paquette, Organometallics, 2000, 19, 4169–4172 CrossRef CAS.
- M. Bochmann, S. J. Lancaster, M. B. Hursthouse and K. M. A. Malik, Organometallics, 1994, 13, 2235–2243 CrossRef CAS.
- M. Bochmann and S. J. Lancaster, Organometallics, 1993, 12, 633–640 CrossRef CAS.
- J. Sassmannshausen, A. Track and F. Stelzer, Organometallics, 2006, 25, 4427–4432 CrossRef CAS.
- S. R. Golisz and J. E. Bercaw, Macromolecules, 2009, 42, 8751–8762 CrossRef CAS.
- R. C. Klet, C. N. Theriault, J. Klosin, J. A. Labinger and J. E. Bercaw, Macromolecules, 2014, 47, 3317–3324 CrossRef CAS.
- E. Despagnet-Ayoub, L. M. Henling, J. A. Labinger and J. E. Bercaw, Dalton Trans., 2013, 42, 15544–15547 RSC.
- Y.-X. Chen, P.-F. Fu, C. L. Stern and T. J. Marks, Organometallics, 1997, 16, 5958–5963 CrossRef CAS.
- C. P. Casey and D. W. Carpenetti Ii, J. Organomet. Chem., 2002, 642, 120–130 CrossRef CAS.
- N. A. Petasis and D. K. Fu, J. Am. Chem. Soc., 1993, 115, 7208–7214 CrossRef CAS.
- B. J. J. v. de Heisteeg, G. Schat, O. S. Akkerman and F. Bickelhaupt, Tetrahedron Lett., 1987, 28, 6493–6496 CrossRef.
- J. A. v. Doorn, H. v. d. Heijden and A. G. Orpen, Organometallics, 1995, 14, 1278–1283 CrossRef.
- L. R. Gilliom and R. H. Grubbs, Organometallics, 1986, 5, 721–724 CrossRef CAS.
- J. D. Meinhart, E. V. Anslyn and R. H. Grubbs, Organometallics, 1989, 8, 583–589 CrossRef CAS.
- H. van der Heijden and B. Hessen, J. Chem. Soc., Chem. Commun., 1995, 145–146 RSC.
- W. Weng, L. Yang, B. M. Foxman and O. V. Ozerov, Organometallics, 2004, 23, 4700–4705 CrossRef CAS.
- I. Furuta, S.-I. Kimura and M. Iwama, Physical Constants of Rubbery Polymers, in Polymer Handbook, ed. E. A. Grulke, 1999, vol. 1 Search PubMed.
- B. C. Bailey, F. Basuli, J. C. Huffman and D. J. Mindiola, Organometallics, 2006, 25, 3963–3968 CrossRef CAS.
- B. C. Bailey, H. Fan, J. C. Huffman, M.-H. Baik and D. J. Mindiola, J. Am. Chem. Soc., 2007, 129, 8781–8793 CrossRef CAS PubMed.
- B. C. Bailey, J. C. Huffman, D. J. Mindiola, W. Weng and O. V. Ozerov, Organometallics, 2005, 24, 1390–1393 CrossRef CAS.
- C. M. Brammell, E. J. Pelton, C.-H. Chen, A. A. Yakovenko, W. Weng, B. M. Foxman and O. V. Ozerov, J. Organomet. Chem., 2011, 696, 4132–4137 CrossRef CAS.
- A. R. Fout, J. Scott, D. L. Miller, B. C. Bailey, M. Pink and D. J. Mindiola, Organometallics, 2009, 28, 331–347 CrossRef CAS.
- M. D. Fryzuk, P. B. Duval, B. O. Patrick and S. J. Rettig, Organometallics, 2001, 20, 1608–1613 CrossRef CAS.
- L. R. Gilliom and R. H. Grubbs, J. Am. Chem. Soc., 1986, 108, 733–742 CrossRef CAS.
- M. Kamitani, B. Pinter, C.-H. Chen, M. Pink and D. J. Mindiola, Angew. Chem., Int. Ed., 2014, 53, 10913–10915 CrossRef CAS PubMed.
- R. F. Jordan, C. S. Bajgur, R. Willett and B. Scott, J. Am. Chem. Soc., 1986, 108, 7410–7411 CrossRef CAS.
- X. Yang, C. L. Stern and T. J. Marks, J. Am. Chem. Soc., 1991, 113, 3623–3625 CrossRef CAS.
- M. Bochmann and M. J. Sarsfield, Organometallics, 1998, 17, 5908–5912 CrossRef CAS.
- K. Liu, Q. Wu, W. Gao and Y. Mu, Dalton Trans., 2011, 40, 4715–4721 RSC.
- K. Liu, Q. Wu, X. Luo, W. Gao and Y. Mu, Dalton Trans., 2012, 41, 3461–3467 RSC.
- Y. Luo, X. Feng, Y. Wang, S. Fan, J. Chen, Y. Lei and H. Liang, Organometallics, 2011, 30, 3270–3274 CrossRef CAS.
- N. Naga and K. Mizunuma, Polymer, 1998, 39, 5059–5067 CrossRef CAS.
- B. Wang, T. Tang, Y. Li and D. Cui, Dalton Trans., 2009, 8963–8969 RSC.
- X.-Y. Wang, Y.-X. Wang, Y.-S. Li and L. Pan, Macromolecules, 2015, 48, 1991–1998 CrossRef CAS.
- T. Xiao, J. Wang, Y. Zhang, W. Gao and Y. Mu, J. Organomet. Chem., 2008, 693, 1195–1202 CrossRef CAS.
- X. Li, X. Wang, X. Tong, H. Zhang, Y. Chen, Y. Liu, H. Liu, X. Wang, M. Nishiura, H. He, Z. Lin, S. Zhang and Z. Hou, Organometallics, 2013, 32, 1445–1458 CrossRef CAS.
- M. Nabika, H. Katayama, T. Watanabe, H. Kawamura-Kuribayashi, K. Yanagi and A. Imai, Organometallics, 2009, 28, 3785–3792 CrossRef CAS.
- P. Sudhakar, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 444–452 CrossRef CAS.
- I. Tritto, L. Boggioni, C. Zampa and D. R. Ferro, Macromolecules, 2005, 38, 9910–9919 CrossRef CAS.
- S. M. Baldwin, J. E. Bercaw, L. M. Henling, M. W. Day and H. H. Brintzinger, J. Am. Chem. Soc., 2011, 133, 1805–1813 CrossRef CAS PubMed.
- P. D. Bolton, E. Clot, A. R. Cowley and P. Mountford, Chem. Commun., 2005, 3313–3315 RSC.
- P. D. Bolton, E. Clot, A. R. Cowley and P. Mountford, J. Am. Chem. Soc., 2006, 128, 15005–15018 CrossRef CAS PubMed.
- G. Theurkauff, A. Bondon, V. Dorcet, J.-F. Carpentier and E. Kirillov, Angew. Chem., Int. Ed., 2015, 54, 6343–6346 CrossRef CAS PubMed.
- S. Hermanek, Chem. Rev., 1992, 92, 325–362 CrossRef CAS.
- F. Teixidor, M. A. Flores, C. Viñas, R. Sillanpää and R. Kivekäs, J. Am. Chem. Soc., 2000, 122, 1963–1973 CrossRef CAS.
- Y.-L. Rao, H. Amarne, S.-B. Zhao, T. M. McCormick, S. Martić, Y. Sun, R.-Y. Wang and S. Wang, J. Am. Chem. Soc., 2008, 130, 12898–12900 CrossRef CAS PubMed.
- T. Shiono, S. Yoshida, H. Hagihara and T. Ikeda, Appl. Catal., A, 2000, 200, 145–152 CrossRef CAS.
- S. S. Reddy, G. Shashidhar and S. Sivaram, Macromolecules, 1993, 26, 1180–1182 CrossRef CAS.
- H. Hammawa, T. M. Mannan, D. T. Lynch and S. E. Wanke, J. Appl. Polym. Sci., 2004, 92, 3549–3560 CrossRef CAS.
- G. M. Sheldrick, Program package SHELXTL V.5.1, Bruker Analytical X-Ray Instruments Inc, Madison, USA, 1997 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H-, 13C-NMR spectra and crystallographic data for L′Zr(CH3)2, L′Zr(Bn)2 and L′Zr(CH2SiMe3)2, 1H-, 13C-, 19F-NMR spectra of [L′Zr(CH3)]+[B(C6F5)4]−, [L′Zr(Bn)]+[B(C6F5)4]− and 1H-NMR spectra of [L′Zr(CH2SiMe3)]+[B(C6F5)4]− and of homo- and copolymers. CCDC 1440878 (L′Zr(CH3)2), 1440879 (L′Zr(Bn)2) and 1440880 (L′Zr(CH2SiMe3)2). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6py00078a |
| This journal is © The Royal Society of Chemistry 2016 |