
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Maleimide-functionalized poly(2-ethyl-2-oxazoline): synthesis and reactivity†
Felix
Wendler
ab,
Tobias
Rudolph
ab,
Helmar
Görls
c,
Nils
Jasinski
de,
Vanessa
Trouillet
f,
Christopher
Barner-Kowollik
de and
Felix H.
Schacher
*ab
aInstitute of Organic and Macromolecular Chemistry (IOMC), Friedrich Schiller University, Jena, Humboldtstr. 10, 07743 Jena, Germany. E-mail: felix.schacher@uni-jena.de
bJena Center for Soft Matter (JCSM), Friedrich Schiller University Jena, Philosophenweg 7, 07743 Jena, Germany
cInstitute for Inorganic and Analytical Chemistry (IAAC), Friedrich Schiller University Jena, Humboldtstrasse 8, D-07743 Jena, Germany
dPreparative Macromolecular Chemistry, Institut für Technische Chemie und Polymerchemie, Karlsruhe Institute of Technology (KIT), Engesserstrasse 18, 76128 Karlsruhe, Germany
eInstitut für Biologische Grenzflächen, Karlsruhe Institute of Technology (KIT), Hermann-von-Helmholtz-Platz 1, 76344 Eggenstein-Leopoldshafen, Germany
fInstitut für Angewandte Materialien (IAM) and Karlsruhe Nano Micro Facility (KNMF), Karlsruhe Institute of Technology (KIT), Hermann-von-Helmholtz-Platz 1, 76344 Eggenstein-Leopoldshafen, Germany
First published on 7th March 2016
Abstract
Poly(2-ethyl-2-oxazoline)s (PEtOx23 – Mn = 2300 g mol−1, Đ = 1.07; PEtOx46 – Mn = 4400 g mol−1, Đ = 1.06) end-functionalized with a maleimide moiety were prepared from azide-terminated PEtOxx-N3via copper-catalyzed azide–alkyne cycloaddition (CuAAC) with an alkyne-bearing maleimide (MI). The latter has been synthesized in a three-step procedure, including protection of the maleimide double bond prior to the modification of PEtOx. PEtOxx-MI was characterized by NMR (1H, 13C), SEC, FT-IR, and MALDI-ToF MS and, after deprotection of the maleimide, used in nitrile imine-mediated tetrazole-ene cycloaddition (NITEC) processes for covalent attachment to silicon surfaces in a grafting-to approach.
Introduction
Both the manipulation and the understanding of soft materials at the nanoscale are improving significantly1 and the field of nanotechnology in conjunction with soft matter and/or functional polymers, surfaces, nanoparticles, and hybrid materials is of interest regarding applications in biomedicine2–5 and optoelectronics.6–8 Ideally, such materials are synthesized in high yields and using straightforward chemistry.9,10 In case of low-yield and unspecific reactions ill-defined materials are obtained, which might be of limited functionality or contain significant amounts of unwanted byproducts.11 Monomers featuring additional (protected) functional groups which are unaffected by the polymerization method of choice are arguably the most important cornerstone for the preparation of polymeric materials with precisely controlled architecture, functionality, and addressability.12–14 Hereby, the respective polymerization technique of choice and its tolerance towards the presence of functional groups limits the range of accessible monomers. In that respect, the development of controlled radical polymerizations with their most important representatives atom-transfer radical polymerization (ATRP),15,16 reversible addition–fragmentation transfer polymerization (RAFT),17,18 and nitroxide-mediated polymerization (NMP)19,20 was a very significant step forward to overcome some of the limitations of traditional living ionic techniques.12However, some functional monomers typically cannot be polymerized in a controlled manner due to potential side reactions (e.g. recombination reactions between the desired functional group and the active polymer chain leading to termination, cross-linking, or branching). In that case, post-polymerization modification represents an elegant alternative to introduce functionalities that are not compatible with polymerization strategies and in this way many interesting macromolecular architectures such as end- and sidechain functionalized linear polymers, block copolymers, star polymers or graft copolymers can be prepared.9,10,12,13,21
Post-polymerization modifications and, in general, polymer/material science have been strongly influenced by the introduction of click chemistry.22–28 For an overview on click chemistry with regard to polymer modifications9,11,13,29,30 or reactions beyond classical click chemistry12,31–33 the reader is directed to recent review articles. Thereby, a conscious use of the term “click chemistry” is required with regard to the specifics of polymer synthesis and a clear distinction from merely “efficient” and “successful” polymer analogous reactions needs to be made.34
In the current study, well-defined poly(2-ethyl-2-oxazoline)s (PEtOx) with a terminal maleimide functionality were prepared and used for surface modification. Therefore, azide-terminated PEtOxx-N3 was synthesized via cationic ring-opening polymerization (CROP),35,36 followed by the conjugation with an alkyne-functionalized maleimide (MI) via copper catalyzed azide–alkyne cycloaddition (CuAAC).28,37–40 The maleimide functionality of MI and, respective, PEtOxx-MI had to be protected and, thus, its deprotection procedure was also investigated. Selected Diels–Alder reactions (DA) fulfill the click criteria and especially reactions of certain dienes with maleimide containing materials renders them an appropriate tool for polymer post-polymerization functionalization,33,41–45 protein conjugation, or the synthesis of block copolymers. In that respect, only few examples for hydrophilic maleimide-functionalized polymeric materials have been reported so far. Prominent examples are poly(ethylene oxide) (PEO)46,47 or poly(ethylene glycol) methacrylate (PEGMA).48,49
After the characterization via NMR, SEC, FT-IR, and MALDI-ToF MS, PEtOxx-MI was used for surface functionalization in light-induced nitrile imine-mediated tetrazole-ene cycloaddition (NITEC) processes.50–54 Furthermore, poly(2-oxazoline)s (POx) coated surfaces open up promising alternatives to PEGylated surfaces for biomedical applications e.g. as antifouling coatings or for DNA binding and release.55–57 In “grafting to” approaches POx have already been attached to glass substrates using photoimmobilization,58–62 but the herein presented NITEC strategy offers several benefits like high selectivity, bio-orthogonality, and fast reaction times.52,63,64
Experimental section
Materials
Acetonitrile (ACN) was purified using a Solvent Purification System (SPS, Innovative Technology, PM-400-3-MD) equipped with two activated alumina columns. 2-Ethyl-2-oxazoline (EtOx) and methyl tosylate (Aldrich) were distilled over barium oxide under reduced pressure before polymerization and stored under argon. N-(3-Dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC-HCl), 4-dimethylaminopyridine (DMAP) and prop-2-yn-1-ol were purchased from Sigma-Aldrich. All other chemicals were used as received if not otherwise mentioned. 4-Maleimidobutyric acid65 and compound 166 were synthesized as reported in the literature.Instruments
Nuclear Magnetic Resonance (NMR) proton and carbon spectroscopy ( 1 H and 13 C) were measured on a 250 MHz or a 300 MHz Bruker AC spectrometer at 298 K using the residual solvent resonance as an internal standard. The chemical shifts are given in ppm.
Size-Exclusion Chromatography (SEC) measurements were performed on a Shimadzu system equipped with a SCL-10A system controller, a LC-10AD pump, a RID-10A refractive index detector and a PSS-SDV-linear S column at 40 °C using a chloroform, triethylamine and 2-propanol (94![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4:2) mixture as eluent at a flow rate of 1 mL min−1. The system was calibrated with polystyrene (100–100000 g mol−1) standards.
:4:2) mixture as eluent at a flow rate of 1 mL min−1. The system was calibrated with polystyrene (100–100000 g mol−1) standards.
Fourier Transform Infrared Spectroscopy (FT-IR) was performed on a FT-IR spectrometer FT-IR Affinity-1 from Shimadzu and measured in the range of 4000–600 cm−1.
For the measurements of the Matrix-Assisted Laser Desorption/Ionization (MALDI) spectra, an Ultraflex III ToF/ToF instrument (Bruker Daltonics, Bremen, Germany) was used. The instrument is equipped with a Nd-YAG laser and a collision cell. All spectra were measured in positive and reflector mode. The instrument was calibrated prior to each measurement with an external PMMA standard from PSS. For the MALDI-ToF-MS sample preparation, separate solutions of polymer (10 mg mL−1 in chloroform), trans-2-[3-(4-tert-butylphenyl)-2-methyl-2-propenylidene] (DCTB) (30 mg mL−1 in chloroform), or 2,5-dihydroxybenzoic acid (DHB) and doping of sodium chloride (NaCl), (100 mg mL−1 in acetone) were prepared and mixed following the dried droplet spotting technique. 0.5 μL of the mixture was spotted onto the target plate.
Elemental Analyses were carried out on a EuroVector EuroEA3000 elemental analyzer.
For crystal structure determination intensity data were collected on a Nonius KappaCCD diffractometer, using graphite-monochromated Mo-Kα radiation. Data were corrected for Lorentz and polarization effects; absorption was taken into account on a semi-empirical basis using multiple-scans.67–69 The structure was solved by direct methods (SHELXS)70 and refined by full-matrix least squares techniques against Fo2 (SHELXL-97).70 All hydrogen atoms were located by difference Fourier synthesis and refined isotropically.
The polymerizations were performed in a Biotage Initiator Sixty microwave synthesizer equipped with a non-invasive FT-IR sensor (accuracy: 2%) in capped vials.
X-Ray Photoelectron Spectroscopy (XPS) was performed in a K-Alpha+ spectrometer (ThermoFisher Scientific, East Grinstead, UK) using a microfocused, monochromated Al Kα X-ray source (400 μm spot size). The K-Alpha charge compensation system was employed during analysis, using electrons of 8 eV energy, and low-energy argon ions to prevent any localized charge build-up. The kinetic energy of the electrons was measured by a 180° hemispherical energy analyzer operated in the constant analyzer energy mode (CAE) at 50 eV pass energy for elemental spectra. Data acquisition and processing using the Thermo Avantage software is described elsewhere.71 The spectra were fitted with one or more Voigt profiles (BE uncertainty: ±0.2 eV) and Scofield sensitivity factors were applied for quantification.72 All spectra were referenced to the C 1s peak (C–C, C–H) at 285.0 eV binding energy controlled by means of the well-known photoelectron peaks of Cu, Ag and Au respectively.
Synthesis
1 H NMR (300 MHz, CDCl3, δ): 3.6–3.2 (br, –N–CH2–CH2–), 2.5–2.2 (br, CO–CH2–CH3), 1.2–1.0 (br, CO–CH2–CH3) ppm.
13
C-NMR (75 MHz, CDCl3, δ): 175–173 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 50–42 (–N–CH2–CH2–), 27–25 (CO–CH2–CH3), 10–8 (CO–CH2–CH3) ppm.
O), 50–42 (–N–CH2–CH2–), 27–25 (CO–CH2–CH3), 10–8 (CO–CH2–CH3) ppm.
PEtOx 23 -N 3 : SEC (CHCl3/i-PrOH/Et3N): Mn = 1900 g mol−1, Đ = 1.10 (PS-calibration), MS (MALDI-ToF MS, DCTB/NaCl): Mn = 1915 g mol−1, Đ = 1.08, yield: 56%.
PEtOx 46 -N 3 : SEC (CHCl3/i-PrOH/Et3N): Mn = 4200 g mol−1; Đ = 1.05 (PS-calibration), MS (MALDI-ToF MS, DCTB/NaCl): Mn = 4636 g mol−1, Đ = 1.03, yield: 75%.
FT-IR:
ν [cm−1] = 2976 and 2932 (CH), 2100 (N3), 1627 (CO), 1429 (CH2/CH3), 1371 (CH3), 1194 (C–N).
:ethyl acetate 7:1). 2 was obtained as white crystals. Yield: 47% (of theory).
1
H NMR (300 MHz, CDCl3, δ): 6.51 (s, 2 H, 2 CH), 5.26 (s, 2 H, 2 CH), 4.67 (d, 2H, OCH2), 3.55 (t, 2H, CH2), 2.84 (s, 2 H, 2 CH), 2.45 (t, 1H, C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH), 2.35 (t, 2 H, CH2), 1.93 (m, 2 H, CH2) ppm.
CH), 2.35 (t, 2 H, CH2), 1.93 (m, 2 H, CH2) ppm.
13
C-NMR (75 MHz, CDCl3, δ): 176.96 (O–CO), 172.51 (N–CO), 137.21 (CC), 81.52 (Cfuran–O), 75.56 (CH2–CC–H), 52.67 (O–CH2), 47.99 (CH–CH), 38.73 (N–CH2), 31.63 (CH2–CO), 23.37 (CH2) ppm.
FT-IR: ν [cm−1] = 3282 (CH), 3015 (CH), 2987 and 2937 (CH), 2361 (CC), 1739 (O–CO), 1689 (N–CO), 1445 (CH2/CH3), 1396 (CH3), 1154 (C–O–C).
MS (MALDI-ToF MS, DHB): m/z calculated for [C15H15NO5]Na+: 312.08; found: 312.08 [M + Na]+.
EA: 62.46% C, 5.62% H, 5.15% N, (calc.: 62.28% C, 5.23% H, 4.84% N).
Crystal data: C15H15NO5, Mr = 289.28 g mol−1, colourless prism, size 0.046 × 0.042 × 0.040 mm3, monoclinic, space group C2/c, a = 28.1106(7), b = 5.8720(2), c = 21.0927(6) Å, β = 130.543(1)°, V = 2645.79(13) Å3, T = −140 °C, Z = 8, ρcalcd = 1.452 g cm−3, μ(Mo-Kα) = 1.1 cm−1, multi-scan, transmin: 0.6986, transmax: 0.7456, F(000) = 1216, 9256 reflections in h(−36/36), k(−7/7), l(−25/27), measured in the range 2.54° ≤ Θ ≤ 27.49°, completeness Θmax = 99.3%, 3026 independent reflections, Rint = 0.0334, 2663 reflections with Fo > 4σ(Fo), 250 parameters, 0 restraints, R1obs = 0.0379, wRobs2 = 0.0925, R1all = 0.0448, wRall2 = 0.0968, GOOF = 1.057, largest difference peak and hole: 0.330/−0.208 e Å−3.
1 H NMR (300 MHz, CDCl3, δ): 6.51 (s, 2 CH), 3.75 (t, CH2), 3.6–3.3 (br, –N–CH2–CH2–), 2.85 (s, 2 CH), 2.5–2.2 (br, CO–CH2–CH3), 1.86 (m, CH2), 1.2–1.0 (br, CO–CH2–CH3) ppm.
13
C-NMR (75 MHz, CDCl3, δ): 176–172 (CO), 136.24 (CC), 80.66 (Cfuran–O), 48–43 (–N–CH2–CH2–), 26–24 (CO–CH2–CH3), 22.40 (–CH2–), 10–8 (CO–CH2–CH3) ppm.
FT-IR: ν [cm−1] = 2980 and 2939 (CH), 1740 (O–CO), 1635 (N–CO), 1426 (CH2/CH3), 1374 (CH3), 1201 (C–N).
PEtOx 23 -MI: SEC (CHCl3/i-PrOH/Et3N): Mn = 2250 g mol−1, Đ = 1.07 (PS-calibration), MS (MALDI-ToF MS, DCTB/NaCl): Mn = 2727 g mol−1, Đ = 1.04, yield: 22%;
PEtOx 46 -MI: SEC (CHCl3/i-PrOH/Et3N): Mn = 4350 g mol−1, Đ = 1.06 (PS-calibration), MS (MALDI-ToF MS, DCTB/NaCl): Mn = 4764 g mol−1, Đ = 1.02, yield: 73%.
Deprotection procedure of PEtOxx-MI
20 mg of PEtOxx-MI were dissolved in 500 μL of deuterated dimethylformamide and the resulting solution was heated to 130 °C for different reaction times. After the desired reaction time the degree of cleavage was determined by 1H NMR spectroscopy (300 MHz, DMF-d6). Alternatively, PEtOxx-MI was deprotected in the bulk at 100 °C in vacuo prior to photografting experiments.Functionalization of silicon wafers with tetrazole (Sur 1)
Silicon wafers were functionalized with tetrazole (Sur 1) according to previous reports.73Polymer photografting onto silicon wafers (Sur 2)
A tetrazole functionalized silicon wafer (Sur 1) was immersed in a solution of deprotected PEtOxx-MI in dry dichloromethane (c = 6 mg mL−1). The solution was degassed with an argon stream for 10 min, while cooled in an ice bath and the surface was irradiated with an Arimed B6 lamp for 24 h. The surface Sur 2 was removed from the solution and washed extensively with dichloromethane. X-ray photoelectron spectroscopy (XPS) was performed before and subsequently to the photo-ligation of deprotected PEtOxx-MI onto the tetrazole surface. The results are summarized in Fig. 4 and the full table of atomic percents (at%) is given in the ESI.†Results and discussion
Maleimide-functionalized poly(2-ethyl-2-oxazoline)s (PEtOxx-MI) with two different chain lengths was prepared via copper catalyzed azide–alkyne cycloaddition (CuAAC). Azide-functionalized poly(2-ethyl-2-oxazoline)s (PEtOxx-N3) of different molar mass and with narrow molecular weight distributions (Đ < 1.2) were synthesized according to literature36,74–76 (Scheme 1) and combined with an alkyne-functionalized maleimide, which has been synthesized in a three-step procedure (Fig. 1A).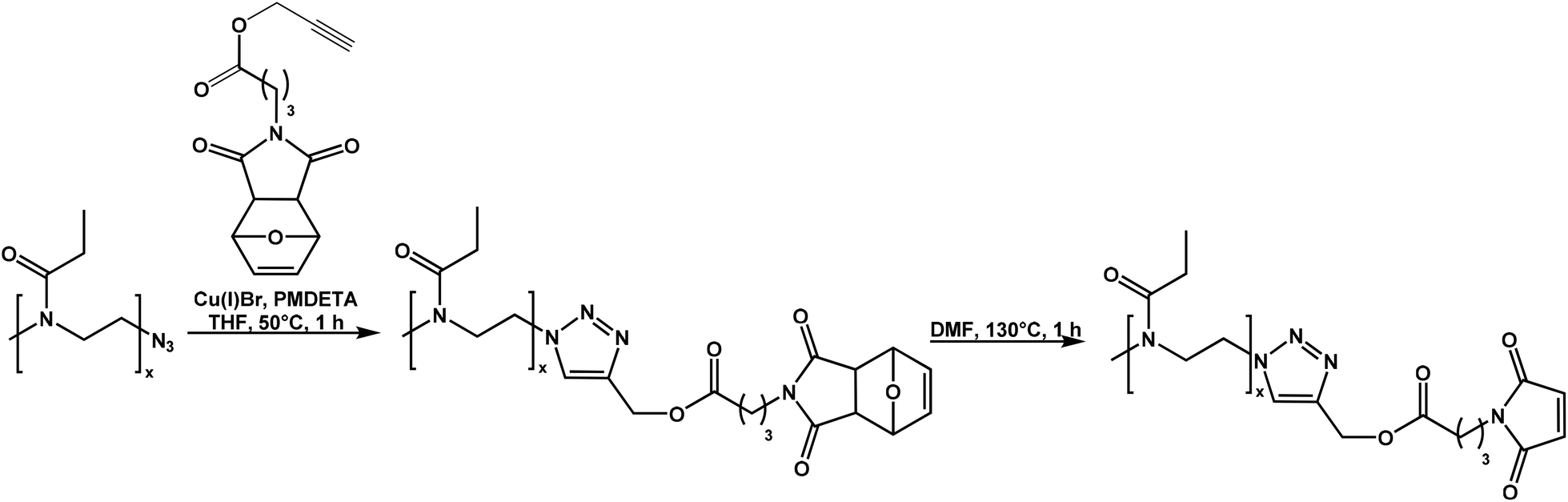 | ||
| Scheme 1 Synthesis of PEtOxx-MI via CuAAC and subsequent deprotection. | ||
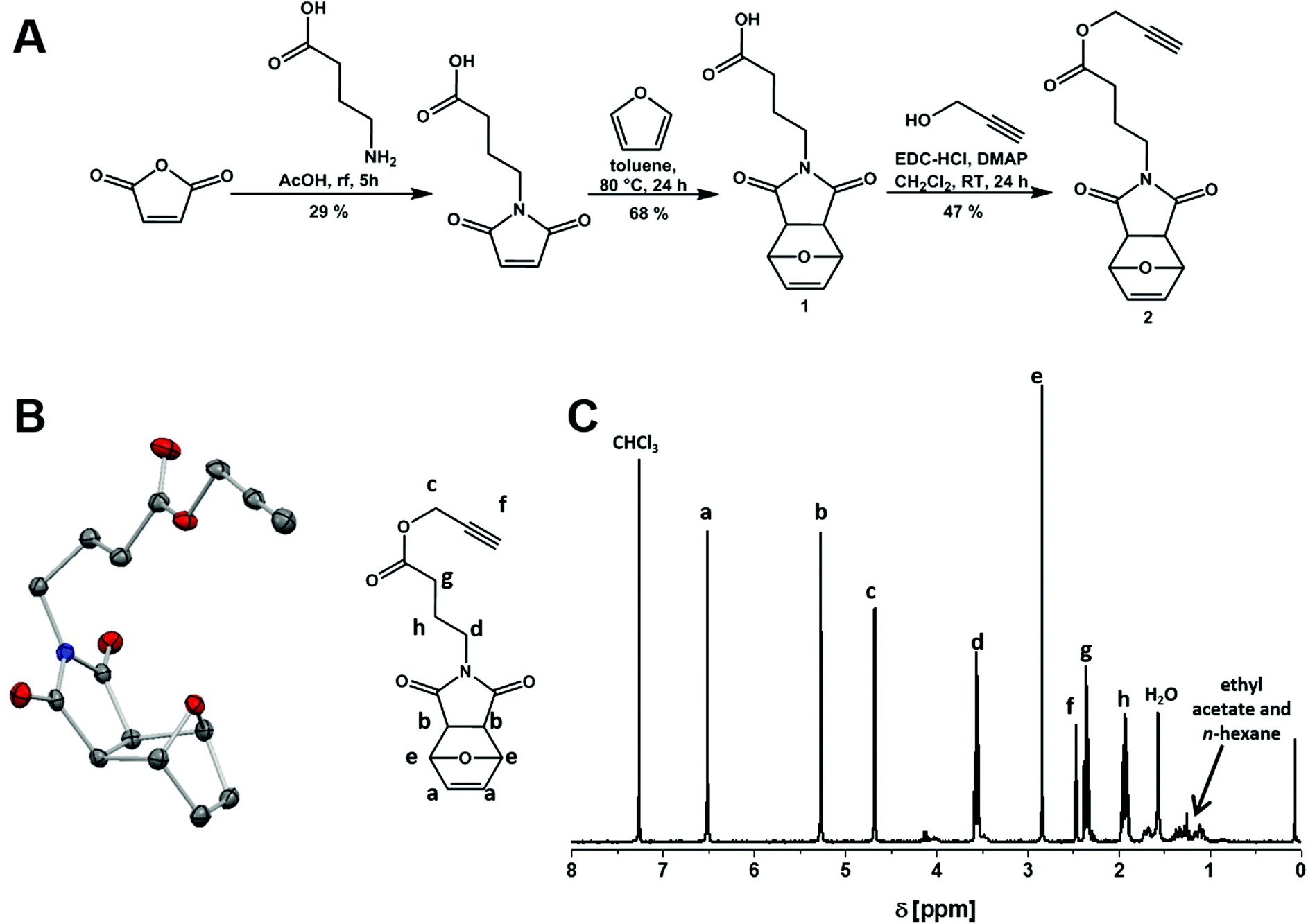 | ||
| Fig. 1 Synthetic strategy for the preparation of the alkyne-functionalized maleimide (A), including the molecule structure (B, 50% probability level, hydrogens omitted, dark grey: carbon, blue: nitrogen, red: oxygen), and a 1H-NMR (300 MHz, CDCl3) spectrum of the alkyne-functionalized maleimide (C). | ||
The protection of the maleimide double bond using furan was necessary to make it less prone against nucleophilic attack.66 In that respect, the actual PEtOxx-MI can be achieved via a subsequent deprotection step (Scheme 1). Besides the characterization (NMR: Fig. 1C, FT-IR: Fig. S1,† MALDI: Fig. S2,† EA) the compound could be crystallized und enabled X-ray structure analysis (Fig. 1B for the respective crystal structure image).
The following CuAAC between the maleimide and PEtOxx-N3 was carried out using copper(I) bromide (CuBr) and N,N,N′,N′′,N′′′-pentamethyldiethylenetriamine (PMDETA) as a catalyst-ligand system.38,40 The temperature was set to 50 °C for one hour to prevent the retro Diels–Alder (retro-DA) reaction with respect to the maleimide-furan Diels–Alder (DA) adduct and afterwards stirred at ambient temperature overnight to ensure full conversion. For purification, copper was removed by a short aluminum oxide (AlOx) column and the final polymer was isolated by precipitation in cold diethyl ether. Characterization was carried out using SEC, NMR (Fig. 2), MALDI (Fig. S3†), and FT-IR (Fig. S4†).
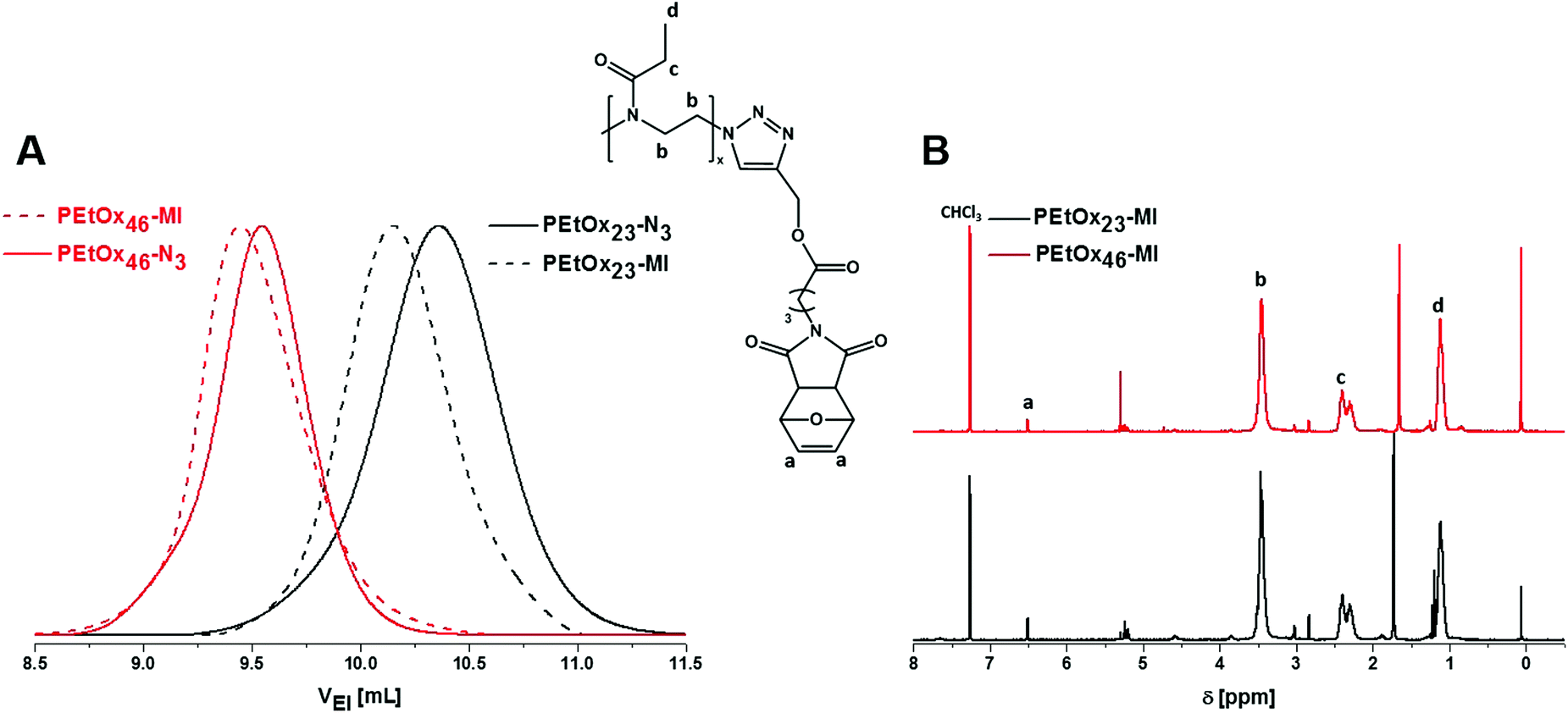 | ||
| Fig. 2 Comparison of the SEC traces for PEtOxx-N3 (straight line)/PEtOxx-MI (dashed line) (A) and characterization via NMR spectroscopy (300 MHz, CDCl3) for a degree of polymerization of: 23 (black traces) and 46 (red traces, B). | ||
An increase of the molar mass for both PEtOx23-MI and PEtOx46-MI by comparison of the respective SEC traces was observed (see Table 1 for detailed information).
The NMR spectrum shows resonances associated with the PEtOx backbone peak at 3.5, 2.3 and 1.2 ppm as well as the furan protection group at 6.5 ppm. The degree of functionalization of PEtOxx-MI was determined to be nearly quantitative within the range of the error of the method (NMR). Subsequently, the deprotection procedure of PEtOxx-MI was investigated in detail. The polymer was dissolved in deuterated dimethylformamide (DMF-d6) and heated to 130 °C for different reaction times in an oil bath. The same approach was carried out for the pristine maleimide for comparison. The resonance at 6.5 ppm was used to determine the degree of functionalization by comparing it to the signal in the range of 2.3 ppm of the PEtOxx-MI. The disappearance of the furan resonance was observed, showing clearly the deprotection of the maleimide with time (Fig. 3). The degree of cleavage was determined and illustrated in a deprotection vs. time plot showing close to 50% of cleavage after only five minutes and complete deprotection within one hour under these conditions for all systems investigated.
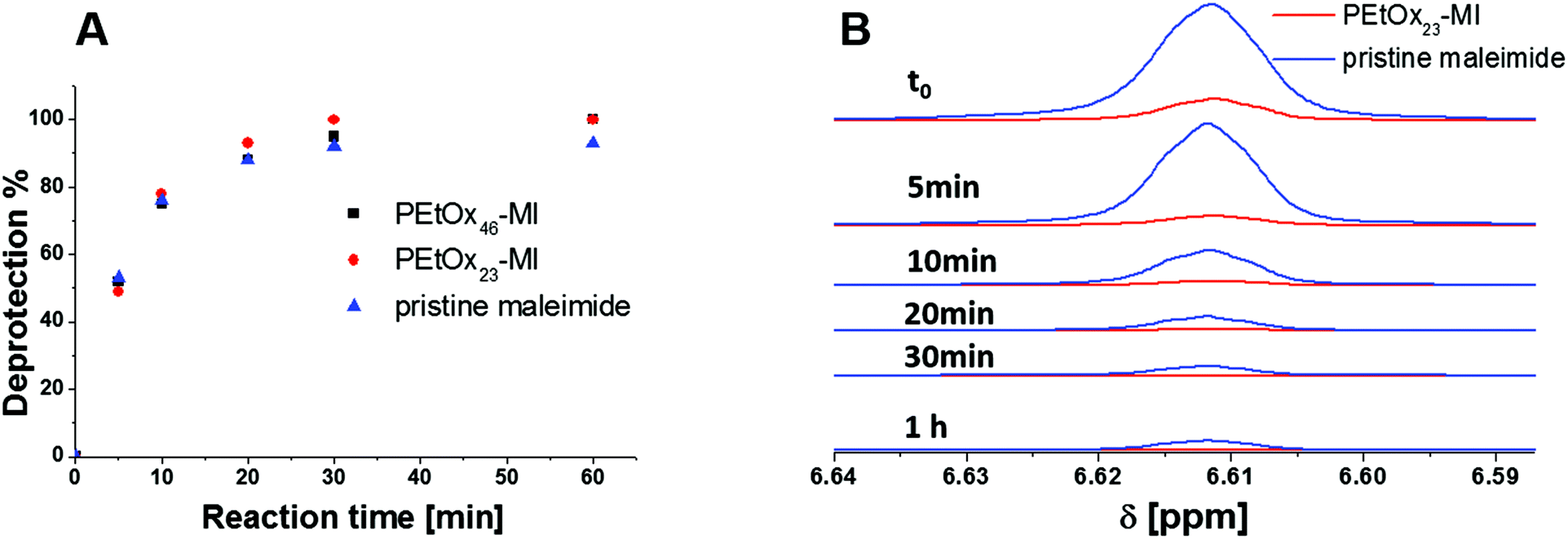 | ||
| Fig. 3 Time dependent deprotection of PEtOx23-MI (red line) and the pristine maleimide (blue line, A) and comparison of the respective 1H-NMR spectra (300 MHz, DMF-d6) at different reaction times (B). | ||
Subsequently, the reactivity of the terminal maleimide was investigated by immobilization of the deprotected PEtOxx-MI on a tetrazole functionalized silicon wafer (Sur 1) via photochemical NITEC reaction (Sur 2, Fig. 4). The tetrazole functionalization was carried out according to a previous report.73 For the NITEC reaction the tetrazole surface (Sur 1) was immersed in a degassed and dry solution of polymer in dichloromethane and exposed to UV-irradiation for 24 h using an Arimed B6 lamp. Unreacted material was removed from the surface by extensive washing with solvent, and subsequently the surface was dried in an argon stream and analyzed via X-ray photoelectron spectroscopy (XPS). The surface with immobilized PEtOxx-MI (Sur 2) was compared to a neat tetrazole functionalized silicon wafer (Sur 1) and the data is shown in Fig. 4.
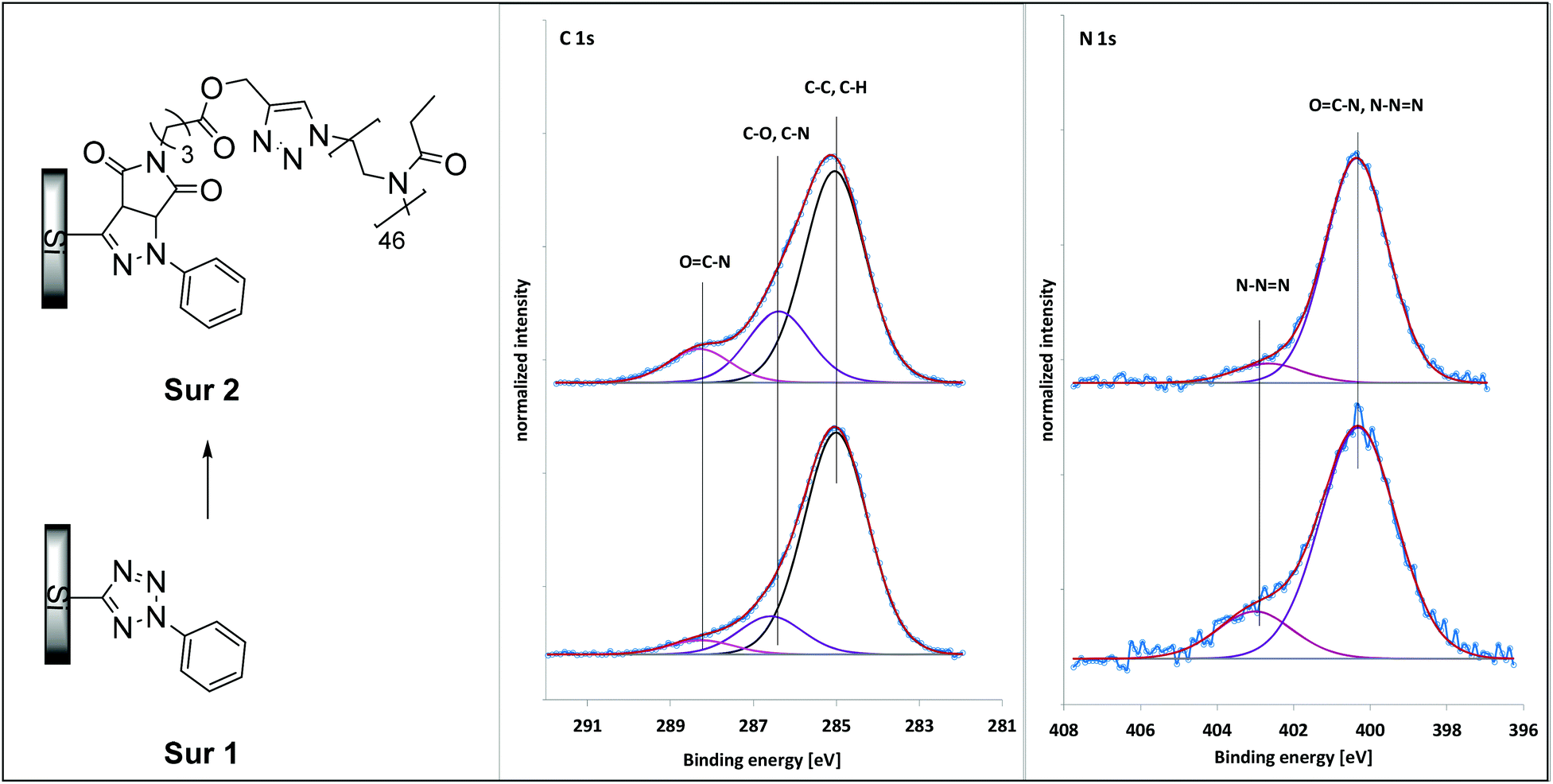 | ||
| Fig. 4 XPS characterization of the PEtOx46-MI functionalized silicon wafer (Sur 2) in comparison to the tetrazole functionalized silicon wafer (Sur 1). | ||
The XPS signals at 286.6 eV and 288.4 eV correspond to C–O, C–N and amide carbons and increase from 3.4 at% to 6.3 at% and 0.9 at% to 3.0 at% for the functionalization with PEtOx46-MI.54 The increase in the signal at 400.3 eV from 1.7 at% to 3.1 at% additionally indicates the presence of amide nitrogen atoms on the surface. A further proof for the formation of a PEtOx layer on the surface is the decrease in signals for silicon and oxygen as evident from Table S3.†
Conclusions
In summary, the preparation and the reactivity of maleimide-functionalized PEtOxx-MI is presented, starting from PEtOxx-N3 and an alkyne-functionalized maleimide. Two degrees of polymerization (23 and 46) for PEtOxx were chosen and the covalent attachment of the maleimide functionality was realized using a CuAAC approach. Due to its high reactivity the maleimide had to be protected with furan prior to the coupling and we could show that complete deprotection within one hour at 130 °C can be achieved afterwards. PEtOxx-MI was subsequently immobilized on a tetrazole functionalized silicon wafer via a photochemical NITEC reaction and this was confirmed by XPS analysis. Such surfaces might be of interest for layer-by-layer approaches or where control over cell adhesion on surfaces is desired – in both cases the deprotection of PEtOxx to linear PEI would be necessary, as we have recently demonstrated for SiO2@P(EtOx-stat-EI) hybrid nanoparticles.77 As an alternative strategy, in case of hydrophobic poly(2-oxazoline)s a maleimide endgroup has been introduced via direct endcapping very recently by Luxenhofer et al.78 These materials were then used to modify elastin-like proteins.Acknowledgements
F. W. and F. H. S. gratefully acknowledged funding by the Thuringian Ministry for Economy, Science, and Digital Society (TMWWDG). C. B.-K. acknowledges continued funding from the Karlsruhe Institute of Technology (KIT) via the BioInterfaces in Technology and Medicine (BIFTM) program. The authors thank Beate Lentvogt and Sandra Köhn for elemental analyses. The authors also thank Bruker Daltonics for their help and support and Sarah Crotty for the MALDI measurements. Florian Kretschmer is acknowledged for help during initial experiments. F. H. S. is further grateful to Ulrich S. Schubert for continuous support.References
- I. W. Hamley, Angew. Chem., Int. Ed., 2003, 42, 1692–1712 CrossRef CAS PubMed.
- M. Ferrari, Nat. Rev. Cancer, 2005, 5, 161–171 CrossRef CAS PubMed.
- E. Katz and I. Willner, Angew. Chem., Int. Ed., 2004, 43, 6042–6108 CrossRef CAS PubMed.
- O. C. Farokhzad and R. Langer, ACS Nano, 2009, 3, 16–20 CrossRef CAS PubMed.
- M. Sarikaya, C. Tamerler, A. K. Y. Jen, K. Schulten and F. Baneyx, Nat. Mater., 2003, 2, 577–585 CrossRef CAS PubMed.
- A. N. Shipway, E. Katz and I. Willner, ChemPhysChem, 2000, 1, 18–52 CrossRef CAS PubMed.
- W. Lu and C. M. Lieber, Nat. Mater., 2007, 6, 841–850 CrossRef CAS PubMed.
- Y. Xia, P. Yang, Y. Sun, Y. Wu, B. Mayers, B. Gates, Y. Yin, F. Kim and H. Yan, Adv. Mater., 2003, 15, 353–389 CrossRef CAS.
- R. K. Iha, K. L. Wooley, A. M. Nyström, D. J. Burke, M. J. Kade and C. J. Hawker, Chem. Rev., 2009, 109, 5620–5686 CrossRef CAS PubMed.
- C. J. Hawker and K. L. Wooley, Science, 2005, 309, 1200–1205 CrossRef CAS PubMed.
- J.-F. Lutz and H. Schlaad, Polymer, 2008, 49, 817–824 CrossRef CAS.
- M. A. Gauthier, M. I. Gibson and H.-A. Klok, Angew. Chem., Int. Ed., 2009, 48, 48–58 CrossRef CAS PubMed.
- B. S. Sumerlin and A. P. Vogt, Macromolecules, 2010, 43, 1–13 CrossRef CAS.
- T. Rudolph, P. Espeel, F. E. Du Prez and F. H. Schacher, Polym. Chem., 2015, 6, 4240–4251 RSC.
- K. Matyjaszewski and J. Xia, Chem. Rev., 2001, 101, 2921–2990 CrossRef CAS PubMed.
- K. Matyjaszewski, Macromolecules, 2012, 45, 4015–4039 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Polymer, 2008, 49, 1079–1131 CrossRef CAS.
- L. Barner, T. P. Davis, M. H. Stenzel and C. Barner-Kowollik, Macromol. Rapid Commun., 2007, 28, 539–559 CrossRef CAS.
- J. Nicolas, Y. Guillaneuf, C. Lefay, D. Bertin, D. Gigmes and B. Charleux, Prog. Polym. Sci., 2013, 38, 63–235 CrossRef CAS.
- L. Tebben and A. Studer, Angew. Chem., Int. Ed., 2011, 123, 5138–5174 CrossRef.
- T. Rudolph, A. Nunns, A. M. Schwenke and F. H. Schacher, Polym. Chem., 2015, 6, 1604–1612 RSC.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS.
- J. E. Moses and A. D. Moorhouse, Chem. Soc. Rev., 2007, 36, 1249–1262 RSC.
- W. Xi, T. F. Scott, C. J. Kloxin and C. N. Bowman, Adv. Funct. Mater., 2014, 24, 2572–2590 CrossRef CAS.
- H. Nandivada, X. Jiang and J. Lahann, Adv. Mater., 2007, 19, 2197–2208 CrossRef CAS.
- C. J. Hawker, V. V. Fokin, M. G. Finn and K. B. Sharpless, Aust. J. Chem., 2007, 60, 381–383 CrossRef CAS.
- W. H. Binder and R. Sachsenhofer, Macromol. Rapid Commun., 2008, 29, 952–981 CrossRef CAS.
- W. H. Binder and R. Sachsenhofer, Macromol. Rapid Commun., 2007, 28, 15–54 CrossRef CAS.
- P. L. Golas and K. Matyjaszewski, QSAR Comb. Sci., 2007, 26, 1116–1134 CAS.
- P. L. Golas and K. Matyjaszewski, Chem. Soc. Rev., 2010, 39, 1338–1354 RSC.
- K. A. Günay, P. Theato and H.-A. Klok, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 1–28 CrossRef.
- A. J. Inglis and C. Barner-Kowollik, Macromol. Rapid Commun., 2010, 31, 1247–1266 CrossRef CAS PubMed.
- A. S. Goldmann, M. Glassner, A. J. Inglis and C. Barner-Kowollik, Macromol. Rapid Commun., 2013, 34, 810–849 CrossRef CAS PubMed.
- C. Barner-Kowollik, F. E. Du Prez, P. Espeel, C. J. Hawker, T. Junkers, H. Schlaad and W. Van Camp, Angew. Chem., Int. Ed., 2011, 50, 60–62 CrossRef CAS PubMed.
- M. Cortez and S. M. Grayson, Polym. Mater. Sci. Eng., 2008, 98, 436–437 CAS.
- G. Volet, T.-X. Lav, J. Babinot and C. Amiel, Macromol. Chem. Phys., 2011, 212, 118–124 CrossRef CAS.
- J.-F. Lutz, Angew. Chem., Int. Ed., 2007, 46, 1018–1025 CrossRef CAS PubMed.
- D. Fournier, R. Hoogenboom and U. S. Schubert, Chem. Soc. Rev., 2007, 36, 1369–1380 RSC.
- J. E. Hein and V. V. Fokin, Chem. Soc. Rev., 2010, 39, 1302–1315 RSC.
- M. Meldal and C. W. Tornøe, Chem. Rev., 2008, 108, 2952–3015 CrossRef CAS PubMed.
- D. J. Hall, H. M. Van Den Berghe and A. P. Dove, Polym. Int., 2011, 60, 1149–1157 CrossRef CAS.
- A. Gandini, Prog. Polym. Sci., 2013, 38, 1–29 CrossRef CAS.
- M. A. Tasdelen, Polym. Chem., 2011, 2, 2133–2145 RSC.
- G. Hizal, U. Tunca and A. Sanyal, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 4103–4120 CAS.
- M. Billing, T. Rudolph, E. Täuscher, R. Beckert and F. Schacher, Polymers, 2015, 7, 2478–2493 CrossRef CAS.
- M. Li, P. De, H. Li and B. S. Sumerlin, Polym. Chem., 2010, 1, 854–859 RSC.
- B. Iskin, G. Yilmaz and Y. Yagci, Polym. Chem., 2011, 2, 2865–2871 RSC.
- G. Mantovani, F. Lecolley, L. Tao, D. M. Haddleton, J. Clerx, J. J. L. M. Cornelissen and K. Velonia, J. Am. Chem. Soc., 2005, 127, 2966–2973 CrossRef CAS PubMed.
- C. Da Pieve, P. Williams, D. M. Haddleton, R. M. J. Palmer and S. Missailidis, Bioconjugate Chem., 2010, 21, 169–174 CrossRef CAS PubMed.
- M. Dietrich, G. Delaittre, J. P. Blinco, A. J. Inglis, M. Bruns and C. Barner-Kowollik, Adv. Funct. Mater., 2012, 22, 304–312 CrossRef CAS.
- E. Blasco, M. Pinol, L. Oriol, B. V. K. J. Schmidt, A. Welle, V. Trouillet, M. Bruns and C. Barner-Kowollik, Adv. Funct. Mater., 2013, 23, 4011–4019 CrossRef CAS.
- C. Rodriguez-Emmenegger, C. M. Preuss, B. Yameen, O. Pop-Georgievski, M. Bachmann, J. O. Mueller, M. Bruns, A. S. Goldmann, M. Bastmeyer and C. Barner-Kowollik, Adv. Mater., 2013, 25, 6123–6127 CrossRef CAS PubMed.
- A. de Los Santos Pereira, N. Y. Kostina, C. Rodriguez-Emmenegger, M. Bruns, C. Barner-Kowollik and C. Barner-Kowollik, Langmuir, 2015, 31, 5899–5907 CrossRef CAS PubMed.
- T. Tischer, C. Rodriguez-Emmenegger, V. Trouillet, A. Welle, V. Schueler, J. O. Mueller, A. S. Goldmann, E. Brynda and C. Barner-Kowollik, Adv. Mater., 2014, 26, 4087–4092 CrossRef CAS PubMed.
- L. Tauhardt, K. Kempe, M. Gottschaldt and U. S. Schubert, Chem. Soc. Rev., 2013, 42, 7998–8011 RSC.
- M. N. Leiske, M. Hartlieb, C. Paulenz, D. Pretzel, M. Hentschel, C. Englert, M. Gottschaldt and U. S. Schubert, Adv. Funct. Mater., 2015, 25, 2458–2466 CrossRef CAS.
- K. Knop, R. Hoogenboom, D. Fischer and U. S. Schubert, Angew. Chem., Int. Ed., 2010, 49, 6288–6308 CrossRef CAS PubMed.
- N. Adden, A. Hoffmann, G. Gross, H. Windhagen, F. Thorey and H. Menzel, J. Biomater. Sci., Polym. Ed., 2007, 18, 303–316 CrossRef CAS PubMed.
- B.-J. Chang, O. Prucker, E. Groh, A. Wallrath, M. Dahm and J. Rühe, Colloids Surf., A, 2002, 198–200, 519–526 CrossRef CAS.
- M. Dahm, B.-J. Chang, O. Prucker, M. Pierkes, T. Alt, E. Mayer, J. Rühe and H. Oelert, Ann. Thorac. Surg., 2001, 71, S437–S440 CrossRef CAS PubMed.
- H. Murata, B. J. Chang, O. Prucker, M. Dahm and J. Rühe, Surf. Sci., 2004, 570, 111–118 CrossRef CAS.
- O. Prucker, C. A. Naumann, J. Rühe, W. Knoll and C. W. Frank, J. Am. Chem. Soc., 1999, 121, 8766–8770 CrossRef CAS.
- Q. Yu, Y. Zhang, H. Wang, J. Brash and H. Chen, Acta Biomater., 2011, 7, 1550–1557 CrossRef CAS PubMed.
- Y. Wang, W. J. Hu, W. Song, R. K. V. Lim and Q. Lin, Org. Lett., 2008, 10, 3725–3728 CrossRef CAS PubMed.
- R. Marcia De Figueiredo, P. Oczipka, R. Fröhlich and M. Christmann, Synthesis, 2008, 1316–1318 Search PubMed.
- T. Rudolph, M. J. Barthel, F. Kretschmer, U. Mansfeld, S. Hoeppener, M. D. Hager, U. S. Schubert and F. H. Schacher, Macromol. Rapid Commun., 2014, 35, 916–921 CrossRef CAS PubMed.
- COLLECT, Data collection software, Nonius B.V., The Netherlands, 1998 Search PubMed.
- Z. Otwinowski and W. Minor, in Methods Enzymol., ed. C. W. Carter and R. M. Sweet, Academic Press, San Diego, USA, 1st edn, 1997, vol. 276, pp. 307–326.
- SADABS 2.10, Bruker-AXS inc., Madison, WI, U.S.A, 2002 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- K. L. Parry, A. G. Shard, R. D. Short, R. G. White, J. D. Whittle and A. Wright, Surf. Interface Anal., 2006, 38, 1497–1504 CrossRef CAS.
- J. H. Scofield, J. Electron Spectrosc. Relat. Phenom., 1976, 8, 129–137 CrossRef CAS.
- Y. Sugawara, N. Jasinski, M. Kaupp, A. Welle, N. Zydziak, E. Blasco and C. Barner-Kowollik, Chem. Commun., 2015, 51, 13000–13003 RSC.
- A. Makino and S. Kobayashi, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 1251–1270 CrossRef CAS.
- F. Wiesbrock, R. Hoogenboom and U. S. Schubert, Macromol. Rapid Commun., 2004, 25, 1739–1764 CrossRef CAS.
- R. Hoogenboom, Macromol. Chem. Phys., 2007, 208, 18–25 CrossRef CAS.
- O. Eckardt, C. Pietsch, O. Zumann, M. von der Lühe, D. S. Brauer and F. H. Schacher, Macromol. Rapid Commun., 2016, 37, 337–342 CrossRef CAS PubMed.
- J. F. Nawroth, J. R. McDaniel, A. Chilkoti, R. Jordan and R. Luxenhofer, Macromol. Biosci., 2016 DOI:10.1002/mabi.201500376.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1434099. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6py00033a |
| This journal is © The Royal Society of Chemistry 2016 |