
Toughening of photo-curable polymer networks: a review†
Samuel Clark
Ligon-Auer
ab,
Martin
Schwentenwein
c,
Christian
Gorsche
ab,
Jürgen
Stampfl
bd and
Robert
Liska
*ab
aInstitute of Applied Synthetic Chemistry, Technische Universität Wien, Getreidemarkt 9/163, 1060 Vienna, Austria. E-mail: robert.liska@tuwien.ac.at; Fax: +43-1-58801-163302; Tel: +43-1-58801-163614
bChristian Doppler Laboratory for Digital and Restorative Dentistry, Technische Universität Wien, Vienna, Austria
cLithoz GmbH, Mollardgasse 85a/2/64-69, 1060 Vienna, Austria
dInstitute of Materials Science and Technology, Technische Universität Wien, Getreidemarkt 9 E308, 1060 Vienna, Austria
First published on 20th November 2015
Abstract
Photo-curable resins based on multifunctional acrylate monomers are commonly applied as thin films (e.g. protective coatings, printing inks, etc.) and in recent years are also used for the fabrication of bulk objects such as dental fillings and 3D-printed parts. While rapid curing and good spatial resolution are advantages to these systems, brittleness and poor impact resistance due to inhomogeneous polymer architecture and high crosslink density are serious drawbacks. By comparison, epoxy thermoset resins suffered many years ago from similar problems, but since then are found in ever demanding applications thanks to a variety of approaches to increase polymer toughness. Based on these successes, researchers have tried to translate strategies for toughening epoxy resins to photopolymer networks. Therefore, this review surveys relevant scientific papers and patents on the development of crosslinked epoxy-based polymers and also photo-curable polymers based on multifunctional acrylates with improved toughness. Strategies developed to reduce brittleness include working with monomers, which intrinsically give tougher polymers, particulate additives, and alternate forms of polymerization and polymer architecture (e.g., dual-cure networks, interpenetrating networks, thiol–ene chemistry). All of these strategies have advantages and yet application specific rigours must also be considered before and during formulation development.
Introduction
Epoxy-based thermosetting resins were first developed in the 1940s and are today found in a range of applications such as metal can coatings, printed circuit boards and aerospace composites. While these crosslinked resins have always been renowned for good chemical resistance and electrical passivity, brittleness was historically a problem. In the 1960s and 1970s, however, techniques were developed for toughening these plastics allowing them to be used more and more in structural applications.By comparison, due to limitations of light penetration, photo-curable multifunctional acrylates have traditionally been applied in thin film applications such as inks and coatings and not as engineering plastics.1,2 The advancement of photo-curable dental restoratives and additive manufacturing (or 3D-printing) based on layer by layer assembly3 have changed the role of photopolymers though, and thus mechanical properties, especially toughness, have taken increasing importance.4 To reduce brittleness, researchers try to translate toughening strategies from epoxy-based systems to acrylate-networks.
Borrowing largely from techniques developed for toughening of industrial epoxy and vinyl ester thermosets (typically but not specifically cured by heat), bending and impact strength of photopolymer networks has improved greatly over the last two decades. This can be attributed to proper selection of monomers and additives as well as layering and curing conditions. To better understand toughening, we first present the commonly used experimental techniques to measure mechanical properties related to toughness. We then introduce some of the fundamental terms used to describe material failure. Next we overview some of the more important toughening techniques as they were first applied to thermosets. Additional toughening techniques developed specifically for photo-curable polymers will be described as well. Focus here is chiefly on acrylate-based monomers, with mention also of photo-curable epoxy and oxetane resins as they are commonly found in 3D-printing resins. It should also be noted that the term “acrylate-based monomer” refers here to methacrylates as well. While methacrylates are less reactive and much less common in coatings, they are of significant importance in bulk applications such as dental fillings and 3D-printed parts. The use of chain transfer agents with acrylate resins is explored fairly in depth and is one of the most promising strategies to improve material properties including toughness. In addition, photo-curable interpenetrating polymer networks (IPNs) and photo-induced phase separation (PIPS) are investigated. In closing, application and processing specific concerns shall be considered.
Techniques for measuring toughness
Toughness is a material property related to the resistance against crack growth. Typically, toughness decreases if strength, stiffness or hardness is increased. For many engineering applications, though, high values for strength, stiffness AND toughness are desired. It is therefore essential to develop efficient toughening mechanisms, which keep toughness at reasonably high levels, even in the case of strong and stiff materials. The stiffness of the sample describes its ability to withstand elastic (reversible) deformation and is determined by the geometry of the sample and the elastic modulus of the utilized material. Strength describes the ability of the sample to withstand plastic (irreversible) deformation. Strength and modulus of a material can be determined by applying tensile testing.Tensile testing
Tensile testing is described as a quasi-static testing method since the experiment is performed with a very slow strain-rate.5 For example, recommended crosshead speeds for common polymers are 20–100 mm min−1 for HDPE, nylon, and PP and 5–20 mm min−1 for ABS, PMMA and polyester.6 With a normal universal testing machine, loading is applied electrically or electro-hydraulically which is essentially shock-free, steadily increasing until ultimate failure of the specimen occurs. The applied tensile load or force induces a uniaxial loading state on the specimen, which results in a homogeneous and evenly distributed axial stress over the sample cross-section. The area of the cross-section is assumed to be constant during the experiment.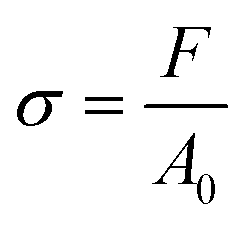 | (1) |
The stress during the measurement is calculated via the equation above while the strain can be determined by the following equation.
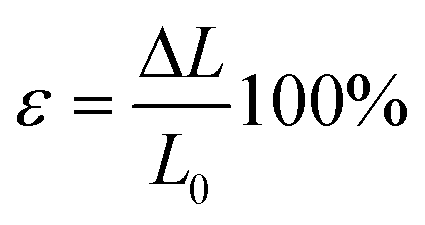 | (2) |
By monitoring the strain with increasing stress, a stress–strain curve is obtained. A typical curve is depicted in Fig. 1 showing the different stages observed when testing polymers. Independent of the type of polymer, the linear regime at the onset of the stress–strain curve can be used to determine the Young's modulus (E), which corresponds to the slope of the curve. When the stress is increased, the curve becomes non-linear, and depending on the type of tested polymer various behaviours can be observed.
 | ||
| Fig. 1 Exemplary stress–strain curves for various polymers: brittle materials (a), tough materials with yield point (b), tough materials without yield point (c) and elastomers (d).7 | ||
Photo-cured acrylates are typically rather brittle and therefore fall into category (a) in Fig. 1: the strain at break (εB) is small, and no yield point with yield strength (σy) can be observed. The maximum stress (σm) occurs at the point of failure and maximum stress is therefore the stress at break (σB). If the cross-link density is sufficiently low, the photo-cured polymer will exhibit elastomeric behaviour, corresponding to curve (d) in Fig. 1. In toughened photo-curable polymers and thermoplastic polymers, behaviour as depicted in (b) and (c) might be observed.
Deformation energy (UT), which is related to the toughness of a material, is defined as the area under the stress (σ)–strain (ε) curve:
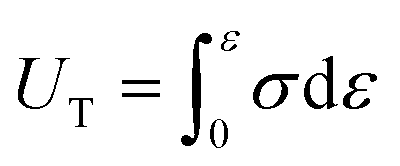 | (3) |
High deformation energy indicates good resistance against crack growth, since crack propagation will require a larger amount of energy in these materials.
Impact testing
Although tensile testing does provide values for strength and for toughness, these values are quasi-static and additional dynamic techniques may be required for polymers intended for real-world applications. For example, resistance to projectiles such as hailstones or gravel is essential for exterior automobile components. To determine if a new material is suitable for such an application, the loading must be applied quickly. High-rate loading or impact can be applied in a number of manners with pendulums being commonly used due to accuracy and reproducibility. For example, two of the most common standardized methods for measuring impact strength (those of Izod and Charpy) are both based on pendulums. While the sample dimensions are different for the Izod and Charpy tests, the basic setup is comparable. In both cases notched as well as unnotched samples can be used. Notches are used in both cases to provide an intentional ‘weak point’ where the sample should break upon impact. A schematic comparison of Izod and Charpy methods is shown in Fig. 2. | ||
| Fig. 2 Schematics of Izod and Charpy impact testing setups. | ||
The most obvious difference in the two methods is that the sample is supported on one end for the Izod test and on both ends for the Charpy test. There are also differences in the relation of the point of impact of the pendulum and of the notch. ISO 180 defines the Izod notch impact strength (aiN) as the absorbed impact work of the fractured notched sample divided by the initial cross section area of the sample under the notch tip.8
 | (4) |
Impact energy can be determined as the loss in potential energy (ΔPE) measured from the difference in release height (h0) and maximum height attained for the pendulum hammer after impact (hfinal).
| ΔPE = Wi = mg(h0 − hfinal) | (5) |
This equation assumes an ideal pendulum where force is based on the gravity (g) driven mass (m) alone. Friction can be accounted for by swinging the hammer without a sample. In comparison to the ISO method, ASTM D256 calculates Izod impact strength by dividing impact energy by the thickness of the specimen (eqn (6)). The result in this case has the dimensions of J m−1.
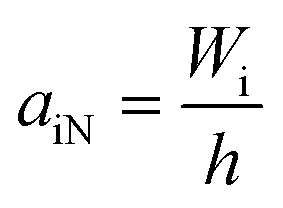 | (6) |
Dynamic mechanical analysis (DMA)
Mechanical properties of polymers are dependent on temperature and on the frequency of an applied load. When polymers are subjected to a sufficiently high mechanical load, they tend to yield. The stresses which lead to yielding are usually strain-rate dependent. This strain-rate-dependent behaviour is called viscoelasticity.9Dynamic Mechanical Analysis (DMA) is a powerful technique to study the viscoelastic nature of polymers. The technique applies a periodic force to the sample and measures the response. The applied dynamic stress (σ) can be described as:
| σ = σ0sin(ωt + δ) | (7) |
The resulting dynamic strain (ε) has a similar equation:
ε = ε0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) sin(ωt) sin(ωt) | (8) |
Dividing these two terms provides modulus as a complex number where the real (or in-phase portion) describes the storage modulus (E′):
 | (9) |
And the imaginary (or out-of-phase portion) describes the loss modulus:
 | (10) |
The storage modulus describes the elastic response of the material and is related to the Young's modulus, E. The loss modulus relates to the viscous properties of the material and is sensitive to different kinds of molecular motions, phase transitions, and other structural heterogeneities.10 Both storage and loss modulus are dependent on temperature although this dependence is not always linear. Non-linear or in some cases abrupt changes in modulus will occur at or near the glass transition temperature (Tg) of a material. Specifically the loss factor (or tanδ), which is the ratio of the loss modulus to the storage modulus, will increase and reach a maximum at the Tg.11 Thus DMA (or DMTA) is a much more sensitive method for measuring Tg than Differential Scanning Calorimetry (DSC). DMA can also be used to investigate the frequency- and time-dependent nature of this transition and is often able to resolve sub-Tg transitions, like beta-, gamma- and delta-transitions that DSC is typically not sensitive enough to measure. Since mechanical properties can change drastically at or near the Tg, this measurement will often partially define the useful temperature range of a polymer. As an example, Young's modulus of crosslinked rubber can increase by a factor of more than 1000 below its Tg, resulting in a hard but also very brittle material. Elastomers with even lower Tg values were developed throughout the twentieth century and have allowed vehicles to operate in extreme arctic or high altitude conditions not possible before. Although photopolymers are not intended for such applications, temperature is an important consideration and the lessons learned in material development are still relevant.
Hardness
Hardness is a material property describing the resistance of a material against penetration. Hardness is usually related to the yield strength of a material, although owing to the differences in measuring techniques it is not possible to give a simple conversion factor applicable for all materials. The two most commonly used methods for measuring the hardness of photo-curable coatings are those of Persoz and of König.12 Both methods are based on pendulums supported by balls resting on top of the sample material. The pendulum is set into motion and the time required for the angle of the pendulum to decrease from 12° to 4° (Persoz) or from 6° to 3° (König) is recorded in seconds.Nanoindentation is a slightly newer and yet well-established method for measuring the hardness of coatings and thin films. With nanoindentation, a small pyramidal shaped diamond tip is pressed into the sample, while the load and displacement are recorded.13 The slope of the load–displacement curve gives the stiffness (S) which can be used with other terms to determine a reduced Young's modulus (Er) for the material.
 | (11) |
Fracture and crack propagation
Polymeric materials intended for use in engineering applications are expected to withstand high levels of stress without failing.14 Fractures are one of the principal causes of catastrophic material failure, which warrants a description of how cracks arise and how they propagate. Fractures originate from defects (cracks, internal voids, inhomogeneities of the microstructure, etc.) within the material or on its surface. The toughness of a material is then defined by its ability to withstand crack growth. Impact strength as well as the area under the stress–strain curve are commonly used metrics for the toughness of a material. Other measurements for material toughness include the critical stress intensity factor (KIC), critical J-integral (JIC) and the crack tip opening displacement (CTOD).Depending on the applied load, a crack can propagate by three principal modes (Fig. 3). In mode I, a tensile force is applied normal to the crack plane. Mode II and mode III propagation are driven by shear forces.
 | ||
| Fig. 3 Modes of crack propagation. | ||
Fractures may be classified as brittle or ductile, where in the first case the principle driving force for crack propagation is the release of elastic energy of the propagating crack which is counteracted by the consumption of surface energy from the two newly created surfaces adjacent to the crack. With ductile materials such as metals and polymers, surface energy plays a lesser role. Instead, a plastic zone develops around the crack tip, and depending on the size of the plastic zone and the level of the yield stress large amounts of energy can be dissipated. In the case of brittle fracture, the energy release rate of the material (G) is related to the Young's modulus (E) by the following equation:
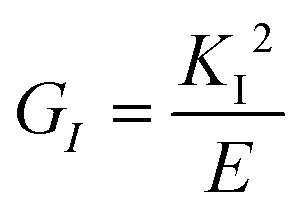 | (12) |
The critical stress intensity factor (KIC) is the minimal value for propagating a crack under mode I tensile forces, and is commonly referred to as the fracture toughness of the material.
Considerations for the mechanical testing of photopolymers
The geometry of the sample and the nature of the testing apparatus have great bearing on results and generally standardized mechanical testing methods are applied. Some materials, including photopolymers printed by layer by layer assembly, can be anisotropic with strength normal to and axial to the build direction being sometimes different.15 Photopolymer samples intended for mechanical testing are commonly cured in moulds and then cured a second time on the back side. For additive manufacturing, thermal post curing is also quite common. While sample geometry is normally defined by a standard, curing conditions (intensity, time, wavelength, and O2 concentration) are not. Such parameters must be considered when comparing mechanical testing results of one polymer sample with those of another.Polymer architecture
The molecular architecture of a polymer has an obvious influence on the mechanical properties of the bulk material. Two important classes of highly crosslinked thermosets are those based on epoxies and those based on acrylates. Epoxy resins are typically cured in the presence of amines or anhydrides in a step growth manner. Acrylate monomers, on the other hand, undergo radical chain growth polymerization. Fig. 4 illustrates schematically how networks develop from both of these monomer systems. While the epoxy-amine system evolves in a fairly regular manner, the acrylate system will polymerize and crosslink inhomogenously leading to a highly irregular network. As the modes of polymerization are fundamentally different, they are thus treated separately. First we would like to consider typical monomers and hardeners and their impact on the polymer architecture. | ||
| Fig. 4 Network formation via step-growth polymerization of epoxy thermosets (above) and radical chain growth of acrylates (below). | ||
Epoxy resins
Epoxy monomers may with appropriate additives be cured by heat or with light. Thermally cured epoxies (also commonly referred to as epoxy thermosets) are by far more commonly used and due to their industrial importance are the subject of hundreds of papers and patents concerning methods to improve polymer toughness.16 The most popular family of monomers are the digylcidyl ether derivatives of bisphenol A (DGEBA in Fig. 5) with varying molecular weight. Other epoxy monomers of commercial use include epoxides of phenol-formaldehyde Novolac resins, hydrogenated DGEBA (H-DGEBA) and glycidyl imides including triglycidyl isocyanurate (TGIC). The viscosity of resins consisting of these structural monomers can be reduced with reactive diluents including alkyl glycidyl ethers (AGE) and epoxidized plant oils such as soy bean oil (ESBO). | ||
| Fig. 5 Epoxy structural monomers and reactive diluents for thermoset resins. | ||
With DGEBA based resins, the distance between crosslinks will be strongly dependent on the average value of n (Fig. 5) and on the concentration and degree of substitution of the curing agent. Amines are most commonly used as curing agents in two-component systems (Fig. 6), where primary aliphatic amines are low in viscosity and have the highest reactivity. To reduce odour and premature gelation in such systems, primary amines such as diethylenetriamine (DETA) can be reacted with ethylene oxide and water to give aliphatic secondary amines endcapped with hydroxyethyl moieties. Aromatic amines such as m-phenylenediamine (MPD) are commonly used as curing agents due to the good thermal properties of the cured polymer but may be substituted with less toxic cycloaliphatic amines (such as PACM) in applications where the extra cost can be justified. Polyether amines (PEA) are claimed to reduce resin viscosity better than traditional polyalkylamines and to improve cured material toughness.17 In comparison to two-component systems, one component epoxy resins are based on less reactive curing agents such as ketamines, which form amines when exposed to air moisture.18
 | ||
| Fig. 6 Commonly used amine curing agents for epoxy resins. | ||
In thermosets, a primary amine can react with two epoxy groups (Scheme 1). When both the amine and epoxy monomer are difunctional, a crosslinked network will steadily arise with numerous hydroxy groups. These hydroxy groups can also react at elevated temperature with epoxy rings and will do so if the amine concentration is kept low.19
 | ||
| Scheme 1 Crosslinking reaction of epoxides with primary amine. | ||
While amines are by far the most commonly used curing agents for epoxy resins, acid anhydrides are the second most utilized class of curing agents and deserve some recognition. Commonly used acid anhydrides for epoxy curing include phthalic anhydride (PA) and methyl tetrahydrophthalic anhydride (MTHPA), the latter of which is used in filament winding composites. Epoxy-acid anhydride resins are much more stable than primary amine based resins and require high temperatures (>200 °C) to crosslink.20 The mechanism of crosslinking is proposed to commence via reaction of the anhydride with secondary alcohols of the epoxy monomer (Scheme 2). The resultant carboxylic acid will then open the epoxy and form new secondary alcohol to propagate the reaction.
 | ||
| Scheme 2 Crosslinking of epoxy thermosets with acid anhydrides.20 | ||
Lewis bases, including tertiary amines and imidazoles, are also used to cure epoxy resins. They can be used alone, where they function as nucleophilic catalytic curing agents for epoxy homopolymerization, or alternately as co-curing agents with primary amines and polyamides and as catalysts for anhydrides. The preferred reaction mechanism of tertiary amines with epoxies will depend on the presence or absence of proton donating moieties.21 In the absence of protons, the tertiary amine will act as a nucleophile and ring open the epoxide to form a zwitterion (with the positive charge on the nitrogen countering a hydroxyl anion). This hydroxyl anion can then ring open another epoxide to form a new anion and thus propagate polymerization. In the presence of weak proton donators such as alcohols, chain transfer will occur from the zwitterion. In the presence of stronger proton donators (phenols and thiols), a proton is donated already to the amine, and the anion (phenyl or thiyl) serves as the nucleophile to open the epoxide ring. Commonly used tertiary amines include 2-dimethylaminomethylphenol (DMAMP) and 2,4,6-tris(dimethylaminomethyl)phenol (TDMAMP), which both contain phenolic hydroxyl groups and can be used as catalysts and co-curing agents for room temperature curing of epoxies.20
Photo-curing epoxy resins
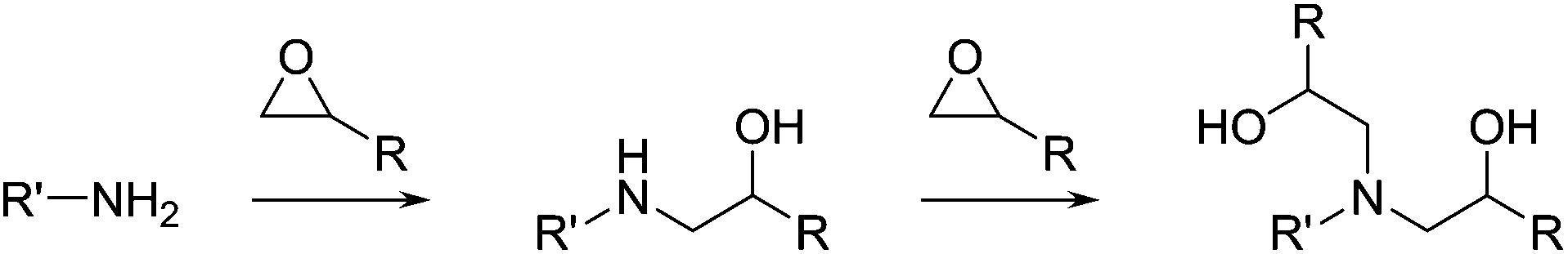
Epoxy monomers can also be cured with light, where a photolabile cationic initiator (typically an aryl sulfonium or iodonium salt)22 is used instead of the traditional amine or anhydride coreactant. Polyols are widely used to improve the toughness of photo-curable DGEBA based resins23,24 and in resins based on more flexible cycloaliphatic epoxides such as 3,4-epoxycyclohexyl-methyl-3,4-epoxycyclohexane carboxylate (ECC in Fig. 7).25 The differences between thermal and photo based epoxy resins are actually rather significant. With photo-curable epoxy resins, the photoacid generator or PAG (shown in Fig. 7 as an aryl iodonium salt) decomposes to give reactive intermediates which can abstract hydrogen atoms from solvent or monomer to give a Bronsted-acid. This acid then propagates the cationic ring opening polymerization.26 In addition to epoxide monomers, oxetane monomers (i.e. DSO in Fig. 7) may also be polymerized with PAGs. Oxetanes are commonly used in commercial additive manufacturing resins since they react fast and give polymers with low shrinkage and reduced moisture uptake relative to analogous polymers based on epoxies.27 | ||
| Fig. 7 Monomers, initiators and fundamental reactions of photo-curable epoxy resins. | ||
Although less commonly used than PAGs, photobase generators (PBGs) can also be used to cure epoxides.28–31 An early class of PBGs was based on [(benzoinyl)oxy]carbonyl protected amines, which release the free amine when exposed to UV irradiation.32 The quantum yield of these early PBGs was often low, leading Arimitsu et al. to study quartenary ammonium salts of ketoprofen and of xanthone acetic acid.33 Curing of aliphatic epoxides was conducted with a thiol coreagent, due to sterical hindrance and thus poor nucleophilicity of the generated tertiary amine. Allonas et al. used quarternary ammonium salts of phenylglyoxylic acid as PBG in combination with thiols to crosslink DGEBA resins. pKa of the ammonium counterion was varied from 8.2 (DABCO) to 13.6 (tetramethylguanidine) which tended to increase the rate of cure as witnessed by disappearance of the epoxy signal in RTIR.34 Addressing the issue of gas formation from decarboxylation of this class of PBG, Arimitsu et al. have also investigated ortho-hydroxy derivatives of cinammic acid.35 Network formation with PBGs will depend on the presence of a coreactant such as a thiol but should be primarily step-growth as is the case with the traditional epoxy/amine thermoset (Fig. 4). Depending on stoichiometry and on the presence of latent moisture, a degree of anionic ring opening polymerization can also be expected.
Acrylates
Typical photoresins consist of a combination of structural monomers, photoinitiator, stabilizers and other additives such as dye or filler all dissolved or dispersed within a reactive diluent.36,37 Acrylate-based monomers undergo radical chain growth polymerization even at room temperature at a rate much faster than that of epoxy polymerization and are thus more readily applied in photo-curable coatings.4,38 A typical acrylate-based resin will consist of oligomeric polyester, polyurethane or DGEBA based diacrylates dissolved in a low viscosity reactive diluent such as dipropylene glycol diacrylate (DPGDA), trimethyloylpropane triacrylate (TMPTA) or pentaerythritol tetraacrylate (PETA).39 Polymerization is triggered by a photoinitiator (either Norrish Type I or Type II).40 Type I initiators are single molecules which decompose into radical fragments when exposed to light of an appropriate wavelength. Benzoin esters (such as that portrayed in Scheme 3) are commonly used. Benzyl acyl phosphine oxides, which can absorb above 400 nm, have become increasingly popular in the last twenty years. Type II photoinitiation systems consist of a light absorber or sensitizer (i.e. benzophenone as shown in the following figure) along with a coinitiator, which donates a hydrogen atom to the sensitizer and in the process provides the initiating radical. Tertiary amines are the most commonly used coinitiators. | ||
| Scheme 3 Photoinduced radical generation, initiation and propagation. | ||
Type I initiators are faster and can be used at a low concentration since both radical fragments are capable of initiating polymerization. Type II systems are still very common since they are typically less expensive and can be less sensitive to oxygen. Oxygen inhibition is a serious problem with acrylate polymerizations,41,42 and additives such as tertiary amines (such as the Type II coinitiator depicted in the figure above) can be used to at least partially mitigate this.43 The benefit of using multiple photoinitiators is exploited with broad wavelength light sources, where the lower wavelength absorbing initiator provides good surface cure and the longer wavelength initiator improves through cure.44
Methacrylates are less reactive than acrylates but are also much less toxic and are thus preferred in photo-curable dental resins.45 Di-, tri- and tetramethacrylate monomers are formed from the same alcohols used to produce acrylates (i.e. pentaerythritol) using methacrylic acid instead of acrylic acid as feed stock. Example monomers are indicated in Fig. 8. Due to high modulus and low shrinkage, the dimethacrylate of DGEBA (Bis GMA) is readily used. Dental restorative resins also contain fairly high loadings of inorganic fillers (typically 70–80 wt%), which has great bearing on resultant optical and mechanical properties.46
 | ||
| Fig. 8 Common acrylate (R = H) and methacrylate (R = CH3) reactive diluents. | ||
Typical photo-curable acrylate resins consist mainly of multifunctional monomers (i.e. diacrylates and triacrylates) where termination reactions are mobility restricted and thus autoacceleration is observed in the early stages of polymerization.47 Usually, the kinetic chain length in early stages of polymerization is rather high, which contributes greatly to the formation of inhomogeneous polymer networks. Reduced termination causes an increased population of propagating radical chains, which are principally tethered to the polymer network. As the polymerization progresses, the majority of acrylate groups are tethered to the network making propagation diffusion controlled and autodeceleration is observed (Fig. 4).48 Ultimately due to occlusion of both radicals and unreacted acrylate moieties, the final double bond conversion (DBC) for a “fully cured” acrylate system will always be some fraction of 100%. Formation of macrocycles and polymer backbiting are yet other phenomenon which will have bearing on mechanical properties and are both found to be more prevalent with flexible as opposed to rigid monomers.49
The addition of chain transfer agents to a radical curing system (e.g., thiols, addition fragmentation chain transfer AFCT agents) can change the uncontrolled radical chain growth mechanism to a step-growth type polymerization process, enabling good regulation over the final polymer architecture by tuning crosslinking density, average kinetic chain length, and distribution of crosslinks alongside the polymer backbone. Hybrid systems consisting of both epoxy and acrylate monomers are also quite common (i.e., interpenetrating networks; IPNs) and will be discussed in the IPN section.
Additives for toughening polymer networks
While photo-curing of thin coatings has been in industrial use since the early 1950s, crosslinking of polymers with heat has been known much longer. For a variety of reasons, organic and inorganic additives have essentially always been used to some extent in these polymer networks. Already in 1907 Baekeland recognized that phenol-formaldehyde resins were much tougher when they were compounded with wood fibres, rubber, or mineral powders.50 Since then the development of thermosets has run hand in hand with the development of polymer composites. Acrylate-based coatings emerged in the early 1930s and by the mid-1940s lightly crosslinked acrylate polymers were being proposed as synthetic rubbers.51 Bisphenol-A based epoxy resins were first developed in the 1940s.52 In the mid-1960s McGarry and coworkers began examining methods to modify epoxy and polyester resins in order to improve the mechanical performance of the materials.53 The mechanical properties of epoxy and acrylate based networks have improved tremendously over the last 50 years thanks to proper use of additives. Some of the more important classes of additives for thermosets are considered here.Inorganic nanoparticles
While polymer composites based on macroscopic and microscopic inorganic particles have been in use for more than 100 years, the systematic use of inorganic nanoparticles in polymers is relatively new. In the early 1990s, Usuki and Kojima54,55 reported a tremendous increase in the tensile strength of nylon 6 (by a factor of 5) with the addition of montmorillonite. It has been recognized since then that nanocomposites can show sometimes significantly altered electrical, optical or mechanical properties from the parent polymer. While this can also be said with most “non-nano” composites, change in properties can often be accomplished with a much lower loading of the inorganic component. Important for photo-curable coatings, particle size is much less than typical layer thicknesses and in some cases below the diffraction limit of the utilized light.Although not fully understood, it is assumed when there are strong interactions between the nano-sized filler and the matrix that the polymer chains adjacent to the nanoparticles will behave differently from those surrounded by other polymer chains alone. Due to the very high surface area provided by nanoparticles, the portion of polymer in the vicinity of the nanoparticle surface can make up the majority of the matrix polymer already at moderate filler loadings and hence, novel properties can arise.56 If adhesion between the filler particle and the matrix is high, debonding is restricted when stress is applied which results in increased modulus and strength (Fig. 9).
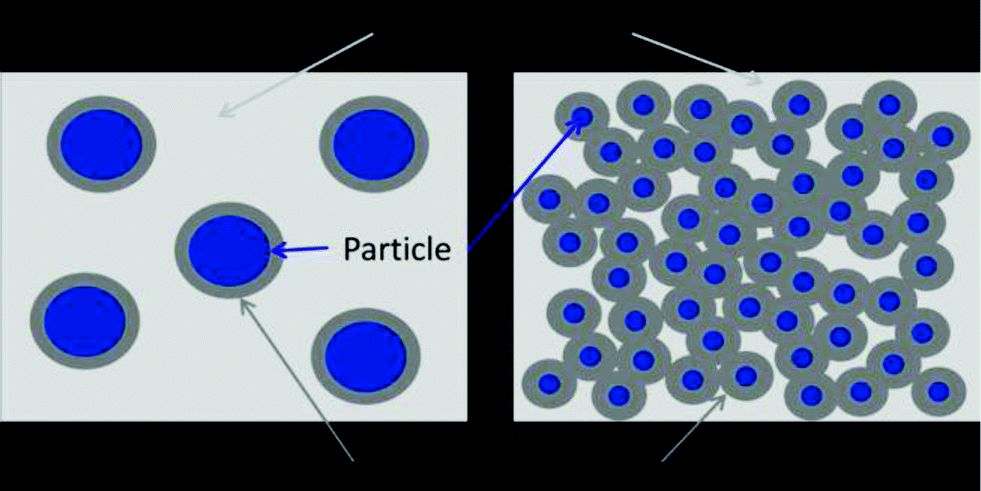 | ||
| Fig. 9 Schematic comparison of interfacial polymer in conventional composites with micron-sized filler (left) and in nanocomposites (right). | ||
Another explanation for the sometimes unexpected thermomechanical properties of nanocomposites is chain confinement.57,58 In this case when little or no interfacial interaction between filler and matrix is present but the interparticle distance is small enough, the polymer chains between two adjacent nanoparticles will be confined in their motion and thus start to act like they would in a thin film.59 Numerous studies have demonstrated that particle size as well as the morphology and the filler–matrix interaction play crucial roles in this process, but elucidation of the toughening mechanism has yet to be fully realized.57,60
Some of the better studied inorganic nanoparticle polymer additives include silica, alumina and CaCO3. Zuiderduin et al. found that sub-micron CaCO3 particles could increase impact strength of polypropylene from 2 to 50 kJ m−2 at high loadings.61 The shift in the brittle-ductile transition temperature was found to shift by up to 50 °C with dependence on both particle size and the presence of surfactant.62,63 Similar improvements have also been found with polyethylene.
Although the toughening effects of inorganic nanoparticles in thermosetting resins is not as remarkable as the results obtained for thermoplastics, improvements in fracture toughness have at least been made.64,65 For example, a number of studies have been conducted using inorganic nanoparticles to improve the toughness of epoxy-based thermosets. The usual base material in these works is DGEBA or a related diepoxide.66 A popular choice of inorganic filler for epoxy resins is surface modified silica nanoparticles, also known as fumed silica. Fumed silica typically has a mean diameter of approximately 20 nm and in some cases has a narrow particle-size distribution. In one study, epoxy formulations cured using different agents (i.e. methylhexahydrophthalic acid anhydride (AD600), polyether-amine (J230) and 4,4′-diaminodiphenyl sulfone (DDS)) were tested with different particle loading. Addition of up to 20 wt% of silica nanoparticles was found to increase the Young's modulus by 50% while maintaining the original Tg of the material. The fracture toughness was increased by a factor between 1.7 and 2.4. Sangermano et al. found that the addition of 4 vol% 150 nm Al2O3 NPs improved both toughness and scratch resistance of photocured cycloaliphatic epoxies.67
Other thermosetting materials successfully toughened with fumed silica include tetraglycidyl-4,4′-diaminodiphenylmethane65 (fracture toughness KIC increased by a factor of 2.2), 3,4-epoxycyclohexyl-methyl-3,4-epoxycyclohexane carboxylate (ECC)68 and diglycidyl ether of bisphenol AF (DGEBF).69 The results of the latter two works regarding toughness improvements are summarized in Fig. 10. Similar to Zhang, Mahrholz et al. also reported an increase by a factor of 1.5 in the DGEBA-SNP system.70
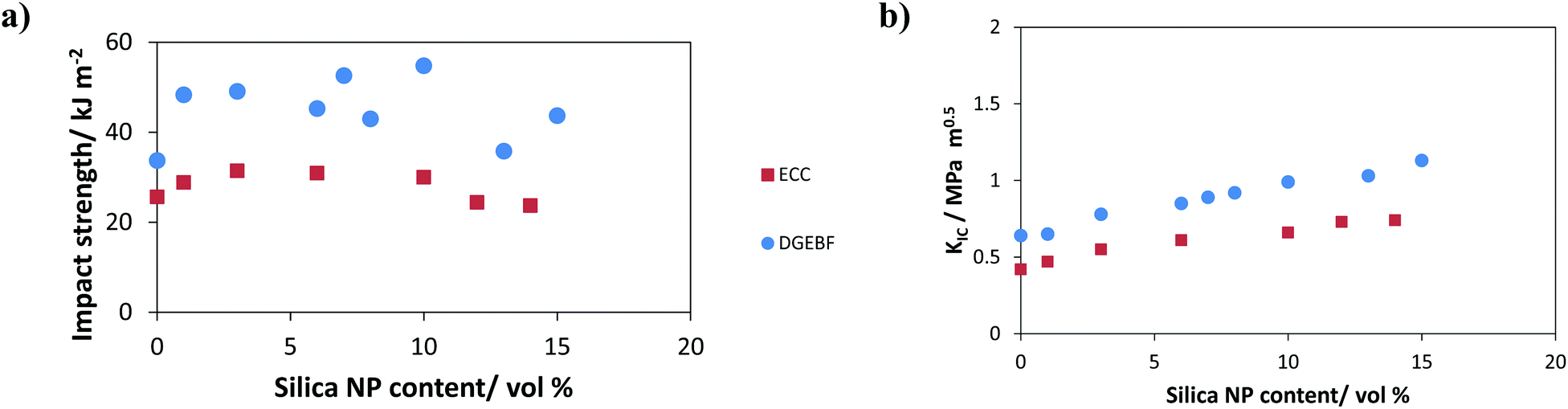 | ||
| Fig. 10 Toughening effects of SNPs on epoxy thermosets. | ||
Rubber
Natural and synthetic rubbers have long been used to improve the mechanical properties (and particularly the impact strength) of a variety of organic polymers and inorganic materials.71 An early example is provided by Aylsworth who in 1914 added natural rubber and sulphur to the polymerization of phenol and formaldehyde producing a tougher plastic that was used for early phonographic discs (before vinyl).72 In the late 1940s Dow introduced high impact strength polystyrene (HIPS) formed by adding polybutadiene to styrene monomer and stirring vigorously during polymerization.73 Not long afterwards acrylonitrile was added to the mixture which provides ABS plastic. Impact strength of HIPS is 2 to 4 times higher than polystyrene and ABS can be more than 8 times as strong (Izod impact >400 J m−1).74 The morphology of ABS and HIPS are not homogenous but consist of micron sized butadiene or styrene-butadiene rubber (SBR) particles dispersed within a continuous phase (polystyrene or polystyrene–acrylonitrile).Not long after this early work with thermoplastics, rubber began to be tested as a toughening additive for epoxy-based thermosets. Epoxy resins, and more specifically those based on DGEBA, find widespread application and one principal reason for this is their high modulus. This modulus arises principally from the rigid and highly crosslinked architecture of the polymer, which is also responsible for its major drawback: poor impact strength.16 Rubber additives thus tend to be used sparingly since they should increase toughness and at the same time not lower the modulus or effect the Tg. Recognizing proper phase separation as an integral component to rubber toughening in epoxy resins, two strategies have developed both to control particle size and to regulate the rubber–matrix interface. The first strategy is based on low molecular weight “liquid rubbers” and the second is based on preformed core–shell particles.75
Liquid rubber
The term “liquid rubbers” is commonly used to refer to elastomeric polymers which have a sufficiently low molecular weight in order to maintain low viscosities to enable homogeneous mixing with the epoxy resin. The most commonly used liquid rubbers are based on polybutadiene or acrylonitrile–polybutadiene copolymers with varying acrylonitrile-content. To improve interfacial adhesion to the polymer matrix, liquid rubbers are usually endcapped with functional groups capable of reacting with it. The most common modification is a carboxyl-endgroup giving carboxyl-terminated butadiene (CTB) or carboxyl-terminated butadiene-acrylonitrile (CTBN), but amino- (ATB/ATBN), epoxy- (ETB/ETBN) or vinyl-modified (VTB/VTBN) compounds are commercially available as well (Fig. 11). The first attempt to utilize this approach in order to improve the toughness of brittle epoxies was made by Sultan and McGarry using butadiene–acrylonitrile rubber.66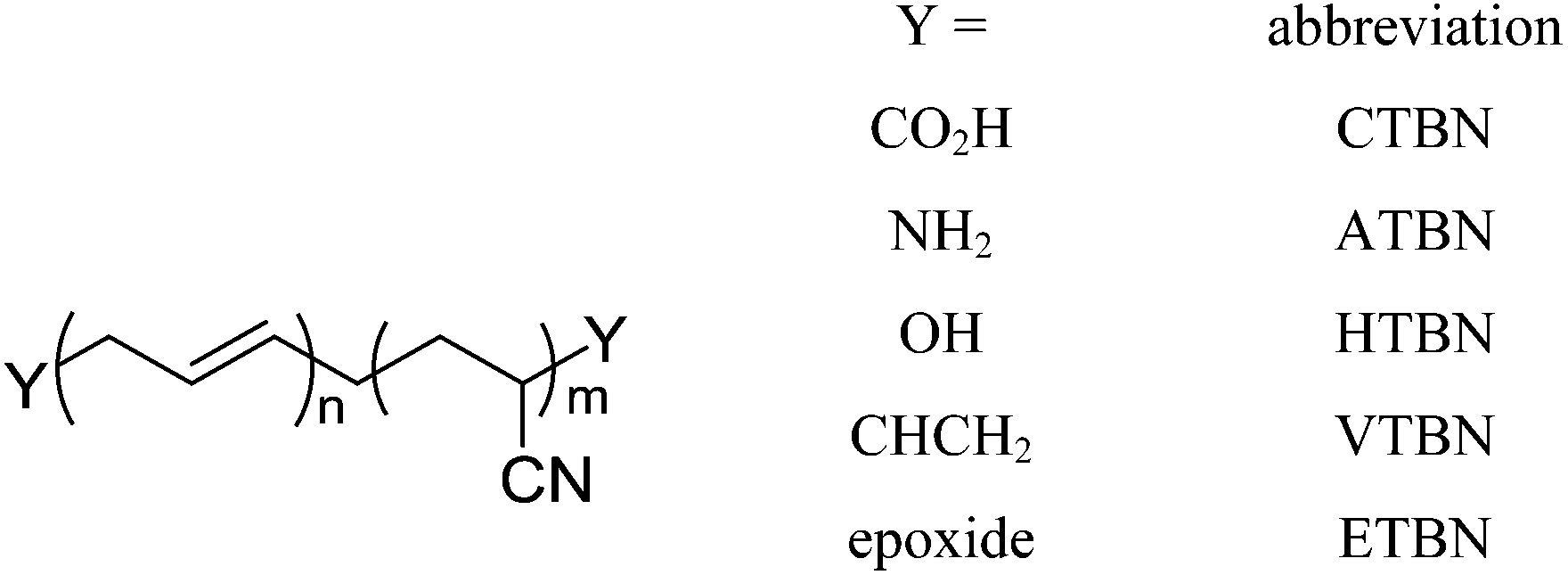 | ||
| Fig. 11 End-capped liquid rubbers for toughened thermoset resins. | ||
Russell and Chartoff showed that the fracture toughness of DGEBA-based thermosets could be improved by a factor of 2 by adding CTBN. They also showed that the volume fraction of the phase separated domains were up to twice as large as the amount of utilized liquid rubber.76 It was then shown that the modulus of the phase separated domains had a greater influence on the toughness of the material than the volume fraction. Bagheri et al. also tested CTBN in DGEBA and reported a size-dependence of the phase-separated domains with respect to the rubber loading. The higher the rubber content, the larger the average domain size (from 0.3 μm for 1 wt% to 0.7 μm for 15 μm). Resultantly the fracture toughness could be increased by a factor of 2.3 at 5 wt% CTBN.77 Kunz et al. also tested the CTBN/DGEBA system and showed a 2.5-fold increase of the fracture toughness at 5 wt%. The same increase was obtained at higher rubber loadings while the modulus was linearly decreasing with increasing rubber content (from 2.96 to 2.25 GPa for 15 wt% CTBN). ATBN-rubber additives were also tested and found to provide almost identical results.78 Chikhi et al. obtained different results when using the same ATBN-rubber in DGEBA-resin, when they showed a fracture toughness improvement by a factor of 1.5 with a distinct maximum at 12.5 wt% additive. The impact strength however, could be enhanced by up to 3 times its original value at the same rubber concentration.79 Thomas et al. employed hydroxyl terminated polybutadiene (HTPB) to toughen DGEBA. Addition of 10 wt% of HTPB caused a 1.5-fold increase in impact strength from 9.2 to 13.6 J m−2 while fracture toughness was 5 times higher than that of the neat epoxy-resin.80 Interesting results were published by Kong et al., who synthesized a liquid rubber possessing pendant epoxy groups. This modifier increased the impact strength by a factor of 1.75 at a rubber loading of 5 wt% with only a moderate decrease of the modulus and heat resistance of the toughened material.81
Although the application of liquid rubbers has been predominantly studied for epoxy systems, there are a few publications dealing with the usage of these modifiers for other thermosetting materials including unsaturated polyester resins and vinyl ester resins.82 It should be mentioned that the term vinyl ester in this context actually refers to methacrylic monomers, usually the products of reacting DGEBA with methacrylic acid to yield the respective dimethacrylate. Due to its high viscosity this compound is usually cured with the addition of approximately 50 wt% of styrene to simplify handling. Robinette et al. has tested the usage of ETBN and VTBN while Auad et al. compared VTBN and CTBN.83,84 The impact on modulus and fracture toughness (quantified as critical stress intensity factor KIC) is depicted in Fig. 12.
 | ||
| Fig. 12 Effect of LRs on Young's modulus (a) and fracture toughness (b) of vinyl ester resins. | ||
It can be seen that an improvement in fracture toughness was achieved although at the expense of the stiffness of the material. Also, distinct differences in performance are noted for the different end groups of the tested liquid rubbers. CTBN exhibits a distinct maximum in toughness at around 5 wt% loading while the copolymerizable VTBN has to be incorporated in larger amounts. The difficulties in toughening of these systems can be attributed to the differences in network formation. While traditional epoxy systems cure via a step growth mechanism, vinyl esters polymerize by a cationic or free radical chain growth mechanism. Celikbilek et al. evaluated the usage of endcapped polydimethylsiloxane (PDMS) in DGEBA resins but were not able to observe any improvement in terms of impact strength or fracture toughness.85
Preformed particles
Not long after the first experiments with liquid rubbers, a related technique was introduced, based not on liquid oligomers but on solid, preformed particles of an elastomer.86 These particles are often termed “core/shell-particles” (CSPs) since the actual toughening part – the elastomeric core – is usually coated with a thin layer of a glassy shell. In the absence of this shell, the elastomeric particles are not stable and stick together. The shell thus prevents agglomeration and allows the particles to hold their size and shape well above the Tg of the rubbery core.87 While standard CSPs consist of an elastomeric core and a glassy shell, there are other variations mentioned in the literature. One example is a multi-layer CSP with a glassy core as well as a glassy shell with a rubbery interlayer.88 The toughening mechanism is similar to conventional CSPs.Marouf et al. used CSPs consisting of a styrene/butadiene core with a PMMA-shell to improve the mechanical properties of a DGEBA-based resin. They reported an increase of the fracture toughness by a factor of 2.3 with the addition of 5 wt% CSP.89 Similar results for this system have been published by other authors.75,77,90,91Fig. 13 provides a comparison of a selection of this data. Generally speaking, fracture toughness could be improved significantly, and the effect was noticeable already at comparably low loadings.
 | ||
| Fig. 13 Effect of CSPs in DGEBA epoxy resins on Young's modulus (a) and fracture toughness (b). Note: Bagheri et al. provide values for toughness of BS/PMMA CSPs but do not give values for modulus.77 | ||
Jingqiang et al. synthesized CSPs based on a soft polyurethane-core and were able to increase the impact toughness of the DGEBA resin by a factor larger than 3 (from 2.25 to 7.6 kJ mol−1 with 8 wt% filler).92 Subramaniyan et al. used commercial CSPs in a vinyl ester/styrene matrix but they were not able to show toughness improvements at loadings of 5 wt%.93 Lin and Shieh carried out a thorough study investigating optimum particle size and modifier loading as well as the influence of copolymerizable groups in the shell. The best thermomechanical properties were obtained with average CSP-diameters around 300 nm; the optimum loading for high fracture toughness was around 20 wt%. The presence of reactive groups in the shell was also found to have a positive effect on the toughness of the composite.94 Naguib et al. have more recently confirmed this finding by adding up to 15 wt% PBA/PMMA-PGMA CSPs to cycloaliphatic epoxy resins.95 Noting reaction of GMA epoxy units with the matrix, impact strength was improved at all tested concentrations and Tg was not affected.
In addition to the previously discussed CSPs, another type of preformed rubber particle has recently been commercialized. Similar to silica nanoparticles, these rubber particles are prepared in the resin monomer. Since the particles are suspended in the resin as a master batch, there is no need for drying and the problem of agglomeration is avoided. He et al. and Le et al. both employed this type of modifier in DGEBA. Since the type of rubber and the particle sizes were not the same in these 2 publications, the observed behaviour was also slightly different.96,97 He et al. reports a 4-fold increase of the fracture toughness with 500 nm particles at 12.5 wt% loading while Le et al. found a 3-fold increase for the 55 nm particles at 5 wt%. In addition to conventional dispersions of rubber-particles, preformed particles based on polydimethylsiloxane (PDMS) are also commercially available. Due to their production process these are, however, only available as master batches in a liquid monomer. PDMS has other advantages for this application since it provides an extremely low Tg and is very hydrophobic. Rey et al. used larger 4 μm particles and were able to achieve a moderate increase of the fracture toughness of DGEBA by a factor of 1.5; however, the Young's modulus was also significantly reduced.98 Bittmann and Ehrenstein used smaller PDMS-particles with an average diameter around 0.5 μm in ECC and reported a slight improvement in toughness. The influence on the stiffness of the material was not disclosed.99
Self-assembly of block copolymers
Jordan et al. stated in their review on trends in nanocomposite technology that for most toughening agents, the efficiency of the filler in the composite is inversely proportional to its size but directly proportional to its surface/volume ratio.59 This concept has been well exploited with the inorganic nanoparticles mentioned earlier. By comparison, the application of rubber nanoparticles did not receive its due notice until more recently. The preparation of rubber nanoparticles as toughening agents is rather demanding since conventional techniques require either the subsequent addition of a rigid shell or the preparation of the particles within the resin to prohibit agglomeration of the particles. A very elegant method to circumvent these restrictions is provided by the self-assembly of block copolymers (BCs). In 1997 Hillmyer et al. were the first to report the formation of nanostructured aggregates (micelles) upon the addition of BCs into epoxy resin.100 After this, numerous publications investigating the behaviour of amphiphilic BCs in thermosetting matrices with emphasis on their effect on the mechanical properties of epoxy resins have been released. Ruiz-Pérez et al. give a compact summary of some of the endeavours to toughen epoxy resin with the aid of BCs.101 They suggest a differentiation between three categories of BCs:• Non-reactive BCs that self-assemble in the uncured state due to the presence of an epoxy-miscible and an epoxy-immiscible block. This order in the uncured state is subsequently fixed during the polymerization.100,102–105
• Non-reactive BCs where both blocks are initially miscible and one of them undergoes microphase separation during the crosslinking reaction of the matrix. In this case, the morphology is generated during the curing process.106–109
• BCs where the resinophilic block contains reactive groups enabling copolymerization with the surrounding matrix. For this group, the other block is immiscible and thus the final morphology is established prior to cure.
Diblock- or triblock copolymers of poly(ethylene oxide) (PEO) and poly(propylene oxide) (PPO) (i.e. PEO-b-PPO or PEO-b-PPO-b-PEO) have been investigated by multiple groups since they are commercially available and applied as non-ionic surfactant in detergents.110,111 These publications primarily focused on the microphase morphologies and polymerization kinetics and not so much on the mechanical properties of the resulting material. Since PEO and PPO both are relatively hydrophilic polymers, phase separation does not occur until the polymerization stage. This results in microphase-separation similar to that exhibited by the PS-PEO block copolymer system (Fig. 14) where in both cases the resinophilic PEO block maximizes contact with the DGEBA-matrix. Other examples for reaction induced phase separation include poly(ε-caprolactone)-b-polybutadiene-b-poly(ε-caprolactone) (PCL-PB-PCL) dispersed in DGEBA.109,112,113
 | ||
| Fig. 14 Reaction-induced microphase separation in BC-toughened thermosets. Reprinted with permission from Macromolecules.109 Copyright 2006 American Chemical Society. | ||
BCs which undergo self-assembly in the uncured state typically consist of blocks of polymers with a more pronounced difference in polarity. Examples of such block copolymers include PEO-b-polyethylene (PE) or PEO-b-poly(ethylene-alt-propylene) (PEP). PEO-PEP copolymers in DGEBA have been extensively investigated in the work by Dean et al. and Liu et al.114,115 With this system, parameters such as block ratio, BC molecular weight and the crosslink density of the matrix are shown to have a strong impact on the self-assembled structures. Not too surprisingly, the morphology itself was found to be a crucial parameter contributing to bulk mechanical properties. An improvement in fracture toughness by a factor of almost 3 was reported by Liu,116 which is an exceptional result compared to others where toughness was enhanced only to values twice as large as that of the neat resin. With only 5 wt% of the PEO-PEP modifier, Young's modulus and heat deflection temperature remain almost unchanged. Ocando et al. used PS-PB with different degrees of epoxidized double bonds in the PB-block also in a DGEBA-based resin. They reported less significant improvements in toughness and also observed significant lowering of stiffness and heat resistance.102
Bates and coworkers also used diblock copolymers of PEO with poly(butylene oxide) (PBO)117 and poly(hexylene oxide) (PHO)118 in DGEBA. Since the contrast of their small angle X-ray apparatus was too low, no information on the behaviour of these BCs in the uncured state is given. Nevertheless the fracture toughness could be increased by a factor of 4 for PEO-PBO and by a factor of 6 for PEO-PHO where the block ratio used resulted in the formation of cylindrical structures. The Young's modulus was reduced by approximately 20% and, most interestingly, the Tg was significantly increased. Similar observations have been reported elsewhere;117 however, no explanation for this surprising behaviour is provided.
Another popular type of BC that has been employed for the toughening of thermosetting materials are compounds based on PDMS. PDMS is inherently incompatible with almost every standard resin system, which necessitates other additives or clever synthesis to avoid macrophase separation. Riffle et al. were the first to propose the preparation of linear block copolymers to fulfil this requirement.119 The behaviour of silicone-based BCs of this type in thermosetting matrices was then tested by various researchers.120,121 The evaluation of their potential as toughening agents for DGEBA was conducted by Könczöl and Mülhaupt while Bittmann and Ehrenstein used a similar silicone BC additive in a cycloaliphatic epoxy resin (ECC).122–124Fig. 15 shows the impact of the silicone BCs on fracture toughness (KIC) and glass transition temperature (Tg) of these two epoxy resin systems.
 | ||
| Fig. 15 Effect of CSPs in DGEBA epoxy resins on toughness (a) and glass transition temperature (b). | ||
Fig. 15 well illustrates how sensitive the application of BCs can be depending on the compatibility of the modifier and the matrix. For the system investigated by Könczöl, microphase separation of 20 nm silicone domains was observed leading to significant improvements in toughness without inadvertently affecting the thermal stability of the material.122 By comparison, Bittmann did not report the formation of any nanostructures.99 It was also noted though that the glass transition temperature decreased with higher BC-loadings while fracture toughness reached a maximum with 5% additive. This indicates that the modifier remains dissolved even after curing.
A very interesting feature of using BC additives is the possibility of simultaneously increasing toughness and heat resistance,125 a phenomenon which appears somewhat contradictory and is still not well understood. Toughness enhancement is dependent on both domain size and on morphology of the self-assembled BCs (i.e. micelles, vesicles or cylinders).126 Liu et al. found that cylindrical micelles of PEP-PEO provide improved toughness in DGEBA-based resins in comparison to resins with spherical micelles.116 Wu et al. synthesized block copolymers of partially sulfonated PS and poly(ethylene-co-butylene) and used these to modify DGEBA based resins. Fracture toughness was increased with the addition of the BCs and was found to reach a maximum where degree of sulfonation was equal to 5.8%.127 TEM show that the particles were flattened, but with higher degree of sulfonation the resinophilic block penetrates into the epoxy matrix making phase separation less apparent.
Special monomers and curing agents for tough epoxy polymers
 | ||
| Fig. 16 Alternative epoxy monomers reported to improve polymer toughness. | ||
 | ||
| Fig. 17 Amine and anhydride curing agents claimed to give tougher epoxy polymers. | ||
In photo-curable DGEBA based resins, aliphatic polyols are commonly used. In combination with cationic initiators, aliphatic polyols are found to react significantly faster than phenolic additives.136 Lützen et al. examined polycaprolactone (PCL) diols of different molecular weights in copolymerization with ECC.137 In comparison to the neat epoxy, the ECC-PCL copolymers have greater fracture toughness and lowers Tg values. The shift in both values is dependent on wt% of PCL and on molecular weight, where lower molecular weight PCL has a more dramatic effect.
In comparison to linear polyols, dendritic and hyperbranched polyols are found also to improve toughness without significant reduction in modulus.138 Manjula Dhevi et al. used hexamethylene di-isocyanate to couple hyperbranched polyesters into DGEBA-based resins.139 The resultant IPN polymers were tougher than the neat DGEBA polymer toughened with a comparable weight of linear polyurethane diol. Thermal stability and Tg were however lowered. Foix et al. have studied hydroxy terminal hyperbranched polyesters as additives for epoxy resins and seen an accelerating effect during cure followed by phase separation.140,141 Improvement in mechanical properties was however most promising when the hyperbranched additives are epoxy terminal.131
Considerations for radical chain growth
Interestingly, several attempts by the authors of this review using different particle toughening strategies suitable for epoxy thermoset resins have not been forthcoming when applied to acrylate networks.142 Differences in the manner in which the epoxy and acrylate networks form provide clarification.The absence of a significant change of polarity during network formation in radical photopolymerization seriously hinders the efficient use of polymerization-induced phase separation, which has been extensively used in thermosetting epoxy systems.21,32,35,143 This means a system comprising of two or more phases must already be present in the uncured polymerization mixture.
Liquid rubbers above a certain concentration improve the toughness of the rigid photopolymers both under low and high strain rates. However, especially at higher contents of LR, one has to sacrifice the stiffness of the material to a certain extent. Moreover, high concentrations of LRs tend to significantly increase the viscosity of the respective formulations, which can become a limiting factor for many applications since highly viscous resins often cannot be processed properly.
Block copolymers also exhibit the potential to modify the mechanical properties of photopolymers (in particular flexibility and impact toughness). While improvements in terms of toughness can be made, this is generally accompanied by a reduction in polymer modulus.142 This indicates that the BCs at least partially remain dissolved in the matrix and in this manner act as plasticizers similar to LRs.
For CSPs to act as toughening agents in thermosetting materials, good adhesion between the dispersed particles and the polymer matrix is required. However, due to high shrinkage, which is typically associated with the photopolymerization of acrylates, good adhesion is difficult to obtain with conventional CSPs. Sandmann et al. used particles with EGDMA/PBA cores and PMMA shells to improve the mechanical properties of urethane dimethacrylate/TEGDMA based dental composites.144 Best results were found with a core–shell ratio of 30/70, which showed a fracture toughness of 1.21 MPa m1/2 (an improvement of 65%), a flexural modulus of 1.90 GPa (an increase of 18%) and a flexural strength of 79 MPa (also an increase of 18%). Success in this case is likely due to good compatibility and thus adhesion of the PMMA shell with the TEGDMA matrix and reduced polymerization shrinkage of the urethane based monomer.
Silica nanoparticles on their own provide no significant improvement in the toughening of the tested acrylate-based polymer networks.142 However, they have the potential to improve other mechanical properties such as stiffness or hardness without affecting any of the other material properties. This behaviour would make SNPs an interesting candidate for a “co-modifier” since they obviously have the ability to counterbalance reduced stiffness caused by other additives.
In general the proven concepts for toughening of epoxy thermosets could not be directly transferred to photopolymerized acrylate-networks. Consequently, in order to fabricate tough photopolymers, the simple use of an additive seems not to be sufficient. Instead, alternative monomers or monomer systems may be required to change the underlying network structure. An optimized architecture of the photopolymer can not only exhibit a better toughness but can also support the ‘toughenability’ of the resulting material, which would allow conventional additives to have a more pronounced effect.16,32,134
The photoinduced polymerization of multi-functional acrylates is a complex process where the ultimate polymer architecture will be dependent on monomer composition, photoinitiator concentration and curing conditions. Generally Type I photoinitiators have higher quantum yields than Type II systems and will thus be used in a lower concentration.30 Kinetic chain length is proportional to the rate of propagation and inversely proportional to rates of initiation and termination.145 Values calculated for photo-cured acrylates at low conversions were from 2000 to 10000 monomer units per radical depending on whether the reaction was performed in air or in nitrogen. Formation of inactive peroxy radicals is the principal culprit for the lower value.42 In a multifunctional acrylate system, the average kinetic chain length will decrease dramatically as gelation occurs and ultimately at high conversions approaches zero. The implications on polymer architecture are that the distance between crosslinks will be highly irregular. Resulting inhomogeneously crosslinked photopolymer networks tend to be very brittle and show low toughness.
Where photo-curing of even a single difunctional monomer leads to complex architecture, the situation does not get simpler with a more typical formulation consisting of 3 or 4 different monomers with different structures, molecular weights, and number of pendant photopolymerizable moieties. Touching on the first of these subjects, Jansen et al. found that acrylate monomers that form hydrogen bonds or that are more polar tend to polymerize at higher rates. This can be ascribed to monomer preorganization and possible cage effects with the propagating radical.146 Hydrogen bonding also has a strong effect on the mechanical properties of the crosslinked polymer. Torres-Filho and Neckers added different diacrylate monomers to a resin consisting of tri- and pentaacrylates and found that with butanediol and short PEG diacrylates, Young's modulus and tensile strength both tended to decrease.15 By comparison, addition of 10 wt% of a hydrogen bond forming polyurethane diacrylate increased both values. Polyurethane diacrylates are cited in a number of patents for improving mechanical properties.147,148 The polyurethane can vary in molecular weight and composition, where one popular class is based on “soft” polyether diols copolymerized with “hard” aromatic diisocyanates. Crystalline “hard” domains tend to phase separate from the “soft” domains which helps to improve toughness and in some cases provide materials with unusually long elongation at break.149
Photo-curing conditions and photoinitiator concentration also contribute to toughness.150 For practical reasons, it can be difficult to find tensile or impact testing results for incompletely cured polymer samples. Real time FTIR (RTIR) and photorheology, however, provide equally useful insight into the process of network formation.151 With the first technique, the integral of the acrylate C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2 stretch is followed and referenced to its starting value to provide double bond conversion (DBC).41 Generally the hardness of a material will increase with DBC, but is also heavily influenced by monomer structure. For example, TMPTA cured with a medium pressure Hg lamp for 60 seconds will have a DBC of approximately 60% and a Persoz hardness of 280 s while DPGDA under the same conditions will have a DBC of 90% and a Persoz hardness of only 120 s.152 Irradiance conditions are also important and particularly in the presence of oxygen. Decker et al. cured DGEBA diacrylate resins and found DBC after 10 seconds to be only 26% when cured with a 20 mW cm−2 source versus 50% with a 55 mW cm−2 source. For laminated samples, the difference is much less: 93% with 20 mW cm−2versus 95% with 55 mW cm−2 irradiance.153
CH2 stretch is followed and referenced to its starting value to provide double bond conversion (DBC).41 Generally the hardness of a material will increase with DBC, but is also heavily influenced by monomer structure. For example, TMPTA cured with a medium pressure Hg lamp for 60 seconds will have a DBC of approximately 60% and a Persoz hardness of 280 s while DPGDA under the same conditions will have a DBC of 90% and a Persoz hardness of only 120 s.152 Irradiance conditions are also important and particularly in the presence of oxygen. Decker et al. cured DGEBA diacrylate resins and found DBC after 10 seconds to be only 26% when cured with a 20 mW cm−2 source versus 50% with a 55 mW cm−2 source. For laminated samples, the difference is much less: 93% with 20 mW cm−2versus 95% with 55 mW cm−2 irradiance.153
While high intensity light and adequate concentration of photoinitiator are both needed to provide polymer hardness, too much of either of these can be detrimental. Using a resin consisting of MMA with TMPTA (Fig. 8), Cunico and Carvalho found that photoinitiator (PI) concentration had a tremendous effect on both modulus and tensile strength.154 Thus polymers cured for 20 minutes with 5 wt% PI (Omnirad 2500 from IGM) were on average 40% stiffer and half as strong as those cured with 2 wt% PI. Interestingly DBC for the polymer with 2 wt% PI was 90% after 60 seconds while the value was only 65% for the same polymer with 5 wt% PI.
Chain transfer
With recognition that rapid gelation (frequently as low as 20% double bond conversion) and high crosslinking density are prime factors contributing to the brittleness of photo-cured systems, coreactants that can regulate network formation have been investigated. Chain transfer agents, which have been used for many decades to regulate the molecular weight of linear polymers, began to be tested for regulating photo-curable resins as well.155 Amines are commonly used as coinitiators in Type II systems and to reduce the effects of oxygen inhibition.156 In both roles they act as hydrogen donors, which in the latter case is also an example of chain transfer. Thiol based chain transfer agents readily react with acrylates and other vinyl compounds and provide a versatile platform for regulating cure kinetics as well as mechanical properties of the final polymer.157A number of other methods for regulating a free radical polymerization are described in literature (e.g., reversible addition fragmentation chain transfer (RAFT)158 and atom-transfer radical polymerization (ATRP)159) but almost exclusively for linear polymers. Irreversible addition fragmentation chain transfer (AFCT)158 reagents also have the potential to regulate radical polymerization reactions. AFCT proceeds similarly to the polyreaction of thiol–ene/yne160–162 systems in a mixed chain growth/step growth-like mechanism. For photopolymerizable crosslinking systems, the thiol–ene and AFCT approaches seem to be more promising today, since RAFT and ATRP reagents tend to have strong UV/vis absorbance and more importantly have strong retardation.
A good measure for the ability of a chain transfer agent to regulate a radical photopolymerization is the chain transfer constant (Ctr = ktr/kp) which is described by the ratio of the reaction constant for chain transfer (ktr) to the reaction constant for monomer propagation (kp). However, this value is greatly influenced by a number of different factors such as monomer reactivity, functionality of the chain transfer agent, reaction temperature, radical initiation, solvent, and conversion.163
Thiol–ene/–yne chemistry
The history of photo-induced thiol–ene chemistry is as old as that of acrylates,164 but has flourished much less primarily due to problems with odour and storage stability.165 Thiols act as potent hydrogen donors in the presence of appropriate sensitization. A photoinitiator (PI) is not essential but allows the radical formation to proceed at a greater rate. The resultant thiyl radical reacts in an anti-Markovnikov manner with an unsaturated carbon–carbon bond (Scheme 4). | ||
| Scheme 4 Radical reactions of double bond in the presence of thiol chain transfer agent. | ||
The rate of addition is highest with electron rich double bonds (norbornenes and vinyl ethers) and much slower with styrene or (meth)acrylates (see Table 1).166 Reactivity with acrylates is intermediate which means that the resulting radical will undergo chain transfer with thiol (ktr) but at a rate significantly less than acrylate propagation (kp). In the presence of oxygen, chain transfer will however become more important since peroxy radicals react several orders of magnitude faster with thiols than with acrylate double bonds. Consequently thiol–ene chemistry is much less sensitive to oxygen inhibition, which is a major topic for the coatings industry.41
Scheme 4 presents chain transfer with a monofunctional thiol. The same cycle of reactions (addition to ene and hydrogen donation) will build polymer networks when the functionality of the thiol and the ene are both more than two. Polymers based on tri- or tetra-thiols with electron rich alkenes such as norbornenes or allyl monomers follow step-growth kinetics to give architectures different from those formed via radical chain growth. In this manner, gelation is delayed allowing a more uniform crosslink density and a higher final double bond conversion. Thiol–ene polymers tend to exhibit much sharper glass transitions at lower temperatures compared to acrylate networks. Thiol–ene polymers also tend to have lower hardness but better impact properties.169 The limited hardness is due in large part to the flexible thioether linkages. With the help of thiol–ene chemistry, high energy absorbing materials can be formed. Due to their homogeneous network structure and very sharp glass transitions, energy absorption values of >90%170,171 and high fracture toughness of 0.6–0.9 MPa m1/2 can be reached.172
Thiols may also undergo photo-induced addition to carbon–carbon triple bonds, allowing thiol–yne chemistry.173 The product of a thiol–yne addition is a vinyl sulphide which can undergo a second reaction with available thiol to give a dithioether. With a terminal alkyne and an excess of thiol under UV irradiation conditions, the preferred product is almost exclusively the 1,2 dithioether.174 The reaction is regiospecific and high yielding, which provide a good platform for building high molecular weight polymers. Thiols and yne monomers should ideally be reacted with a ratio of 2 to 1, where the arising network structure will be more crosslinked and tend to have a higher glass transition and higher modulus compared to a thiol–ene system with comparable degree of functionality.175 Curable formulations containing urethane oligomers with alkyne groups and polythiols can be photo-cured into hard, heat-resistant and tough films, which are promising for coatings and adhesives.176
Thiol–ene/–yne chemistry is described as a “click” process, with advantages of high conversion, fast reaction rates, low oxygen inhibition, and good tunability of the resulting polymer networks formed via both radical step-growth polymerization as well as base-catalysed Michael-addition reactions.161,162,177
Thiols are often used with monomers like (meth)acrylates that prefer to undergo homopolymerization. In this case, polymer will grow via a mixed chain growth/step growth-like manner (Scheme 4), where the induced chain transfer (CT) reaction moves the gel point to a higher conversion (e.g. from 25 to 50%). With less double bonds remaining after gelation, a subsequent reduction in shrinkage stress is observed. In addition, the more homogeneous polymer networks exhibit sharper and more defined thermal glass transitions. The less stiff networks with flexible thioether bridges tend to have improved impact resistance. Bowman et al. have studied the use of thiols including pentaerythritol tetra(3-mercaptopropionate) (PETMP) in composites for dentistry,178,179 claiming methacrylate conversion of >90% which is rare for highly crosslinked photopolymerized networks and should lead to improved long-term biocompatibility. Moreover, the thiol–ene-based networks exhibited better flexural strength, increased depth of cure, and reduced shrinkage stress compared to standard composite formulations for dental applications. Another trending application for regulated photopolymerization using thiol–ene chemistry is the field of tissue engineering (e.g.: shape memory polymers,180 hydrogels181–183). Depending on the thiol used (e.g. trimethylolpropane tris(3-mercaptopropionate) (TMPMP) or an isophorone diurethane based hexathiol (IPDUT)), the thermomechanical and mechanical properties can be tailored over a wide range suitable for biomedical applications. Moreover, the shape memory response and shape recovery of thiol-based networks is more rapid and more pronounced compared to standard acrylic-based shape memory systems. This is due to sharper thermal phase transitions, which arise from the more homogeneous network structures. For biocompatible hydrogels, PETMP184 or dithiolthreitol (DTT)185–187 are commonly used as water compatible thiols. Alternatively the thiol functionality can be incorporated into a water soluble biomacromolecule (e.g., reduced bovine serum albumin BSA,188 hemicellulose,189 cyclodextrin190). These thiol–ene systems are amenable to lithographic processing188,191 and may also be of interest as smart coatings.192
The most commonly referenced thiols are esters derived from 3-mercaptopropanoic acid. Depending on the required network properties, thiols with different functionalities (e.g., di-, tri-, tetra-, hexafunctional) and functional groups (e.g., urea, ester, ether) can be used (Fig. 18). The company Bruno Bock is a reliable source for many of the commercially available thiols.
 | ||
| Fig. 18 Examples of thiols typically used as additives in photopolymerizable formulations. | ||
The approach of using multifunctional thiols as CT reagents in diacrylate systems is very promising, but unpleasant odour,193 tendency to colorize the materials, and storage stability issues194 have hindered development. While toughness of photo-cured thiol–ene/yne based materials can be quite high, other mechanical properties such as hardness and storage modulus are compromised due to the flexibility of the thioether bridges, reduced crosslink density and shorter kinetic chain lengths.195,196 Moreover, for thiol-acrylate systems the acrylate homo-polymerization is greatly preferred to reaction with thiol (Ctr is significantly <1) thus a fraction of thiol groups will remain following polymerization.167,168,197
Thiols have also been used in photopolymerization with vinyl ether monomers. In the presence of a photoacid generator, the vinyl ether can undergo both radical thiol–ene reactions as well as cationic homopolymerization. In a 50:50 trithiol/trivinyl ether system, polymerization is almost exclusively thiol–ene, but in a 25:75 trithiol/trivinyl ether system homopolymerization will predominate after the thiol is exhausted. The resultant mechanical properties (tensile strength and deformation energy) of this polymer are higher than the 50:50 polymer and much higher than the trivinyl ether homopolymer (0:100). Young's modulus of the 25:75 polymer was intermediate to the two other polymers.198
AFCT
Addition fragmentation chain transfer (AFCT) reagents were first reported to modify radical polymerization in the late 1980s199,200 and since then more potent reagents have been developed with reactivity dependent on the monomer undergoing polymerization.201,202 In comparison to thiols which cause chain transfer via hydrogen transfer, AFCT reagents add to propagating radicals in a manner similar to that of the utilized monomer. Research on AFCT reagents has over the years mainly focused on molecular weight control of linear polymers (e.g., styrene203 and methyl methacrylate204), for the synthesis of hyperbranched polymers,205 or end-group functionalization.203,205–208 Only recently AFCT reagents including ethylenically unsaturated acrylates209 and β-allyl sulfides205,210 have been mentioned for the modification of photo-crosslinked functional materials (Fig. 19). In comparison to radical thiol–ene polymerization (Scheme 4), the AFCT mechanism can also regulate the radical polymerization reaction in a mixed chain growth/step growth-like manner (Scheme 5).125,126 | ||
| Fig. 19 AFCT reagents used in photopolymerized crosslinked networks. | ||
 | ||
| Scheme 5 General mechanism for addition–fragmentation chain transfer (AFCT). | ||
Where thiols donate hydrogen atoms to propagating radicals, addition–fragmentation chain transfer (AFCT) reagents allow a slightly different method for modifying the growth of the polymer network.211 An AFCT reagent158,159 (Scheme 5) consists usually of a reactive double bond that allows radical attack and a leaving group L that forms a reactive radical after fragmentation. In between the double bond and the leaving group can be usually either a CH2–group or an oxygen (X). The reactivity of the double bond depends on the activating group A. Commonly used A groups include C6H5, CN and CO2R′. After radical attack of the double bond, an intermediate radical (INT) is formed which can either undergo further propagation, reaction back to the starting components (both not beneficial) or the desired fragmentation via β-scission to form the radical of the leaving group, which should have high reactivity to initiate a new radical chain.201 The stability of INT is influenced by the activating group A and usually propagation of INT is precluded due to steric hindrance. The more stable the radical INT, the longer it takes for the fragmentation step. This could ultimately lead to retardation of the radical reaction which is an undesired side effect of the AFCT approach.
Scheme 5 is drawn in a very general manner. In the case where the propagating radical is carbon centred and X = O, the reaction will be irreversible, where in the case of X = CH2 a terminal vinyl group is formed that can form additional crosslinks by subsequent radical addition. The reaction mechanism proceeds similarly to thiol–ene chemistry and this could potentially lead to materials with comparable properties.158,159 By adding AFCT reagents to a radical polymerizable crosslinking monomer mixture, the uncontrolled radical chain growth mechanism can be altered to provide materials with a more defined and homogeneous network structure and, consequently, tunable mechanical properties (Fig. 20).
 | ||
| Fig. 20 AFCT regulated radical network modification giving access to tunable thermo-mechanical properties. | ||
To determine the ideal AFCT reagent for (meth)acrylate monomers, the kinetic rate constants need to be taken into consideration. Most importantly, the rate constant of fragmentation for the AFCT reagent needs to be significantly higher than the rate for self-propagation. For comparison of different AFCT reagents, the chain transfer constant Ctr can be seen as a measure for the compatibility of an AFCT reagent with a certain monomer.
• When Ctr < 1: chain growth is the dominating reaction step yielding inhomogeneous polymers with broad molecular weight distribution.
• When Ctr > 1: the chain transfer reaction dominates and the molecular weight of the polymer is lower at the beginning of the reaction, until the AFCT reagent is exhausted. After consumption of the AFCT reagent, the radical reaction proceeds in a chain growth manner leading to high and broad distributed molecular weights.
• When Ctr ∼ 1: chain transfer and propagation happen with the same probability indicating a homogeneous consumption of AFCT reagent and monomer. The resulting polymers are more homogeneous and can have well defined thermal-mechanical properties.
Generally, Ctr for AFCT reagents with acrylates, styrenes or vinyl esters can range from 0.5–2, sometimes even higher (up to >80), depending on the reagent.158 Finding an AFCT reagent with a Ctr close to 1 allows a more homogeneous copolymerization reaction by comparison to the thiol–acrylate system. In general, highly crosslinked polymers with AFCT reagents have more homogeneous polymer architectures, which paves the way to potential applications such as shape memory polymers180,212 and covalent adaptable networks.213
Bowman et al. have reported the first use of AFCT with allyl sulfides for acrylate214 and methacrylate205 photopolymerization. In the case of acrylate systems, the photopolymerization induced shrinkage stress could be greatly reduced by tuning the base formulations. For methacrylate systems, the allyl sulfide AFCT reagent is not ideal and stress reduction could only be achieved in combination with thiol–ene chemistry (Fig. 21). Nevertheless, the approach with methacrylic monomers has great potential for in the field of dentistry.215 In both systems the network architecture of the polymer is regulated, which indicates that the resulting materials could exhibit improved toughness. Unfortunately, some significant delay in curing has to be accepted.
 | ||
| Fig. 21 Stress relaxation via β-allyl sulfides in thiol-norbornene-based systems.216 | ||
A similar study of methacrylate network regulation with AFCT reagents has recently been published by Liska et al. Here β-allyl sulfones were used in methacrylate photopolymerization to regulate final properties such as thermal polymer phase transition, network density, and network homogeneity.215 By providing regulation over the radical photopolymerization process, the resulting networks exhibit reduced shrinkage stress, improved conversion and higher toughness (Fig. 22).217
 | ||
| Fig. 22 β-Allyl sulfones as AFCT reagents in methacrylate-based networks. | ||
Mono- and difunctional β-allyl sulfones (MAS and DAS in Fig. 23) regulate the photopolymerization of dimethacrylates and exhibit significant advantages over thiols. In addition to the notable issues of storage stability and odour, alkyl thiol chain transfer agents do not homogeneously react with methacrylates as methacrylate homopolymerization is greatly preferred with a Ctr < 1. By comparison, β-allyl sulfones react homogeneously with methacrylates (Ctr ∼ 1)218 which allows gelation times at higher double bond conversions compared to the thiol/methacrylate system. This corresponds with a reduction of shrinkage stress, which is significantly lowered for β-allyl sulfone systems. Moreover, materials with increased toughness (factor ∼5) are obtained while keeping the storage modulus and indentation hardness unaffected or even improved.
 | ||
| Fig. 23 Mono-(MAS) and difunctional β-allyl sulfones (DAS) as AFCT reagents. | ||
More recently, additional AFCT reagents for regulation of methacrylate network formation have been developed. Generally substitution of the vinyl activating group allows for modification of material properties with lower concentration of additive. AFCT reagents with appropriate substitution of the leaving group, on the other hand, provide higher conversion at gelation with no delay in polymerization speed.219 All in all, the effectiveness of an AFCT reagent is dependent on the utilized monomer system, and solubility and side reactions such as H-transfer must be considered as well.220
Photo-induced living radical polymerization
One of the more exciting recent developments in the field of photopolymer chemistry has been the introduction of photo mediated living radical polymerization techniques.221 Reversible-addition fragmentation chain transfer (RAFT) is a special case of AFCT where either R˙ or X is not carbon. Two of the more commonly utilized classes of RAFT agents are the dithioesters (where X and A = S) and the trithiocarbonates (where X, A and Z = S).222 Recently Boyer et al. have established photoinduced electron transfer (PET) RAFT, which uses an Ir or Ru catalyst to regulate propagation by reducing the thiocarbonylthio group on the growing polymer chain.223 Photochemical atom transfer radical polymerization (ATRP)224 and photoinduced iodine-transfer polymerization (ITP)224 methods have also been developed. The principal benefit of these living radical polymerization techniques is the linear relation of conversion and molecular weight which lends to polydispersity values approaching 1.225 A practical disadvantage is that the rate of reaction is orders of magnitude lower than typical photopolymerizations. Living radical techniques are readily used to provide well defined block copolymers. Although it is hard to draw a general trend between mechanical properties and polydispersity, Morales et al. found that PS/PB BC additives with PS blocks with higher PDI values (1.6) were 50% more effective in improving the impact resistance of HIPS versus additives with PS PDI values of 1.1.226Interpenetrating networks (IPNs)
Mixed modes of polymerization
Acrylate and epoxide monomers do not readily copolymerize either under radical or cationic conditions. Mixtures containing both acrylates (with radical photoinitiators) and epoxides (with cationic photoinitiators) can however undergo polymerization simultaneously (although with different rate constants) to form an interpenetrating network (IPN).153 In a system of miscible monomers, where the functionality of both acrylate and epoxide systems is greater than 2, crosslinking will lock the acrylate and epoxide polymers together non-covalently but also irreversibly. Practically co-continuous phases can be observed with different degrees of interpenetration between the two networks.227 Decker et al. found that with the hybrid system consisting of DGEBA diacrylate with ECC, the acrylate component polymerized much faster than the epoxide under laminated conditions but that the order was reversed in air. The Persoz hardness of the hybrid polymer was higher (360 s) than that of the neat epoxide (340 s) and the acrylate polymer (250 s). Dean and Cook used a mixture of methacrylate monomer with radical photoinitiator and epoxy with thermally labile anhydride to test the influence of curing order.228 When the mixture was exposed first to UV irradiation and then heated, the methacrylate portion reached a higher conversion and when the order of cure was reversed the epoxide reached a higher conversion. In the first case, two Tg's are observed (105 °C and 196 °C) and in the second case just the lower transition is seen (and sharpened). Jansen et al. prepared IPNs of PPO-based epoxides with MMA.229 In the presence of a dimethacrylate crosslinker a full-IPN was formed and without it a semi-IPN. A third system containing the epoxy methacrylate monomer GMA to provide a covalently linked copolymer was also prepared. Of the three epoxy/methacrylate systems, the semi-IPN was found to have the highest toughness, reaching a maximum with an epoxy ratio of 30% for tensile toughness and 70% for the impact test.While mixed epoxide/acrylate polymers are more common, other mixed mode photo-curable polymer systems have also been investigated. Ortiz et al. investigated the effect of thiol–ene monomers (a di-tertiary amine tetra allyl ene and a trithiol) added to DGEBA based resins.230 With this system, the tertiary amine served the dual purpose of curing the epoxide. Using RTIR, conversion of thiol was found to increase even after the allyl had been consumed which the authors clarify as evidence that the thiol is also acting as an epoxy curing agent. DMTA of these hybrid copolymers shows a sharpening and decrease of the Tg from 93 °C for the epoxy cured with diethylenetriamine to 64 °C for the polymer with 40% thiol–ene monomers. Interestingly low-temperature storage modulus was about the same for those two polymers and the highest for the polymer with 20% thiol–ene additive.
Polymerization induced phase separation
It was earlier explained in the section on rubber additives, that a degree of heterogeneity between the rubber and bulk polymer phases is required to efficiently dissipate potential crack propagating forces and thus increase toughness. Heterogeneous materials consisting of two distinct phases can be prepared by blending of partially compatible polymers at high temperature,231via self-assembly of block copolymers,101 or by polymerization induced phase separation (PIPS).232 Photo-PIPS with acrylate monomers has been used to lock liquid crystalline domains within a bulk matrix for electro-optic applications233 and more recently to reduce shrinkage stress and improve mechanical properties of crosslinked polymers.234 Crawford et al. determined that a sufficiently low irradiance is needed to limit the rate of polymerization to ensure the desired spinodal decomposition mode of phase separation. Szczepanski and Stansbury have studied the phase separation behaviour of different methacrylate prepolymers (PMMA, PEMA, PBMA) dissolved in TEGDMA monomer and subjected to UV irradiation.234 Phase separation was efficient with both PMMA and PEMA but not with PBMA presumably due to its sub-ambient Tg, meaning that the polymer was in the rubbery stage throughout polymerization. Phase separation generally slightly precedes gelation and allows ultimate monomer conversions higher than in homogenous monomer systems due to extended auto-acceleration. Using PMMA prepolymers of different molecular weight, it was found that 120 kDa PMMA could induce spinodal decomposition already at 5 wt% loading, while PMMA of 11 kDa did so at 20 wt% loading and that of 1 kDa remained homogenous at all tested concentrations.235 The resin with 20 wt% 120 kDa PMMA exhibited half the shrinkage stress of the neat TEGDMA system with no significant reduction in modulus. The best results were obtained when the polymer was cured to 50% conversion with low irradiance (300 µW cm2) followed by higher intensity (20 mW cm2) curing.Self-healing
Although not specifically defined as toughening, recent developments of self-healing polymeric materials do relate to the prevention of crack propagation and thus may be worth mentioning. Self-healing methods can be broadly classified as either intrinsic of extrinsic.236 Intrinsic self-healing is based on thermally or photolytic bond formation of moieties within the polymer network. Chen et al. introduced self-healing thermosets using reversible Diels–Alder cycloaddition of furans and maleimides.237 The material can be cracked into two separate pieces and then mended by heating to 120–150 °C. While strength of the mended piece was first reported to be compromised by close to 50%, further system refinements have been introduced allowing full fracture toughness recovery even after multiple cycles.238 Among the more commonly used photolytic self-healing methods are the reversible 2 + 2 cycloaddition of coumarin and cinammic acid based derivatives (Scheme 6).239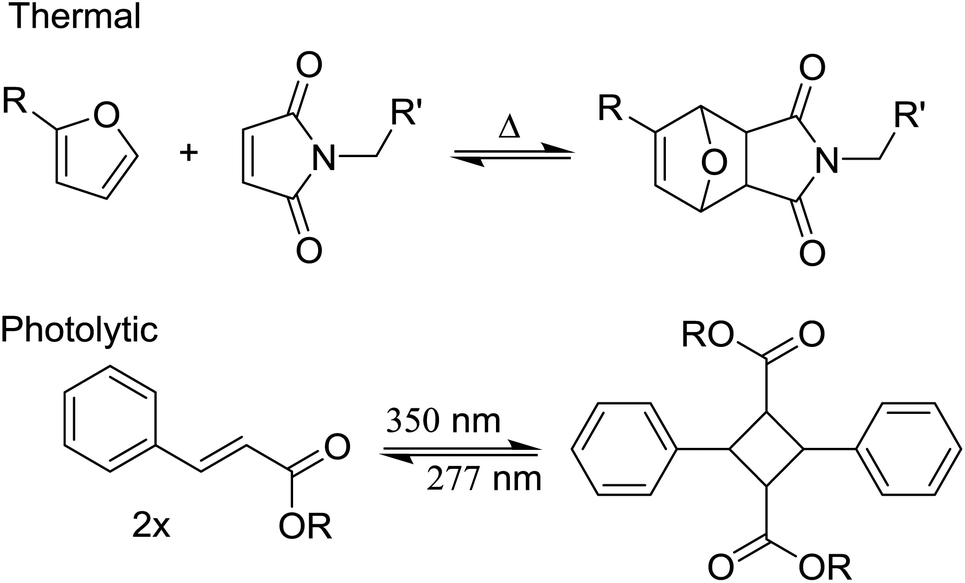 | ||
| Scheme 6 Thermal and photolytic methods of intrinsic self-healing. | ||
Extrinsic self-healing is based on additives such as hollow fibres, microcapsules, or vascular systems containing a self-healing agent which should release during crack propagation.236 Self-healing agents include mixtures of dicyclopentadiene with Grubb's catalyst, siloxanes with tin catalysts, eopoxy/amine systems and organic solvents. Yao et al. added microcapsules containing GMA monomer to PS grown via RAFT polymerization.240 Upon fracture, the GMA released and continued to propagate chain growth from the trithiocarbonate end groups. Impact strength of specimens with 15 wt% microcapsules was actually higher after fracture and healing. An example of extrinsic self-healing particularly pertinent to the photopolymer chemist is provided by Leigh et al. where they use a photoresin with a high concentration of dye as the hydraulic fluid for a micro-actuated forcep.241 Due to the high concentration of dye, the resin remains liquid. Puncture to the housing membrane, however, secretes a small volume of resin to the upper surface where it then cures in ambient light. The cured resin seals the hole while the bulk remains liquid as desired for the application.
Applications
Coatings
Coatings have always been one of the principal applications for photo-curable polymers. Protective coatings for metal, wood,242 plastics and concrete243 are intended to be durable and resist scratches, tears, punctures or other disturbances which may expose the underlying material surface. Typical thicknesses for photo-curable coatings are largely application dependent. As an example, wood is commonly coated with waterborne acrylate coatings which photocure to a thickness from 50–100 µm and generally the thicker the coating the better the impact resistance.244Photo-curable coatings are also used for optical fibres, where one of the principal purposes is to improve flexural strength. The coating can be applied in two layers, where the bottom layer in contact with the glass should be soft and adhere well.245 The top layer, which is commonly based on urethane or epoxy acrylates is comparatively harder (ideally >400 MPa)246 and provides the desired resistance. Both layers are applied and cured during fibre drawing, which is one of the appeals of photo-curing over slower thermal curing. Static charge on the surface of the glass fibre during the coating process can be a problem and has been addressed with additives such as nonionic amines.247 To lower the coefficient of friction of the outer layer and prevent snagging of adjacent fibres, a silicone based acrylate may be added to the formulation.246 Silicone epoxy monomers are also used as optical cladding material, where high UV transparency of the cured clad allows direct laser patterning of Braggs gratings within the core. In this special case, concentration of iodonium photoacid is very low (>0.001 wt%) allowing transparency at 254 nm of more than 85% (Fig. 24).248
Adhesives
Photo-curable adhesives are used to bind glass or plastics to another surface (glass, plastic, metal, or wood). Transparency of the top layer is generally a prerequisite, and the UV cutoff of the material is also to be considered: roughly 320 nm for glass depending on type and 385 nm for polycarbonate.250 Plastic additives, in particular UV stabilizers, will also affect light penetration.251 For these reasons, photoinitiators that absorb at or above 400 nm may be needed. To ensure adhesion in shadow regions, a second slower mode of curing (based for instance on heat or moisture) may be used. Tensile adhesion strength can be assessed by using a Universal tester to pull two adhered pieces apart. Recognizing that adhesion can be reduced by moisture, Kim et al. used fluorinated silanes and colloidal silica to improve the adhesion strength by 40% of an epoxy acrylate between two glass plates submerged in boiling water.251 On polycarbonate surfaces, polycarbonate based acrylates (0.92 kgf m−1) adhere better than urethane acrylates (0.46 kgf m−1) or epoxy acrylates (0.0576 kgf m−1).252Additive manufacturing
The term “additive manufacturing” (AM) is that preferred by the ASTM to collectively refer to automated “additive” techniques such as stereolithography (SLA) and fused deposition modelling (FDM) used for building well-defined 3D structures.253 “Additive” is thus used to contrast traditional “subtractive” techniques such as milling254 and stamping.255 AM tends to be more precise, less energy intensive,256 and produces no waste from residual unused build material.257 AM has long been considered “rapid” in the prototyping stage of product development, and yet more recently as new and specialized applications have arisen “rapid manufacturing” is now a viable option. AM methods are available for building objects with metals, plastics and other materials. Of these methods, FDM with acrylonitrile butadiene styrene (ABS) is one of the best established AM techniques and stands as a good benchmark. ABS is a reasonably tough polymer (Izod impact strength of 340 J m−1),258 but it does have its shortfalls. Most notably, ABS is optically opaque and its surface properties are inappropriate for most biomedical applications.259 The high temperature of the FDM extruder head is also a limiting factor. By comparison, photo-based AM techniques such as SLA operate at lower temperature and allow feature resolution at least one to two orders smaller than that allowed by FDM. Commercially available resins for photo-based AM allow finished objects with mechanical properties varying from soft and flexible to stiff. Table 2 demonstrates the range of mechanical properties from the three major resin manufacturers. 3D Systems and Stratasys both package their resins in cartridges for material jetting AM devices.260,261 A major advantage of jetting versus vat photopolymerization (i.e. traditional SLA) is that multiple materials can be printed during the same build. Stratasys describes their “Digital Materials” as composites of two or more photoresins with optical and mechanical properties controlled by the ratio of the different resins. While the composition of commercial AM photoresins is proprietary, patents and MSDS sheets indicate that both acrylate and epoxy monomers are used. The advantage of a mixed monomer system in AM is that the acrylate component will cure quickly and define the 3D structure, while the epoxy component can be thermally post cured and limit shrinkage. It is also worth noting that photo-based AM is a layer by layer process, which has consequences on spatial resolution and can impart anisotropic mechanical properties. Particulate additives which settle during the build or which increase viscosity may not be appropriate for AM and particularly with jetting processes.| Material | Manufacturer | Tensile modulus/MPa | Tensile strength/MPa | Izod impact/J m−1 | Elongation/% |
|---|---|---|---|---|---|
| Visijet SL Impact | 3D Systems | 2626 | 48 | 65 | 14 |
| Visijet SL Tough | 3D Systems | 1890 | 41 | 44 | 18 |
| Somos 8110 | DSM | 317 | 26 | 87 | 27 |
| Somos 8120 | DSM | 690 | 26 | 59 | 26 |
| Somos 14120 | DSM | 2460 | 46 | 24 | 7.9 |
| Endur RGD450 | Stratasys | 1700 | 20 | 30 | 20 |
| Digital ABS | Stratasys | 2600 | 55 | 65 | 25 |
| TangoPlus FLX930 | Stratasys | <100 | 0.8 | — | 170 |
Dental/medical
Photo-curable polymers have been in use in dentistry for more than 40 years. With direct dental restoratives, the prepared tooth surface consisting of enamel and dentin is first etched (commonly with 30–40% phosphoric acid) to remove the dentin smear layer.263 Next a thin primer layer is added to serve as an intermediary between hydrophilic dentin and the hydrophobic composite. These two-steps can alternately be combined into one step by using self-etching primers containing acidic methacrylate monomers.46 After curing the primer, the bulk restorative is applied and cured. Photo-curable restoratives are composites consisting of typically 70–90 wt% (30–55 vol%) inorganic filler within a dimethacrylate resin.264 The high filler content is necessary for aesthetics, to increase modulus and strength, and to reduce polymerization shrinkage and associated stress.265 Enamel is indeed the hardest material in the human body but is also notoriously brittle and can crack from shrinkage forces which develop during and after photo-curing. Light scattering in dental composites limits depth of cure meaning that deep carries will require multiple coat-and-cure cycles. Commonly used methacrylate monomers for dentistry include 2,2-bis[4-(2-hydroxy-3-methacryloyloxypropyl)phenyl]propane (Bis-GMA), urethane dimethacrylate (UDMA) and decanediol dimethacrylate (D3MA).266 The filler consists principally of micro- and nano-particulate silicates with heavier elements such as barium, zirconium and strontium added to improve radio-opacity.267 Filler particles are treated with silane coupling agents (such as 3-(methacryloyloxypropyltrimethoxysilane), which links the particles within the polymer matrix and helps in the transfer of stress. In addition to substantial masticatory cyclic stresses (chewing), dental composites are exposed to an aqueous environment with regular temperature variations. Dental restoratives must not only be tough they must also stay tough quite literally a lifetime.The addition of chain transfer agents to dental formulations has been established as such reagents not only increase the toughness but also limit shrinkage stress while increasing conversion and adhesion. Stansbury and Bowman have published a number of patents on the use of thiol–ene formulations for dental applications.193,268,269 Cramer and Bowman have added to this work,270,271 using allyl sulfides272,273 and trithiocarbonates274 as chain transfer agents. The company 3M has also claimed patents that focus on the benefits of chain transfer agents for self-adhesive compositions,275 high refractive index formulations (for increased curing depth),276 or dental formulations in general,277–279 using monomeric209 or oligomeric280 AFCT reagents based on crosslinkers with cleavable ethylenically unsaturated polymerizable groups.
In addition to dentistry, photo-curable polymers are finding use in a variety of other biomedical applications. Using tomographic images (from MRI or X-ray) to create an STL file, photo-based AMT is more and more regularly used for the production of patient specific surgical guides, which saves operational time and reduces associated trauma.281 While the mechanical demands for plastic guides are low, this is by no means the case for many other AM medical applications. Notably CAD-CAM can also be used to produce anatomically correct implants to serve as replacements for damaged human tissue.282,283 The demands for materials used in tissue engineering are high. Notably a very high degree of biocompatibility is required for a long-term implant, which warrants the study of low toxic alternatives to acrylates.284,285 Among these alternative monomers, vinyl esters have been successfully used to 3D print a porous bone graft. Six months after implanting the graft into a rabbit femur, histology indicated growth of new bone tissue with good osteoconductivity. Vinyl esters have good photoreactivity, which can be increased by addition of thiol comonomers.195 Thiol-vinyl ester based networks also have substantially improved impact strength, with modulus adjusted by choice of thiol functionality. Thiols have also been used to improve the tear strength (as assessed with a PLA surgical thread) of photo-cured urethane-acrylate networks intended for use as vascular grafts.286 For vascular grafts, the material compliance (the inverse modulus) should match that of the native vessel to avoid causing disturbances in blood flow which can lead to stenosis.287 While biodegradability was traditionally considered a weakness for a long term implant, more recently biodegradable polymers have been shown to help in the harbouring of native cells and in the regeneration of new tissue.288 In this case, surface degradation is essential to maintain the structure and mechanical properties of the graft.289 As more fresh tissue grows around and into the graft, the mechanical properties of the tissue develop.290 The initial stages of tissue growth within the graft can be performed within or outside of the patient's body by culturing native cells in a laboratory.291 Nutrients and growth factors are in this case provided until the tissue graft can be transplanted. The intention is that native tissue gradually becomes the principal component while the graft degrades. At the same time, the collective mechanical properties of tissue and the artificial graft should remain fairly constant. These principals have indeed been demonstrated in animal models, and clinical tests in human are within the foreseeable future.
Hydrogels
Hydrogels are swollen polymer networks where the principal component by weight is water.292 Crosslinking can be based on covalent or non-covalent bonding or even a combination of the two. Due to rapid cure and to the relatively benign nature of light, photocrosslinking of water soluble acrylate monomers based on PEG,293 PVA, or natural polymers such as gelatine294 provides a versatile platform for forming hydrogels. The resultant mechanical properties of such hydrogels, however, are in often poor with typical fracture energy values only 0.1–1 J m−2.295 The Young's modulus and stress–strain curve of a hydrogel will be largely dependent on molecular structure, crosslink density and water concentration. The copolymer system based on polyglycerol sebacate and PEG shows a decreasing elastic modulus from 2.2 MPa to 13 kPa and a 6 fold increase in elongation with increasing PEG content.296In 2005 Tanaka et al. classified classes of hydrogels with distinctly improved mechanical properties.297 First among these are topological gels where crosslinks are based on non-covalent rotaxane linkages which provide hydrogels capable of being stretched by 20 times their original length. The second category of mechanically improved gels is the nanocomposite (NC) gel. The addition of clay to hydrogels is here found to increase modulus and tensile toughness. The final class of improved hydrogel is the double network (DN) gel.298 DN gels are formed in two steps, where first a highly crosslinked rigid gel is formed and second a loosely cross-linked gel is formed within the first network.299 The mechanical properties of DN gels are tuneable and can vastly exceed those of traditional hydrogels: elastic modulus from 0.1–1 MPa, compressive fracture stress of 20–60 MPa, elongation to 2000% and fracture energy values up to 1000 J m−2.300
Spearheaded principally by the groups of Bowman, Anseth and Hoyle, great advances have been made in toughening of hydrogels via thiol–ene and thiol–yne chemistry.301,302 Linear and 4-arm PEG oligomers end capped with norbornene or allyl ethers can be copolymerized with hydrophilic multifunctional thiols to give hydrogels with significantly higher strength and elongation versus traditional PEG acrylate based hydrogels.303,304 The more regular networks formed via step-growth polymerization of complimentary monomers are apparently more efficient at dissipating fracture stresses than the more random networks formed via radical chain growth of acrylates.305 Thiol-norbornene photo-crosslinking has also been used to build PEG-PDMS hydrogels where critical strain energy under compression (Gc) could be increased from 7 J m−2 for the pure PEG hydrogel to 100 J m−2 where the volume fraction of PDMS was 3.6 times greater than PEG.306 Photoinduced thiol–yne crosslinking has also been applied to the synthesis of hydrogels with the benefit that higher modulus gels are also possible.174 More recently tetrazole-alkene photo click chemistry has been used to give low modulus (0.65–25 kPa) hydrogels.307
Conclusions and outlook
Photo-curable polymers provide unique solutions to a broad range of industrial problems. Fast cure kinetics has always been a driving force and yet, with the expansion in applications from mostly coatings to bulk materials as well, mechanical properties have grown in importance. Heavily crosslinked networks based on acrylates and epoxides can often be quite brittle but fortunately strategies have been developed to counter these effects. Some monomers such as those based on urethanes and other moieties which form interchain hydrogen bonds give networks with intrinsically higher tensile strength. Since photocurable epoxy resins intrinsically form more highly crosslinked networks than conventional epoxy thermosets, polyol CTAs are imperative for materials with good mechanical properties. Inorganic and organic particulate additives, which have long been used to improve toughness in thermoplastics and thermosets, prove advantageous in photo-curable networks as well. To prevent light scattering which can occur with larger (>1 µm) particles, block copolymers and nanoparticles are perhaps more appropriate. Resins based on oligomeric and hyperbranched acrylates exhibit less polymerization shrinkage versus those based on smaller molecule monomers and concurrently provide materials with better impact strength. Also alternate styles of polymerization and particularly those based on thiol–ene/yne and other CTAs tend to provide networks with higher toughness and provide a versatile platform for modifying mechanical properties. AFCT agents have been shown to regulate photo-crosslinking of (meth)acrylates very well due to their near unity chain transfer constants. This allows more homogenous network formation and materials with high impact strength and high modulus. IPNs based on monomers or monomer systems that react simultaneously and separately allow mechanical properties often times superior to those of the individual components.While many different strategies for toughening of photo-curable materials have been developed and improvements have been made, there is still great potential in the combination of successful strategies. In particular, chain transfer and other methods for better regulating network ought to be tested in combination with traditional particle and nanoparticle additives. Combined strategy approaches seem well-suited in cases where not just toughness, but strength and hardness should be high. Ultimately the intended application of the polymer network will dictate the choice of the monomer system and additives. While fast cure and good mechanical properties alone may suffice in rapid prototyping, medical implants garner additional requirements including excellent biocompatibility and other properties specific to the effected tissue. In the case of biodegradable tissue grafts, toughness of the polymer alone may be transcendental although that of the graft and tissue collectively should be maintained for a lifetime.
Acknowledgements
Thanks go to the Christian Doppler Society and the company Ivoclar Vivadent for establishing the Laboratory for Digital and Restorative Dentistry and to the Austrian Science Foundation FWF (project P27059) for additional financial support.References
- J. P. Fouassier, X. Allonas and D. Burget, Prog. Org. Coat., 2003, 47, 16–36 CrossRef CAS.
- R. Bongiovanni, F. Montefusco, A. Priola, N. Macchioni, S. Lazzeri, L. Sozzi and B. Ameduri, Prog. Org. Coat., 2002, 45, 359–363 CrossRef CAS.
- S. Lin, Q. Cai, J. Ji, G. Sui, Y. Yu, X. Yang, Q. Ma, Y. Wei and X. Deng, Compos. Sci. Technol., 2008, 68, 3322–3329 CrossRef CAS.
- R. Liska, M. Schuster, R. Inführ, C. Turecek, C. Fritscher, B. Seidl, V. Schmidt, L. Kuna, A. Haase, F. Varga, H. Lichtenegger and J. Stampfl, J. Coat. Technol. Res., 2007, 4, 505–510 CrossRef CAS.
- B. I. Sandor, R. Roloff and I. K. Glasgow, in Mechanical Engineering Handbook, ed. F. Kreith, CRC Press LLC, Boca Raton, FL, 1999 Search PubMed.
- W. F. Hosford, in Tensile Testing, ed. P. Han, ASM International, Materials Park, OH, 1992, pp. 1–24 Search PubMed.
- Kerbschlagbiegeversuch, http://wiki.polymerservice-merseburg.de/index.php/Kerbschlagbiegeversuch, Accessed 15 August 2015.
- ISO180:2000, Plastics - Determination of Izod impact strength, International Organization for Standardization, Geneva, CH, 2000 Search PubMed.
- F. Liu, Y. Zhong, C. Jiang, D. Wang, Z.-K. Wang and M.-T. Run, Polym. Compos., 2012, 33, 999–1006 CrossRef CAS.
- J. D. Ferry, Viscoelastic Properties of Polymer, John Wiley Interscience, New York, 1970 Search PubMed.
- L. Pan and B. Zhang, J. Sound Vib., 2009, 319, 822–831 CrossRef.
- A. Mhanna, F. Sadaka, G. Boni, C.-H. Brachais, L. Brachais, J.-P. Couvercelle, L. Plasseraud and L. Lecamp, J. Am. Oil Chem. Soc., 2014, 91, 337–348 CrossRef CAS.
- K. Cicha, T. Koch, J. Torgersen, Z. Li, R. Liska and J. Stampfl, J. Appl. Phys., 2012, 112, 94906 CrossRef.
- J. Scheirs, Compositional and Failure Analysis of Polymers, John Wiley & Sons, Ltd., Chichester, 2000 Search PubMed.
- A. Torres-Filho and D. C. Neckers, Chem. Mater., 1995, 7, 744–753 CrossRef CAS.
- R. Bagheri, B. T. Marouf and R. A. Pearson, Polym. Rev., 2009, 49, 201–225 CrossRef CAS.
- H. G. Waddill (Texaco Chemical Company), US5053465, 1989 Search PubMed.
- L. O. Cummings, P. A. Terrizzi and N. A. Gac (Air Products and Chemicals, Inc.), US6121350, 1997 Search PubMed.
- S. Corezzi, D. Fioretto, G. Santucci and J. M. Kenny, Polymer, 2010, 51, 5833–5845 CrossRef CAS.
- H. Q. Pham and M. J. Marks, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim, 2012, pp. 155–244 Search PubMed.
- B. A. Rozenberg, in Epoxy Resins and Composites II, Advances in Polymer Science 75, ed. K. Dusek, Springer Verlag, Berlin, 1986, pp. 146–156 Search PubMed.
- J. V. Crivello, J. Photopolym. Sci. Technol., 2009, 22, 575–582 CrossRef CAS.
- C. A. George and C. W. I. V. Stobbie (Minnesota Mining and Manufacturing Co.), EP620259A2, 1994 Search PubMed.
- Y. Orikasa, S. Sakazume, S. Nishimura and Y. Maki (Nippon Petrochemicals Co., Ltd.; Nippon Oils & Fats Co., Ltd.), EP314188A2, 1989 Search PubMed.
- D. A. Tilbrook, R. L. Clarke, N. E. Howle and M. Braden, Biomaterials, 2000, 21, 1743–1753 CrossRef CAS PubMed.
- J. V. Crivello and K. Dietliker, in Chemistry & Technology of UV & EB Formulation for Coatings, Inks & Paints, ed. P. K. T. Oldring, SITA Technology Ltd., London, 1991, pp. 327–478 Search PubMed.
- J. Xu (DSM IP Assets B.V.), U.S. Patent9090020, 2015 Search PubMed.
- H. Salmi, X. Allonas, C. Ley, D. Marechai and A. Ak, J. Photopolym. Sci. Technol., 2012, 25, 147–151 CrossRef CAS.
- M. Sangermano, A. Vitale and K. Dietliker, Polymer, 2014, 55, 1628–1635 CrossRef CAS.
- K. Dietliker, R. Hüsler, J. L. Birbaum, S. Ilg, S. Villeneuve, K. Studer, T. Jung, J. Benkhoff, H. Kura, A. Matsumoto and H. Oka, Prog. Org. Coat., 2007, 58, 146–157 CrossRef CAS.
- K. Dietliker, T. Jung, J. Benkhoff, H. Kura, A. Matsumoto, H. Oka, D. Hristova, G. Gescheidt and G. Rist, Macromol. Symp., 2004, 217, 77–98 CrossRef CAS.
- J. F. Cameron, C. G. Willson and J. M. J. Fréchet, J. Am. Chem. Soc., 1996, 118, 12925–12937 CrossRef CAS.
- K. Arimitsu and R. Endo, Chem. Mater., 2013, 25, 4461–4463 CrossRef CAS.
- X. Allonas, H. Salmi and C. Ley, in Rad Tech, Chicago, IL, 2014 Search PubMed.
- K. Arimitsu, Y. Takemori, A. Nakajima, A. Oguri, M. Furutani, T. Gunji and Y. Abe, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 1174–1177 CrossRef CAS.
- J. P. Fouassier, in Photochemistry and UV Curing: New Trends, ed. J. P. Fouassier, Research Signpost, Kerala, 2006, pp. 1–8 Search PubMed.
- J. V. Crivello and K. Dietliker, Photoinitiators for Free Radical Cationic & Anionic Photopolymerisation, John Wiley & Sons Ltd, Chichester, 1998 Search PubMed.
- R. Chandra, B. P. Thapliyal, B. Sehgal and R. K. Soni, Polym. Int., 1992, 29, 185–190 CrossRef CAS.
- K. Oguni (Sony Corp.), US7727610, 2006 Search PubMed.
- C. Decker, Prog. Polym. Sci., 1996, 21, 593–650 CrossRef CAS.
- B. Husár, S. C. Ligon, H. Wutzel, H. Hoffmann and R. Liska, Prog. Org. Coat., 2014, 77, 1789–1798 CrossRef.
- S. C. Ligon, B. Husar, H. Wutzel, R. Holman and R. Liska, Chem. Rev., 2014, 114, 557–589 CrossRef CAS PubMed.
- J. R. Wade, J. R. Burch and R. M. Potts (E.I. DuPont de Nemours), EP0275147, 1993 Search PubMed.
- R. S. Kosiorek and L. R. Gatechair (Ciba-Geigy Corp.), US4649062, 1985 Search PubMed.
- N. Moszner and U. Salz, Prog. Polym. Sci., 2001, 26, 535–576 CrossRef CAS.
- J. Kong, R. Ning and Y. Tang, J. Mater. Sci., 2006, 41, 1639–1641 CrossRef CAS.
- T. M. Lovestead, A. K. O'Brien and C. N. Bowman, J. Photochem. Photobiol., A, 2003, 159, 135–143 CrossRef CAS.
- M. D. Goodner and C. N. Bowman, Chem. Eng. Sci., 2002, 57, 887–900 CrossRef CAS.
- J. E. Elliott, L. G. Lovell and C. N. Bowman, Dent. Mater., 2001, 17, 221–229 CrossRef CAS PubMed.
- L. K. Baekeland, US942699, 1909 Search PubMed.
- A. F. Clarke (National Dairy Products Company), US2418688, 1947 Search PubMed.
- P. Castan, US2324483, 1943 Search PubMed.
- J. N. Sultan and F. J. McGarry, Polym. Eng. Sci., 1973, 13, 29–34 CAS.
- Y. Kojima, A. Usuki, M. Kawasumi, A. Okada, Y. Fukushima, T. Kurauchi and O. Kamigaito, J. Mater. Res., 1993, 8, 1185–1189 CrossRef CAS.
- A. Usuki, Y. Kojima, M. Kawasumi, A. Okada, Y. Fukushima, T. Kurauchi and O. Kamigaito, J. Mater. Res., 1993, 8, 1179–1184 CrossRef CAS.
- C. M. Chan, J. Wu, J. X. Li and Y. K. Cheung, Polymer, 2002, 43, 2981–2992 CrossRef CAS.
- Y. Termonia, J. Polym. Sci., Part B: Polym. Phys., 2010, 48, 687–692 CrossRef CAS.
- P. Rittigstein and J. M. Torkelson, J. Polym. Sci., Part B: Polym. Phys., 2006, 44, 2935–2943 CrossRef CAS.
- J. Jordan, K. I. Jacob, R. Tannenbaum, M. A. Sharaf and I. Jasiuk, Mater. Sci. Eng., A, 2005, 393, 1–11 CrossRef.
- W. Gacitua, A. Ballerini and J. Zhang, Maderas: Cienc. Tecnol., 2005, 7, 159–178 Search PubMed.
- W. C. J. Zuiderduin, C. Westzaan, J. Huétink and R. J. Gaymans, Polymer, 2003, 44, 261–275 CrossRef CAS.
- T. Guo, L. Wang, A. Zhang and T. Cai, J. Appl. Polym. Sci., 2005, 97, 1154–1160 CrossRef CAS.
- Z. H. Liu, K. W. Kwok, R. K. Y. Li and C. L. Choy, Polymer, 2002, 43, 2501–2506 CrossRef CAS.
- B. Wetzel, F. Haupert and M. Q. Zhang, Compos. Sci. Technol., 2003, 63, 2055–2067 CrossRef CAS.
- G. Ragosta, M. Abbate, P. Musto, G. Scarinzi and L. Mascia, Polymer, 2005, 46, 10506–10516 CrossRef CAS.
- S. Bhuniya and B. Adhikari, J. Appl. Polym. Sci., 2003, 90, 1497–1506 CrossRef CAS.
- M. Sangermano, M. Naguib and M. Messori, Macromol. Mater. Eng., 2013, 298, 1184–1189 CrossRef CAS.
- H. Zhang, Z. Zhang, K. Friedrich and C. Eger, Acta Mater., 2006, 54, 1833–1842 CrossRef CAS.
- H. Zhang, L. C. Tang, Z. Zhang, K. Friedrich and S. Sprenger, Polymer, 2008, 49, 3816–3825 CrossRef CAS.
- T. Mahrholz, J. Stängle and M. Sinapius, Composites, Part A, 2009, 40, 235–243 CrossRef.
- L. H. Lewandowski and D. F. Klemmensen (The Goodyear Tire & Rubber Company), US6136899, 1999 Search PubMed.
- J. W. Aylsworth, US1111284, 1914 Search PubMed.
- J. L. Amos, M. J. Lloyd and M. O. Ray (Dow Chemical Co.), US2694692, 1954 Search PubMed.
- G. H. Fremon and W. N. Stoops (Union Carbide), US3168593, 1965 Search PubMed.
- R. A. Pearson and A. F. Yee, J. Mater. Sci., 1991, 26, 3828–3844 CrossRef CAS.
- B. Russell and R. Chartoff, Polymer, 2005, 46, 785–798 CrossRef CAS.
- R. Bagheri and R. A. Pearson, Polymer, 1996, 37, 4529–4538 CrossRef CAS.
- S. C. Kunz, J. A. Sayre and R. A. Assink, Polymer, 1982, 23, 1897–1906 CrossRef CAS.
- N. Chikhi, S. Fellahi and M. Bakar, Eur. Polym. J., 2001, 38, 251–264 CrossRef.
- R. Thomas, D. Yumei, H. Yuelong, Y. Le, P. Moldenaers, Y. Weimin, T. Czigany and S. Thomas, Polymer, 2008, 49, 278–294 CrossRef.
- J. Kong, R. Ning and Y. Tang, J. Mater. Sci., 2006, 41, 1639–1641 CrossRef CAS.
- J. S. Ullett and R. P. Chartoff, Polym. Eng. Sci., 1995, 35, 1086–1097 CAS.
- M. L. Auad, P. M. Frontini, J. Borrajo and M. I. Aranguren, Polymer, 2001, 42, 3723–3730 CrossRef CAS.
- E. J. Robinette, S. Ziaee and G. R. Palmese, Polymer, 2004, 45, 6143–6154 CrossRef CAS.
- C. Celikbilek, G. Akovali and C. Kaynak, Polym. Bull., 2004, 51, 429–435 CrossRef CAS.
- Y. Ozari, S. B. Nelsen and A. Klein (GAF Corp.), US4448932A, 1984 Search PubMed.
- P. Hazot, C. Pichot and A. Maazouz, Macromol. Chem. Phys., 2000, 201, 632–641 CrossRef CAS.
- P. A. Lovell, J. McDonald, D. E. J. Saunders and R. J. Young, Polymer, 1993, 34, 61–69 CrossRef CAS.
- B. T. Marouf, R. A. Pearson and R. Bagheri, Mater. Sci. Eng., A, 2009, 515, 49–58 CrossRef.
- H. R. Azimi, R. A. Pearson and R. W. Hertzberg, J. Mater. Sci., 1996, 31, 3777–3789 CrossRef CAS.
- A. Maazouz, H. Sautereau and J. F. Gérard, Polym. Bull., 1994, 33, 67–74 CrossRef CAS.
- S. Jingqiang, Z. Yafeng, Q. Jindong and K. Jianzheng, J. Mater. Sci., 2004, 39, 6383–6384 CrossRef.
- A. K. Subramaniyan and C. T. Sun, Composites, Part A, 2007, 38, 34–43 CrossRef.
- K. F. Lin and Y. D. Shieh, J. Appl. Polym. Sci., 1998, 70, 2313–2322 CrossRef CAS.
- M. Naguib, S. Grassini and M. Sangermano, Macromol. Mater. Eng., 2013, 298, 106–112 CrossRef CAS.
- J. He, D. Raghavan, D. Hoffman and D. Hunston, Polymer, 1999, 40, 1923–1933 CrossRef CAS.
- Q. H. Le, H. C. Kuan, J. B. Dai, I. Zaman, L. Luong and J. Ma, Polymer, 2010, 51, 4867–4879 CrossRef CAS.
- L. Rey, N. Poisson, A. Maazouz and H. Sautereau, J. Mater. Sci., 1999, 34, 1775–1781 CrossRef CAS.
- E. Bittmann and G. W. Ehrenstein, Angew. Makromol. Chem., 1998, 258, 93–98 CrossRef CAS.
- M. A. Hillmyer, P. M. Lipic, D. A. Hajduk, K. Almdal and F. S. Bates, J. Am. Chem. Soc., 1997, 119, 2749–2750 CrossRef CAS.
- L. Ruiz-Pérez, G. J. Royston, J. P. A. Fairclough and A. J. Ryan, Polymer, 2008, 49, 4475–4488 CrossRef.
- C. Ocando, A. Tercjak, M. D. Martín, J. A. Ramos, M. Campo and I. Mondragon, Macromolecules, 2009, 42, 6215–6224 CrossRef CAS.
- S. Ritzenthaler, F. Court, L. David, E. Girard-Reydet, L. Leibler and J. P. Pascault, Macromolecules, 2002, 35, 6245–6254 CrossRef CAS.
- V. Rebizant, V. Abetz, F. Tournilhac, F. Court and L. Leibler, Macromolecules, 2003, 36, 9889–9896 CrossRef CAS.
- T. J. Hermel-Davidock, H. S. Tang, D. J. Murray and S. F. Hahn, J. Polym. Sci., Part B: Polym. Phys., 2007, 45, 3338–3348 CrossRef CAS.
- M. Larrañaga, N. Gabilondo, G. Kortaberria, E. Serrano, P. Remiro, C. C. Riccardi and I. Mondragon, Polymer, 2005, 46, 7082–7093 CrossRef.
- M. Larrañaga, M. D. Martin, N. Gabilondo, G. Kortaberria, A. Eceiza, C. C. Riccardi and I. Mondragon, Colloid Polym. Sci., 2006, 284, 1403–1410 Search PubMed.
- F. Meng, Z. Xu and S. Zheng, Macromolecules, 2008, 41, 1411–1420 CrossRef CAS.
- F. Meng, S. Zheng, H. Li, Q. Liang and T. Liu, Macromolecules, 2006, 39, 5072–5080 CrossRef CAS.
- J. Mijovic, M. Shen, J. W. Sy and I. Mondragon, Macromolecules, 2000, 33, 5235–5244 CrossRef CAS.
- Q. Guo, K. Wang, L. Chen, S. Zheng and P. J. Halley, J. Polym. Sci., Part B: Polym. Phys., 2006, 44, 975–985 CrossRef CAS.
- F. Meng, S. Zheng, W. Zhang, H. Li and Q. Liang, Macromolecules, 2005, 39, 711–719 CrossRef.
- H. Duran, S. Meng, N. Kim, J. Hu, T. Kyu, L. V. Natarajan, V. P. Tondiglia and T. J. Bunning, Polymer, 2008, 49, 534–545 CrossRef CAS.
- J. M. Dean, P. M. Lipic, R. B. Grubbs, R. F. Cook and F. S. Bates, J. Polym. Sci., Part B: Polym. Phys., 2001, 39, 2996–3010 CrossRef CAS.
- J. Liu, H.-J. Sue, Z. J. Thompson, F. S. Bates, M. Dettloff, G. Jacob, N. Verghese and H. Pham, Acta Mater., 2009, 57, 2691–2701 CrossRef CAS.
- J. Liu, Z. J. Thompson, H.-J. Sue, F. S. Bates, M. A. Hillmyer, M. Dettloff, G. Jacob, N. Verghese and H. Pham, Macromolecules, 2010, 43, 7238–7243 CrossRef CAS.
- J. Wu, Y. S. Thio and F. S. Bates, J. Polym. Sci., Part B: Polym. Phys., 2005, 43, 1950–1965 CrossRef CAS.
- Y. S. Thio, J. Wu and F. S. Bates, Macromolecules, 2006, 39, 7187–7189 CrossRef CAS.
- J. S. Riffle, W. P. Steckle Jr., K. A. White and R. S. Ward, Polym. Prepr. (Am. Chem. Soc., Div. Polym. Chem.), 1985, 26, 251–257 CAS.
- Q. Guo, F. Chen, K. Wang and L. Chen, J. Polym. Sci., Part B: Polym. Phys., 2006, 44, 3042–3052 CrossRef CAS.
- Z. Xu and S. Zheng, Polymer, 2007, 48, 6134–6144 CrossRef CAS.
- L. Könczöl, W. Döll, U. Buchholz and R. Mülhaupt, J. Appl. Polym. Sci., 1994, 54, 815–826 CrossRef.
- U. Buchholz and R. Mülhaupt, Polym. Prepr. (Am. Chem. Soc., Div. Polym. Chem.), 1992, 33, 205–206 CAS.
- R. Mülhaupt and U. Buchholz, Adv. Chem. Ser., 1996, 252, 75–94 CrossRef.
- K. Mimura, H. Ito and H. Fujioka, Polymer, 2001, 42, 9223–9233 CrossRef CAS.
- C. Declet-Perez, E. M. Redline, L. F. Francis and F. S. Bates, ACS Macro Lett., 2012, 1, 338–342 CrossRef CAS.
- S. Wu, Q. Guo, M. Kraska, B. Stühn and Y.-W. Mai, Macromolecules, 2013, 46, 8190–8202 CrossRef CAS.
- K. L. Hawthorne and F. C. Henson, in Epoxy Resin Chemistry II, ACS Symposium Series 221, ed. R. S. Bauer, American Chemical Society, Washington D.C., 1983 Search PubMed.
- Y. Murata, Y. Nakanishi and M. Yosumura (Shell Internationale Research B. V.), JP3315436, 2002 Search PubMed.
- H. J. Sue, J. D. Earls, R. E. Hefner Jr., M. I. Villarreal, E. I. Garcia-Meitin, P. C. Yang, C. M. Cheatham and C. J. G. Plummer, Polymer, 1998, 39, 4707–4714 CrossRef CAS.
- D. Foix, A. Serra, L. Amparore and M. Sangermano, Polymer, 2012, 53, 3084–3088 CrossRef CAS.
- F. Hofflin, L. Konczol, W. Doll, J. Morawiec and R. Mulhaupt, J. Appl. Polym. Sci., 2000, 76, 623–634 CrossRef CAS.
- A. Lutz and B. Steiner (Dow Global Technologies LLC), US8278398, 2012 Search PubMed.
- B. Burton, D. Alexander, H. Klein, A. Garibay-Vasquez, A. Pekarik and C. Henkee, Epoxy formulations using Jeffamine® polyetheramines, Huntsman Corp., The Woodlands, TX, 2005 Search PubMed.
- Y.-G. Hsu, K.-H. Lin, T.-Y. Lin, Y.-L. Fang, S.-C. Chen and Y.-C. Sung, Mater. Chem. Phys., 2012, 132, 688–702 CrossRef CAS.
- R. M. Bymark, A. L. Griggs, J. S. Mchattie and T. L. Mckenzie (Minnesota Mining and Manufacturing Company), US5407978, 1993 Search PubMed.
- H. Lützen, P. Bitomsky, K. Rezwan and A. Hartwig, Eur. Polym. J., 2013, 49, 167–176 CrossRef.
- S. Ramesh (BASF Corp.), US6569956, 1999 Search PubMed.
- D. Manjula Dhevi, S. N. Jaisankar and M. Pathak, Eur. Polym. J., 2013, 49, 3561–3572 CrossRef CAS.
- D. Foix, X. Ramis, M. Sangermano and A. Serra, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 1133–1142 CrossRef CAS.
- D. Foix, X. Fernández-Francos, X. Ramis, A. Serra and M. Sangermano, React. Funct. Polym., 2011, 71, 417–424 CrossRef CAS.
- M. Schwentenwein, Ph.D. Thesis, Vienna University of Technology, 2012.
- K. H. Chae, C. S. Lee and J. H. Kim, Polym. Adv. Technol., 2011, 22, 1427–1433 CrossRef CAS.
- B. Sandmann, B. Happ, I. Perevyazko, T. Rudolph, F. H. Schacher, S. Hoeppener, U. Mansfeld, M. D. Hager, U. K. Fischer, P. Burtscher, N. Moszner and U. S. Schubert, Polym. Chem., 2015, 6, 5273–5280 RSC.
- C. Decker, Macromolecules, 1990, 23, 5217–5220 CrossRef CAS.
- J. F. G. A. Jansen, A. A. Dias, M. Dorschu and B. Coussens, Macromolecules, 2003, 36, 3861–3873 CrossRef CAS.
- S. Kozakai, M. Asano, S. Konishi and T. Ohba (Shin-Etsu Chemical Co., Ltd.), US6668122 B2, 2002 Search PubMed.
- K. Yamazaki, M. Nishimura, T. Uemura, A. Yamamoto, S. Kozakai and M. Asano (Takeda Chemical Industries, Ltd.; Shin-Etsu Chemical Co., Ltd.), US6075065, 1997 Search PubMed.
- K.-Y. Lee, K.-Y. Kim, I.-R. Hwang, Y.-S. Choi and C.-H. Hong, Polym. Test., 2009, 29, 139–146 CrossRef.
- M. A. Zumbrum, G. L. Wilkes and T. C. Ward, in Radiation Curing in Polymer Science and Technology, ed. J. P. Fouassier and J. F. Rabek, Elsevier, London, 1993, pp. 101–151 Search PubMed.
- S. M. Gasper, D. N. Schissel, L. S. Baker, D. L. Smith, R. E. Youngman, L.-M. Wu, S. M. Sonner, R. R. Hancock, C. L. Hogue and S. R. Givens, Macromolecules, 2006, 39, 2126–2136 CrossRef CAS.
- C. Decker and K. Moussa, Eur. Polym. J., 1991, 27, 881–889 CrossRef CAS.
- C. Decker, T. Nguyen Thi Viet, D. Decker and E. Weber-Koehl, Polymer, 2001, 42, 5531–5541 CrossRef CAS.
- M. Wesley Machado Cunico and J. de Carvalho, Rapid Prototyping J., 2014, 20, 121–132 CrossRef.
- C. Plessis, G. Arzamendi, J. R. Leiza, H. A. S. Schoonbrood, D. Charmot and J. M. Asua, Macromolecules, 1999, 33, 4–7 CrossRef.
- R. S. Davidson, in Radiation Curing in Polymer Science and Technology, ed. J. P. Fouassier and J. F. Rabek, Elsevier, London, 1993, pp. 153–176 Search PubMed.
- O. D. McNair, D. P. Brent, B. J. Sparks, D. L. Patton and D. A. Savin, ACS Appl. Mater. Interfaces, 2014, 6, 6088–6097 CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Polymer, 2008, 49, 1079–1131 CrossRef CAS.
- Y. Yagci and I. Reetz, React. Funct. Polym., 1999, 42, 255–264 CrossRef CAS.
- C. N. Bowman, B. D. Fairbanks, N. B. Cramer and K. S. Anseth, Polym. Prepr. (Am. Chem. Soc., Div. Polym. Chem.), 2010, 51, 703–704 CAS.
- C. E. Hoyle and C. N. Bowman, Angew. Chem., Int. Ed., 2010, 49, 1540–1573 CrossRef CAS PubMed.
- C. E. Hoyle, T. Y. Lee and T. Roper, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 5301–5338 CrossRef CAS.
- Polymer Handbook, ed. J. Brandrup and E. H. Immergut, Wiley, Hoboken, NJ, 4th edn, 1998 Search PubMed.
- C. R. Morgan (W.R. Grace & Co.), US3784524, 1974 Search PubMed.
- P. Esfandiari, S. C. Ligon, J. J. Lagref, R. Frantz, Z. Cherkaoui and R. Liska, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 4261–4266 CrossRef CAS.
- C. E. Hoyle, T. Y. Lee and T. Roper, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 5301–5338 CrossRef CAS.
- N. B. Cramer, S. K. Reddy, A. K. O'Brien and C. N. Bowman, Macromolecules, 2003, 36, 7964–7969 CrossRef CAS.
- L. Lecamp, F. Houllier, B. Youssef and C. Bunel, Polymer, 2001, 42, 2727–2736 CrossRef CAS.
- O. D. McNair, A. P. Janisse, D. E. Krzeminski, D. E. Brent, T. E. Gould, J. W. Rawlins and D. A. Savin, ACS Appl. Mater. Interfaces, 2013, 5, 11004–11013 CAS.
- A. F. Senyurt, H. Wei, B. Phillips, M. Cole, S. Nazarenko, C. E. Hoyle, S. G. Piland and T. E. Gould, Macromolecules, 2006, 39, 6315–6317 CrossRef CAS.
- A. F. Senyurt, H. Wei, C. E. Hoyle, S. G. Piland and T. E. Gould, Macromolecules, 2007, 40, 4901–4909 CrossRef CAS.
- A. F. Senyurt, C. E. Hoyle, H. Wei, S. G. Piland and T. E. Gould, Macromolecules, 2007, 40, 3174–3182 CrossRef CAS.
- A. B. Lowe, Polymer, 2014, 55, 5517–5549 CrossRef CAS.
- B. D. Fairbanks, E. A. Sims, K. S. Anseth and C. N. Bowman, Macromolecules, 2010, 43, 4113–4119 CrossRef CAS.
- J. W. Chan, J. Shin, C. E. Hoyle, C. N. Bowman and A. B. Lowe, Macromolecules, 2010, 43, 4937–4942 CrossRef CAS.
- E. Takiyama and S. Takano (Showa Highpolymer Co., Ltd.), JP62257935A, 1987 Search PubMed.
- M. J. Kade, D. J. Burke and C. J. Hawker, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 743–750 CrossRef CAS.
- H. Lu, J. A. Carioscia, J. W. Stansbury and C. N. Bowman, Dent. Mater., 2005, 21, 1129–1136 CrossRef CAS PubMed.
- J. E. Boulden, N. B. Cramer, K. M. Schreck, C. L. Couch, C. Bracho-Troconis, J. W. Stansbury and C. N. Bowman, Dent. Mater., 2011, 27, 267–272 CrossRef CAS PubMed.
- D. P. Nair, N. B. Cramer, T. F. Scott, C. N. Bowman and R. Shandas, Polymer, 2010, 51, 4383–4389 CrossRef CAS PubMed.
- J. D. McCall and K. S. Anseth, Biomacromolecules, 2012, 13, 2410–2417 CrossRef CAS PubMed.
- D.-C. Wu, X. J. Loh, Y.-L. Wu, C. L. Lay and Y. Liu, J. Am. Chem. Soc., 2010, 132, 15140–15143 CrossRef CAS PubMed.
- M. C. Cushing and K. S. Anseth, Science, 2007, 316, 1133–1134 CrossRef CAS PubMed.
- R. T. C. Cleophas, M. Riool, H. C. Quarles van Ufford, S. A. J. Zaat, J. A. W. Kruijtzer and R. M. J. Liskamp, ACS Macro Lett., 2014, 3, 477–480 CrossRef CAS.
- T.-Y. Lin, C. S. Ki and C.-C. Lin, Biomaterials, 2014, 35, 6898–6906 CrossRef CAS PubMed.
- H. Shih and C.-C. Lin, Biomacromolecules, 2012, 13, 2003–2012 CrossRef CAS PubMed.
- X.-H. Qin, P. Gruber, M. Markovic, B. Plochberger, E. Klotzsch, J. Stampfl, A. Ovsianikov and R. Liska, Polym. Chem., 2014, 5, 6523–6533 RSC.
- X.-H. Qin, J. Torgersen, R. Saf, S. Muehleder, N. Pucher, S. C. Ligon, W. Holnthoner, H. Redl, A. Ovsianikov, J. Stampfl and R. Liska, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 4799–4810 CrossRef CAS.
- L. Maleki, U. Edlund and A.-C. Albertsson, Biomacromolecules, 2015, 16, 667–674 CrossRef CAS PubMed.
- M. Arslan, T. N. Gevrek, R. Sanyal and A. Sanyal, Eur. Polym. J., 2015, 62, 426–434 CrossRef CAS.
- L. M. Campos, I. Meinel, R. G. Guino, M. Schierhorn, N. Gupta, G. D. Stucky and C. J. Hawker, Adv. Mater., 2008, 20, 3728–3733 CrossRef CAS.
- T. S. Clark, C. E. Hoyle and S. Nazarenko, J. Coat. Technol. Res., 2008, 5, 345–351 CrossRef CAS.
- C. N. Bowman, J. Carioscia, H. Lu and J. W. Stansbury (The Regents of the University of Colorado), WO2005086911 A2, 2005 Search PubMed.
- P. Esfandiari, S. C. Ligon, J. J. Lagref, R. Frantz, Z. Cherkaoui and R. Liska, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 4261–4266 CrossRef CAS.
- A. Mautner, X. Qin, H. Wutzel, S. C. Ligon, B. Kapeller, D. Moser, G. Russmueller, J. Stampfl and R. Liska, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 203–212 CrossRef CAS.
- A. Mautner, X. Qin, B. Kapeller, G. Russmueller, T. Koch, J. Stampfl and R. Liska, Macromol. Rapid Commun., 2012, 33, 2046–2052 CrossRef CAS PubMed.
- N. B. Cramer and C. N. Bowman, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 3311 CrossRef CAS.
- H. Wei, Q. Li, M. Ojelade, S. Madbouly, J. U. Otaigbe and C. E. Hoyle, Macromolecules, 2007, 40, 8788–8793 CrossRef CAS.
- G. F. Meijs and E. Rizzardo, Makromol. Chem., Rapid Commun., 1988, 9, 547–551 CrossRef CAS.
- G. F. Meijs, E. Rizzardo and S. H. Thang, Macromolecules, 1988, 21, 3122–3124 CrossRef CAS.
- D. Colombani, Prog. Polym. Sci., 1999, 24, 425–480 CrossRef CAS.
- D. Colombani and P. Chaumont, Prog. Polym. Sci., 1996, 21, 439–503 CrossRef CAS.
- L. An, C. Gao, X. Yan, Z. Fu, W. Yang and Y. Shi, Colloid Polym. Sci., 2012, 290, 719–729 CAS.
- L. Hutson, J. Krstina, C. L. Moad, G. Moad, G. R. Morrow, A. Postma, E. Rizzardo and S. H. Thang, Macromolecules, 2004, 37, 4441–4452 CrossRef CAS.
- E. Sato, I. Uehara, H. Horibe and A. Matsumoto, Macromolecules, 2014, 47, 937–943 CrossRef CAS.
- R. Popielarz, J. Polym. Sci., Part A: Polym. Chem., 1996, 34, 3471–3484 CrossRef CAS.
- K. Tanaka and B. Yamada, Macromol. Chem. Phys., 2000, 201, 1565–1573 CrossRef CAS.
- E. Sato, P. B. Zetterlund and B. Yamada, Macromolecules, 2004, 37, 2363–2370 CrossRef CAS.
- G. D. Joly, L. R. Krepski, A. R. Fornof, S. Yurt, B. N. Gaddam, A. S. Abuelyaman and M. A. Kropp (3M Innovative Properties Company), WO2012112304 A1, 2012 Search PubMed.
- H. Y. Park, C. J. Kloxin, M. F. Fordney and C. N. Bowman, Macromolecules, 2012, 45, 5647–5652 CrossRef CAS PubMed.
- G. Moad, E. Rizzardo and S. H. Thang, Polymer, 2008, 49, 1079–1131 CrossRef CAS.
- C. M. Yakacki, R. Shandas, D. Safranski, A. M. Ortega, K. Sassaman and K. Gall, Adv. Funct. Mater., 2008, 18, 2428–2435 CrossRef CAS PubMed.
- C. J. Kloxin and C. N. Bowman, Chem. Soc. Rev., 2013, 42, 7161–7173 RSC.
- C. R. Fenoli, J. W. Wydra and C. N. Bowman, Macromolecules, 2014, 47, 907–915 CrossRef CAS.
- C. Gorsche, M. Griesser, G. Gescheidt, N. Moszner and R. Liska, Macromolecules, 2014, 47, 7327–7336 CrossRef CAS.
- H. Y. Park, C. J. Kloxin, A. S. Abuelyaman, J. D. Oxman and C. N. Bowman, Macromolecules, 2012, 45, 5640 CrossRef CAS PubMed.
- C. Gorsche, T. Koch, N. Moszner and R. Liska, Polym. Chem., 2015, 6, 2038–2047 RSC.
- T. Sato, M. Seno, M. Kobayashi, T. Kohno and H. Tanaka, Eur. Polym. J., 1995, 31, 29–34 CrossRef CAS.
- R. Liska, C. Gorsche, K. Seidler and N. Moszner (Ivoclar Vivadent AG), EP15166848, 2015 Search PubMed.
- S. C. Ligon, K. Seidler, C. Gorsche, M. Griesser, N. Moszner and R. Liska, J. Polym. Sci., Part A: Polym. Chem., 2015 DOI:10.1002/pola.27788.
- F. A. Leibfarth, K. M. Mattson, B. P. Fors, H. A. Collins and C. J. Hawker, Angew. Chem., Int. Ed., 2013, 52, 199–210 CrossRef CAS PubMed.
- C. Boyer, V. Bulmus, T. P. Davis, V. Ladmiral, J. Liu and S. Perrier, Chem. Rev., 2009, 109, 5402–5436 CrossRef CAS PubMed.
- J. Xu, K. Jung and C. Boyer, Macromolecules, 2014, 47, 4217–4229 CrossRef CAS.
- T. G. Ribelli, D. Konkolewicz, S. Bernhard and K. Matyjaszewski, J. Am. Chem. Soc., 2014, 136, 13303–13312 CrossRef CAS PubMed.
- H. Fischer, Chem. Rev., 2001, 101, 3581–3610 CrossRef CAS PubMed.
- G. Morales, R. Díaz de León, P. Acuña, R. Flores Flores and A. Montalvo Robles, Polym. Eng. Sci., 2006, 46, 1333–1341 CAS.
- B. J. P. Jansen, S. Rastogi, H. E. H. Meijer and P. J. Lemstra, Macromolecules, 1999, 32, 6283–6289 CrossRef CAS.
- K. Dean and W. D. Cook, Macromolecules, 2002, 35, 7942–7954 CrossRef CAS.
- B. J. P. Jansen, S. Rastogi, H. E. H. Meijer and P. J. Lemstra, Macromolecules, 1999, 32, 6290–6297 CrossRef CAS.
- R. Acosta Ortiz, A. García Valdez, A. Navarro Tovar, A. Hilario de la Cruz, L. González Sánchez, J. García Trejo, J. Espinoza Muñoz and M. Sangermano, J. Polym. Res., 2014, 21, 1–11 CAS.
- T. Inoue, Prog. Polym. Sci., 1995, 20, 119–153 CrossRef CAS.
- Y. Liu, J. Appl. Polym. Sci., 2013, 127, 3279–3292 CrossRef CAS.
- N. J. Crawford, M. D. Dadmun, T. J. Bunning and L. V. Natarajan, Polymer, 2006, 47, 6311–6321 CrossRef CAS.
- C. R. Szczepanski, C. S. Pfeifer and J. W. Stansbury, Polymer, 2012, 53, 4694–4701 CrossRef CAS PubMed.
- C. R. Szczepanski and J. W. Stansbury, Polymer, 2015, 70, 8–18 CrossRef CAS PubMed.
- E. B. Murphy and F. Wudl, Prog. Polym. Sci., 2010, 35, 223–251 CrossRef CAS.
- X. Chen, F. Wudl, A. K. Mal, H. Shen and S. R. Nutt, Macromolecules, 2003, 36, 1802–1807 CrossRef CAS.
- T. A. Plaisted and S. Nemat-Nasser, Acta Mater., 2007, 55, 5684–5696 CrossRef CAS.
- C.-M. Chung, Y.-S. Roh, S.-Y. Cho and J.-G. Kim, Chem. Mater., 2004, 16, 3982–3984 CrossRef CAS.
- L. Yao, Y. C. Yuan, M. Z. Rong and M. Q. Zhang, Polymer, 2011, 52, 3137–3145 CrossRef CAS.
- S. J. Leigh, J. Bowen, C. P. Purssell, J. A. Covington, D. R. Billson and D. A. Hutchins, Bioinspir. Biomim., 2012, 7, 044001 CrossRef CAS PubMed.
- S. C. Ligon, B. Husár, H. Wutzel, H. Hoffmann, J. Torgersen and R. Liska, in RadTech Asia, Shanghai, CN, 2013 Search PubMed.
- S. Mauro, in RadTech Europe, Basel, CH, 2011, p. id 6.4 Search PubMed.
- M. Pavlič, K. Borut and M. Petrič, in PRA's 8th International Woodcoatings Congress, Amsterdam, NL, 2012.
- I. V. Khudyakov, M. B. Purvis and R. J. Overton (Alcatel), US6489376, 2002 Search PubMed.
- P. J. Shustack and D. A. Wilson (Borden Chemical, Inc.), US6048911, 2000 Search PubMed.
- D. S. Lee, K. B. Min, J. M. Ryu, H. W. Choi and Y. M. Kim (Luvantix Co., Ltd.), WO2005 0715053, 2005 Search PubMed.
- M. Bahadur, S. O. n. Glover and A. W. Norris (Dow Corning Corporation), US8861916, 2014 Search PubMed.
- T. K. Hilty, H. M. Bank, A. Revis and D. P. Jones (Dow Corning), EP0424000 A2, 1991 Search PubMed.
- B. Goss, Int. J. Adhes. Adhes., 2002, 22, 405–408 CrossRef CAS.
- H. K. Kim, J. G. Kim, J. D. Cho and J. W. Hong, Polym. Test., 2003, 22, 899–906 CrossRef CAS.
- T. Billiet, M. Vandenhaute, J. Schelfhout, S. Van Vlierberghe and P. Dubruel, Biomaterials, 2012, 33, 6020–6041 CrossRef CAS PubMed.
- M. H. Gabr, M. A. Elrahman, K. Okubo and T. Fujii, J. Mater. Sci., 2010, 45, 3841–3850 CrossRef CAS.
- D. Mao, H. Zou, M. Liang, S. Zhou and Y. Chen, J. Appl. Polym. Sci., 2014, 131, 39797 Search PubMed.
- S. A. Xu, G. T. Wang and Y. W. Mai, J. Mater. Sci., 2013, 48, 3546–3556 CrossRef CAS.
- R. Thomas, C. Sinturel, J. Pionteck, H. Puliyalil and S. Thomas, Ind. Eng. Chem. Res., 2012, 51, 12178–12191 CAS.
- W. Zhou and J. Cai, J. Appl. Polym. Sci., 2012, 124, 4346–4351 CrossRef CAS.
- R. Thomas, A. Boudenne, L. Ibos, Y. Candau and S. Thomas, J. Appl. Polym. Sci., 2010, 116, 3232–3241 CAS.
- R. Thomas, S. Durix, C. Sinturel, T. Omonov, S. Goossens, G. Groeninckx, P. Moldenaers and S. Thomas, Polymer, 2007, 48, 1695–1710 CrossRef CAS.
- ProJet® 6000 & 7000 Professional 3D Printers, 3DSystems Inc., Rock Hill, SC, 2015.
- PolyJet™ Materials Data Set, Stratasys Ltd., Rehovot, IL, 2014.
- Somos® Stereolithography Materials Selector Guide, DSM Desotech, Elgin, IL, 2015.
- G. Gao, G. Du, Y. Cheng and J. Fu, J. Mater. Chem. B, 2014, 2, 1539–1548 RSC.
- B. De and N. Karak, J. Appl. Polym. Sci., 2014, 131, 40327 CrossRef.
- D. Ratna and A. K. Banthia, Polym. Int., 2000, 49, 281–287 CrossRef CAS.
- E. E. La Fleur (Rohm and Haas Company), US20030050397 A1, 2003 Search PubMed.
- A. R. Curtis, W. M. Palin, G. J. P. Fleming, A. C. C. Shortall and P. M. Marquis, Dent. Mater., 2009, 25, 180–187 CrossRef CAS PubMed.
- J. W. Stansbury, C. N. Bowman and M. Trujillo (The Regents of the University of Colorado), WO2005107626 A1, 2005 Search PubMed.
- C. N. Bowman, H. Lu and J. W. Stansbury (The Regents of the University of Colorado), WO2005041807 A1, 2005 Search PubMed.
- C. N. Bowman and N. B. Cramer (University of Colorado), WO2010114760 A1, 2010 Search PubMed.
- C. N. Bowman and N. B. Cramer (University of Colorado), US20130123381 A1, 2013 Search PubMed.
- C. N. Bowman, C. Kloxin, H. Y. Park and D. Leung (University of Colorado), US20120202917 A1, 2012 Search PubMed.
- A. S. Abuelyaman, J. D. Oxman, Y. Wang, C. N. Bowman, H. Y. Park and C. J. Kloxin (3M Innovative Properties Company, The Regents of the University of Colorado), US20120295228A1, 2012 Search PubMed.
- C. Bowman and D. Leung (University of Colorado), US20130096219 A1, 2013 Search PubMed.
- G. D. Joly, A. S. Abuelyaman, B. D. Craig, A. Falsafi, J. D. Oxman, L. R. Krepski, W. H. Moser and S. Yurt (3M Innovative Properties Company), WO2013028397 A2, 2013 Search PubMed.
- W. H. Moser, G. D. Joly, A. R. Fornof, A. S. Abuelyaman, A. Falsafi and L. R. Krepski (3M Innovative Properties Company), WO2014151363 A1, 2014 Search PubMed.
- G. D. Joly, A. S. Abuelyaman, A. R. Fornof, B. D. Craig, L. R. Krepski, W. H. Moser, S. Yurt and J. D. Oxman (3M Innovative Properties Company), WO2014074373 A1, 2014 Search PubMed.
- G. D. Joly, A. S. Abuelyaman, A. R. Fornof, B. D. Craig, L. R. Krepski, W. H. Moser, S. Yurt and J. D. Oxman (3M Innovative Properties Company), WO2014074427 A1, 2014 Search PubMed.
- G. D. Joly, L. R. Krepski, B. N. Gaddam, A. S. Abuelyaman, B. D. Craig, T. D. Dunbar, C. Cao, J. D. Oxman, A. Falsafi, W. H. Moser and H. T. Bui (3M Innovative Properties Company), WO2012112350 A2, 2012 Search PubMed.
- A. R. Fornof, W. H. Moser, L. R. Krepski, G. D. Joly, A. S. Abuelyaman, A. Falsafi and B. N. Gaddam (3M Innovative Properties Company), WO2014099317A1, 2014 Search PubMed.
- R. Bibb, D. Eggbeer, P. Evans, A. Bocca and A. Sugar, Rapid Prototyping J., 2009, 15, 346–354 CrossRef.
- F. P. W. Melchels, M. A. N. Domingos, T. J. Klein, J. Malda, P. J. Bartolo and D. W. Hutmacher, Prog. Polym. Sci., 2012, 37, 1079–1104 CrossRef CAS.
- F. P. W. Melchels, J. Feijen and D. W. Grijpma, Biomaterials, 2010, 31, 6121–6130 CrossRef CAS PubMed.
- B. Husár, C. Heller, M. Schwentenwein, A. Mautner, F. Varga, T. Koch, J. Stampfl and R. Liska, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 4927–4934 CrossRef.
- C. Heller, M. Schwentenwein, G. Russmüller, T. Koch, D. Moser, C. Schopper, F. Varga, J. Stampfl and R. Liska, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 650–661 CrossRef CAS.
- S. Baudis, F. Nehl, S. C. Ligon, A. Nigisch, H. Bergmeister, D. Bernhard, J. Stampfl and R. Liska, Biomed. Mater., 2011, 6, 055003 CrossRef PubMed.
- J. Iqbal, Y. Onuma, J. Ormiston, A. Abizaid, R. Waksman and P. Serruys, Eur. Heart J., 2014, 35, 765–776 CrossRef PubMed.
- S. Baudis, S. C. Ligon, K. Seidler, G. Weigel, C. Grasl, H. Bergmeister, H. Schima and R. Liska, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 1272–1280 CrossRef CAS.
- S. A. Guelcher, Tissue Eng., Part B, 2008, 14, 3–17 CrossRef CAS PubMed.
- M. Yeganegi, R. A. Kandel and J. P. Santerre, Acta Biomater., 2010, 6, 3847–3855 CrossRef CAS PubMed.
- P. M. Crapo, T. W. Gilbert and S. F. Badylak, Biomaterials, 2011, 32, 3233–3243 CrossRef CAS PubMed.
- P. Martens and K. S. Anseth, Polymer, 2000, 41, 7715–7722 CrossRef CAS.
- K. A. Davis, J. A. Burdick and K. S. Anseth, Biomaterials, 2003, 24, 2485–2495 CrossRef CAS PubMed.
- X.-H. Qin, J. Torgersen, R. Saf, S. Muehleder, N. Pucher, S. C. Ligon, W. Holnthoner, H. Redl, A. Ovsianikov, J. Stampfl and R. Liska, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 4799–4810 CrossRef CAS.
- Y. Tanaka, K. Fukao and Y. Miyamoto, Eur. Phys. J. E, 2000, 3, 395–401 CrossRef CAS.
- N. Annabi, A. Tamayol, J. A. Uquillas, M. Akbari, L. E. Bertassoni, C. Cha, G. Camci-Unal, M. R. Dokmeci, N. A. Peppas and A. Khademhosseini, Adv. Mater., 2014, 26, 85–124 CrossRef CAS PubMed.
- Y. Tanaka, J. P. Gong and Y. Osada, Prog. Polym. Sci., 2005, 30, 1–9 CrossRef CAS.
- M. A. Haque, T. Kurokawa and J. P. Gong, Polymer, 2012, 53, 1805–1822 CrossRef CAS.
- J. P. Gong, Y. Katsuyama, T. Kurokawa and Y. Osada, Adv. Mater., 2003, 15, 1155–1158 CrossRef CAS.
- Y. Na, Korea-Aust. Rheol. J., 2013, 25, 185–196 CrossRef.
- C. E. Hoyle and C. N. Bowman, Angew. Chem., Int. Ed., 2010, 49, 1540–1573 CrossRef CAS PubMed.
- M. A. Azagarsamy and K. S. Anseth, ACS Macro Lett., 2012, 2, 5–9 CrossRef PubMed.
- J. D. McCall and K. S. Anseth, Biomacromolecules, 2012, 13, 2410–2417 CrossRef CAS PubMed.
- B. D. Fairbanks, M. P. Schwartz, A. E. Halevi, C. R. Nuttelman, C. N. Bowman and K. S. Anseth, Adv. Mater., 2009, 21, 5005–5010 CrossRef CAS PubMed.
- M. Malkoch, R. Vestberg, N. Gupta, L. Mespouille, P. Dubois, A. F. Mason, J. L. Hedrick, Q. Liao, C. W. Frank, K. Kingsbury and C. J. Hawker, Chem. Commun., 2006, 2774–2776 RSC.
- J. Cui, M. A. Lackey, G. N. Tew and A. J. Crosby, Macromolecules, 2012, 45, 6104–6110 CrossRef CAS.
- Y. Fan, C. Deng, R. Cheng, F. Meng and Z. Zhong, Biomacromolecules, 2013, 14, 2814–2821 CrossRef CAS PubMed.
Footnote |
| † This paper is dedicated to our friend and colleague Dr Branislav Husár who passed away in the autumn of 2014. Dr Husár was an innovative scientist with a cool head and a warm heart. He was a recent recipient of a Marie Curie grant with a steadily growing list of publications. We are left to contemplate on what was surely to be a great scientific career that sadly the world will never know. |
| This journal is © The Royal Society of Chemistry 2016 |