
Helical polymer/Fe3O4 NPs constructing optically active, magnetic core/shell microspheres: preparation by emulsion polymerization and recycling application in enantioselective crystallization†
Huaiyu
Chen
ab,
Jinyong
Zhou
ab and
Jianping
Deng
*ab
aState Key Laboratory of Chemical Resource Engineering, Beijing University of Chemical Technology, Beijing 100029, China. E-mail: dengjp@mail.buct.edu.cn
bCollege of Materials Science and Engineering, Beijing University of Chemical Technology, Beijing 100029, China
First published on 14th October 2015
Abstract
Hybrid materials combining chirality and magneticity are stimulating much interest in diverse research areas. This article reports the preparation of a new type of optically active, magnetic Fe3O4 NP@polyacetylene core/shell microspheres (Fe3O4@PA MPs) consisting of magnetic Fe3O4 NPs as the core and helical polyacetylene as the shell. The Fe3O4@PA MPs integrate two significant concepts, “macromolecular helicity-derived chirality” and “magneticity” in one single microsphere entity. The composite MPs were prepared by emulsion polymerization approach and characterized by TEM, XRD, FT-IR, VSM, TGA, circular dichroism and UV-vis absorption spectroscopy techniques. They simultaneously showed fascinating optical activity and considerable magneticity. The MPs were further used as a chiral additive to induce enantioselective crystallization of racemic threonine. L-Threonine was preferentially induced to form rectangular-shaped crystals with an enantiomeric excess up to 90% (after enantioselective crystallization twice). The microspheres can be recycled conveniently with the assistance of an external magnetic field, demonstrating the MPs’ significant potential applications in chiral fields.
Introduction
Core/shell structured composite particles, which excellently combine both the advantages of the core and the shell components, are gathering rapidly increasing interest due to the ready control over their composition, morphology, and properties.1 Recently, extensive efforts have been focused on the design and synthesis of core/shell particles possessing multiple functions, well exemplified by magnetic core/shell particles.2–5 The particles constructed by a magnetic core and a polymer/inorganic shell possess interesting properties, such as superparamagnetic properties and biocompatibility,6 making the particles full of promise for significant applications as isolating materials for biomolecules,3 catalysts,6 magnetic resonance imaging contrast agents,7 targeted drug delivery media,8etc. As a novel kind of magnetic material, composite particles simultaneously showing optical activity and magneticity have found various applications in chiral-related fields. For instance, chiral catalysts were immobilized on magnetic spheres and applied in asymmetric catalyses.9–13 Magnetic particles were chirally functionalized with chiral selectors and used in chiral separation processes.14–16 Regrettably, the chirality in such composite particles was primarily limited to chiral small molecules and biomacromolecules.17–20 In a lot of cases, synthetic helical polymers are more competitive than small chiral compounds for the well-known “chiral amplification” effect.21 In this context, we began our efforts to establish chiral magnetic composite particles by taking advantage of chiral helical substituted polyacetylenes. In the preceding study, we prepared polyacetylene/Fe3O4 composite microspheres by precipitation polymerization and applied them in inducing alanine towards enantioselective crystallization.22 However, the Fe3O4 NPs were randomly distributed in the microspheres (including the surface), so a further extension in applying the chiral composite microspheres is significantly limited. In order to improve the structure, performance, and particularly the potential applications of the optically active, magnetic microspheres, core/shell structured microspheres were designed and successfully synthesized in the present work.Synthetic helical polymers have received a great deal of attention owing to their distinctive helical structures and the corresponding properties that cannot be found in ordinary polymers. Helical polymers have visually elegant structures and possess fascinating optical activity,23 chiral resolution ability,24etc. Over the past two decades, a series of helical polymers were synthesized,25–40 among which helical polyacetylenes are the most intensively explored. Since the pioneering work of Heeger, MacDiarmid, and Shirakawa41–43 on polyacetylenes, considerable efforts have been devoted to the polymer and its derivatives, in particular the helical substituted polyacetylenes in which the conjugated polymer main chains adopt helical conformations while the pendant groups endow the desired functions.44–47 Although helical polyacetylenes possess fascinating properties, they frequently suffer from disadvantages like insufficient solubility, thermal instability and/or unsatisfactory processability.48 Therefore it is of great significance to find effective ways to circumvent the disadvantages. Design and synthesis of acetylene-based nanoparticles (NPs) seem to be an efficient and facile approach to overcome these limitations. The Mecking group49 has made breakthroughs in preparing acetylene-based polymer NPs. We also have prepared optically active helical substituted polyacetylene particles via emulsion polymerization,50 precipitation polymerization,51 and helix-sense-selective polymerizations of achiral monomers.52,53 Particularly, we synthesized core/shell polyacetylene NPs composed of optically active helical polyacetylene cores and different shells.1,54 The helical polyacetylene NPs promisingly find remarkable applications in enantioselective crystallization,54 chirally-controlled release,55 chiral catalysis,56 among the other significant applications. Unfortunately, the recovery of the chiral NPs after use remains challenging, a big issue in terms of practical applications. Endowing the optically active core/shell particles with magnetic properties will undoubtedly help improve their recoverability.
In the present study, we synthesized optically active, magnetic core/shell structured Fe3O4@polyacetylene microspheres (defined as Fe3O4@PA MPs) by taking magnetic Fe3O4 NPs as the core and helical substituted polyacetylene as the shell. The composite MPs integrated the magneticity of the Fe3O4 core and the intriguing optical activity of the helical polymer shell. Accordingly, the core/shell MPs in theory shall possess both rapid magnetic responsivity and optical activity. Excitingly, this hypothesis was validated in the present work. We further employed the resulting MPs for performing enantioselective crystallization by taking rac-threonine as a model compound. L-Threonine was found to be preferentially induced to crystallize. It is worth emphasizing that the composite MPs can be easily recycled with the help of a magnetic field after the enantioselective crystallization process.
Experimental
Materials
FeCl3·6H2O, anhydrous sodium acetate (NaAc), polyethylene glycol, ethylene glycol, anhydrous ethanol, polyethylene glycol tert-octylphenyl ether (denoted as Triton X-100), and N,N-dimethylformamide (DMF) were purchased from Beijing Chemical Reagents Company (China) and used as received. Propargylamine, isobutyl chloroformate (Alfa Aesar), 4-methylmorpholine (Alfa Aesar), and (1S)-(−)-camphanic acid (Aldrich) were used directly. Phosphotungstic acid (Alfa Aesar) was used to prepare 1.5% aqueous solution (pH = 6.4). O-Propargyloxy-N-triethoxysilylpropyl urethane (denoted as OPNTU silane) was purchased from Gelest Inc. (nbd)Rh+B−(C6H5)4 was prepared as reported.57 A substituted acetylene monomer (M1, as structurally presented in Scheme 1) was synthesized by following a procedure in the literature.58 Dibutynyl adipate (M2, Scheme 1) was synthesized according to the literature and used as a cross-linking agent.59D- and L-Threonine (Aladdin) were used without further purification. The solvents were purified by distillation under reduced N2 pressure. Freshly deionized water was used in all experiments. | ||
| Scheme 1 Schematic illustration for preparing the optically active, magnetic Fe3O4@PA core/shell microspheres (Fe3O4@PA MPs). | ||
Measurements
Fourier transform infrared (FT-IR) spectra were recorded with a Nicolet NEXUS 670 spectrophotometer (KBr tablet). Circular dichroism (CD) and UV-vis absorption spectra were recorded on a JASCO J-810 spectropolarimeter. Powder X-ray diffraction (XRD) patterns were recorded on a D/max2500 VB2+/PC X-ray diffractometer (Rigaku) using Cu Kα radiation. Transmission electron microscopy (TEM) images were obtained on an H-800 (Hitachi) transmission electron microscope at an accelerating voltage of 200 kV. Aqueous phosphotungstic acid (1.5%) was used to stain the microspheres. The morphology of the threonine crystals was observed with a Hitachi S-4800 scanning electron microscope (SEM). Thermogravimetric analysis (TGA) was carried out with a Mettler Toledo TGA at a scanning rate of 10 °C min−1 in air. Magnetic characterization was carried out on a vibrating sample magnetometer (VSM, LakeShore 7410 VSM) at room temperature. Specific rotations were measured on a JASCO P-1020 digital polarimeter with a sodium lamp (λ = 589 nm) as the light source at room temperature.Synthesis and modification of Fe3O4 nanoparticles (Fe3O4 NPs)
The magnetic Fe3O4 NPs were synthesized through a modified solvothermal reaction according to the reported method.60 Briefly, 1.35 g of FeCl3·6H2O was dissolved in 40 ml of ethylene glycol. Subsequently, 3.6 g of NaAc and 1.0 g of polyethylene glycol were added, and then the mixture was stirred vigorously. One hour later, the mixture was transferred into a Teflon lined stainless steel autoclave (50 ml capacity), heated to 200 °C and then maintained at this temperature for 15 h. After the autoclave was cooled to room temperature, the Fe3O4 NPs were isolated and rinsed successively with deionized water and ethanol three times with the help of a magnet. After washing and separating, the Fe3O4 NPs were dried under vacuum at 60 °C for 24 h. Afterwards, the Fe3O4 NPs were treated with OPNTU silane as follows. 0.3 g of Fe3O4 NPs was dispersed homogeneously in 50 ml of toluene assisted with ultrasound, followed by addition of 1 ml of OPNTU silane. After mechanically stirring at 70 °C for 24 h, OPNTU silane modified Fe3O4 NPs (alkynyl-Fe3O4 NPs) were harvested. The NPs were rinsed with ethanol three times and then dried under vacuum at 60 °C for 24 h.Synthesis of Fe3O4@polyacetylene core/shell microspheres (Fe3O4@PA MPs)
The Fe3O4@PA core/shell MPs were prepared via a seeded emulsion polymerization approach. The major procedure is as follows. The polymerization was performed in a three-necked flask equipped with a N2 inlet, a mechanical stirrer, and a condenser. In the flask, 3.50 g (5.4 mmol) of Triton X-100 was dissolved completely in 17 ml of deionized water, followed by the addition of 0.012 g of alkynyl-Fe3O4 NPs. The mixture dispersion was mechanically stirred and deoxygenated by pursing with N2 for 30 min. Then M1 (0.105 g, 0.45 mmol per 1 ml DMF) and M2 (0.0225 g, 0.09 mmol per 1 ml DMF) were added dropwise in the dispersion, followed by the addition of (nbd)Rh+B−(C6H5)4 catalyst (0.0032 g, 0.0063 mmol per 1 ml DMF). After the charge of the catalyst solution, polymerization proceeded at 30 °C for 3 h. After that, the Fe3O4@PA core/shell MP emulsion was formed. In order to acquire pure Fe3O4@PA MPs, the microspheres were isolated and washed with deionized water three times with the help of a magnet. The microspheres were re-dispersed into a certain amount of deionized water. For preparation of pure substituted polyacetylene nanoparticles (PA NPs), only acetylene monomer (M1) was added into the polymerization system, i.e. without adding alkynyl-Fe3O4 NPs and M2 (the crosslinking agent), while keeping the other conditions unchanged. In order to acquire pure particles from the original polymer emulsions, Triton X-100 was repeatedly removed by centrifugation (20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm).
000 rpm).
Enantioselective crystallization by using Fe3O4@PA MPs
Enantioselective crystallization experiments were performed as follows.61 A supersaturated solution of racemic threonine was prepared by dissolving D- and L-threonine (each 2.7 g) in water (20 ml), and the solution was heated to 70 °C under stirring till complete dissolution. Afterwards, 20 mg of Fe3O4@PA MPs was added into the above racemic threonine supersaturated solution and stirred for about 20 min. The solution was then left for ambient cooling to room temperature (approximately 25 °C) in 2 h. To monitor the crystallization process within 24 h, a small amount of the formed crystals was taken out at certain intervals. After drying, the crystals were subjected to XRD and CD spectroscopy analyses. Meanwhile, about 0.5 mL solution was taken from the residual threonine solution at the same intervals, then dried under vacuum at 40 °C for 24 h, and subjected to measurements. 0.06 g of the threonine crystals acquired at different times was re-dissolved in water (10 ml) to form an aqueous solution of the same concentration (0.05 mol L−1). Through optical rotation measurement of the residual solution samples, we determined their specific rotation ([α]). The enantiomeric excess (ee) was calculated according to: ee (%) = [α]/[αmax] × 100%, where [α] is the measured specific rotation of the sample and [αmax] is the specific rotation of the enantiopure threonine measured under the same conditions (0.05 mol L−1, in water).62 It was found that in the first cycle of crystallization, L-threonine was preferentially induced to crystallize (ee = 25%, more details are presented below).After enantioselective crystallization, threonine crystals filtered from the residual solution were re-dissolved in water. The Fe3O4@PA MPs originally coated by the resulting crystals were separated by a magnet, washed with water three times, and dried at 40 °C for 24 h. The collected Fe3O4@PA MPs were reused again for conducting a second cycle of enantioselective crystallization. In the second enantioselective crystallization, a predetermined amount of L-threonine crystals obtained in the first cycle was dissolved in water to form a threonine supersaturated aqueous solution while the other experimental conditions remained the same as in the first cycle. For comparison, achiral alkynyl-Fe3O4 NPs were also utilized as additives instead of chiral Fe3O4@PA MPs for crystallization of racemic threonine in the same manner.
Results and discussion
Preparation of Fe3O4@PA core/shell MPs
The synthesis procedure for the optically active, magnetic Fe3O4@PA core/shell MPs (Fe3O4@PA MPs) is illustrated in Scheme 1. The procedure involves two major steps. Firstly, Fe3O4 NPs were synthesized and then modified by OPNTU silane, offering triple bond modified Fe3O4 NPs (alkynyl-Fe3O4 NPs). Secondly, Fe3O4@PA MPs were prepared via a seeded emulsion polymerization route by using the alkynyl-Fe3O4 NPs as seeds, Triton X-100 as the emulsifier, M1 as the substituted acetylene monomer, and dibutynyl adipate (M2) as the crosslinking agent. The practice successfully provided the anticipated core/shell MPs, as reported below. The resulting Fe3O4 NPs, OPNTU silane-modified Fe3O4 NPs (alkynyl-Fe3O4 NPs), and optically active, magnetic Fe3O4@PA MPs were characterized by TEM, SEM, XRD, FTIR, VSM, TG, CD and UV-vis absorption measurements. All the characterization will be reported in the following sections.The morphology and the size of the Fe3O4 NPs and the Fe3O4@PA MPs were characterized by TEM (Fig. 1) and SEM (Fig. S1 in the ESI†). Fig. 1a displays the typical TEM images of Fe3O4 NPs. The average diameter is ca. 280 nm. The morphology and size of the Fe3O4@PA MPs are shown in Fig. 1b, in which the average diameter of the composite microspheres is approximately 1050 nm. More noticeably, the core/shell structures can be observed. To make the core/shell structure more clear, phosphotungstic acid was used to stain the polyacetylene shell, so the PA shells seem to be much darker than the Fe3O4 cores. In contrast, the average diameter of the pure polyacetylene nanoparticles (PA NPs), which were prepared under similar conditions and taken as a control sample, was only about 680 nm (Fig. 1c). Similar results were obtained in SEM images (Fig. S1†). A comparison of the TEM and SEM images definitely demonstrates that in the Fe3O4@PA core/shell MPs, the substituted polyacetylene was successfully attached onto the surface of the Fe3O4 NPs.
 | ||
| Fig. 1 Typical TEM images of (a) Fe3O4 NPs, (b) optically active, magnetic Fe3O4@PA core/shell composite MPs, stained by aqueous phosphotungstic acid solution (conc., 1.5%) (inset, enlarged one microsphere) and (c) pure substituted polyacetylene NPs. | ||
Fig. 2 shows the XRD patterns of (a) Fe3O4 NPs, (b) optically active, magnetic Fe3O4@PA core/shell MPs, and (c) pure polyacetylene NPs as a reference. Diffraction peaks (111), (200), (311), (222), (400), (422), (511), and (440) shown in Fig. 2a, and can be indexed as face centered cubic Fe3O4 (the Joint Committee on Powder Diffraction Standards (JCPDS) reference (no. 19-0629)). Fig. 2b shows the XRD pattern of Fe3O4@PA MPs. All the diffraction peaks of Fe3O4 NPs shown in Fig. 2a can be found in Fig. 2b. The XRD pattern of the pure PA NPs has a wider peak at about 2θ = 19.20 in Fig. 2c. Accordingly, the wider peak at 2θ = 19.26 shown in Fig. 2b originates from amorphous substituted polyacetylene shells. The observations further show us that Fe3O4@PA MPs were successfully prepared.
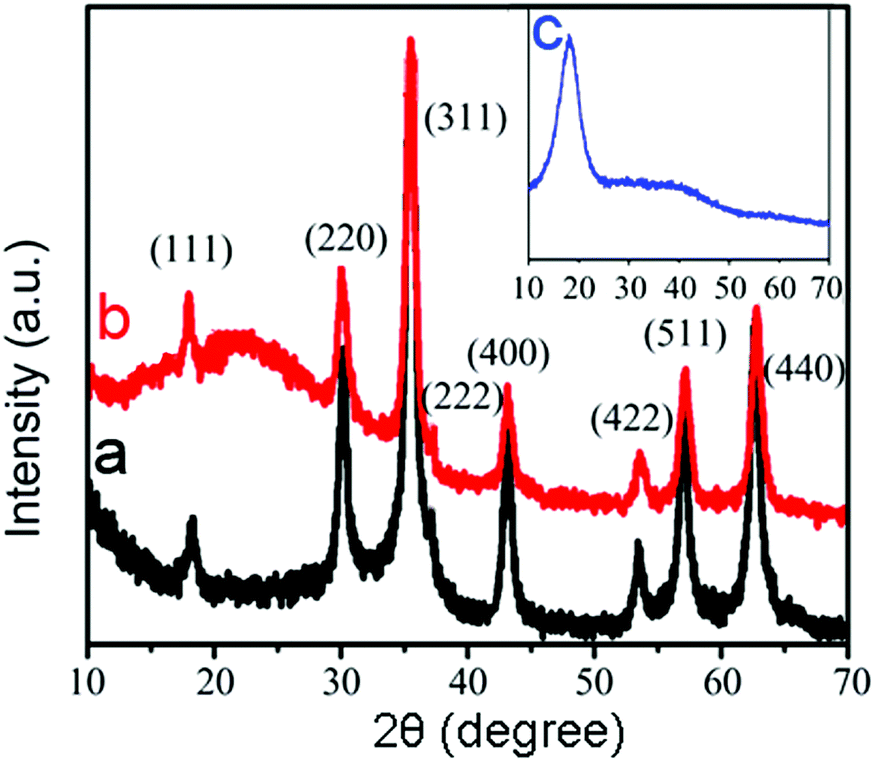 | ||
| Fig. 2 XRD patterns of (a) Fe3O4 NPs, (b) optically active, magnetic Fe3O4@PA core/shell MPs, and (c) pure substituted polyacetylene NPs. | ||
Next, the products were subjected to FT-IR spectra measurements, as shown in Fig. 3. The vibrational absorption peak at 588 cm−1 (Fig. 3a) can be assigned to the characteristic of the Fe–O stretching vibration.63 The absorption peaks corresponding to Si–O (1128 cm−1) and C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C (2103 cm−1) can be found in the spectrum of alkynyl-Fe3O4 NPs (Fig. 3b), indicating that the Fe3O4 NPs were successfully functionalized by alkynyl moieties. The FT-IR spectrum of the Fe3O4@PA core/shell MPs is presented in Fig. 3c, in which the new peaks at 1525, 1665, and 1790 cm−1 are attributed to the amide I band (C
C (2103 cm−1) can be found in the spectrum of alkynyl-Fe3O4 NPs (Fig. 3b), indicating that the Fe3O4 NPs were successfully functionalized by alkynyl moieties. The FT-IR spectrum of the Fe3O4@PA core/shell MPs is presented in Fig. 3c, in which the new peaks at 1525, 1665, and 1790 cm−1 are attributed to the amide I band (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching), amide II band (N–H bending), and CO stretching of the lactone group (in the M1 units). The peak at 1740 cm−1 corresponds to the ester group in both M1 units and the cross-linking agent (M2). The peaks provide evidence for the presence of M1 units and cross-linking agent units in the composite microspheres.57 The FT-IR spectra further prove that magnetic Fe3O4 NPs and helical substituted polyacetylene chains are both present in the composite microspheres. For this conclusion, CD and UV-vis spectra will offer more information, and will be discussed later.
O stretching), amide II band (N–H bending), and CO stretching of the lactone group (in the M1 units). The peak at 1740 cm−1 corresponds to the ester group in both M1 units and the cross-linking agent (M2). The peaks provide evidence for the presence of M1 units and cross-linking agent units in the composite microspheres.57 The FT-IR spectra further prove that magnetic Fe3O4 NPs and helical substituted polyacetylene chains are both present in the composite microspheres. For this conclusion, CD and UV-vis spectra will offer more information, and will be discussed later.
 | ||
| Fig. 3 FT-IR spectra of (a) Fe3O4 NPs, (b) alkynyl-Fe3O4 NPs, (c) optically active, magnetic Fe3O4@PA core/shell MPs, and (d) pure polyacetylene NPs (KBr tablet). | ||
The thermal properties of the Fe3O4@PA MPs and the PA NPs were measured by the TGA technique, as illustrated in Fig. 4. Compared to the original Fe3O4 NPs, the TGA curve of the alkynyl-Fe3O4 NPs showed a weight loss of 9.6% (Fig. 4b). The weight loss is due to OPNTU silane, which further confirms that the Fe3O4 NPs were successfully functionalized by alkynyl moieties. Fe3O4@PA MPs showed a weight loss of 85% when the temperature was increased from 100 to 700 °C (Fig. 4c). So the residual amount of Fe3O4@PA MPs was about 15%. This value is reasonably much higher than that of the pure PA NPs (Fig. 4d).
 | ||
| Fig. 4 TGA curves of (a) Fe3O4 NPs, (b) alkyne-Fe3O4 NPs, (c) Fe3O4@PA MPs, and (d) pure substituted polyacetylene NPs. All the TGA curves were measured at a scanning rate of 10 °C min−1 in air. | ||
Our previous investigations demonstrate that circular dichroism (CD) and UV-vis absorption spectroscopy are effective techniques for identifying the helical structures and optical activity of substituted polyacetylenes and the particles thereof.50–54,58 The optically active, magnetic Fe3O4@PA core/shell MPs and the pure PA NPs were thus subjected to CD and UV-vis spectrum measurements. To characterize the optical activity of the Fe3O4@PA MPs, we obtained pure microspheres by isolating them from the emulsions and washed them three times with H2O with the aid of a magnet. The Fe3O4@PA MPs were dispersed again in water and then subjected to measurements. The CD spectra of the Fe3O4@PA MPs dispersion in water are presented in Fig. 5a, while the corresponding UV-vis spectra are shown in Fig. 5b. The CD signals and UV-vis absorption of Fe3O4@PA MPs can be observed around at 340 nm, quite similar to the CD signal and UV-vis absorption of the pure PA NPs. This further identifies that the helical polyacetylene was successfully coated on Fe3O4 NPs. According to our earlier studies dealing with helical substituted polyacetylenes,22 we conclude that the Fe3O4@PA MPs were composed of helical polymers with predominantly one-handed screw sense and accordingly the MPs possessed considerable optical activity. We further infer that the presence of Fe3O4 cores did not affect the polymers in terms of forming helical structures of preferential helicity. It should be pointed out that in Fig. 5, both the CD signal and UV-vis absorption of the chiral composite NPs are weaker relative to the pure polymer NPs. This results from two aspects: the relatively low content of helical polymers in the composite MPs and the spectra were just qualitatively measured.
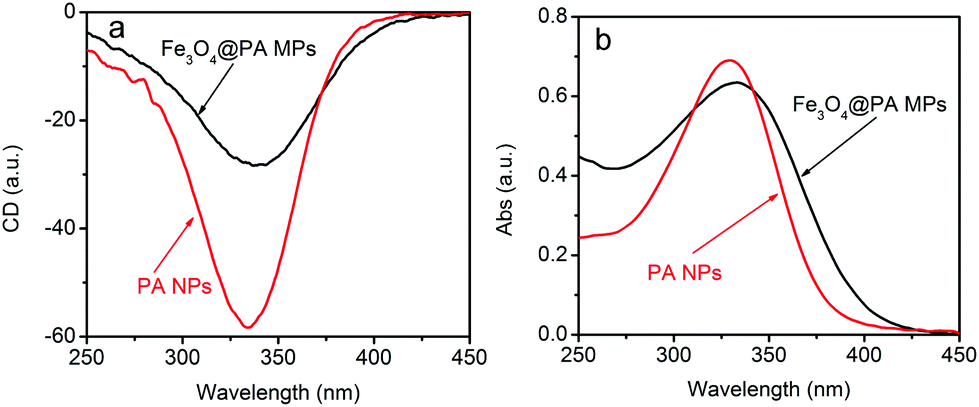 | ||
| Fig. 5 CD and UV-vis absorption spectra of the optically active, magnetic Fe3O4@PA core/shell MPs and pure polyacetylene nanoparticles (PA NPs) in aqueous dispersions. | ||
The CD spectrum measurement clearly shows the optical activity of the composite MPs. Besides, the composite microspheres are also expected to show interesting magneticity. To make this point clear, the magnetic properties of the Fe3O4@PA MPs were examined by using a VSM at 300 K. The relevant results are illustrated in Fig. 6. Fig. 6 shows the hysteresis loops of the Fe3O4 NPs (a) and the Fe3O4@PA MPs (b), indicating that their saturation magnetization is 66.85 and 23.59 emu g−1, respectively. A certain difference was observed between the magnetization value measured and the corresponding theoretical value, most likely due to the fact that some Fe3O4 NPs were not completely coated by polyacetylene. The VSM results convincingly show that the Fe3O4@PA MPs possessed remarkable magnetic properties. The high saturation magnetization of the microspheres afforded them excellent magnetic responsivity. As the insets in Fig. 6 show, the time from the dispersion state (b) to the aggregation state (c) was achieved within 11 s under an external magnetic field. When the external magnet was removed, the state (b) can be completely recovered from the state (c) just by shaking. Therefore the optically active, magnetic core/shell MPs can be easily recycled with the help of a magnet. Herein it is important to note that photos of the pure Fe3O4 NPs (Fig. 6a inset) and the optically active, magnetic Fe3O4@PA MP (Fig. 6b) dispersions were taken. The color of the pure Fe3O4 NPs was black, while the Fe3O4@PA MPs exhibited a yellow-brown color because of the presence of yellow helical substituted polyacetylene. Helical substituted polyacetylenes always show a light yellow color (or yellowish colors) in both solution and particulate states.58
 | ||
| Fig. 6 The hysteresis loops of Fe3O4 NPs and optically active, magnetic Fe3O4@PA MPs. The inset shows the photos of (a) Fe3O4 NPs and (b) optically active, magnetic Fe3O4@PA MP aqueous dispersions; (c) separability of the optically active, magnetic Fe3O4@PA MPs by placing an external magnetic field. The time from state (b) to state (c) is within 11 s. | ||
Enantioselective crystallization by using the core/shell microspheres
With the optically active, magnetic composite MPs in hand, we next examined their utility as chiral additives for enantioselective crystallization. The Fe3O4@PA MPs’ shell is constructed by using chirally helical polymer chains, so they are expected to undergo chiral interaction preferentially with one of the enantiomers, by which enantioselective crystallization takes place.64 In the present study, racemic threonine is taken as a model compound for exploring the inducing effects of the composite MPs. Thanks to the convenient isolation ability of the present magnetic MPs, we performed a two-cycle enantioselective crystallization process to improve the ee of the induced crystals. The major procedure for the two-cycle enantioselective crystallization of threonine by using Fe3O4@PA MPs in aqueous solution is illustratively presented in Scheme 2. | ||
| Scheme 2 A schematic for illustrating the two-cycle enantioselective crystallization of racemic threonine by using Fe3O4@PA MPs. | ||
In Scheme 2, in the first enantioselective crystallization cycle, Fe3O4@PA MPs were added into the racemic D,L-threonine supersaturated aqueous solution, as detailed in the Experimental section. L-Threonine crystals (ee approx. 25%) were more favorably induced in the first cycle of enantioselective crystallization. After magnetic separation, Fe3O4@PA MPs were restored and re-used for the second cycle of enantioselective crystallization, wherein the L-threonine crystals induced in the first cycle were used, aiming at further improving the ee. Excitingly, the ee of the L-threonine crystals after the second cycle of crystallization increased up to about 90%.
The SEM technique was used to observe the L-threonine crystals induced in the first cycle as a function of crystallization time. Fig. 7 shows the relevant SEM images. In Fig. 7a, clusters of crystals commenced to form and could be clearly viewed (after crystallization for six hours). Twelve hours later (Fig. 7b), rectangular-shaped crystals were observed more clearly. In Fig. 7c and d, the crystals continuously grew larger but still well maintained the rectangular shape; moreover, the crystals became progressively more regular. The SEM images clearly show the appearance and the growth of the induced crystals. To acquire deeper insights into the growth of the crystals in the course of enantioselective crystallization in the presence of Fe3O4@PA MPs as chiral seeds, the yield of the crystals against crystallization time is presented in Fig. 8. At the early stage, no crystals were formed (within the first four hours). From then on, crystals began to form and the yield of the crystals drastically increased with prolonging crystallization time (4–12 h, the second stage). After that, the yield of the crystals increased slowly and then was nearly constant (the third stage). The maximum yield of the crystals could be as high as 40%. The observation is in agreement with the SEM images (Fig. 7). The L-threonine crystals in the second crystallization cycle showed the same crystal morphology as observed in Fig. S2 (ESI†). However, the crystals yield increased more rapidly at the second stage relative to that in the first cycle of enantioselective crystallization (Fig. 8).
 | ||
| Fig. 7 SEM images of the L-threonine crystals induced by Fe3O4@PA MPs. | ||
 | ||
| Fig. 8 Yield of L-threonine crystals induced by Fe3O4@PA MPs as a function of the crystallization time. | ||
To further validate our conclusion, the crystals induced in the two cycles and the residual solutions were measured by CD spectra, as shown in Fig. 9. Just like pure L-threonine, the threonine crystals obtained in both the 1st and 2nd crystallization cycles showed a positive CD signal around 210 nm. The threonine crystals obtained in the 2nd crystallization cycle had a stronger CD signal than that from the 1st cycle. This observation is in accordance with the ee values of the induced L-threonine crystals (25% and 90% in the 1st and 2nd cycles, respectively). In sharp contrast to the induced L-threonine crystals, the corresponding residual solutions showed a negative CD signal at the same wavelength (210 nm). From the CD spectra, we know that L-threonine was preferentially induced to crystallize, while D-threonine predominantly remained in the residual solutions. To further justify the effects of Fe3O4@PA MPs in the enantioselective crystallization process, we measured the CD spectra of the threonine crystals induced by using achiral alkynyl-Fe3O4 NPs instead of chiral Fe3O4@PA MPs. The induced crystals assumed a similar morphology to that induced by using chiral composite MPs, but they did not show a CD signal in the wavelength range of interest (ee, about zero in this case). These results demonstrate that Fe3O4@PA MPs play essential roles in inducing the enantioselective crystallization of L-threonine.
 | ||
| Fig. 9 CD spectra of (a) Fe3O4@PA MPs, (b) pure L-threonine, (c) L-threonine crystals induced by Fe3O4@PA MPs and (d) the residual solution after removing the L-threonine crystals in the 1st crystallization; (e) L-threonine crystals induced by Fe3O4@PA MPs and (f) the residual solution after removing the L-threonine crystals in the 2nd crystallization; (g) threonine crystals induced by alkynyl-Fe3O4 NPs. All of the CD measurements were conducted in aqueous solution (or dispersion) at room temperature. | ||
In order to obtain a better understanding of the enantioselective crystallization, the ee of the threonine crystals (L-threonine in excess in the threonine crystals induced by Fe3O4@PA MPs, as discussed above) and the corresponding residual solutions were plotted against crystallization time. As Fig. 10a illustrates the ee of both the threonine crystals and the residual solutions increased as a function of the crystallization time. For the L-threonine crystals, a maximum ee of 25% was achieved around 18 h. From then on, ee decreased slightly, due to the fact that D-threonine began to crystallize faster than L-threonine. So the optimal crystallization time is determined to be 18 h. The changing trend in the ee of the residual solution is consistent with the above observation. Nonetheless, the ee value of the induced crystals is not high enough from the viewpoint of practical applications. To further improve the purity of the L-threonine crystals induced in the first enantioselective crystallization cycle, we performed the second cycle of the enantioselective crystallization by using the recycled Fe3O4@PA MPs after the first cycle use. As shown in Fig. 10b, after ca. 12 h, the maximum ee in the second crystallization cycle excitingly increased up to 90%, showing the significant potential applications of the Fe3O4@PA MPs as chiral additives. The Fe3O4@PA MPs recycled after the first two-cycle enantioselective crystallization were re-used for another “two-cycle” crystallization process in the same manner. The ee of the obtained crystals in this case was found to be up to 84%. This demonstrates that the Fe3O4@PA MPs were highly efficient in inducing enantioselective crystallization even after a “two-cycle” process.
 | ||
| Fig. 10 ee as a function of the crystallization time (L-threonine in excess) by using Fe3O4@PA MPs (a) in the 1st and (b) 2nd enantioselective crystallization cycles. | ||
XRD analyses were further performed on the L-threonine crystals obtained in the 2nd enantioselective crystallization cycle (Fig. 11). When compared to pure L-threonine, the XRD of L-threonine crystals induced by Fe3O4@PA MPs showed nearly the same XRD patterns, only with a slight difference in the diffraction intensity. The XRD measurement further demonstrates the high purity of the L-threonine crystals after the 2nd enantioselective crystallization cycle.
 | ||
| Fig. 11 Typical X-ray diffraction patterns of (a) pure L-threonine and (b) L-threonine crystals induced by Fe3O4@PA MPs after the 2nd crystallization. | ||
Conclusion
We successfully prepared a novel type of optically active, magnetic Fe3O4@PA core/shell structured microsphere in which helical polyacetylene constituted the shell and Fe3O4 NPs constituted the core. The composite microspheres were prepared readily through a seeded emulsion polymerization approach. The core/shell microspheres demonstrated both magneticity originated from the Fe3O4 NP core and optical activity derived from the chirally helical substituted polyacetylene shell. The microspheres could be separated conveniently with the help of an external magnetic field. The optically active, magnetic microspheres efficiently induced enantioselective crystallization of L-threonine from racemic threonine, forming rectangular-shaped crystals. Due to the presence of magnetic cores, the microspheres can be easily restored after use. After enantioselectively crystallizing twice, the ee of the induced L-threonine increased up to 90%. In the following studies, we will further optimize the composition and structure of the novel core/shell optically active, magnetic microspheres. Apart from being used as a chiral additive toward enantioselective crystallization, they are also expected to serve as chiral catalysts, chiral carriers for delivering chiral drugs, among the other potential uses.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21474007, 21274008, 21174010), the Funds for Creative Research Groups of China (51221002), and the “Specialized Research Fund for the Doctoral Program of Higher Education” (SRFDP 20120010130002).References
- B. Chen, J. P. Deng, X. Q. Liu and W. T. Yang, Macromolecules, 2010, 43, 3177 CrossRef CAS.
- W. Li and D. Y. Zhao, Adv. Mater., 2013, 25, 142 CrossRef CAS PubMed.
- H. M. Chen, C. H. Deng and X. M. Zhang, Angew. Chem., Int. Ed., 2010, 49, 607 CrossRef CAS PubMed.
- W. H. Feng, H. X. Dong, L. B. Niu, X. Wen, L. Huo and G. Y. Bai, J. Mater. Chem. A, 2015, 3, 19807 CAS.
- X. L. Jia, J. Y. Wang, K. Wang and J. T. Zhu, Langmuir, 2015, 31, 8732 CrossRef CAS PubMed.
- Q. M. Kainz and O. Reiser, Acc. Chem. Res., 2014, 47, 667 CrossRef CAS PubMed.
- J. Gallo, N. Kamaly, I. Lavdas, E. Stevens, Q.-D. Nguyen, M. Wylezinska-Arridge, E. O. Aboagye and N. J. Long, Angew. Chem., Int. Ed., 2014, 53, 9550 CrossRef CAS PubMed.
- G. N. Wang, L. Jin, Y. K. Dong, L. Niu, Y. X. Liu, F. Ren and X. G. Su, New J. Chem., 2014, 38, 700 RSC.
- O. Gleeson, G.-L. Davies, A. Peschiulli, R. Tekoriute, Y. K. Gun'ko and S. J. Connon, Org. Biomol. Chem., 2011, 9, 7929 CAS.
- B. Panella, A. Vargas and A. Baiker, J. Catal., 2009, 261, 88 CrossRef CAS.
- A. G. Hu, G. T. Yee and W. B. Lin, J. Am. Chem. Soc., 2005, 127, 12486 CrossRef CAS PubMed.
- T. Q. Zeng, L. Yang, R. Hudson, G. H. Song, A. R. Moores and C.-J. Li, Org. Lett., 2011, 13, 442 CrossRef CAS PubMed.
- Y. Q. Sun, G. H. Liu, H. Y. Gu, T. Z. Huang, Y. L. Zhang and H. X. Li, Chem. Commun., 2011, 47, 2583 RSC.
- S. Ghosh, A. Z. M. Badruddoza, M. S. Uddin and K. Hidajat, J. Colloid Interface Sci., 2011, 354, 483 CrossRef CAS PubMed.
- Y. T. Liu, A. Tian, X. Wang, J. Qi, F. K. Wang, Y. Ma, Y. Ito and Y. Wei, J. Chromatogr., A, 2015, 1400, 40 CrossRef CAS PubMed.
- H. J. Choi and M. H. Hyun, Chem. Commun., 2009, 6454 RSC.
- X. X. Zheng, L. Zhang, J. Y. Li, S. Z. Luo and J.-P. Cheng, Chem. Commun., 2011, 47, 12325 RSC.
- J. Chen, R.-P. Liang, X.-N. Wang and J.-D. Qiu, J. Chromatogr., A, 2015, 1409, 268 CrossRef CAS PubMed.
- J. Huang, P. Su, B. J. Zhao and Y. Yang, Anal. Methods, 2015, 7, 2754 RSC.
- M. B. Gawande, P. S. Branco and R. S. Varma, Chem. Soc. Rev., 2013, 42, 3371 RSC.
- M. M. Green, J.-W. Park, T. Sato, A. Teramoto, S. Lifson, R. L. B. Selinger and J. V. Selinger, Angew. Chem., Int. Ed., 1999, 38, 3139 CAS.
- H. Y. Chen, L. Li, D. Liu, H. J. Huang, J. P. Deng and W. T. Yang, RSC Adv., 2014, 4, 63611 RSC.
- E. Yashima, K. Maeda and Y. Furusho, Acc. Chem. Res., 2008, 41, 1166 CrossRef CAS PubMed.
- Y. Okamoto, Adv. Polym. Sci., 2013, 261, 391 CrossRef CAS.
- J. Z. Liu, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2009, 109, 5799 CrossRef CAS PubMed.
- E. Yashima, K. Maeda, H. Iida, Y. Furusho and K. Nagai, Chem. Rev., 2009, 109, 6102 CrossRef CAS PubMed.
- Y. Y. Xu, H. Jiang, Q. J. Zhang, F. Wang and G. Zou, Chem. Commun., 2014, 50, 365 RSC.
- V. Percec, J. G. Rudick, M. Peterca and P. A. Heiney, J. Am. Chem. Soc., 2008, 130, 7503 CrossRef CAS PubMed.
- J. F. Reuther and B. M. Novak, J. Am. Chem. Soc., 2013, 135, 19292 CrossRef CAS PubMed.
- L. J. Liu, T. Namikoshi, Y. Zang, T. Aoki, S. Hadano, Y. Abe, I. Wasuzu, T. Tsutsuba, M. Teraguchi and T. Kaneko, J. Am. Chem. Soc., 2013, 135, 602 CrossRef CAS PubMed.
- C. H. Zhang, H. L. Wang, Q. Q. Geng, T. T. Yang, L. J. Liu, R. Sakai, T. Satoh, T. Kakuchi and Y. Okamoto, Macromolecules, 2013, 46, 8406 CrossRef CAS.
- J. X. Cui, J. Zhang and X. H. Wan, Chem. Commun., 2012, 48, 4341 RSC.
- S. Mayer and R. Zentel, Prog. Polym. Sci., 2001, 26, 1973 CrossRef CAS.
- N. Suzuki, M. Fujiki, R. Kimpinde-Kalunga and J. R. Koe, J. Am. Chem. Soc., 2013, 135, 13073 CrossRef CAS PubMed.
- M. Shiotsuki, F. Sanda and T. Masuda, Polym. Chem., 2011, 2, 1044 RSC.
- K. Maeda, S. Wakasone, K. Shimomura, T. Ikai and S. Kanoh, Macromolecules, 2014, 47, 6540 CrossRef CAS.
- Y. Yoshida, Y. Mawatari, A. Motoshige, R. Motoshige, T. Hiraoki, M. Wagner, K. Müllen and M. Tabata, J. Am. Chem. Soc., 2013, 135, 4110 CrossRef CAS PubMed.
- A. Pietropaolo and T. Nakano, J. Am. Chem. Soc., 2013, 135, 5509 CrossRef CAS PubMed.
- C. R. Boehm and E. M. Terentjev, Macromolecules, 2014, 47, 6086 CrossRef CAS.
- S. Arias, F. Freire, E. Quiñoá and R. Riguera, Angew. Chem., Int. Ed., 2014, 53, 13720 CrossRef CAS PubMed.
- A. J. Heeger, Angew. Chem., Int. Ed., 2001, 40, 2591 CrossRef CAS.
- A. G. MacDiarmid, Angew. Chem., Int. Ed., 2001, 40, 2581 CrossRef CAS.
- H. Shirakawa, Angew. Chem., Int. Ed., 2001, 40, 2574 CrossRef.
- J. Q. Qu, T. Katsumata, M. Satoh, J. Wada and T. Masuda, Macromolecules, 2007, 40, 3136 CrossRef CAS.
- Z. L. Tang, H. Iida, H.-Y. Hu and E. Yashima, ACS Macro Lett., 2012, 1, 261 CrossRef CAS.
- C. K. W. Jim, J. W. Y. Lam, C. W. T. Leung, A. Qin, F. Mahtab and B. Z. Tang, Macromolecules, 2011, 44, 2427 CrossRef CAS.
- L. Ding, Y. Y. Huang, J. P. Deng and W. T. Yang, Macromolecules, 2011, 44, 736 CrossRef CAS.
- X. F. Luo, X. Q. Liu, B. Chen, J. P. Deng and W. T. Yang, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 5611 CrossRef CAS.
- S. Mecking, Colloid Polym. Sci., 2007, 285, 605 CAS.
- W. F. Li, X. Liu, G. Y. Qian and J. P. Deng, Chem. Mater., 2014, 26, 1948 CrossRef CAS.
- H. J. Huang, Y. B. Yuan and J. P. Deng, Macromolecules, 2015, 48, 3406 CrossRef CAS.
- X. F. Luo, L. Li, J. P. Deng, T. T. Guo and W. T. Yang, Chem. Commun., 2010, 46, 2745 RSC.
- X. F. Luo, J. P. Deng and W. T. Yang, Angew. Chem., Int. Ed., 2011, 50, 4909 CrossRef CAS PubMed.
- B. Chen, J. P. Deng, L. Y. Tong and W. T. Yang, Macromolecules, 2010, 43, 9613 CrossRef CAS.
- J. Y. Liang, C. Song and J. P. Deng, Appl. Mater. Interfaces, 2014, 6, 19041 CrossRef CAS PubMed.
- D. Y. Zhang, H. Y. Zhang, C. Song, W. T. Yang and J. P. Deng, Synth. Met., 2012, 162, 1858 CrossRef CAS.
- R. R. Schrock and J. A. Osborn, Inorg. Chem., 1970, 9, 2339 CrossRef CAS.
- J. Tabei, R. Nomura and T. Masuda, Macromolecules, 2002, 35, 5405 CrossRef CAS.
- R. Y. Liu, F. Sanda and T. Masuda, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 4175 CrossRef CAS.
- H. Deng, X. L. Li, Q. Peng, X. Wang, J. P. Chen and Y. D. Li, Angew. Chem., Int. Ed., 2005, 44, 2782 CrossRef CAS PubMed.
- D. D. Medina, A. Gedanken and Y. Mastai, Chem. – Eur. J., 2011, 17, 11139 CrossRef CAS PubMed.
- L. C. Preiss, L. Werber, V. Fischer, S. Hanif, K. Landfester, Y. Mastai and R. Muñoz-Espí, Adv. Mater., 2015, 27, 2728 CrossRef CAS PubMed.
- Q. Gao, F. H. Chen, J. L. Zhang, G. Y. Hong, J. Z. Ni, X. Wei and D. J. Wang, J. Magn. Magn. Mater., 2009, 321, 1052 CrossRef CAS.
- B. Chen, J. P. Deng and W. T. Yang, Adv. Funct. Mater., 2011, 21, 2345 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5py01549a |
| This journal is © The Royal Society of Chemistry 2016 |