DOI:
10.1039/C5PY01488C
(Paper)
Polym. Chem., 2016,
7, 235-243
Thiophene-fused isoindigo based conjugated polymers for ambipolar organic field-effect transistors†
Received
15th September 2015
, Accepted 23rd October 2015
First published on 23rd October 2015
Abstract
A series of donor–acceptor (D–A) type conjugated polymers based on novel thiophene-fused isoindigo (TII) were designed and synthesized via palladium catalyzed Stille copolymerization. We found that the redesign of the synthesis of brominated TII was necessary, and the α-bromination had to be completed at the very beginning, or β-brominated TII was obtained, which only led to cross conjugated polymers. Once the bromination was introduced at the correct position, fully conjugated D–A type polymers could be obtained. A series of fully conjugated polymers were obtained by Stille coupling polymerization of α-brominated TII with different donors, and among them, copolymers with thiophene (T) and (E)-1,2-bis(3-octylthiophen-2-yl)ethene (TVT-8) showed acceptable solubility and were suitable to fabricate solution-processable organic field-effect transistors (OFETs). Top-gate/bottom-contact (TG/BC) devices were constructed for the polymers to test their OFET performances. Both fully conjugated polymers exhibit two-orders of magnitude higher charge carrier mobilities than the cross conjugated ones, and PTII-T shows balanced electron and hole mobilities and PTII-TVT-8 is a p-type dominated semiconductor. These observations indicated that the developed TII unit that has improved coplanarity can be a promising building block for the construction of highly efficient conjugated polymers for OFET applications.
Introduction
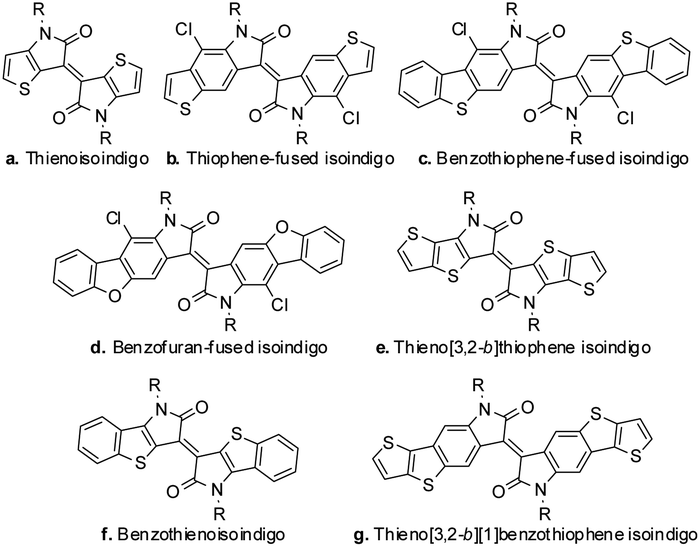
Organic semiconducting materials have received increasing attention in recent years because of their promising applications in various areas such as organic field-effect transistors (OFETs), organic light-emitting diodes (OLEDs), organic solar cells (OSCs), and sensors.1–5 In the past few years, the need for better performing materials in the organic electronics field has inspired a resurgence of natural dyes and pigments, such as diketopyrrolopyrrole (DPP), naphthalene diimide (NDI) and indigo. Isoindigo is a well-known structural motif for the design of such OFET materials. Its electron-deficient core as well as its off axis dipole moment promote intermolecular interactions and ambient stability is imparted through the generally low-lying HOMO levels.6–12
The modification of the isoindigo core plays an important role in the manipulation of the properties of the resulting materials, since it could alter the HOMO/LUMO energy levels, change the strength of π–π interaction and even decrease the steric hindrance in some cases. The modification of the isoindigo core was firstly reported by Ashraf and coworkers, in which the benzene ring in isoindigo was replaced with a thiophene ring. The resultant compound, namely thienoisoindigo (Scheme 1a), was used as a novel building block for polymeric OFETs, and balanced hole and electron mobilities up to 0.1 cm2 V−1 s−1 were reported.13 Inspired by this report, we synthesized thiophene-fused isoindigo (as shown in Scheme 1b) and disclosed its photophysical and electrochemical properties, showing that the intramolecular charge transfer was enhanced due to the extension of the π-conjugated system.14 Further extension of the π-conjugated system (benzothiophene-fused isoindigo and benzofuran-fused isoindigo, Scheme 1c and d) led to small molecular OFETs with electron mobility up to 0.074 cm2 V−1 s−1 under ambient conditions.15 More recently, similar modifications on the isoindigo core were also reported (Scheme 1e–g) including thieno[3,2-b]thiophene isoindigo,16 benzothienoisoindigo,17 and thieno[3,2-b][1]benzothiophene isoindigo.18 OFET devices fabricated with small molecules and polymers based on these structures all showed promising performances.16–18 All this progress indicates the great potential of isoindigo core modification in improving OFET properties, which deserves further exploration.
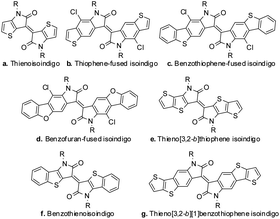 |
| | Scheme 1 The reported manipulation on the isoindigo core. | |
As our continuous effort in the design of small molecules and macromolecules based on a modified isoindigo core, we herein wish to report the synthesis of polymers based on thiophene-fused isoindigo building blocks and the investigation of their performance as OFET materials.
Results and discussion
Polymer synthesis and properties
Recently, we reported a new way to synthesize TII with a linear alkyl chain on a nitrogen atom.14 It is observed from the single crystal X-ray diffraction study that in 1,1′-dimethyl-isoindigo, the dihedral angle between the two oxindole rings is around 22.3(2)°,19 which is destructive for long range conjugation. While for TII, the dihedral angle is close to 0° (see ESI, Fig. S1†), which we believe is beneficial to form good intermolecular π–π stacking. Furthermore, we anticipate that when TII is copolymerized with other co-monomers, the steric hindrance between the two building blocks will be further decreased due to the less-hindered terminal thiophene rings on TII. Thus, the corresponding copolymers of TII will show better planarity than that of isoindigo, and lead to better OFET performance.
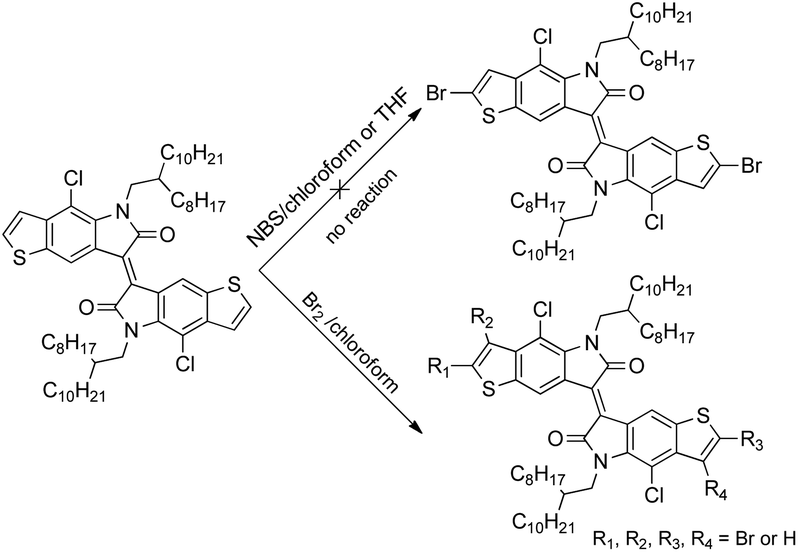
Bromination of TII was necessary to obtain the corresponding monomer for Stille coupling polymerization. However, the direct bromination on TII turned out to be rather difficult (Scheme 2): no reaction was observed in THF and chloroform from 0 °C to reflux using N-bromosuccinimide (NBS) as the bromination reagent, even when acetic acid was added to promote the reaction. Bromination using liquid bromine under various conditions also failed, yielding inseparable multi-brominated mixtures, presumably due to the similar reaction activities of α- and β-positions of the outer thiophene ring, caused by electron delocalization in the whole conjugated structure. We have to change our itinerary to introduce the bromine at the earlier stage.
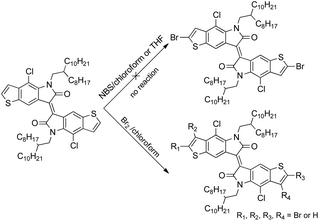 |
| | Scheme 2 Attempts for direct bromination on TII. | |
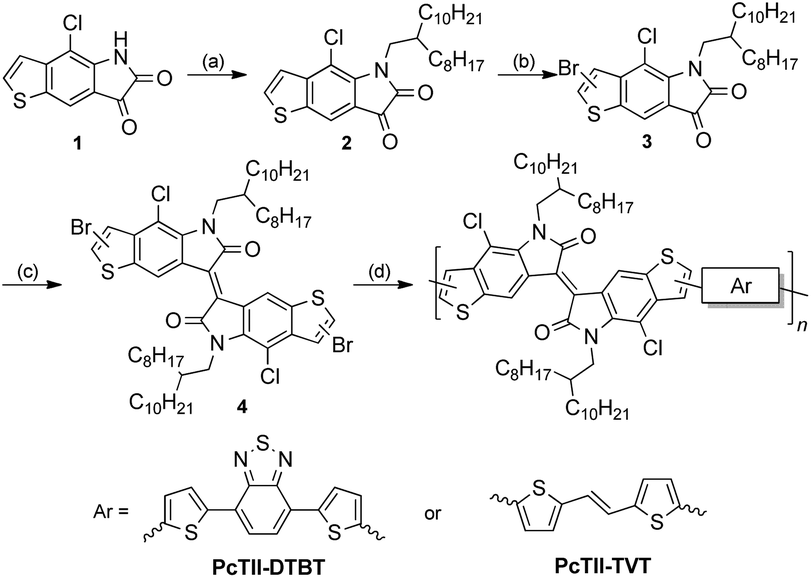
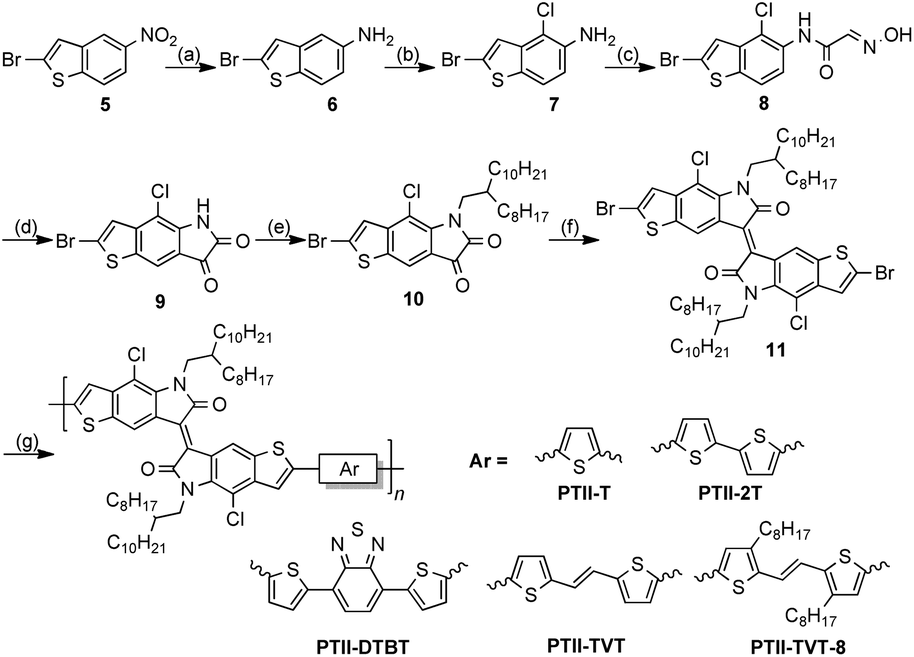
Thus, the synthetic route was revised, as shown in Scheme 3. 4-Chloro-5H-thieno[2,3-f] indole-6,7-dione (1) was alkylated with 9-(bromomethyl)nonadecane using K2CO3, and the reaction proceeded smoothly at 50 °C to give 2 in 71% yield. Due to the electron deficiency of the dione, bromination of 2 only works with liquid bromine in the presence of sodium acetate, giving a mono-brominated compound 3 as a pure single regio-isomer in 70% yield. The dibrominated TII (4) was synthesized in 90% yield according to the method reported by Bogdanov.20
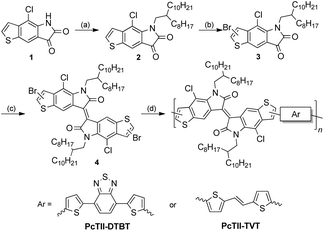 |
| | Scheme 3 Original synthetic route of two TII based polymers. Reagents and conditions: (a) 9-(bromomethyl)nonadecane, K2CO3, DMF, 50 °C, 71%; (b) Br2, AcONa, AcOH, reflux, 2 h, 70%; (c) P(NEt2)3, CH2Cl2, −60 °C, 90%; (d) 4,7-bis(5-(trimethylstannyl)thiophen-2-yl)benzo[c][1,2,5]thiadiazole or (E)-1,2-bis(5-(trimethylstannyl)thiophen-2-yl)ethane, Pd(PPh3)4, toluene, 110 °C, 48 h; for PcTII-DTBT, 85%; for PcTII-TVT, 92%. | |
With the dibrominated TII in hand, Stille-coupling polymerization between 4 and two different monomers, 4,7-bis(5-(trimethylstannyl)thiophen-2-yl)benzo[c][1,2,5]thiadiazole (DTBT) or (E)-1,2-bis(5-(trimethylstannyl)thiophen-2-yl)ethane (TVT) was tested, giving PcTII-DTBT and PcTII-TVT respectively, in high yields. Both polymers were obtained as dark red metallic solids after careful purification and showed excellent solubility in common organic solvents such as chloroform.
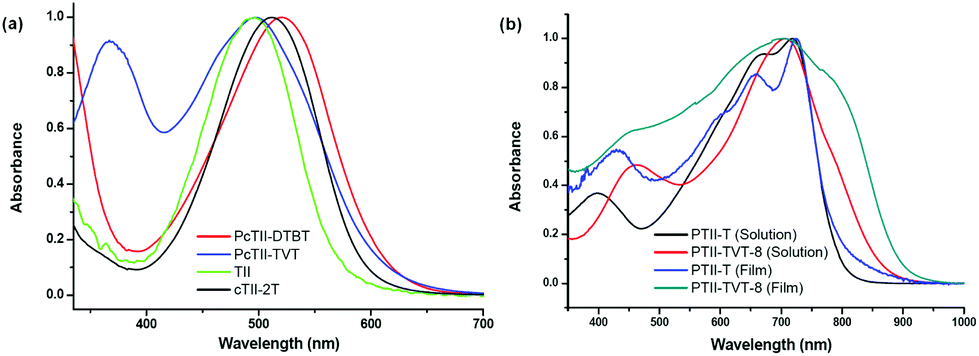
UV-vis absorption spectra of both the polymers in chloroform solution were recorded (Fig. 1a). To our surprise, compared with the TII monomer, the polymers did not show any obvious bathochromic-shifted absorption, which was not in accordance with the literature about the conjugated polymers of isoindigo.7,13,21,22 Reynolds and coworkers have reported two low-energy gap oligothiophenes connected with isoindigo, both of which showed a greater red shift in the UV spectra.6 To rule out the possible problems that might be involved in Stille coupling, a model compound cTII-2T, with two thiophene rings capped at either end of TII, was synthesized in the same way and well characterized (see the Experimental section for details). The same result was obtained: cTII-2T only has a red-shift of 10 nm to TII, which aroused more questions on the regioselectivity of the dibrominated TII (4). Very possibly, dibromination occurred at the β-position of the benzothiophene core, which led to cross conjugation rather than full conjugation.23 To confirm the position of the bromine atom on the thiophene ring, the single crystal of an analogue of compound 3 (alkylated with dodecane chain) was prepared, since the longer branched alkyl chain was detrimental for single crystal growth.
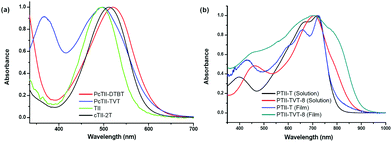 |
| | Fig. 1 (a) Normalized UV-vis absorption spectra of TII, cTII-2T, and two TII based polymers in CHCl3 solution (1 × 10−5 M); (b) normalized UV-vis absorption spectra of two TII based polymers both in chlorobenzene solution (1 × 10−5 M) and in the film. | |
The single structure depicted in Scheme 4 clearly showed that the bromine atom was at the β-position of the thiophene ring, validating our presumption of cross conjugation. By examining the literature in detail, we found out that for benzofused heterocycles such as benzofurans, benzothiophenes, and indoles, treatment of the unsubstituted parent substrate with an electrophilic halogen source typically provides access to β-halogenated substrates.24–28 So, to achieve full conjugation, the bromine atom has to be introduced at the α-position at the very beginning, before the formation of the benzothiophene core. Thus, the synthetic route was further modified and is shown in Scheme 5. The starting material 2-bromo-5-nitrobenzo[b]thiophene (5) was synthesized according to the literature.29,30 Following the similar procedure to previously reported ones,14 α,α′-dibrominated TII (11) was synthesized. Polymers based on α-brominated TII were synthesized via Stille-coupling polymerization, with five different co-monomers, denoted as Ar in Scheme 5. The solubility of the resulting polymer is greatly influenced by the structure of the co-monomers: when TII was copolymerized with 2,5-bis(trimethylstannyl)thiophene, a copolymer with excellent solubility in chloroform was obtained (Scheme 5, PTII-T); when switched to (E)-1,2-bis(3-octyl-5-(trimethylstannyl)-thiophen-2-yl)ethene, the solubility of the copolymer PTII-TVT-8 was acceptable in heated chlorobenzene and o-dichlorobenzene; while for 5,5′-bis(trimethylstannyl)-2,2′-bithiophene, 4,7-bis(5-(trimethylstannyl)thiophen-2-yl)benzo[c][1,2,5]thiadiazole, and (E)-1,2-bis(5-(trimethylstannyl)thiophen-2-yl)ethane, the solubility of the resulting copolymers became very poor in all aromatic solvents, even in heated 1,2,4-trichlorobenzene. Thus, we were unable to fully characterize the properties of PTII-2T, PTII-DTBT, and PTII-TVT. Meanwhile, compared with the cross conjugated polymers we prepared before, these polymers exhibit decreased solubility, which could be explained by the improved rigidity and linearity of the polymer backbone. All the polymers were obtained as dark blue metallic solids after careful purification, presenting a completely different color from cross conjugated polymers due to the longer effective conjugation length and donor–acceptor interactions. Molecular weights of PTII-T and PTII-TVT-8 were evaluated by high temperature gel permeation chromatography (GPC) with 1,2,4-trichlorobenzene (TCB) as the eluent at 150 °C. Both polymers show comparable Mn (PTII-T, 26.6 kDa; PTII-TVT-8, 31.0 kDa). Nonetheless, the PDI of PTII-TVT-8 (3.46) is larger than that of PTII-T (2.41). The large PDI might be the result of strong aggregation of the polymers. We also measured their thermal properties. Both polymers showed decomposition temperatures above 350 °C under a nitrogen atmosphere, and no phase transition was observed before decomposition.
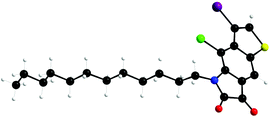 |
| | Scheme 4 Single crystal structure of dodecane substituted compound 3. | |
 |
| | Scheme 5 Modified synthetic route of polymers based on α-brominated TII. Reagents and conditions: (a) SnCl2·H2O, HCl, MeOH, reflux 4 h, 90%; (b) N-chlorosuccinimide, THF, reflux 2 h, 88%; (c) Cl3CCH(OH)2, NH2OH·HCl, Na2SO4, HCl, H2O/EtOH (v/v = 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), 80 °C, 2 h, 91%; (d) H2SO4, 80 °C, 30 min; (e) 9-(bromomethyl)nonadecane, K2CO3, DMF, 50 °C, 12 h, 2 steps, 35%; (f) P(NEt2)3, CH2Cl2, −78 °C to room temperature, 92%; (g) 2,5-bis(trimethylstannyl)thiophene, 5,5′-bis(trimethylstannyl)-2,2′-bithiophene, 4,7-bis(5-(trimethylstannyl)thiophen-2-yl)benzo[c][1,2,5]thiadiazole, (E)-1,2-bis(5-(trimethylstannyl)thiophen-2-yl)ethane, (E)-1,2-bis(3-octyl-5-(trimethylstannyl)thiophen-2-yl)ethene, Pd(PPh3)4, toluene, 110 °C, 48 h; for PTII-2T, PTII-DTBT, and PTII-TVT, ca. 20%; for PTII-T, 92%; for PTII-TVT-8, 71%. :1), 80 °C, 2 h, 91%; (d) H2SO4, 80 °C, 30 min; (e) 9-(bromomethyl)nonadecane, K2CO3, DMF, 50 °C, 12 h, 2 steps, 35%; (f) P(NEt2)3, CH2Cl2, −78 °C to room temperature, 92%; (g) 2,5-bis(trimethylstannyl)thiophene, 5,5′-bis(trimethylstannyl)-2,2′-bithiophene, 4,7-bis(5-(trimethylstannyl)thiophen-2-yl)benzo[c][1,2,5]thiadiazole, (E)-1,2-bis(5-(trimethylstannyl)thiophen-2-yl)ethane, (E)-1,2-bis(3-octyl-5-(trimethylstannyl)thiophen-2-yl)ethene, Pd(PPh3)4, toluene, 110 °C, 48 h; for PTII-2T, PTII-DTBT, and PTII-TVT, ca. 20%; for PTII-T, 92%; for PTII-TVT-8, 71%. | |
Optical properties
Since PTII-T and PTII-TVT-8 exhibit acceptable solubility, their electronic properties and OFET performance were examined in detail. UV-vis absorption spectra of the two polymers both in solution and in a thin film were recorded (Fig. 1b). Compared with their cross conjugated counterparts, a greater red-shift was observed in the UV-vis spectra for both polymers, clearly indicating that they are fully conjugated. This again testified that full conjugation is needed to form a strong donor–acceptor (D–A) intramolecular charge transfer complex, facilitating electron delocalization over the conjugated main chain.31
The UV-vis spectrum of each polymerin solution displays two absorption bands characteristic of D–A systems from 350 to 1000 nm: a high-energy band attributed to the localized π–π* transition, and a low-energy band assigned to excited states with a prevalent highest occupied molecular orbital (HOMO)–lowest unoccupied molecular orbital (LUMO) contribution. Through the increase of the effective conjugation length after the introduction of the TVT unit, a red-shifted absorption band for PTII-TVT-8 relative to PTII-T is predicted; however, note that the absorption maximum of PTII-TVT-8 is blue-shifted vs. that of PTII-T by 16 nm in the film and 8 nm in solution. This may be due to the destabilization of the LUMO with successive expansion of the π-system of the thiophene moieties, which could be supported by the data from CV. While both polymers show similar maximum absorption around 710 nm in solution, the onset of the absorption of PTII-TVT-8 is remarkably red-shifted compared with that of PTII-T (900 nm vs. 800 nm), indicating that the bandgap of PTII-TVT-8 is smaller than that of PTII-T. Optical bandgaps (Eoptg) deduced from the absorption edges of the film spectra are in the same order: PTII-T (1.56 eV) > PTII-TVT-8 (1.35 eV). In the film state, the UV spectrum of PTII-T exhibits little difference from that in the solution state, indicating that the aggregation state of PTII-T in the film is very similar to that in solution. On the other hand, the UV-vis spectrum of PTII-TVT-8 in the film state is quite different from that in the solution state: the localized π–π* transition at 462 nm in the solution state disappears, and a strong and broad absorption ranging from 400 nm to 900 nm appears (with maximum absorption peak at 704 nm). Furthermore, a red-shifted shoulder at 766 nm was observed, indicating that stronger π–π stacking exists in the film of PTII-TVT-8.
Electrochemical properties
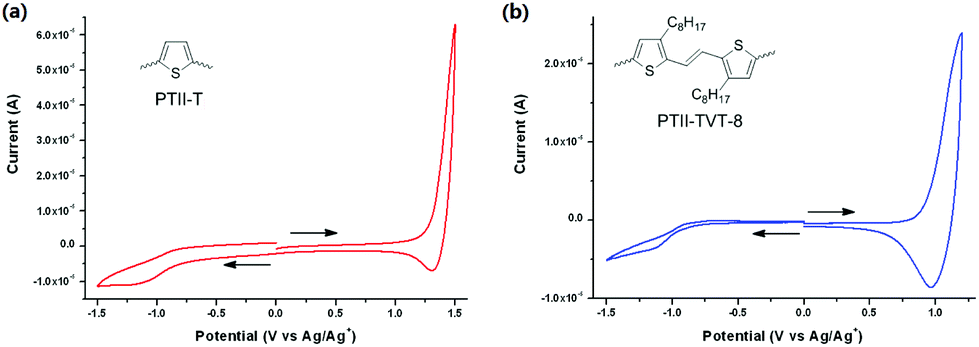
In order to experimentally determine the energy levels of the polymers, we investigated the electrochemical properties of PTII-T and PTII-TVT-8via cyclic voltammetry (CV) (Fig. 2). Thin films of both polymers were drop-cast onto a glassy carbon electrode respectively, and their CV diagrams were recorded in an acetonitrile solution containing 0.1 M n-Bu4NPF6 under an inert atmosphere versus the Ag/AgCl redox couple. The data are summarized in detail in Table 1. The HOMO energy level was calculated using the equation EHOMO = −(Eoxonset − ferrocene) − 4.8 eV, where Eoxonset was the onset of the oxidation potential. The LUMO energy level was deduced from the equation ELUMO = EHOMO + Eoptg.32 The HOMO/LUMO levels of PTII-T are −5.73 and −4.17 eV, and those of PTII-TVT-8 are −5.35/−4.00 eV. Compared with PTII-T, PTII-TVT-8 exhibits a high-lying HOMO level, due to the stronger electron-donating ability of TVT than the thiophene ring. Both polymers belong to low band-gap conducting polymers, and one would like to predict that both will exhibit ambipolar charge transfer properties. In particular, the LUMO level of PTII-T is even lower, which implies that the electron-transfer process in PTII-T will be more facile than that in PTII-TVT-8.
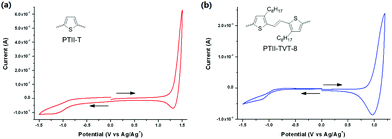 |
| | Fig. 2 Cyclic voltammogram of (a) PTII-T and (b) PTII-TVT-8 in thin film drop-casting on a glassy carbon electrode and tested in n-Bu4NPF6/CH3CN solution (scan rate: 0.1 V s−1). | |
Table 1 Molecular weight, optical (UV-vis) properties and electrochemical data
| Polymers |
M
n (kDa) |
PDI |
λ
max (nm) solution |
λ
max (nm) film |
E
optg (eV) |
LUMO (eV) |
HOMO (eV) |
|
PTII-T
|
26.6 |
2.41 |
715 |
720 |
1.56 |
−4.17 |
−5.73 |
|
PTII-TVT-8
|
31.0 |
3.46 |
707 |
704 |
1.37 |
−4.00 |
−5.35 |
OFET properties
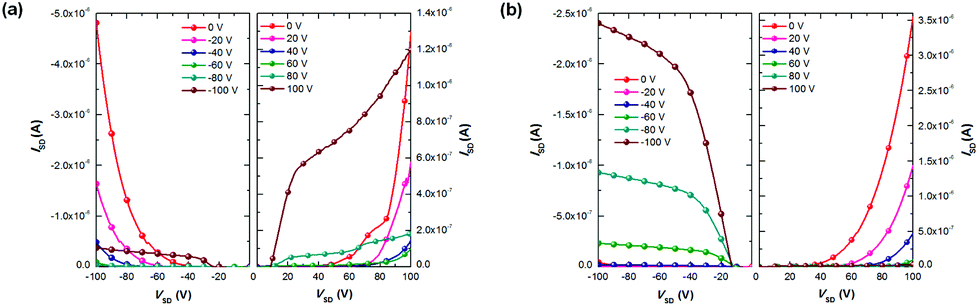
To test their OFET performance, top-gate/bottom-contact (TG/BC) devices were constructed for both polymers to fabricate polymer FETs. This device configuration is preferred for ambient-stable ambipolar or n-type organic materials, because of its better injection characteristics and the encapsulation effect of the top dielectric layer.9 The semiconducting layer was deposited by spin-coating the corresponding polymer solutions (3 mg mL−1 in TCE) on an untreated Au (source–drain)/SiO2/Si substrate. After thermal annealing the film at 180 °C for 5 min, a CYTOP solution was spin-coated on top of the polymer film as the dielectric layer, and an aluminum layer was thermally evaporated as the gate electrode. All devices were tested in ambient conditions on a probe station and exhibited ambipolar properties (Fig. 3). As shown in Table 2, both fully conjugated polymers exhibit electron and hole mobilities in the 10−2 order of magnitude, much higher than the cross conjugated polymers (which show mobilites in 10−4 range or lower, see the ESI†). PTII-T shows balanced electron and hole mobilities of 0.029 and 0.018 cm2 V−1 s−1, respectively. On the other hand, PTII-TVT-8 exhibits better hole mobility (0.037 cm2 V−1 s−1), almost one-order larger than its electron mobility (0.004 cm2 V−1 s−1). These results indicate that both polymers are good p-type semiconductors, but for electron transport, PTII-TVT-8 was not as good as PTII-T. At the first glance, PTII-TVT-8 should also exhibit balanced hole and electron mobilities by judging from its HOMO/LUMO level. However, it is also generally accepted that in D–A type polymers, p-type charge transfer becomes predominant with the size increase of the donor. Thus, PTII-TVT-8 mainly exhibits the hole transfer ability since TVT is a larger and bigger electron donor than the thiophene ring. PTII-TVT-8 shows better hole mobility and smaller threshold voltage than PTII-T, presumably due to incorporation of the highly π-extended TVT unit with a better electron donating ability than the thiophene unit. Compared with other reported isoindigo based polymers, the OFET performances of TII based polymers are lower, presumably due to the molecular weights of PTII-T and PTII-TVT-8 being relatively low. The sufficiently high molecular weight enables polymer chains to form well-interconnected crystalline grains to establish efficient pathways for intergranular charge transport.33 It was reported that the mobility increase upon annealing was observed in other isoindigo based polymers,7,34,35 and we also tried to improve the OFET performance by annealing the film at a higher temperature for a longer time. However, no substantial improvements were observed in comparison with as-spun films, implying that a subtle change in the structure of the polymer backbones can greatly affect the thermodynamic behaviour of the resulting solid-state thin film.
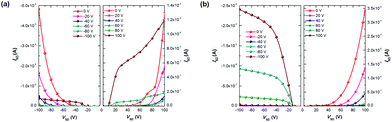 |
| | Fig. 3 Output characteristics of (a) PTII-T and (b) PTII-TVT-8 OFET devices. All OFET devices (W/L = 20) were fabricated with CYTOP under ambient conditions. | |
Table 2 OFET performance of PTII-T and PTII-TVT-8 thin films under ambient conditions
| Polymers |
μ
e (cm2 V−1 s−1) |
V
T (V) |
I
on/Ioff |
μ
h (cm2 V−1 s−1) |
V
T (V) |
I
on/Ioff |
|
PTII-T
|
0.029 |
72 |
538 |
0.018 |
−81 |
103 |
|
PTII-TVT-8
|
0.004 |
83 |
4 × 103 |
0.037 |
−53 |
103 |
Morphology
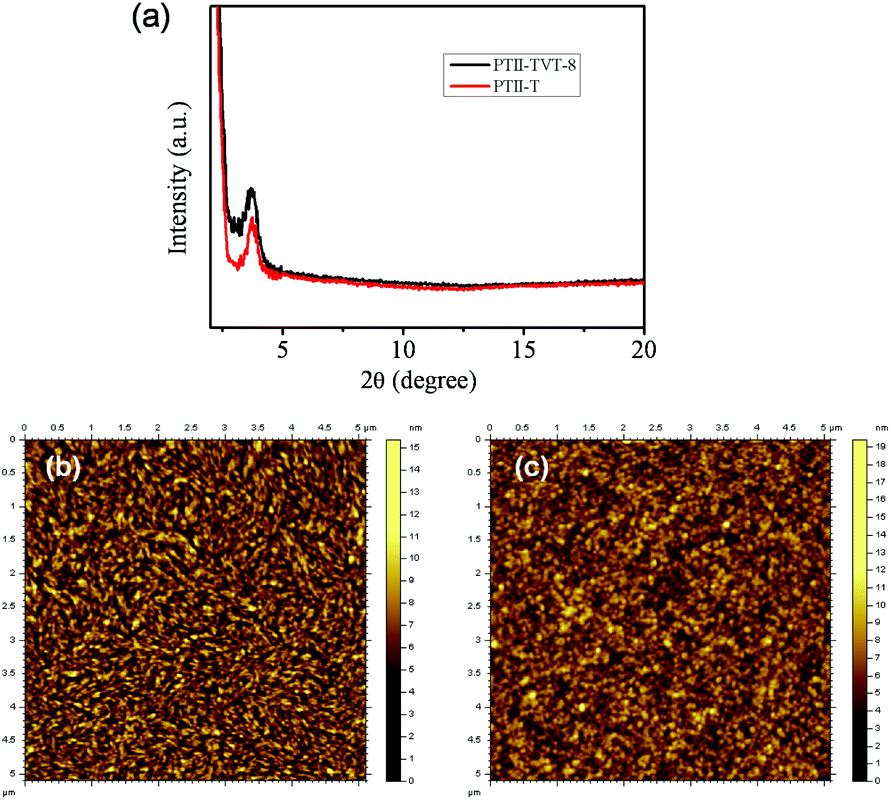
The thin film crystallinity and morphologies of both polymers were investigated by X-ray diffraction (XRD) and tapping-mode atomic force microscopy (AFM) to better understand the relationship between the morphology and device performance. The crystallinity and molecular orientations of thin films were investigated by XRD analysis. The out of plane XRD patterns of the thin film of PTII-T and PTII-TVT-8 are shown in Fig. 4a. The d-spacing of both molecules was calculated from the out-of-plane XRD patterns and Bragg equation. PTII-T and PTII-TVT-8 films exhibited similar primary diffraction peaks at 2θ of 3.75°, corresponding to the d (100)-spacing of 23.55 Å, indicating a good layer-by-layer packed lamellar structure.
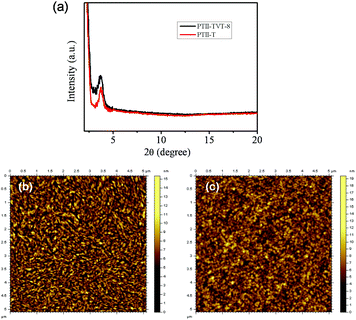 |
| | Fig. 4 (a) Out-of-plane XRD patterns of PTII-T and PTII-TVT-8; tapping-mode AFM height images of (b) PTII-T and (c) PTII-TVT-8 films. Films were spin-coated from the TCE solutions of the polymers (3 mg mL−1). | |
Besides crystallinity, the semiconductor film morphology is another decisive factor that affects charge transport properties. For OFETs to achieve high charge mobility, it is favorable for thin films to have large interconnected domains in order to minimize crystallite grain boundaries.33 As illustrated in Fig. 4, the AFM image of the PTII-TVT-8 film showed almost a smooth and spherical morphology, while the PTII-T film shows relatively fine, highly interconnected fibrillar domains with an obvious reduction of the surface roughness (RMS = 0.707 nm) when compared to PTII-TVT-8 (RMS = 1.48 nm). Crystalline zones of the PTII-T film are formed presumably due to strong intermolecular π–π interactions. We speculate that the more efficient ambipolar charge transport in the PTII-T film is related to more densely interconnected compact domains of bundled nanofibers.
Conclusions
In conclusion, we have presented the synthesis of both cross conjugated and fully conjugated polymers based on thiophene-fused isoindigo. Solution-processed OFETs based on two fully conjugated polymers exhibited stable performance under ambient conditions due to their low-lying LUMO level. Both fully conjugated polymers exhibit better performance than cross conjugated polymers. PTII-T shows balanced electron and hole mobilities of 0.029 and 0.018 cm2 V−1 s−1 respectively, and PTII-TVT-8 exhibits better hole mobility (0.037 cm2 V−1 s−1). Further improvement in OFET performance can be expected by optimizing the electron rich units and improving the molecular weight of TII-based polymers with both good solubility and OFET properties.
Experimental
General procedures and experimental details
All glassware was completely dried before use. Chemicals and solvents were purchased from commercial suppliers or purified by standard techniques. TLC plates were observed by exposure to UV light and/or immersion in a phosphomolybdic acid staining solution followed by drying. Column chromatography was carried out using a silica gel 200–300 mesh. 1H NMR and 13C NMR spectra were recorded on a Bruker Avance-III 600 MHz with tetramethylsilane (TMS) as an internal standard at 298 K and CDCl3 was used as the solvent. The coupling constants J are given in Hz. The IR spectra were observed with a Nicolet 6700 FTIR Spectrometer. Cyclic voltammetry (CV) curves were performed on an electrochemistry workstation (CHI660D, Chenhua, Shanghai) in an anhydrous argon-saturated chloroform solution (10−3 mol L−1) of 0.1 M tetrabutylammonium hexafluorophosphate (n-Bu4NPF6) at room temperature by using a three-electrode system, which utilizes the glassy carbon electrode as the working electrode, Pt as the counter electrode and a Ag/AgCl as the reference electrode at a potential scan rate of 0.1 V s−1. The potential of the reference electrode in chloroform was identified by using ferrocene as the internal standard. The UV-vis spectra were recorded with a Hitachi U-4100 UV-vis spectrophotometer in an anhydrous chloroform solution (10−5 mol L−1). HRMS were investigated on a Bruker Maxis UHR TOF. Gel permeation chromatography (GPC) was performed on a Waters 1151 pump and a UV-vis monitor (700 nm) using 1,2,4-trichlorobenzene (TCB) as the eluent (150 °C). XRD patterns were investigated with a Rigaku D/MAX-2500 X-ray diffractometer. AFM patterns were collected with a Digital Instrument NanoScope IIIa. TGA patterns were collected with a TGA Q50 V20.13 Build 39. DSC patterns were collected with a DSC Q20 V24.11 Build 124.
Device fabrication and characterization
Top-gate/bottom-contact FET devices were fabricated using n++-Si/SiO2 (300 nm) substrates. The gold source and drain bottom electrodes (with Ti as the adhesion layer) were patterned by photolithography on the SiO2 surface. The substrates were treated by using ultrasonication in acetone, cleaning agent, deionized water (twice), and isopropanol. The treated substrates were dried under vacuum at 80 °C. The substrates were used by transferring into a glove box. The thin films of the small molecules were deposited on the substrates by spin-coating using TCE solutions (3 mg mL−1) at 1500 rpm for 60 s. After OFET thin films were deposited, a CYTOP solution (CTL809M:CT-solv180 = 3:1) was spin-coated onto the semiconducting layer at 2000 rpm for 60 s resulting in a dielectric layer of 500 nm thick. The CYTOP layer was then cross-linked at 100 °C for 1 h. A layer of Al (50 nm) was then evaporated onto the dielectric layer by thermal evaporation as gate electrodes. The OFET devices had a channel ratio of width to length (W/L = 20). The performances of the OFETs were investigated on a probe stage using a Keithley 4200 parameter analyzer under an atmosphere (humidity 50–60%). The carrier mobility, μ, was calculated from the data in the saturated regime according to the equation ISD = (W/2L)Ciμ(VG − VT)2, where ISD is the drain current in the saturated regime. W and L are the semiconductor channel width and length, respectively. Ci (Ci = 3.5 nF) is the capacitance per unit area of the gate dielectric layer. VG and VT are the gate voltage and threshold voltage, respectively. VG − VT of the devices was determined from the relationship between the square root of ISD and VG in the saturated regime.
Synthetic procedures
4-Chloro-5H-thieno[2,3-f]indole-6,7-dione (1) and 2-bromo-5-nitrobenzo[b]thiophene (5) were synthesized according to the literature.14,29,30
4-Chloro-5-(2-octyldodecyl)-5H-thieno[2,3-f]indole-6,7-dione (2).
To a solution of 4-chloro-5H-thieno[2,3-f]indole-6,7-dione (238 mg, 1.0 mmol, 1.0 equiv.) and potassium carbonate (207 mg, 1.5 mmol, 1.5 equiv.) in dimethylformamide (DMF) (10 mL), 1-bromo-2-octyldodecane (434 mg, 1.2 mmol, 1.2 equiv.) was added under argon. The mixture was stirred for 24 h at 50 °C and then the solvent was removed under reduced pressure. The residues were purified by silica gel chromatography with eluting (PE:EA = 10:1) to give 4-chloro-5-(2-octyldodecyl)-5H-thieno[2,3-f]indole-6,7-dione as a red solid (337 mg, 65%). 1H NMR (CDCl3, 600 MHz, ppm) δ: 8.06 (s, 1H), 7.85 (d, J = 5.5 Hz, 1H), 7.58 (d, J = 5.5 Hz, 1H), 4.09 (d, J = 7.6 Hz, 2H), 2.06 (m, 1H), 1.39–1.25 (m, 32H), 0.91–0.86 (m, 6H). 13C NMR (CDCl3, 150 MHz, ppm): δ 182.64, 159.67, 145.98, 141.19, 135.10, 135.00, 123.71, 119.83, 118.53, 110.69, 46.32, 37.59, 37.57, 31.91, 31.87, 31.79, 30.86, 30.84, 30.04, 29.73, 29.63, 29.62, 29.58, 29.53, 29.34, 29.29, 26.14, 26.10, 22.70, 22.67, 22.64, 14.13, 14.12, 14.08. ESI-HRMS: Calcd for [M + H]+: 518.2860. Found: 518.2850.
3-Bromo-4-chloro-5-(2-octyldodecyl)-5H-thieno[2,3-f]indole-6,7-dione (3).
A mixture of 4-chloro-5-(2-octyldodecyl)-5H-thieno[2,3-f]indole-6,7-dione (518 mg, 1.0 mmol, 1.0 equiv.), sodium acetate (164 mg, 2.0 mmol, 2.0 equiv.), bromine (206 μL, 4.0 mmol, 4.0 equiv.), and acetic acid (2.0 mL) was heated under reflux for 2 h, and then poured into crushed ice (20 mL). The mixture was extracted with dichloromethane, and then the solvent was removed under reduced pressure. The residues were purified by silica gel chromatography with eluting (PE:EA = 10:1) to give 3-bromo-4-chloro-5-(2-octyldodecyl)-5H-thieno[2,3-f]indole-6,7-dione as a red solid (418 mg, 70%). 1H NMR (CDCl3, 600 MHz, ppm) δ: 8.03 (s, 1H), 7.84 (s, 1H), 4.16 (d, J = 7.6 Hz, 2H), 2.06 (m, 1H), 1.39–1.26 (m, 32H), 0.91–0.87 (m, 6H). 13C NMR (CDCl3, 150 MHz, ppm): δ 182.58, 159.57, 142.65, 138.81, 135.70, 134.20, 119.75, 118.91, 112.24, 107.16, 46.96, 37.66, 37.64, 31.92, 31.88, 31.79, 30.85, 30.83, 30.06, 29.75, 29.65, 29.62, 29.57, 29.52, 29.35, 29.30, 26.12, 26.08, 22.70, 22.68, 22.65, 14.14, 14.12, 14.08. ESI-HRMS: Calcd for [M + H]+: 596.1965. Found: 596.1949.
β-Dibromothiophene fused isoindigo (4, β-Br-TII).
Tris(diethylamino)phosphine (274 μL, 1.0 mmol, 1.0 equiv.) was added dropwise to a solution of 3-bromo-4-chloro-5-(2-octyldodecyl)-5H-thieno[2,3-f]indole-6,7-dione (597 mg, 1.0 mmol, 1.0 equiv.) in dichloromethane (10 mL) at −78 °C under an argon atmosphere. Thereafter, the temperature was gradually increased to room temperature and stirred for another 30 min. The solvent was removed under reduced pressure, and the residue was then purified by column chromatography using petroleum ether as the eluent to give β-Br-TII as a dark-red solid (511 mg, 88%). 1H-NMR (CDCl3, 600 MHz) δ: 9.65 (s, 2H), 7.62 (s, 2H), 4.14 (d, J = 7.6 Hz, 4H), 2.03 (m, 2H), 1.36–1.25 (m, 64H), 0.90–0.84 (m, 12H). 13C-NMR (CDCl3, 150 MHz) δ: 168.32, 138.34, 134.80, 134.52, 132.16, 131.03, 123.40, 123.11, 109.42, 106.27, 46.65, 37.71, 31.93, 31.88, 31.81, 30.90, 30.14, 29.79, 29.72, 29.68, 29.64, 29.62, 29.57, 29.38, 29.33, 26.14, 26.11, 22.71, 22.68, 22.64, 14.15, 14.12, 14.08. APCI-HRMS: Calcd for [M + H]+: 1161.3932. Found: 1161.3932.
cTII-2T
.
β-Br-TII (116 mg, 0.1 mmol, 1.0 equiv.), tributyl(thiophen-2-yl)stannane (78 mg, 0.21 mmol, 2.1 equiv.), Pd(PPh3)4 (2.3 mg, 2 mol%), and 2 mL of THF were added to a Schlenk tube. The tube was charged with argon through a freeze–pump–thaw cycle three times. The mixture was stirred for 24 h at 80 °C. The solvent was removed under reduced pressure, and the residue was then purified by column chromatography using petroleum ether as the eluent to give cTII-2T as a dark-red solid (103 mg, 89%). 1H-NMR (CDCl3, 600 MHz) δ: 9.72 (d, 2H, J = 1.86 Hz), 7.63 (s, 2H), 7.43 (dd, 2H, J1 = 5.10 Hz, J2 = 1.08 Hz), 7.10 (m, 4H), 4.11 (d, J = 7.6 Hz, 4H), 2.01 (m, 2H), 1.29–1.21 (m, 64H), 0.90–0.83 (m, 12H). 13C-NMR (CDCl3, 150 MHz) δ: 168.60, 138.24, 137.61, 137.49, 135.66, 133.58, 132.25, 131.10, 128.91, 126.43, 125.88, 123.43, 122.85, 109.85, 46.55, 37.44, 31.93, 31.88, 31.80, 30.81, 30.08, 29.73, 29.68, 29.64, 29.60, 29.54, 29.37, 29.33, 26.05, 26.02, 22.71, 22.68, 22.66, 14.15, 14.13, 14.09. APCI-HRMS: Calcd for [M + H]+: 1167.5497. Found: 1167.5487.
2-Bromobenzo[b]thiophen-5-amine (6).
To a suspension of 2-bromo-5-nitrobenzo[b]thiophene (1.29 g, 5.0 mmol, 1.0 equiv.) in methanol (15 mL) were added stannous chloride dehydrate (6.77 g, 30.0 mmol, 6.0 equiv.) and a catalytic amount of hydrochloric acid. Then the mixture was heated to reflux and stirred until TLC indicated the consumption of starting materials (up to 4 hours). Then reaction was then cooled to 0 °C in an ice bath, and 2 M aqueous solution of NaOH was added to adjust the pH value of the system to 8–9. The product was extracted with ethyl acetate 5 times and the combined organic phase was dried with anhydrous MgSO4. The solvent was removed under reduced pressure, and the residue was then purified by column chromatography using petroleum ether/ethyl acetate (4:1) as the eluent to give 2-bromobenzo[b]thiophen-5-amine as a white solid (1.02 g, 90%). 1H-NMR (CDCl3, 600 MHz) δ: 7.49 (d, 1H, J = 8.54 Hz), 7.16 (s, 1H), 6.99 (d, J = 2.22 Hz, 1H), 6.76 (dd, J1 = 8.54 Hz, J2 = 2.22 Hz, 1H), 3.73 (br, 2H). 13C-NMR (CDCl3, 150 MHz) δ: 144.00, 140.66, 131.41, 125.92, 122.15, 115.88, 114.64, 107.50. APCI-HRMS: Calcd for [M + H]+: 227.9483, found: 227.9482.
2-Bromo-4-chlorobenzo[b]thiophen-5-amine (7).
To the solution of 2-bromobenzo[b]thiophen-5-amine (2.28 g, 10.0 mmol, 1.0 equiv.) in anhydrous THF (50 mL) was added N-chlorosuccinimide (1.47 g, 10.0 mmol, 1.0 equiv.) in one portion. The mixture was heated to reflux and stirred for 2 hours. The reaction was quenched with water and extracted with dichloromethane 3 times. The combined organic phases were dried with sodium sulfate. The solvent was removed under reduced pressure and the crude product was purified by column chromatography (silica gel; eluent: PE:EA = 10:1) to afford 2-bromo-4-chlorobenzo[b]thiophen-5-amine as a white solid (2.32 g, 88%). 1H-NMR (CDCl3, 600 MHz) δ: 7.38 (m, 2H), 6.81 (d, J = 8.40 Hz, 1H), 4.10 (br, 2H). 13C-NMR (CDCl3, 150 MHz) δ: 140.18, 138.57, 131.43, 124.58, 120.32, 116.81, 114.78, 111.16. APCI-HRMS: Calcd for [M + H]+: 261.9093, found: 261.9090.
(E)-N-(2-Bromo-4-chlorobenzo[b]thiophen-5-yl)-2-(hydroxyimino)acetamide (8).
Solution A: To a 6.0 mL ethanol solution of 2-bromo-4-chlorobenzo[b]thiophen-5-amine (1.45 g, 5.5 mmol, 1.0 equiv.) were added ethanol (35 mL), conc. HCl (2.5 mL), water (130 mL) and anhydrous Na2SO4 (6.25 g, 44.0 mmol, 8.0 equiv.). To aid dissolution, the mixture was heated to approximately 40 °C and stirred vigorously. Solution B: Chloral hydrate (1.36 g, 8.25 mmol, 1.5 equiv.) and hydroxylamine hydrochloride (1.34 g, 19.25 mmol, 3.5 equiv.) were dissolved in 25 mL of water and stirred for 10 min. Solution A was then added in one portion to solution B. The resulting mixture was heated to 80 °C, and stirred for 2 hours. Then the mixture was cooled to room temperature. The precipitate was collected by filtration and washed with water. After drying in a vacuum oven overnight, 1.68 g (91%) of product was obtained. The crude product can be used in the next step without further purification. APCI-HRMS: Calcd for [M + H]+: 332.9100, found: 332.9093.
2-Bromo-4-chloro-5H-thieno[2,3-f]indole-6,7-dione (9).
A 25 mL flask was charged with 5.0 mL of conc. sulfuric acid. After heating to 50 °C in an oil bath, (E)-N-(2-bromo-4-chlorobenzo[b]thiophen-5-yl)-2-(hydroxyimino)acetamide (1.00 g, 3.0 mmol) was added over a period of 10 min. The resulting deep red solution was heated to 80 °C for 30 min and then cooled to room temperature. The mixture was then added rapidly to a vigorously stirred mixture of 30 mL of crashed ice and left undisturbed for an hour. The precipitate was removed by filtration and washed with water. The crude product was dried at low pressure, whereupon 0.91 g (95%) of red powder was obtained. The crude product can be used in the next step without further purification. APCI-HRMS: Calcd for [M + H]+: 315.8835, found: 315.8829.
2-Bromo-4-chloro-5-(2-octyldodecyl)-5H-thieno[2,3-f]indole-6,7-dione (10).
To a solution of 2-bromo-4-chloro-5H-thieno[2,3-f]indole-6,7-dione (0.91 g, 2.9 mmol, 1.0 equiv.) and potassium carbonate (0.80 mg, 5.8 mmol, 2.0 equiv.) in dimethylformamide (DMF) (30 mL), 1-bromo-2-octyldodecane (2.10 g, 5.8 mmol, 2.0 equiv.) was added under argon. The mixture was stirred for 24 h at 50 °C and then the solvent was removed under reduced pressure. The residues were purified by silica gel chromatography with eluting (PE:EA = 10:1) to give 2-bromo-4-chloro-5-(2-octyldodecyl)-5H-thieno[2,3-f]indole-6,7-dione as a red solid (0.63 g, 35% for 2 steps). 1H NMR (CDCl3, 600 MHz, ppm) δ: 7.89 (s, 1H), 7.56 (s, 1H), 4.06 (d, J = 7.6 Hz, 2H), 2.01 (m, 1H), 1.38–1.24 (m, 32H), 0.90–0.87 (m, 6H). 13C NMR (CDCl3, 150 MHz, ppm): δ 182.19, 159.39, 145.85, 141.90, 135.80, 126.53, 124.51, 118.49, 118.11, 109.61, 46.32, 37.59, 31.92, 31.88, 30.81, 30.03, 29.64, 29.62, 29.57, 29.53, 29.35, 29.30, 26.11, 22.70, 22.68, 14.14, 14.12. APCI-HRMS: Calcd for [M + H]+: 596.1965, found: 596.1963.
α-Dibromothiophene fused isoindigo (11, α-Br-TII).
Tris(diethylamino)phosphine (466 μL, 1.7 mmol, 1.0 equiv.) was added dropwise to a solution of 2-bromo-4-chloro-5-(2-octyldodecyl)-5H-thieno[2,3-f]indole-6,7-dione (1.01 g, 1.7 mmol, 1.0 equiv.) in dichloromethane (35 mL) at −78 °C under an argon atmosphere. Thereafter, the temperature was gradually increased to room temperature and stirred for another 30 min. The solvent was removed under reduced pressure, and the residue was then purified by column chromatography using petroleum ether as the eluent to give α-Br-TII as a dark-red solid (0.91 g, 92%). 1H-NMR (CDCl3, 600 MHz) δ: 9.54 (s, 2H), 7.48 (s, 2H), 4.11 (d, J = 7.6 Hz, 4H), 2.02 (m, 2H), 1.32–1.23 (m, 64H), 0.91–0.86 (m, 12H). 13C-NMR (CDCl3, 150 MHz) δ: 168.31, 141.55, 137.27, 135.13, 132.22, 126.02, 122.47, 121.73, 120.89, 107.35, 46.10, 37.69, 31.93, 31.89, 30.94, 30.10, 29.68, 29.64, 29.62, 29.57, 29.38, 29.33, 26.20, 26.19, 22.71, 22.68, 14.15, 14.11. APCI-HRMS: Calcd for [M + H]+: 1161.3932, found: 1161.3950.
Procedures for Stille polymerization and polymer purification
PTII-T
.
α-Br-TII (116 mg, 0.1 mmol, 1.0 equiv.), 2,5-bis(trimethylstannyl)thiophene (41 mg, 0.1 mmol, 1.0 equiv.), Pd(PPh3)4 (2.3 mg, 2 mol%), and 2.5 mL of toluene were added to a Schlenk tube. The tube was charged with argon through a freeze–pump–thaw cycle three times. The mixture was stirred for 48 h at 110 °C. And then the mixture was precipitated into methanol (50 mL). The precipitate was filtered and purified via Soxhlet extraction for 8 h with methanol, 12 h with hexane, and finally was collected with chloroform. The chloroform solution was then concentrated and precipitated into methanol (50 mL) to give a dark blue solid (100 mg, 92%). GPC: Mn = 26.6 kDa; PDI = 2.41.
PTII-TVT-8
.
α-Br-TII (116 mg, 0.1 mmol, 1.0 equiv.), (E)-1,2-bis(3-octyl-5-(trimethylstannyl)thiophen-2-yl)ethene (74 mg, 0.1 mmol, 1.0 equiv.), Pd(PPh3)4 (2.3 mg, 2 mol%), and 2.5 mL of toluene were added to a Schlenk tube. The tube was charged with argon through a freeze–pump–thaw cycle three times. The mixture was stirred for 48 h at 110 °C. And then the mixture was precipitated into methanol (50 mL). The precipitate was filtered and purified via Soxhlet extraction for 8 h with methanol, 12 h with chloroform, and finally was collected with chlorobenzene. The chlorobenzene solution was then concentrated and precipitated into methanol (50 mL) to give a dark blue solid (101 mg, 71%). GPC: Mn = 31.0 kDa; PDI = 3.46.
Acknowledgements
This work is supported by “100 Talents” Program from Chinese Academy of Sciences and National Science Foundation of China (NSFC no. 21174157, 51573204).
Notes and references
- S. Kola, J. Sinha and H. E. Katz, J. Polym. Sci., Part B: Polym. Phys., 2012, 50, 1090–1120 CrossRef CAS
 .
.
- A. Mishra and P. Bauerle, Angew. Chem., Int. Ed., 2012, 51, 2020–2067 CrossRef CAS PubMed .
- J. Mei, Y. Diao, A. L. Appleton, L. Fang and Z. Bao, J. Am. Chem. Soc., 2013, 135, 6724–6746 CrossRef CAS PubMed .
- Y. Zhao, Y. Guo and Y. Liu, Adv. Mater., 2013, 25, 5372–5391 CrossRef CAS PubMed .
- S. Holliday, J. E. Donaghey and I. McCulloch, Chem. Mater., 2014, 26, 647–663 CrossRef CAS .
- J. Mei, K. R. Graham, R. Stalder and J. R. Reynolds, Org. Lett., 2010, 12, 660–663 CrossRef CAS PubMed .
- T. Lei, Y. Cao, Y. Fan, C.-J. Liu, S.-C. Yuan and J. Pei, J. Am. Chem. Soc., 2011, 133, 6099–6101 CrossRef CAS PubMed .
- T. Lei, Y. Cao, X. Zhou, Y. Peng, J. Bian and J. Pei, Chem. Mater., 2012, 24, 1762–1770 CrossRef CAS .
- T. Lei, J.-H. Dou, Z.-J. Ma, C.-H. Yao, C.-J. Liu, J.-Y. Wang and J. Pei, J. Am. Chem. Soc., 2012, 134, 20025–20028 CrossRef CAS PubMed .
- P. Deng and Q. Zhang, Polym. Chem., 2014, 5, 3298–3305 RSC .
- T. Lei, J.-Y. Wang and J. Pei, Acc. Chem. Res., 2014, 47, 1117–1126 CrossRef CAS PubMed .
- R. Stalder, J. Mei, K. R. Graham, L. A. Estrada and J. R. Reynolds, Chem. Mater., 2014, 26, 664–678 CrossRef CAS .
- R. S. Ashraf, A. J. Kronemeijer, D. I. James, H. Sirringhaus and I. McCulloch, Chem. Commun., 2012, 48, 3939–3941 RSC .
- N. Zhao, L. Qiu, X. Wang, Z. An and X. Wan, Tetrahedron Lett., 2014, 55, 1040–1044 CrossRef CAS .
- S. Xu, N. Ai, J. Zheng, N. Zhao, Z. Lan, L. Wen, X. Wang, J. Pei and X. Wan, RSC Adv., 2014, 5, 8340–8344 RSC .
- I. Meager, M. Nikolka, B. C. Schroeder, C. B. Nielsen, M. Planells, H. Bronstein, J. W. Rumer, D. I. James, R. S. Ashraf, A. Sadhanala, P. Hayoz, J.-C. Flores, H. Sirringhaus and I. McCulloch, Adv. Funct. Mater., 2014, 24, 7109–7115 CAS .
- T. Hasegawa, M. Ashizawa and H. Matsumoto, RSC Adv., 2015, 5, 61035–61043 RSC .
- W. Yue, R. S. Ashraf, C. B. Nielsen, E. Collado-Fregoso, M. R. Niazi, S. A. Yousaf, M. Kirkus, H. Y. Chen, A. Amassian, J. R. Durrant and I. McCulloch, Adv. Mater., 2015, 27, 4702–4707 CrossRef CAS PubMed .
- Y. K. Voronina, D. B. Krivolapov, A. V. Bogdanov, V. F. Mironov and I. A. Litvinov, J. Struct. Chem., 2012, 53, 413–416 CrossRef CAS .
- A. V. Bogdanov, V. F. Mironov, L. I. Musin, B. I. Buzykin and A. I. Konovalov, Russ. J. Gen. Chem., 2008, 78, 1977–1979 CrossRef CAS .
- T. Lei, J.-H. Dou, Z.-J. Ma, C.-J. Liu, J.-Y. Wang and J. Pei, Chem. Sci., 2013, 4, 2447–2452 RSC .
- Y. Deng, J. Liu, J. Wang, L. Liu, W. Li, H. Tian, X. Zhang, Z. Xie, Y. Geng and F. Wang, Adv. Mater., 2014, 26, 471–476 CrossRef CAS PubMed .
- N. F. Phelan and M. Orchin, J. Chem. Educ., 1968, 45, 633–637 CrossRef CAS .
- R. P. Dickinson and B. Iddon, J. Chem. Soc. C, 1968, 2733–2737 RSC .
- E. Baciocchi, R. Ruzziconi and G. V. Sebastiani, J. Am. Chem. Soc., 1983, 105, 6114–6120 CrossRef CAS .
- T. Yamamoto, S. Ogawa and R. Sato, Tetrahedron Lett., 2004, 45, 7943–7946 CrossRef CAS .
- T. Kobayashi, M. Nakashima, T. Hakogi, K. Tanaka and S. Katsumura, Org. Lett., 2006, 8, 3809–3812 CrossRef CAS PubMed .
- T. Qi, Y. Guo, Y. Liu, H. Xi, H. Zhang, X. Gao, Y. Liu, K. Lu, C. Du, G. Yu and D. Zhu, Chem. Commun., 2008, 6227–6229 RSC .
- S. G. Newman, V. Aureggi, C. S. Bryan and M. Lautens, Chem. Commun., 2009, 5236–5238 RSC .
- T. T. B. Nguyen, T. Lomberget, N. C. Tran and R. Barret, Tetrahedron, 2013, 69, 2336–2347 CrossRef CAS .
- Y. J. Cheng, S. H. Yang and C. S. Hsu, Chem. Rev., 2009, 109, 5868–5923 CrossRef CAS PubMed .
- J.-L. Bredas, Mater. Horiz., 2014, 1, 17–19 RSC .
- H. N. Tsao and K. Mullen, Chem. Soc. Rev., 2010, 39, 2372–2386 RSC .
- J. Mei, D. H. Kim, A. L. Ayzner, M. F. Toney and Z. Bao, J. Am. Chem. Soc., 2011, 133, 20130–20133 CrossRef CAS PubMed .
- G. Kim, A. R. Han, H. R. Lee, J. Lee, J. H. Oh and C. Yang, Chem. Commun., 2014, 50, 2180–2183 RSC .
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1422575, 1422577. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5py01488c |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.