
Influence of the secondary structure on the AIE-related emission behavior of an amphiphilic polypeptide containing a hydrophobic fluorescent terminal and hydrophilic pendant groups†
Li-Yang
Lin
,
Po-Chiao
Huang
,
Deng-Jie
Yang
,
Jhen-Yan
Gao
and
Jin-Long
Hong
*
Department of Materials and Optoelectronic Science, National Sun Yat-Sen University, Kaohsiung 80424, Taiwan. E-mail: jlhong@mail.nsysu.edu.tw; Tel: +886-7-5252000, ext 4065
First published on 12th October 2015
Abstract
In this study of the amphiphilic polypeptide TP-PPLG-g-MEO2 containing a hydrophobic tetraphenylthiophene (TP) terminal, with aggregation-induced emission (AIE) properties, and hydrophilic ether side groups, it was found that secondary structures (α-helix, β-sheet and random coil) of the peptide chains affected the AIE-related emission of the TP terminal. The amphiphilic TP-PPLG-g-MEO2, prepared from a ring-opening polymerization and the following click reaction, contained a secondary structure mostly composed of α-helical chains. The main α-helical chain nevertheless can be converted into a β-sheet structure by adding NaCl into the aqueous solution of TP-PPLG-g-MEO2 and by doing so, the solution emission can be enhanced. In addition, the coil chain generated in the alkaline solution was found to emit efficiently with the highest emission intensity among all three conformations. The relationship between the AIE property and the secondary structure of the peptide chains can therefore be evaluated. All three secondary structures were also used as luminescent sensors to test their sensitivity for bovine serum albumin (BSA).
Introduction
Since the first-discovered silole-derivative of 2,3,4,5,6-pentaphenyl-1-methylsilole1,2 (PMS) as a luminogen with aggregation-induced emission (AIE) properties, many AIE-active luminogens (AIEgens)3–10 have been prepared and characterized regarding their unique feature that the dilute, non- or weakly-emissive solution of AIEgens can be tuned to emit intensely in the solution aggregated and solid states. Theoretical studies suggested that the restricted intramolecular rotation (RIM) of non-planar, propeller-shaped AIEgens10–12 in the solution aggregated and solid states is the inherent mechanism responsible for the enhanced emission of AIEgens. With regard to the promising emission properties, several AIEgens had been explored for high-tech applications such as optoelectronic materials,13–16 chemical sensors17–19 and biomedical probes.20,21Synthetic polypeptides have been under considerable investigation regarding their potential applications in various scientific fields and their close relationship to proteins.22–32 The secondary structures (α-helix, β-sheet and random coil) of peptide chains are cornerstones for the construction of the well-defined tertiary structure of proteins, therefore, a study on the synthetic polypeptide and its chain conformations in organic solvents illustrated one of the major efforts previously dedicated to understanding the complicated protein–protein interactions. In this respect, several traditional luminogens (such as carbazolyl,33 dansyl34 and pyrene35) had been chemically incorporated with synthetic polypeptides in order to monitor the chain conformation of polypeptides in solution. For example, no excimer emission was detected for the pyrene-labeled polypeptide35 since pyrene groups were well separated from each other by the polypeptide chains.
AIEgens were previously incorporated with synthetic polypetides36,37 with the purpose of detecting conformational changes of the peptide chain by AIE-related emission. An AIEgen of tetraphenylthiophene (TP) served as the terminal and central units of poly(benzyl-L-glutamate)s (PBLGs) in the synthetic polypeptides of TP1PBLG and TP2PBLG,36 respectively. Because the intermolecular aggregation of TP centers in TP2PBLG is sterically blocked by the large α-helical chains of TP2PBLG, a solution of TP2PBLG is lower in emission than a solution of TP1PBLG, whose TP terminal can be readily approached by other TP terminals to result in aggregates with a high emission intensity. Adding trifluoroacetic acid (TFA) to a solution of TP2PBLG converted the rigid α-helical chain into a flexible random coil; after that, the emission intensity of TP2PBLG was greatly increased due to the easy aggregation of the TP centers in the flexible coil chains. In contrast to the hydrophobic TP1PBLG and TP2PBLG, a water-soluble, TP-terminated polypeptide containing ionic sulfonate pendent groups37 was synthesized later and characterized, exhibiting a pH-induced helix-to-coil transition in an alkaline aqueous solution. Again, the emission of this ionic polypeptide in an alkaline solution was enhanced to a great extent due to the easy aggregation of the TP terminals in the coil chains.
The above examples36,37 suggested crucial roles for the random coil and α-helix structures in enhancing and weakening the AIE-related emission, respectively, of synthetic polypeptides. Despite early efforts, a correlation between the β-sheet structure and AIE activity is still in demand due to the lack of a reliable way to generate a peptide chain composed mostly of a β-sheet structure. This question can nevertheless be solved soon since we found that the fraction of β-sheet conformations can be greatly enhanced by adding NaCl salt into an aqueous solution of the amphiphilic polypeptide TP-PPLG-g-MEO2 (Scheme 1). With a hydrophobic TP terminal and hydrophilic methyl bis(ethylene oxide) (MEO2) etheric side groups, the target polypeptide TP-PPLG-g-MEO2 is amphiphilic and can be prepared by a two-step reaction procedure including a ring-opening polymerization (ROP) of the PLG-NCA monomer initiated by the amino-functionalized AIEgen TP-NH2 and the following click reaction for the introduction of the MEO2 side groups. The amphiphilic TP-PPLG-g-MEO2, composed mostly of α-helical chains, can be dispersed in salt and in alkaline aqueous solutions to generate the respective β-sheet and random-coil chains for correlating their AIE-related emission behavior with the secondary structures of the peptide chains. This correlation is meaningful considering that the aggregation tendency and corresponding AIE-related emission of the TP terminal in TP-PPLG-g-MEO2 are essentially controlled by the conformation of the peptide chain connecting to the TP terminal; that is, aggregation of TP terminals is affected by neighboring peptide chains of different chain conformations and so is the aggregation-related AIE emissive behavior. Besides theoretical correlation between the conformational change and AIE activity, all three secondary structures generated in this study were further employed as luminescent sensors for the natural protein bovine serum albumin (BSA). Corresponding luminescence responses can be used to evaluate how well the individual secondary structure complexed to BSA. The complexation tendency is strongly related to the aggregation level of TP terminals in the large peptide matrix of BSA.
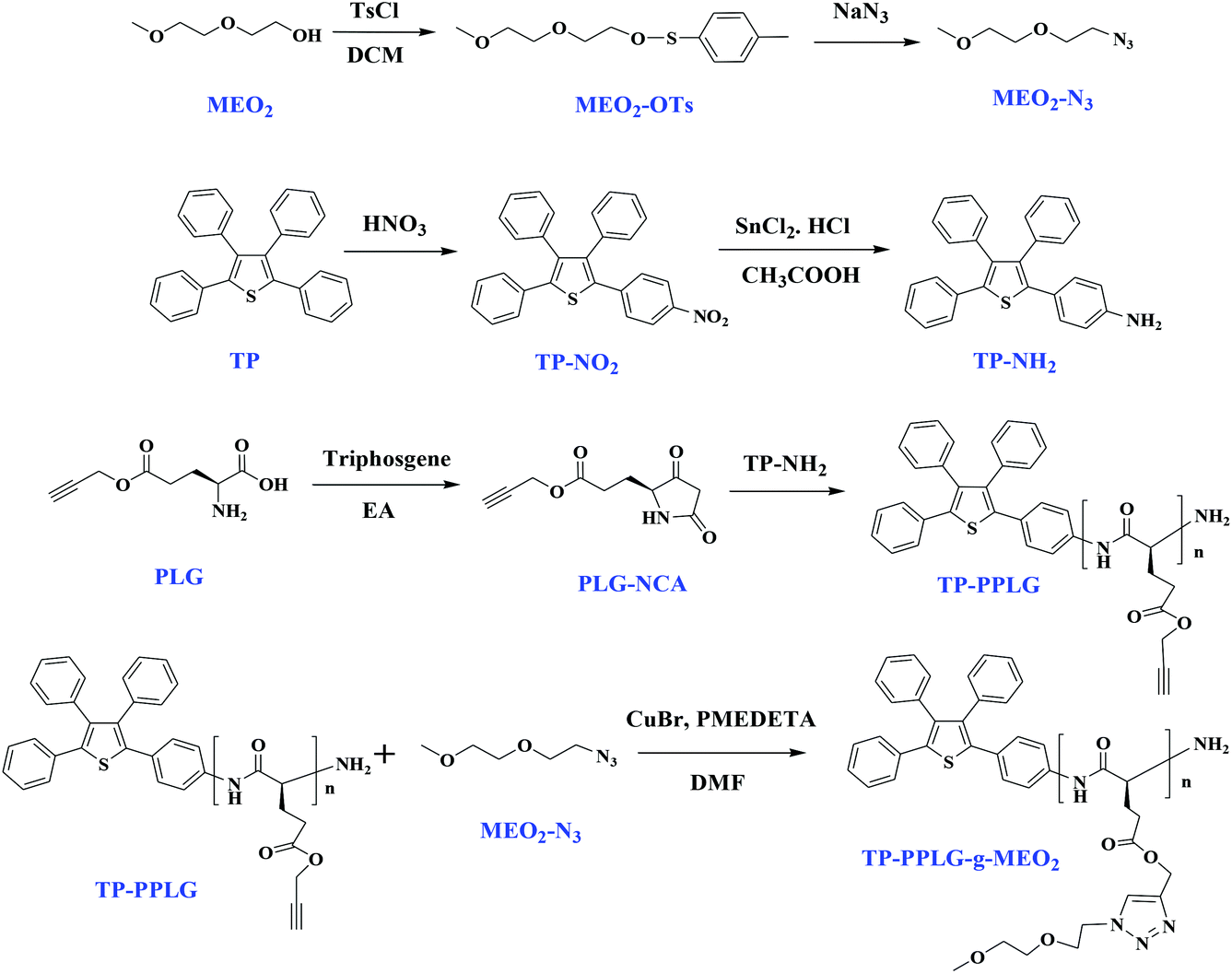 | ||
| Scheme 1 Syntheses of MEO2-N3, TP-NH2, and the PLG-NCA monomer for the ring-opening polymerization by TP-NH2 and the following click reaction to obtain TP-PPLG-g-MEO2. | ||
Experimental section
Materials
DMF (Aldrich) was refluxed and distilled over CaH2 (Aldrich) under nitrogen atmosphere to a flask containing alumina before use. CuBr (98%, Aldrich) was stirred overnight in acetic acid, filtered, washed with ethanol and diethyl ether, and then dried in a vacuum before use. Compounds of TP-NH2,38 MEO2-N3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 39 and γ-propargyl-L-glutamate N-carboxyanhydride (PLG-NCA)40,41 were prepared according to the reported procedures. Polypeptides of TP-PPLG and TP-PPLG-g-MEO2 were synthesized from the procedures given below.
39 and γ-propargyl-L-glutamate N-carboxyanhydride (PLG-NCA)40,41 were prepared according to the reported procedures. Polypeptides of TP-PPLG and TP-PPLG-g-MEO2 were synthesized from the procedures given below.
Ring-opening polymerization of N-carboxyanhydride PLG-NCA to prepare TP-PPLG
A solution of PLG-NCA (3 g, 14.2 mmol) in anhydrous DMF (20 mL) was stirred and bubbled with argon for 20 min before adding a solution of TPNH2 (0.03 mL, 0.48 mmol) in anhydrous DMF (5 mL). The reaction mixture was stirred for 2 days at room temperature and the resulting polymer was precipitated from diethyl ether, and dried in a vacuum oven. 1H NMR (500 MHz, CD2Cl2): δ 8.34 (broad, Hf), 7.5–7.03 (broad, 19H, Hg), 4.68 (s, Hb), 3.98 (broad, He), 2.72 (broad, 2H, Hc), 2.48 (s, 1H, Ha), 2.32–2.19 (broad, 2H, Hd) (Fig. 2a). 13CNMR (125 MHz, CD2Cl2): δ 177.2, 173.0, 132–128, 79.3, 76.0, 57.5, 52.9, 31.9, 26.4. (Fig. S5a†).Preparations of TP-PPLG-g-MEO2 by click reaction
MEO2-N3 (0.15 g, 0.8 mmol), TP-PPLG (0.1 g, 0.6 mmol), and CuBr (0.03 g, 0.12 mmol) were dissolved in DMF (25 mL) in a two-neck flask equipped with a magnetic stirrer and a condenser. After three freeze−thaw−pump cycles, the ligand N,N,N′,N′′,N′′-pentamethyldiethylenetriamine (0.02 mL, 0.12 mmol) was added. Whole reaction mixtures were further subjected to three freeze−thaw−pump cycles before heating at 60 °C for 24 h. After cooling to room temperature, the resulting mixtures were passed through a neutral alumina column to remove the copper catalysts. The solution was then concentrated by rotary evaporation and precipitated from diethyl ether. The precipitate was filtered off and dried under a vacuum at room temperature to obtain the final yellow-white product. TP-PPLG-g-MEO2: 1H NMR (500 MHz, d6-DMSO/DCl (tiny)) 34 (s, Hf′), 8.14 (s, Hh′), 7.22–7.03 (broad, 19H, Hg′), 5.23–5.11 (s, Hb′), 4.50 (t, Hi′), 3.83 (t, Hj′), 3.54 (t, Hk′), 3.44 (t, Hl′), 3.27–3.21 (s, Hm′), 2.56–2.2 (broad, Hc′), 2.12–1.86 (broad, Hd′) (Fig. 2b). 13C NMR (125 MHz, CD2Cl2): δ 177.2, 173.9, 144.0, 129.36, 126.0, 72–70.1, 59.6, 50.8, 30.6, 26.0 (Fig. S5b†).Measurements
1H NMR spectra were recorded at room temperature using a Bruker AM 500 (500 MHz) spectrometer with tetramethylsilane (TMS) as the external standard. 13C CP-MAS NMR spectra were acquired on a Bruker 14.1 T wide-bore Avance III spectrometer equipped with a 4 mm double-resonance magic-angle-spinning (MAS) probe head. The Larmor frequency used for 13C NMR was 150.92 MHz and the samples were spun at 12 kHz. Infrared spectra were recorded using a Bruker Tensor 27 Fourier-transform infrared (FTIR) spectrophotometer; 32 scans were collected at a spectral resolution of 1 cm−1. The solid polymer powders were homogeneously blended with KBr before being pressed to make pellets for measurements. DSC analyses were performed using a TA Q-20 differential scanning calorimeter operated at a heating rate of 10 °C min−1. Relative molecular weights of the polymers were determined through gel permeation chromatography (GPC) using a Waters 510 high performance liquid chromatography (HPLC) system with DMF as the eluent and a flow rate of 0.4 mL min−1. The molecular weight calibration curve was obtained from polystyrene standards. The PL emission spectra were obtained from a LabGuide X350 fluorescence spectrophotometer using a 450 W Xe lamp as the continuous light source. A small quartz cell with dimensions 0.2 × 1.0 × 4.5 cm3 was used to accommodate the solution sample. A “right angle” geometry was employed such that fluorescence was collected at a right angle to the excitation beam used in the spectral measurement. Circular dichroism (CD) spectra were recorded using a JASCO J-810 spectrometer, with the sample in deionized water at a concentration of 10−4 M. The spectra were also curve-fitted using the Spectra Manager program to resolve the fraction of the individual secondary structure. The dimension of the aggregate particles was determined using a DLS instrument on a Malvern ZetaSizer Nano ZS90 spectrometer. A He–Ne laser operating at 633 nm was used as the light source.Results and discussion
Synthesis of the water-soluble, ionic polymer TP-PPLG-MEO2
According to Scheme 1, the click reaction between the alkyne side groups of TP-PPLG and MEO2-N3 (1-(2-methoxyethoxy)-2-azidoethane) is the key step leading to TP-PPLG-g-MEO2. Hence, the two intermediates of TP-PPLG and MEO2-N3, involved in the click reaction needed to be prepared primarily. TP-PPLG was synthesized from the ring-opening polymerization of PLG-NCA40,41 initiated by the amino group of TP-NH2.38 PLG-NCA was prepared from the facile cyclization of propargyl-glutamate (PLG)40,41 by triphosgene whereas TP-NH2 was prepared from a two-step synthesis procedure, including nitration of TP and the subsequent reduction of the nitro group to generate TP-NH2. Another intermediate, MEO2-N3,39 was synthesized by a substitution reaction of sodium azide (NaN3) and tosylated-MEO2 (MEO2-OTs), which was prepared from the reaction of MEO2 and tosyl chloride (TsCl). All intermediates (Fig. S1–4. ESI†) and products were carefully purified and identified. The molecular weight and fraction of the secondary structure of TP-PPLG and TP-PPLG-g-MEO2 are summarized in Table 1 for comparison.| Sample | Molecular weight | Secondary structure | |||||
|---|---|---|---|---|---|---|---|
|
M
na |
PDIa |
M
nb |
DPb |
A
helixc |
A sheet |
A
rodc |
|
| a Obtained from GPC. b Determined from 1H NMR (calculated from the peak ratio between resonances Hb and Hg in TP-PPLG and between Hb′ and Hg′ in TP-PPLG-g-MEO2). c Determined from deconvoluting the infrared absorption peaks in Fig. 4. | |||||||
| TP-PPLG | 53000 |
1.15 | 17838 |
104.5 | 72.7 | 15.2 | 12.1 |
| TP-PPLG-g-MEO2 | 156000 |
1.22 | 32999 |
105.2 | 89.4 | 4.9 | 5.7 |
Primarily, FTIR analysis was applied to confirm the success of the click reaction. Fig. 1 characterizes the absorption changes of the azido and acetylene functions in MEO2-N3 and TP-PPLG, respectively. The signals at 2130 cm−1, representative of the alkyne group of TP-PPLG, and 2105 cm−1, representative of the azido group of MEO2-N3, are absent in the spectrum of TP-PPLG-g-MEO2. A successful click reaction, which is efficient in converting azido and acetylene groups into triazole rings, should take place preferentially.
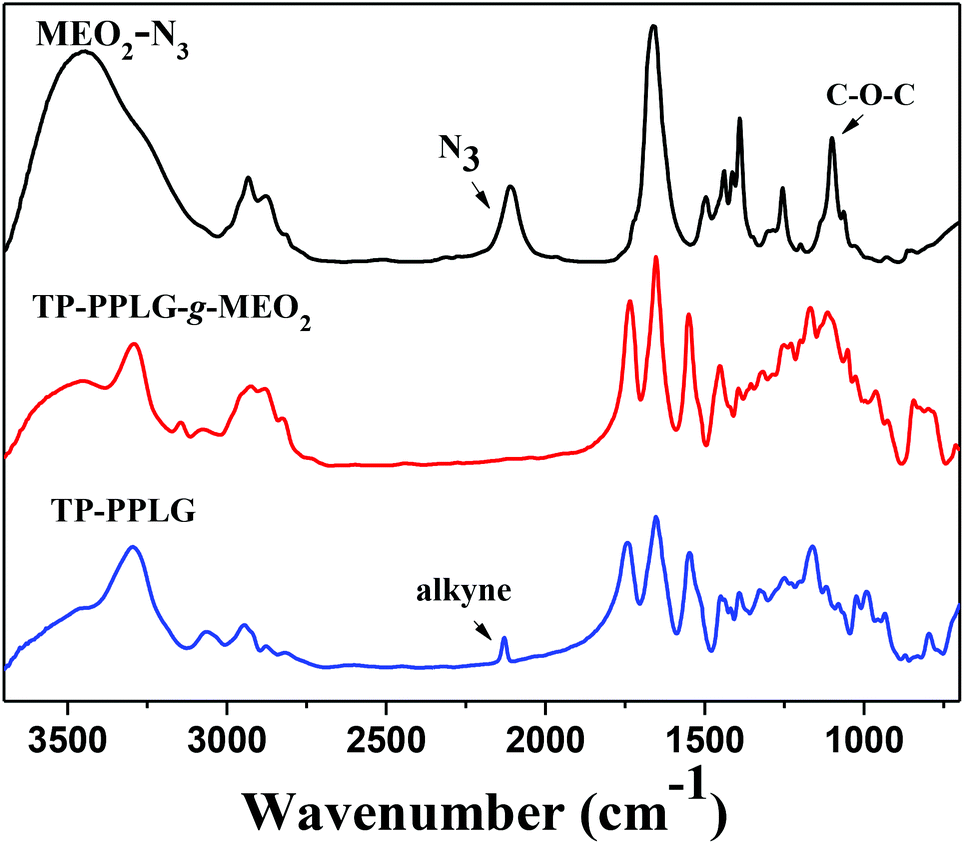 | ||
| Fig. 1 Solid FTIR spectra of MEO2-N3, TP-PPLG and TP-PPLG-g-MEO2. | ||
We further compared 1H NMR spectra of TP-PPLG and TP-PPLG-g-MEO2 in Fig. 2, to demonstrate the success of the click reaction. In the spectrum of TP-PPLG (Fig. 2a), the alkyne protons Ha resonated at 2.47 ppm as a singlet, overlapping with resonances of alkyl protons Hc and Hd, and solvent peaks in the range of 2.17 to 2.73 ppm. The click reaction transformed the alkyne group into triazine and therefore, the corresponding protons Hb′ of TP-PPLG-g-MEO2 resonated at 5.17 ppm (Fig. 2b). The resonance of CH2 protons Hb, neighboring the alkyne group of TP-PPLG, originally were located at 4.70 ppm but after the click reaction, the CH2 protons Hb′ now neighbored the triazole ring of TP-PPLG-g-MEO2 and therefore resonated at a lower field of 5.17 ppm. A successful click reaction was therefore verified from the 1H NMR spectra.
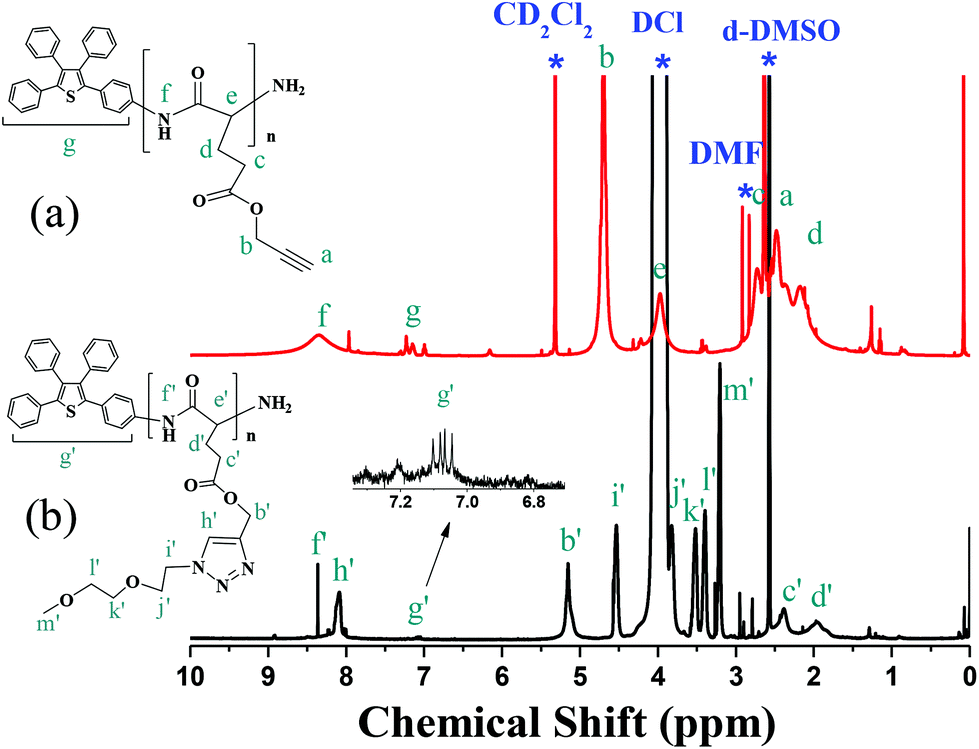 | ||
| Fig. 2 1H NMR spectra of (a) TP-PPLG (in d6-DMSO) and (b) TP-PPLG-g-MEO2 (in d6-DMSO + DCl (tiny)). | ||
By calculating the intensity ratio of resonances Hb to Hg, the average molecular weight (Mn, 17840 g mol−1, Table 1) of TP-PPLG was conveniently evaluated (Table 1) but for TP-PPLG-g-MEO2, the aromatic multiplets of Hg′ cannot be detected in the spectrum (Fig. S4b†) conducted in pure CD2Cl2. Aromatic multiplets can only be resolved in the spectrum (Fig. 2b) conducted in d6-DMSO containing small amounts of DCl. In this case, acidic DCl acted to rupture intramolecular hydrogen bonds (H bonds) existing in the rigid α-helical chains, converting rigid helical chains into flexible random coils. The coil-linked TP terminals of TP-PPLG-g-MEO2 chains were no longer hampered in rotation, as the original helical chains were before adding DCl, therefore rendering detectable NMR signals due to the enhanced molecular motion in acidic solution. Therefore, the molecular weight of TP-PPLG-g-MEO2 can be calculated from the intensity ratio of Hb′ to Hg′ and the result indicated a Mn of 33000 g mol−1. The calculated degrees of polymerization (DPs) of TP-PPLG and TP-PPLG-g-MEO2 are in close proximity to 105; therefore, the conversion of the click reaction must be quantitative in order to result in similar values for the DPs.
The Mn values measured from the GPC analysis are also included in Table 1, which are much higher than the values obtained from 1H NMR. The high value from the GPC analysis is due to the early elution of the rigid polymer chains, which is especially true for the large α-helical chains. With a large diameter, α-helical chains spend little if any time in the pores of the gels packed in GPC columns; therefore, they elute quickly to result in Mn values that are much higher than the polymer standard (polystyrene) of the same Mn value. The early elution also resulted in Mn values much higher than the more realistic Mn values determined from 1H NMR.
Secondary structure of TP-PPLG and TP-PPLG-g-MEO2
FTIR analysis provided information regarding the secondary structures of TP-PPLG and TP-PPLG-g-MEO2. Analysis of these spectra using the second-derivative technique42 revealed that the amide I band at 1655 cm−1 is characteristic of an α-helical chain. For polypeptides possessing a β-sheet conformation, the amide I band appeared at 1627 cm−1 while for random coil or turn populations, a characteristic peak at 1693 cm−1 was resolved. In addition, the free C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O of the peptide side chains provided a signal at 1740 cm−1. Based on the characteristic peaks, absorptions in the range of 1500 to 1800 cm−1 were de-convoluted into a series of Gaussian curves (Fig. 3), to evaluate the fraction of each of the peaks. Table 1 shows the fitting results of the amide I group for α-helix, β-sheet and random coil structures. Clearly, peptide chains of TP-PPLG-g-MEO2 are higher in the content of α-helical conformations compared to TP-PPLG, which demonstrated the role of long MEO2 chains in promoting α-helical chains. The α-helical chain can be regarded as a rigid rod stabilized through intramolecular H bond interactions whereas the β-sheet is a structure stabilized by intermolecular H bond interactions.43 Comparatively, a random coil is a flexible chain with the least stability considering it has fewer and more irregular H bond interactions compared to α-helical and β-sheet structures. The long MEO2 side groups sterically shielded the main chain amides from intermolecular H bonding to other peptide chains and thus favored the formation of an α-helix with predominant intramolecular H bonds. Based on stability, certain fractions of the least stable coil chains of TP-PPLG also became part of the stable α-helical chains in TP-PPLG-g-MEO2.
O of the peptide side chains provided a signal at 1740 cm−1. Based on the characteristic peaks, absorptions in the range of 1500 to 1800 cm−1 were de-convoluted into a series of Gaussian curves (Fig. 3), to evaluate the fraction of each of the peaks. Table 1 shows the fitting results of the amide I group for α-helix, β-sheet and random coil structures. Clearly, peptide chains of TP-PPLG-g-MEO2 are higher in the content of α-helical conformations compared to TP-PPLG, which demonstrated the role of long MEO2 chains in promoting α-helical chains. The α-helical chain can be regarded as a rigid rod stabilized through intramolecular H bond interactions whereas the β-sheet is a structure stabilized by intermolecular H bond interactions.43 Comparatively, a random coil is a flexible chain with the least stability considering it has fewer and more irregular H bond interactions compared to α-helical and β-sheet structures. The long MEO2 side groups sterically shielded the main chain amides from intermolecular H bonding to other peptide chains and thus favored the formation of an α-helix with predominant intramolecular H bonds. Based on stability, certain fractions of the least stable coil chains of TP-PPLG also became part of the stable α-helical chains in TP-PPLG-g-MEO2.
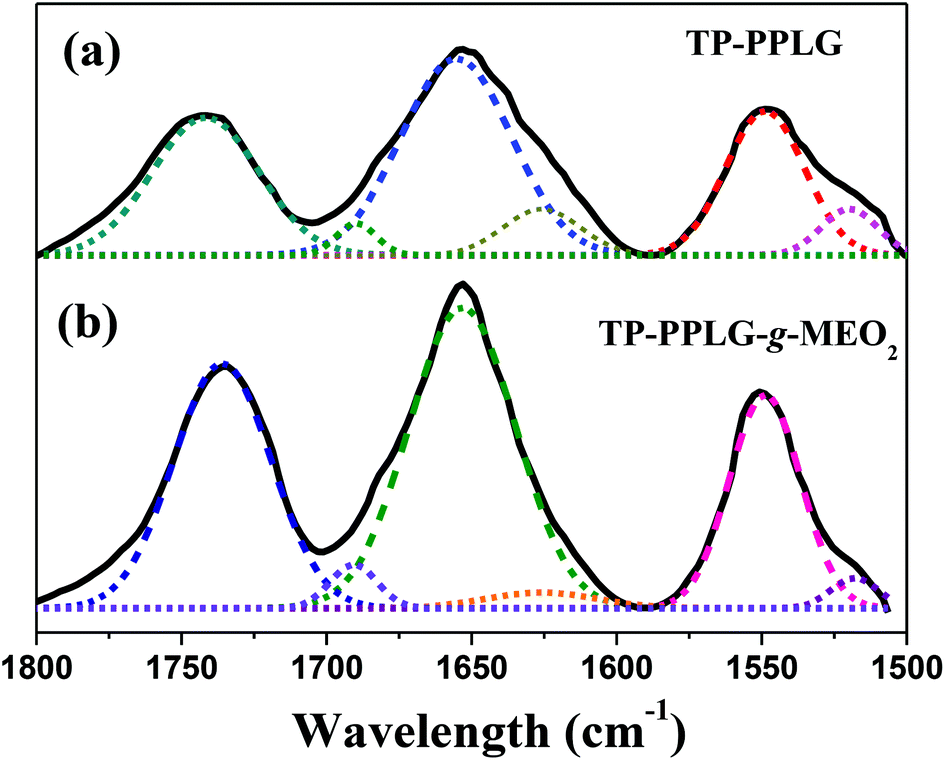 | ||
| Fig. 3 Curve-fitting of the FTIR spectra of (a) TP-PPLG and (b) TP-PPLG-g-MEO2 in the spectral range of 1500 to 1800 cm−1. | ||
AIE character of TP-PPLG-g-MEO2
The AIE character of TP-PPLG-g-MEO2 was primarily evaluated based on the solution emission responses to concentration and aggregation. The effect of concentration was demonstrated by the solution emission spectra of TP-PPLG-g-MEO2 in dichloromethane (Fig. 4a). In contrast to the emission quenching caused by solution thickening, the emission of TP-PPLG-g-MEO2 nevertheless became greater in intensity as the solution concentration of TP-PPLG-g-MEO2 was increased from 2.5 × 10−7 M to 2.5 × 10−5 M (relative to the number of polymer chains). Here, the solution emission spectra in Fig. 4a consist of two overlapping bands, representative of the short-wavelength monomer emission at 420 nm and the long-wavelength aggregate emission at 480 nm, respectively. The fraction of the aggregate emission was actually raised by altering the concentration. A high concentration raised the aggregation level of TP terminals, which directly enhanced the aggregate emission.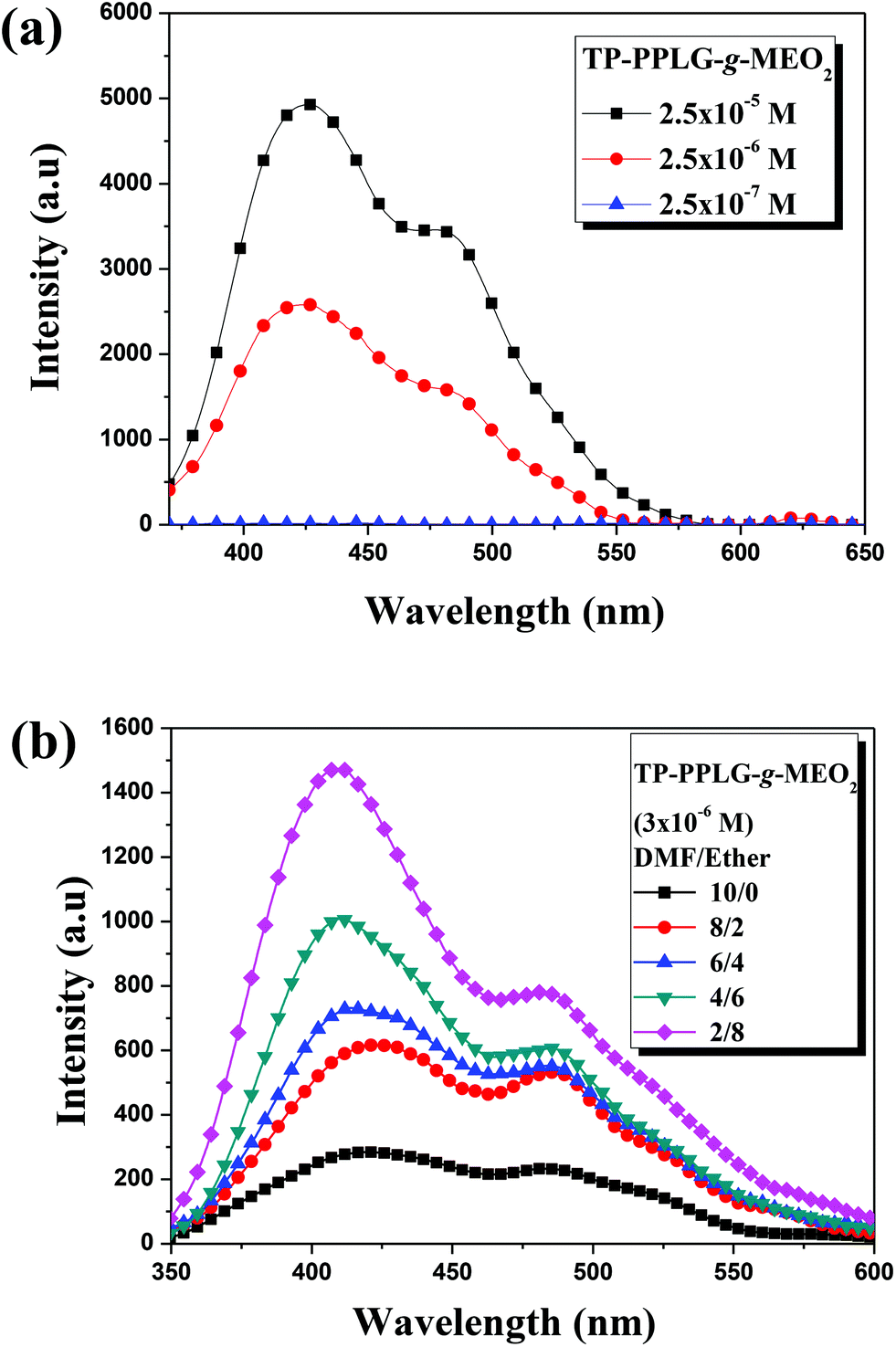 | ||
| Fig. 4 Solution emission spectra of (a) TP-PPLG-g-MEO2 in dichloromethane of different concentrations and in (b) solution mixtures of dimethyl foramide (DMF)/diethyl ether (λex = 340 nm). | ||
The aggregation of TP terminals in solvent/nonsolvent mixtures was also used in this study to identify the AIE effect. In this respect, diethyl ether (Et2O) was employed as a nonsolvent to induce solution aggregation of TP terminals in a good solvent of dimethylforamide (DMF). Upon irradiation at 340 nm, a dilute solution (3 × 10−6 M) of TP-PPLG-g-MEO2 in DMF already emitted with discernible intensity (Fig. 4b) and adding Et2O (while keeping the concentration of TP-PPLG-g-MEO2 at 3 × 10−6 M) resulted in progressive intensity gains in both the monomer and aggregate emissions. Upon adding Et2O, TP terminals tend to be more associated together and therefore are more hampered in rotational motion, thereby promoting the AIE-related emission.
Secondary structure in relation to the AIE-related emission
As illustrated in Scheme 2, peptide chains of TP-PPLG-g-MEO2 are subjected to conformational changes, from the initial α-helix to a β-sheet or to a random coil, under different applied experimental conditions and conformational changes in correlation to the AIE activity are primarily discussed here before entering detailed results. In general, mutual approaches (aggregation) of the TP terminals, as the key to controlling molecular rotation and AIE-related emission, are affected by the dimension and rigidity of the peptide chains connecting to TP units. Concerning chain dimensions, rigid α-helical rods, whose reported diameters (15–26 Å33,44,45) are larger than the molecular width (9 Å) of the TP unit, are effective in blocking the intermolecular approaches of the TP terminals 3-dimensionally. Hampered aggregation of TP terminals in α-helical chains is detrimental for AIE activity, and therefore weakens the luminescence of α-helical peptide chains. Compared to TP terminals in helical chains, β-sheet-linked TP terminals are easier to associate together to form aggregates since the 2D β-sheet chains are not as spatially repulsive to each other as the bulky, 3D helical chains. A higher emission is expected for the TP terminals in the β-sheet compared to the α-helical chains. Among all three secondary structures, the flexible random coil is no doubt the best at moving TP terminals in close proximity due to the free segmental motion of flexible coil chains; therefore, we expected a high aggregate emission for TP-PPLG-g-MEO2 chains in the random coil conformation.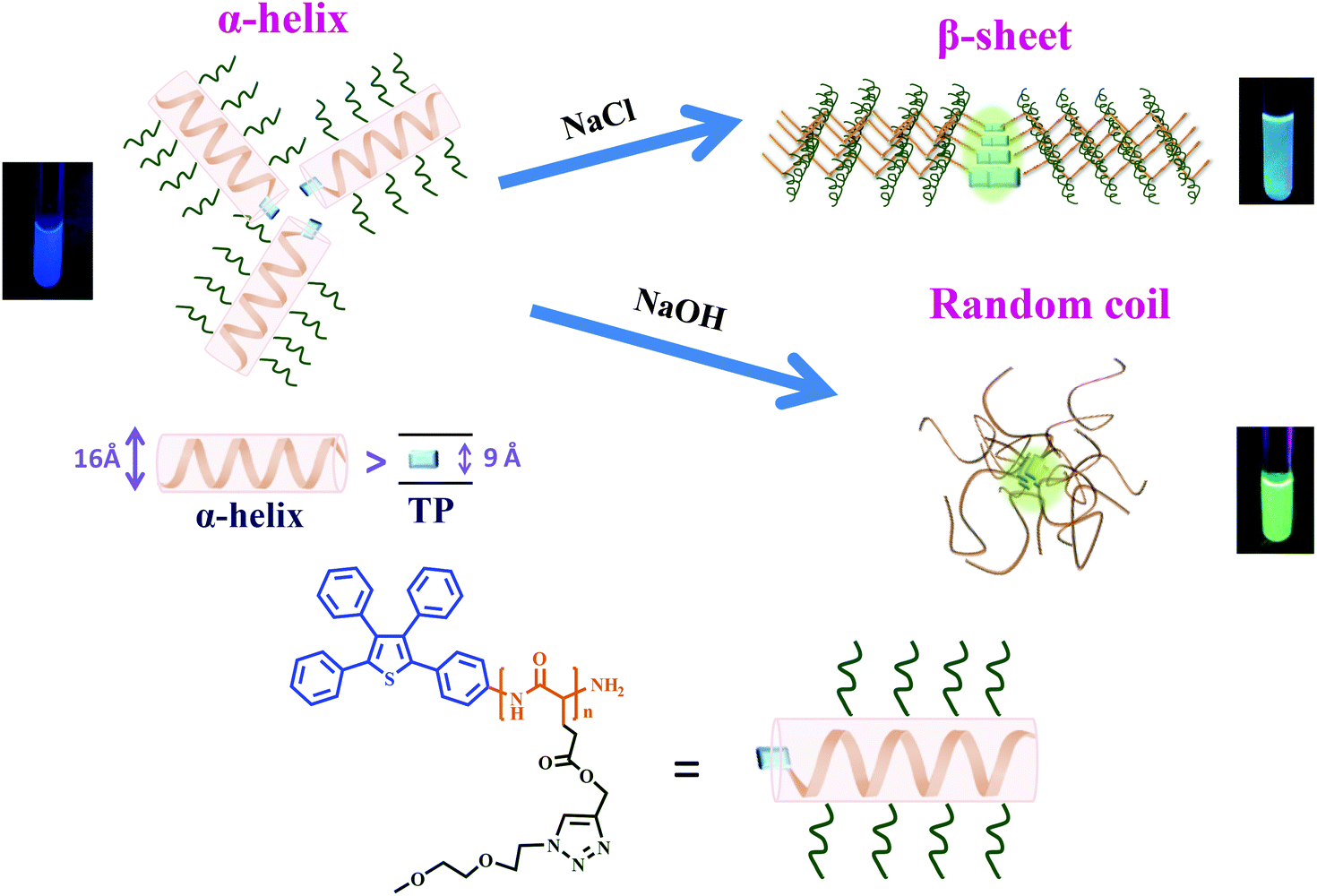 | ||
| Scheme 2 Conformational changes of TP-PPLG-g-MEO2 from an α-helix to a β-sheet in an aqueous salt solution and from an α-helix to a random coil in an alkaline aqueous solution, respectively. | ||
Most of the TP-PPLG-g-MEO2 chains in the aqueous solution are in the α-helical conformation and because of that they can be conveniently converted into β-sheets and random coil chains in NaCl salt and alkaline solutions, respectively. The induced helix-to-sheet and helix-to-coil transitions and the corresponding emission variations will be identified and discussed separately below.
Conformational transformation and emission variation in a salt solution
For amphiphilic polymers in aqueous solutions, adding salts46–50 were reported to affect the solubility of polymers in water and the corresponding lower critical solution temperature (LCST) of salt solutions. Taking the aqueous solution of poly(N-isopropyl acrylamide) (PNIPAM)49 as an example, cations of the salts preferably reacted with the side-chain amide groups of PNIPAM, resulting in the dehydration of amide side groups and depression of LCST with increasing salt content in the aqueous solution. For TP-PPLG-g-MEO2, we also observed a decrease of LCST (“salting-out” effect) with increasing NaCl content in the aqueous solution (Fig. S9†). As it was discussed before, the “salting-out” effect is therefore not the focus of this study; instead, the salt-induced conformational transformation and emission change are more of interest to us.Fig. 5 shows the emission enhancement caused by adding NaCl to an aqueous solution of TP-PPLG-MEO2. With increasing salt content in the solution, both the monomer and aggregate emissions gradually gained in intensity. The intensity ratio of aggregate and monomer emissions (Ia/Im) was then calculated and is summarized in Table 2, which indicates that Ia/Im increased from 0.677 to 0.825 as the salt content was increased from 0 to 0.154 M. A higher value of Ia/Im that correlates with a greater aggregation level of the TP terminals was therefore involved in the newly-developed secondary structure induced by NaCl. The enhanced aggregation is supposed to be responsible for the observed enhanced emission of the aqueous salt solution.
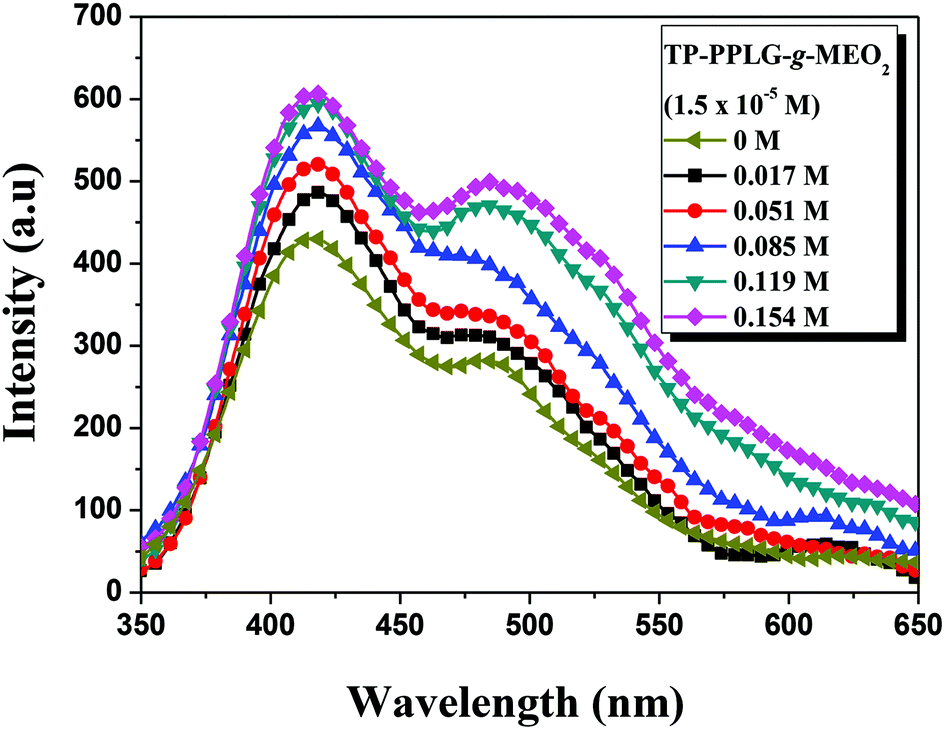 | ||
| Fig. 5 Emission spectra of an aqueous solution of TP-PPLG-g-MEO2 (1.5 × 10−5 M) containing different amounts of NaCl (λex = 340 nm). | ||
We then recorded circular dichroism (CD) spectra (Fig. 6) to characterize the corresponding conformations responsible for the enhanced emissions of the salt solutions. The α-helical structure was characterized by a triply inflected spectrum,39 corresponding to two negative bands at 208 and 222 nm and a strong positive band at 192 nm. The β-sheet structure was characterized by a negative minimum band near 218 nm and a positive maximum near 198 nm. The random coil structure was characterized by a small positive band at 218 nm and a negative band near 200 nm. Fig. 6 shows the gradual transformation of the α-helical conformation, in correlation to the slight change of the ellipticity ratio ([θ]222/[θ]208), upon increasing NaCl content from 0 to 0.154 M. Disregarding the slight change of the ellipticity ratio, a significant spectral variation occurred in the short-wavelength region in which the main band shifted from 192 nm, characteristic of α-helical chains, to 198 nm, typical of β-sheet chains. The spectra were then fitted, using the Spectra Manager program, to resolve the percentages of each secondary structure and the results are illustrated in the inset of Fig. 6, which indicate that the content of α-helical chains decreased from 72.9% to 23.3%, corresponding to an increase in β-sheet chains from 3% to 54.1% (also, refer to the calculated results in Table S1†). Therefore, the high fraction of 3D α-helical chains were transformed into 2D β-sheet chains by NaCl salts added in the solution. The intimately-packed β-sheet chains (Scheme 2) are therefore responsible for the better emission of the salt solution but what is the driving force causing this interesting helix-to-sheet transition? The 1H NMR analysis of the salt solutions may give us a clue to evaluate the potential cause leading to the helix-to-sheet transition.
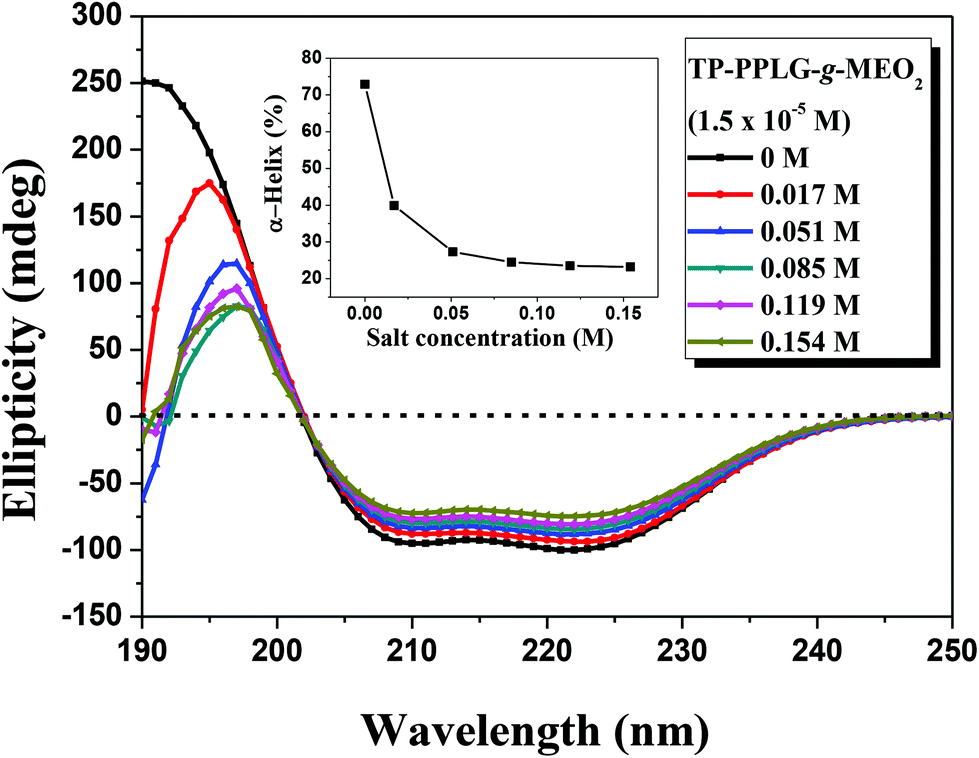 | ||
| Fig. 6 Circular dichroism spectrum of an aqueous solution of TP-PPLG-g-MEO2 (= 1.5 × 10−5 M) containing different amounts of NaCl and the calculated fraction of α-helix vs. the amount of NaCl (inset) in the solution. | ||
It was envisaged that NaCl additives complexed to both amide groups of the main chain and heteroatoms (nitrogen and oxygen) of the side chain, pulling both chain segments close enough to form intimately-packed β-sheet chains. Such a structural transformation was indirectly demonstrated by the 1H NMR band shape analysis of solutions of TP-PPLG-MEO2 in D2O containing different amounts of NaCl (Fig. 7). The band shape analysis was previously performed in a study of rotation-induced conformational changes.51 The main principle behind this analysis is that fast conformational exchanges caused by fast molecular rotations would result in sharp resonance peaks, whereas slower exchanges due to restricted molecular rotations broaden resonance peaks. Moreover, in many polymer systems,52,53 the rotational restriction imposed by polymer chains is so effective that the corresponding resonance peaks become weaker or disappear. In the present study, the concentration of TP-PPLG-g-MEO2 was kept at a constant value of 1.5 × 10−5 M but the resonance peaks due to protons in the main (Hb) and side (Hc–k) groups still broadened and weakened with increasing NaCl from 0 to 0.154 M. Presumably, sodium cations complexed to amide CO groups of the main chains and heteroatoms (nitrogen and oxygen) of the MEO2 side chains and hampered the rotations of the main and side chain groups to result in the broadening and weakening of the resonance peaks, due to the clumsy response of the corresponding chemical bonds towards the stimuli of the external magnetic field.
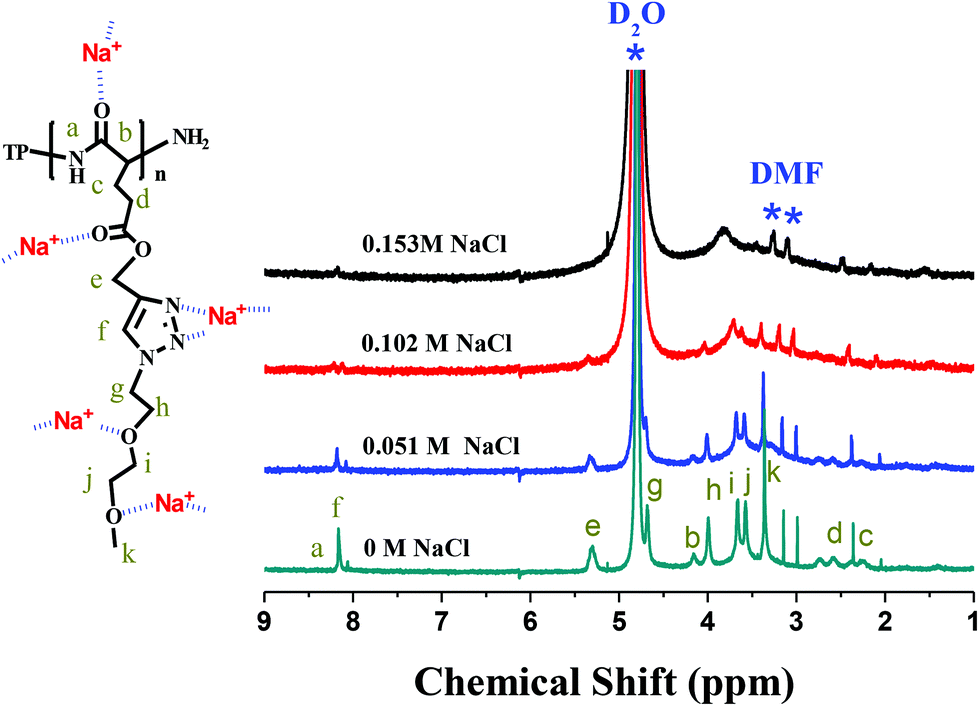 | ||
| Fig. 7 1H NMR spectra of TP-PPLG-g-MEO2 (= 1.5 × 10−5 M) in D2O containing different amounts of NaCl. | ||
In the salt solution, intramolecular H bonds, originally prevalent in α-helical chains, were ruptured and replaced by intermolecular ionic bonds, between sodium cations and amide main chain groups, present in the β-sheet structure. Complexation of sodium cations and MEO2 side groups also functioned to hold the neighbouring MEO2 side groups at closer distances, which is beneficial for the intimately-packed β-sheet structure. When linked by the intimately-packed β-sheet chains, TP terminals associate together more easily, resulting in an emission with a higher efficiency than the helix-linked TP terminals, whose mutual approaches were seriously blocked by large helical rods.
Conformational transformation and emission variation in an alkaline solution
Poly(L-glutamic acid) was reported to undergo a helix-to-coil transition at pH 6.18.53,54 A similar transition was observed in β-cyclodextrin (β-CD)-grafted PPLG,55 however, at a higher pH range in between 11 and 11.5 due to the steric hindrance or the stable helical structure inherited by β-CD grafts. Likewise, the helix-to-coil transition was observed for the TP-PPLG-g-MEO2 system in an alkaline solution, which helps evaluate the effect of chain conformation on AIE activity. As illustrated in Fig. 8, the emission of the aqueous TP-PPLG-g-MEO2 solution indeed depends on the applied pH of the solution. At pH ≤ 7, the aqueous solutions emitted similarly without notable variation. However, when NaOH was gradually added to the solution, the initial monomer emission was continuously transformed into a major aggregate emission with an intensity much higher than the initial monomer emission. The aggregate emission at 520 nm gradually developed in intensity in alkaline solutions and at pH 13.6, the aqueous solution emitted with a large aggregate emission, at an intensity at least 8-fold greater than that at pH 7. Easy intermolecular aggregation of TP terminals chains must be prevalent in the alkaline solution, in order to emit with a major aggregate emission as shown in Fig. 8.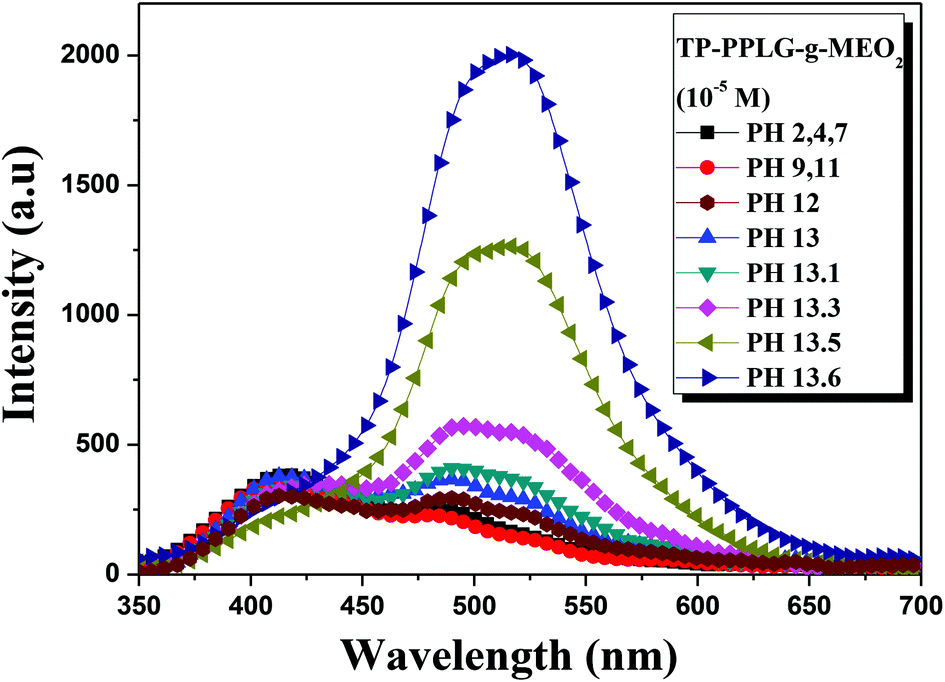 | ||
| Fig. 8 Emission spectra of aqueous TP-PPLG-g-MEO2 (= 10−5 M) solutions at different pH values (λex = 340 nm). | ||
The conformational transformation was also traced by CD spectra (Fig. 9). By increasing the pH from 2 to 11, there are only slight variations in the ellipicity ratio of [θ]222/[θ]208. A major conformational transformation occurred only at pH > 12 according to the spectral change where triply inflected peaks reduced to a small doubly inflected dichroic spectrum. At this stage, the majority of α-helical chains should have converted into random coils by NaOH.
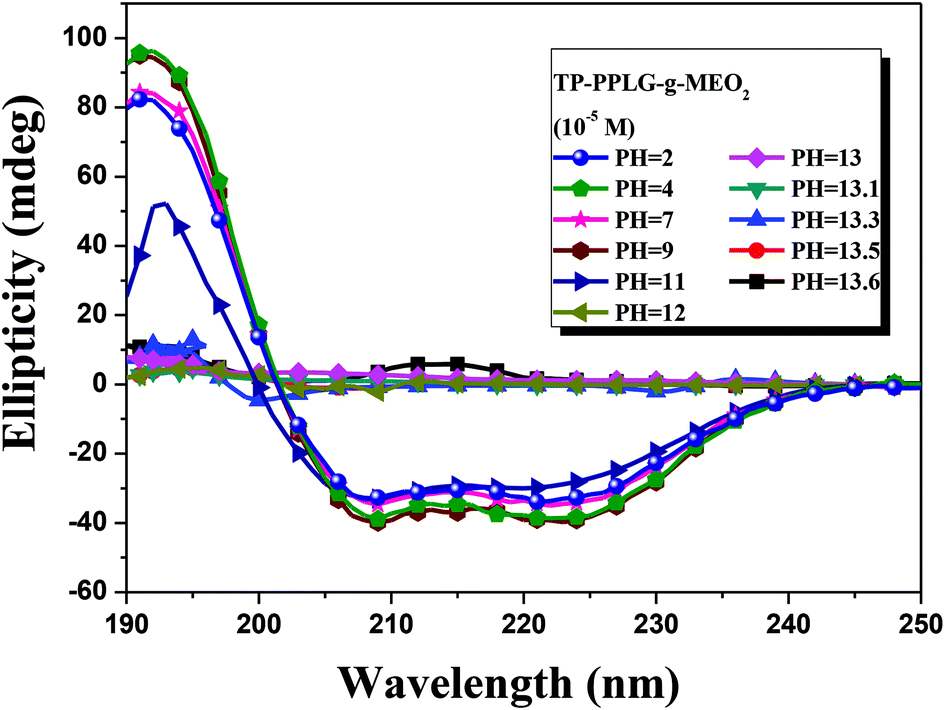 | ||
| Fig. 9 Circular dichroism spectrum of aqueous TP-PPLG-g-MEO2 (= 10−5 M) solutions at different pH values. | ||
Conformational transformation of peptide chains affected the extent of aggregation and the AIE-related emission of peptide-linked TP terminals. As illustrated in Scheme 2, with mobile segmental motion, the flexible peptide chains easily moved their TP terminals to a closer range to become a highly-aggregated TP phase, which emitted with a strong aggregate emission at an intensity much higher than TP units linked by the less-mobile α-helical or β-sheet structure.
Luminescence responses of α-helical, β-sheet and random coil chains toward bovine serum albumin
As a large globular protein (66 kDa) consisting of 583 amino acid residues in a single polypeptide chain, bovine serum albumin (BSA)56 is one of the most intensively-studied proteins in biochemistry or biomedical science due to its structural similarity with human serum albumin (HSA).57 In practice, BSA is often used as a complexing material for certain dyes and probes, in which the emission response58 can be used to study the binding mechanisms. Since we can prepare luminescent polypeptides of three secondary structures, it would be interesting to evaluate the possibility of individual secondary peptide chains acting as luminescent probes for BSA. In particular, the luminescence response of TP-PPLG-g-MEO2 to complexing with BSA is a reflection of the aggregation level of TP terminals linked by peptide chains of different secondary structures.Experimentally, α-helix, random coil and β-sheet chains prepared from the respective neutral, alkaline and salt solutions were used for complexing with different amounts of BSA. The results suggested that solution emissions of random coil (Fig. 10a) and β-sheet chains (Fig. 10b) are both decreased by the inclusion of BSA, which is distinctly different from the intensity gain observed in the emission spectra (Fig. 11) from the α-helical chain. Considering that the AIE-related emission is closely related to the aggregation level of the luminogenic TP, the emission reductions observed in alkaline and salt solutions should be correlated with the dissociation of aggregated TP terminals. Particularly, regarding the large reduction of the aggregate emission (Fig. 10a) in the alkaline solution, dissociation of TP aggregates in random coil chains must be significant in order to correlate with the large spectral change. Comparatively, that the emission loss (Fig. 10b) is minor for the salt solution indicated that there is only a slight dissociation of TP terminals during the complexation of β-sheet chains with BSA. The emission gain (Fig. 11) observed for α-helical chains is therefore correlated with the enhanced rotational restriction of TP terminals.
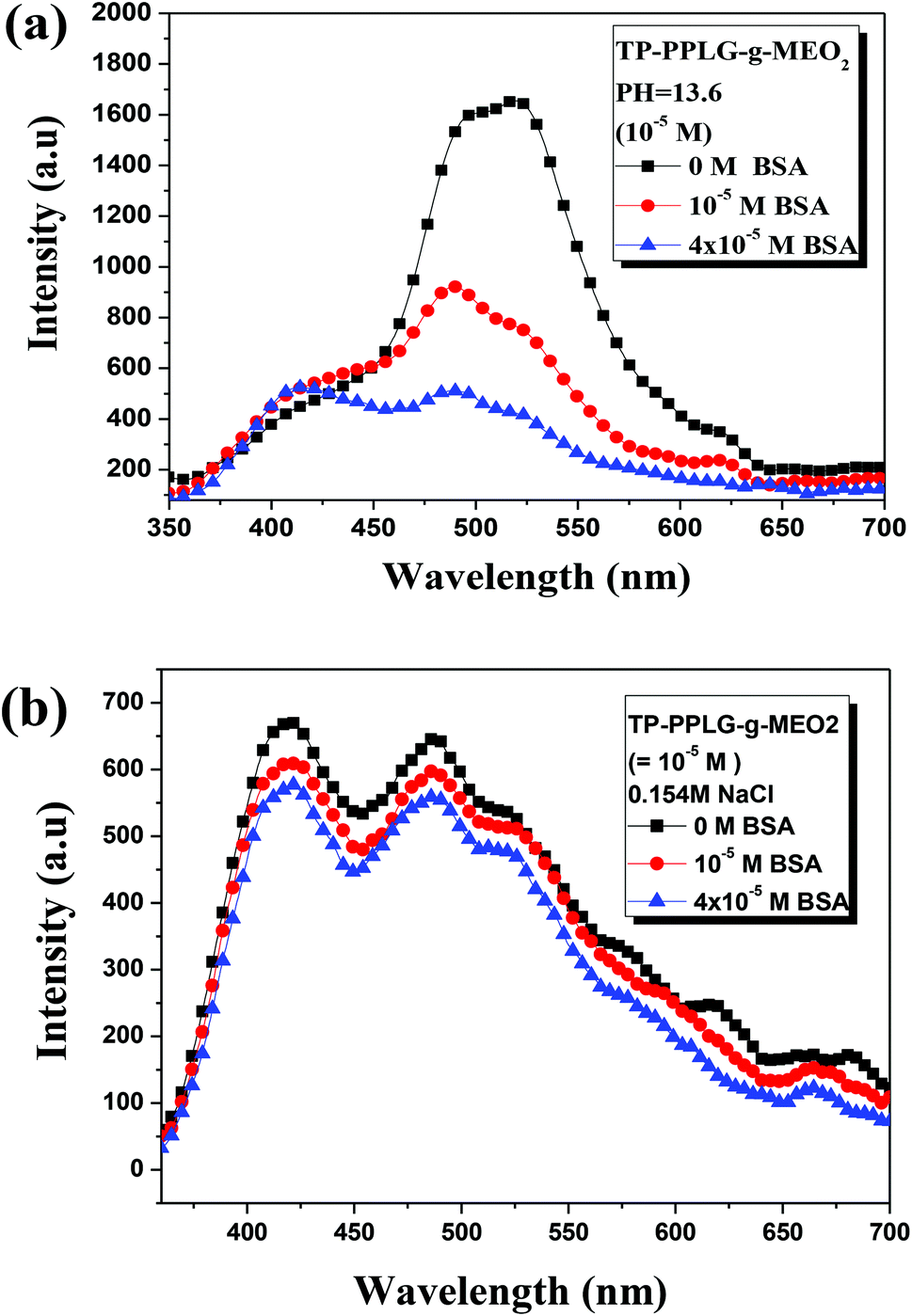 | ||
| Fig. 10 Emission spectral variations of an aqueous TP-PPLG-g-MEO2 (10−5 M) solution in an (a) alkaline solution (pH = 13.6) and in a (b) neutral solution containing NaCl (0.156 M) in response to different amounts of BSA from 0 to 4 × 10−5 M (λex = 340 nm). | ||
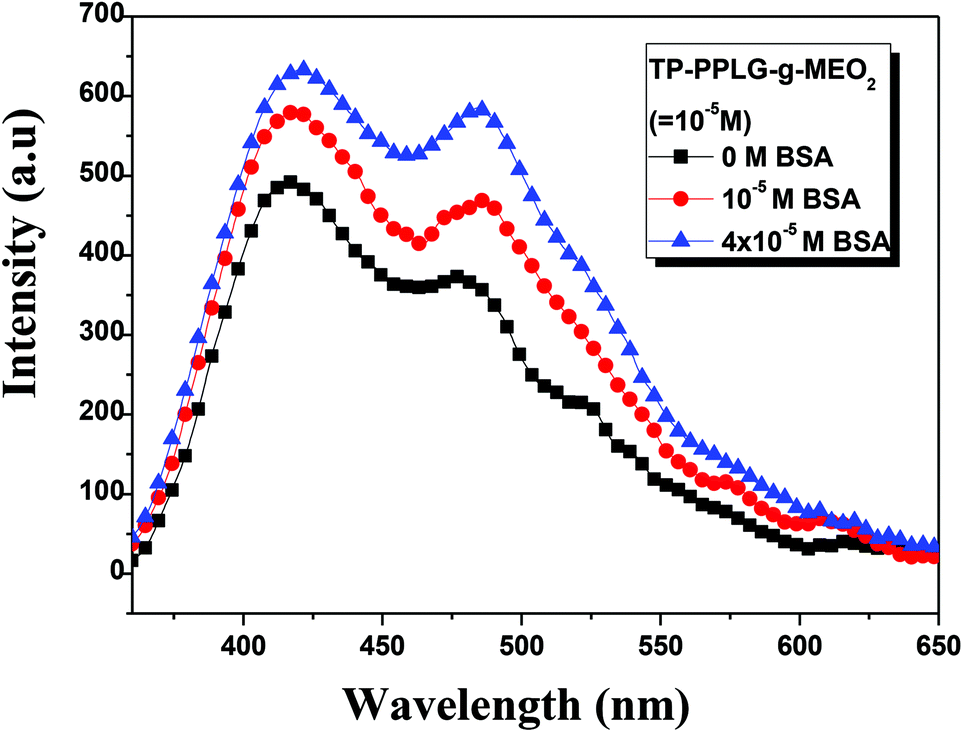 | ||
| Fig. 11 Emission spectral variation of aqueous TP-PPLG-g-MEO2 (10−5 M) solutions in response to different amounts of BSA from 0 to 4 × 10−5 M (λex = 340 nm). | ||
The large emission reduction observed in alkaline solutions suggested a preferable complexation of random coil chains in BSA; most likely, the flexible coil chains readily diffused into the interior domain of the large BSA chains, to result in an exclusive reaction responsible for the large reduction of the aggregate emission shown in Fig. 10a. With low structural stability, flexible coil chains were subjected to a significant conformational change when complexed with BSA. It was then envisaged that stable chains with structural integrity are less prone to undergo conformational change during complexation with BSA. When complexed to BSA, the α-helical chains, with structural integrity maintained by intramolecular H bonds, and the β-sheet conformation, stabilized by intermolecular H bonds, are therefore suggested to undergo minor structural changes and less emission variations compared to the unstable random coil. When in the BSA domain, the stable α-helical rod remained in the rigid conformation and rotational restriction of the TP terminals was further reinforced by the viscous matrix of BSA, thereby resulting in the enhanced emission observed in Fig. 11. According to the small emission variation shown in Fig. 10b, the less stable β-sheet chain may undergo slight conformational change when complexed with BSA. Some intermolecular H bonds in the β-sheet chains may be ruptured, resulting in the dissociation of TP aggregates and the slight emission reduction.
Conclusion
The amphiphilic polypeptide TP-PPLG-g-MEO2 with an AIE-active TP terminal and ether MEO2 side groups was prepared from the ring opening polymerization of a PLG-NCA monomer and the following click reaction. TP-PPLG-g-MEO2 was shown to have active AIE properties according to its emission behavior in response to concentration changes and aggregation.In an aqueous solution, the majority of α-helical chains of TP-PPLG-g-MEO2 can be converted into β-sheet and random coil structures by NaCl salt and NaOH base, respectively. With an improved aggregation tendency, TP terminals linked by 2D β-sheet chains emitted with a higher emission intensity than TP terminals linked by large 3D helical rods. TP terminals linked by flexible random coils are the most mobile ones, rendering highly-aggregated TP terminals with an emission intensity higher than TP terminals linked by less-mobile helix and β-sheet chains. The role of the secondary structure of the peptide chain in determining the aggregation tendency and AIE-related emission behavior was therefore evaluated.
The complexation of BSA resulted in an emission enhancement of helix-linked TP terminals due to the reinforced rotational restriction imposed by BSA. In contrast, a large emission reduction was observed in TP terminals linked by random coil chains and most likely, a significant dissociation of the aggregated TP terminals occurred during the complexation of the coil chains with BSA. Comparatively, a structural change in β-sheet chains is small due to the slight reduction in the emission intensity upon complexing with BSA.
Conflict of interest
The authors declare no competing financial interest.Acknowledgements
We appreciate the financial support from the Ministry of Science and Technology, Taiwan, under the contract no. NSC 102-2221-E-110-084-MY3 and 104-2221-E-110-050.References
- J. Luo, Z. Xie, J. W. Y. Lam, L. Cheng, H. Chen, C. Qiu, H. S. Kwok, X. Zhan, Y. Liu, D. Zhu and B. Z. Tang, Chem. Commun., 2001, 1740 RSC.
- B. Z. Tang, X. Zhan, G. Yu, P. P. S. Lee, Y. Liu and D. Zhu, J. Mater. Chem., 2001, 11, 2974 RSC.
- Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Commun., 2009, 4332 RSC.
- D. Ding, K. Li, B. Liu and B. Z. Tang, Acc. Chem. Res., 2013, 46, 2441 CrossRef CAS PubMed.
- Z. Zhao, J. W. Y. Lam and B. Z. Tang, J. Mater. Chem., 2012, 22, 23726 RSC.
- M. Wang, G. Zhang, D. Zhang, D. Zhu and B. Z. Tang, J. Mater. Chem., 2010, 20, 1858 RSC.
- Z. Zhao, J. W. Y. Lam and B. Z. Tang, Soft Matter, 2013, 9, 4564 RSC.
- Aggregation-Induced Emission: Fundamentals, ed. A. Qin and B. Z. Tang, John Wiley & Sons, Ltd, NY, 2013 Search PubMed.
- J. Mei, Y. Hong, J. W. Y. Lam, A. Qin, Y. Tang and B. Z. Tang, Adv. Mater., 2014, 26, 5429 CrossRef CAS PubMed.
- H. Wang, E. Zhao, J. W. Y. Lam and B. Z. Tang, Mater. Today, 2015, 18, 365 CrossRef CAS.
- Z. Li, Y. Dong, B. Mi, Y. Tang, M. Häussler, H. Tong, Y. Dong, J. W. Y. Lam, Y. Ren, H. H. Y. Sung, K. S. Wong, P. Gao, L. D. Williams, H. S. Kwok and B. Z. Tang, J. Phys. Chem. B, 2005, 109, 10061 CrossRef CAS PubMed.
- J. Chen, C. C. W. Law, J. W. Y. Lam, Y. Dong, S. M. F. Lo, I. D. Williams, D. Zhu and B. Z. Tang, Chem. Mater., 2003, 15, 1535 CrossRef CAS.
- Z. Zhao, C. Y. K. Chan, S. Chen, C. Deng, J. W. Y. Lam, C. K. W. Jim, Y. Hong, P. Lu, Z. Chang, X. Chen, P. Lu, H. S. Kwok, H. Qiu and B. Z. Tang, J. Mater. Chem., 2012, 22, 4527 RSC.
- J. Huang, X. Yang, J. Wang, C. Zhong, L. Wang, J. Qin and Z. Li, J. Mater. Chem., 2012, 22, 2478 RSC.
- J. Huang, N. Sun, J. Yang, R. Tang, Q. Li, D. Ma, J. Qin and Z. Li, J. Mater. Chem., 2012, 22, 12001 RSC.
- J. Huang, N. Sun, Y. Dong, R. Tang, P. Lu, P. Cai, Q. Li, D. Ma, D. Qin and Z. Li, Adv. Funct. Mater., 2013, 23, 2329 CrossRef CAS.
- X. Chen, X. Y. Shen, E. Guan, Y. Liu, A. Qin, J. Z. Sun and B. Z. Tang, Chem. Commun., 2013, 49, 1503 RSC.
- X. Wang, J. Hu, T. Liu, G. Zhang and S. Liu, J. Mater. Chem., 2012, 22, 8622 RSC.
- X. Huang, X. Gu, G. Zhang and D. Zhang, Chem. Commun., 2012, 48, 12195 RSC.
- C. Li, T. Wu, C. Hong, G. Zhang and S. Liu, Angew. Chem., Int. Ed., 2012, 51, 455 CrossRef CAS PubMed.
- Y. Yu, A. Qin, C. Feng, P. Lu, K. M. Ng, K. Q. Luo and B. Z. Tang, Analyst, 2012, 137, 5592 RSC.
- K. Tohyama and W. C. Miller, Nature, 1981, 289, 813 CrossRef CAS PubMed.
- Y. Bae, S. Fukushima, A. Harada and K. Kataoka, Angew. Chem., Int. Ed., 2003, 42, 4640 CrossRef CAS PubMed.
- H. Tang and D. Zhang, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 2340 CrossRef CAS.
- A. Gitsas, G. Floudas, M. Mondeshki, H. W. Spiess, T. Aliferis, H. Iatrou and N. Hadjichristidis, Macromolecules, 2008, 41, 8072 CrossRef CAS.
- H. Tang, L. Yin, H. Lu and J. Cheng, Biomacromolecules, 2012, 13, 2609 CrossRef CAS PubMed.
- Y. Cheng, C. He, C. Xiao, J. Ding, H. Cui, X. Zhuang and X. Chen, Biomacromolecules, 2013, 14, 468 CrossRef CAS PubMed.
- V. K. Kotharangannagari, A. Sanchez-Ferrer, J. Ruokolainen and R. Mezzenga, Macromolecules, 2011, 44, 4569 CrossRef CAS.
- J. Huang and A. Heise, Chem. Soc. Rev., 2013, 42, 7373 RSC.
- Y. Shen, X. Fu, W. Fu and Z. Li, Chem. Soc. Rev., 2015, 44, 611 Search PubMed.
- H. Tian, Z. Tang, X. Zhuang, X. Chen and X. Jing, Prog. Polym. Sci., 2012, 37, 237 CrossRef CAS.
- S. Zhang and Z. Li, J. Polym. Sci., Part B: Polym. Phys., 2013, 51, 546 CrossRef CAS.
- P. Doty, J. H. Bradbury and A. M. Holtzer, J. Am. Chem. Soc., 1956, 78, 947 CrossRef CAS.
- V. Pokorná, D. Výprachtický and J. Pecka, Macromol. Biosci., 2001, 1, 185 CrossRef.
- K. T. Kim, C. Park, G. W. M. Vandermeulen, D. A. Rider, C. Kim, M. A. Winnik and I. Manners, Angew. Chem., Int. Ed., 2005, 117, 8178 CrossRef.
- S. T. Li, Y. C. Lin, S. W. Kuo, W. T. Chuang and J. L. Hong, Polym. Chem., 2012, 3, 2393 RSC.
- K. Y. Shih, T. S. Hsiao, S. L. Deng and J. L. Hong, Macromolecules, 2014, 47, 4037 CrossRef CAS.
- S. T. Li, Y. C. Lin, S. W. Kuo, W. T. Chuang and J. L. Hong, Polym. Chem., 2012, 3, 2393 RSC.
- Y. Cheng, C. He, C. Xiao, J. Ding, X. Zhuang and X. Chen, Polym. Chem., 2011, 2, 2627 RSC.
- Y. C. Lin and S. W. Kuo, Polym. Chem., 2012, 3, 162 RSC.
- Y. C. Lin and S. W. Kuo, Polym. Chem., 2012, 3, 882 RSC.
- Y. Zhang, H. Lu, Y. Lin and J. Cheng, Macromolecules, 2011, 44, 6641 CrossRef CAS PubMed.
- P. Papadopoulous, G. Floudas, H. A. Klok, I. Schnell and T. Pakula, Biomacromolecules, 2004, 5, 81 CrossRef PubMed.
- T. Torii, T. Yamashita, H. Ushiki and K. Horie, Eur. Polym. J., 1993, 29, 465 CrossRef CAS.
- F. A. Bovey and G. V. D. Tiers, Adv. Polym. Sci., 1963, 3, 139 CrossRef CAS.
- J. P. Magnusson, A. Khan, G. Pasparakis, A. Q. Saeed, W. X. Wang and C. Alexander, J. Am. Chem. Soc., 2008, 130, 10852 CrossRef CAS PubMed.
- R. Freitag and F. Garret-Flaudy, Langmuir, 2002, 18, 3434 CrossRef CAS.
- Y. Cheng, C. He, C. Xiao, J. Ding, X. Zhuang and X. Chen, Polym. Chem., 2011, 2, 2627 RSC.
- J. F. Lutz, Ö. Akdemir and A. Hoth, J. Am. Chem. Soc., 2006, 128, 13046 CrossRef CAS PubMed.
- J. Hwang and T. Deming, J. Biomacromolecules, 2001, 2, 17 CrossRef CAS.
- J. Chen, C. C. W. Law, J. W. Y. Lam, Y. Dong, S. M. Lo, I. D. Williams, D. Zhu and B. Z. Tang, Chem. Mater., 2003, 15, 1535 CrossRef CAS.
- C. T. Lai, R. H. Chien, S. W. Kuo and J. L. Hong, Macromolecules, 2011, 44, 6546 CrossRef CAS.
- C. M. Yang, Y. W. Lai, S. W. Kuo and J. L. Hong, Langmuir, 2012, 28, 15725 CrossRef CAS PubMed.
- R. Zimmermann, T. Kratzmu, D. Erickson, D. Li, H. G. Braun and C. Werner, Langmuir, 2004, 20, 2369 CrossRef CAS PubMed.
- Y. C. Lin, P. I. Wang and S. W. Kuo, Soft Matter, 2012, 8, 9676 RSC.
- Y. Moriyama, D. Ohta, K. Hadiya, Y. Mitsui and K. J. Takeda, Protein Chem., 1996, 15, 265 CrossRef CAS PubMed.
- X. M. He and D. C. Carter, Nature, 1992, 358, 209 CrossRef CAS PubMed.
- Y. Z. Zhang, B. Zhou, X. P. Zhang, P. Huang, C. H. Li and Y. Liu, J. Hazard. Mater., 2009, 163, 1345 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Table S1 and Fig. S1–S9. See DOI: 10.1039/c5py01485a |
| This journal is © The Royal Society of Chemistry 2016 |