
Cheap and fast: oxalic acid initiated CO2-based polyols synthesized by a novel preactivation approach†
Shunjie
Liu
ab,
Yusheng
Qin
*b,
Lijun
Qiao
b,
Yuyang
Miao
b,
Xianhong
Wang
*b and
Fosong
Wang
b
aUniversity of Chinese Academy of Sciences, Beijing 100049, People's Republic of China
bKey Laboratory of Polymer Ecomaterials, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Changchun 130022, People's Republic of China. E-mail: ysqin@ciac.ac.cn; Fax: +86 0431 85262252; Tel: +86 0431 85262252; xhwang@ciac.ac.cn; Fax: +86 0431 85689095; Tel: +86 0431 85262250
First published on 12th October 2015
Abstract
Oxalic acid, the cheapest dicarboxylic acid, was used as an effective initiator to synthesize polyols by copolymerization of CO2 and propylene oxide over a zinc–cobalt double metal cyanide catalyst. Generally, reaction times as long as 255 min were observed for complete PO conversion, due to the existence of the free carboxylic acid group of oxalic acid. To overcome this disadvantage, we proposed a novel preactivation approach by formation of oxalic acid based oligo-ether-diol in advance. About 4.75 PO monomers were initiated at 80 °C, which was independent of time and oxalic acid amount; the diol then acted as a chain transfer agent for the following copolymerization. Under the optimal conditions the reaction could proceed to completion in 150 min, which was a remarkable reduction in reaction time compared to the previous reaction time of 255 min. Notably, the resulting CO2-based diol was stable up to 190 °C, indicating that oxalic acid may be applied as an effective initiator for this copolymerization.
Introduction
Polyurethane (PU) is a class of polymer built up of carbamate linkages, which finds versatile applications as foams, adhesives, surface coatings, sealants, thermoplastic elastomers, and so on.1,2 PU is traditionally and most commonly formed by reacting isocyanate with polyols. However, with the precipitous slide of the price of isocyanate (<$2000 per ton since Mar. 30, 2015) in recent years, originally cheap polyols have become the key factor to reduce PU cost. One promising route to reduce the cost of polyols is the introduction of an extremely low-cost feedstock such as carbon dioxide (CO2) into the backbone of polyether polyols, yielding oligo(carbonate-ether) polyols. Until now, it has been confirmed that low molecular weight oligo(carbonate-ether) polyols can afford high-quality polyurethane materials comparable with those from common polyether polyols.3,4As shown in Scheme 1, oligo(carbonate-ether) polyols can be synthesized from the copolymerization of propylene oxide (PO) with CO2 over zinc–cobalt double metal cyanide (Zn–Co–DMC) catalyst in the presence of a proton-containing initiator.5–15 In Scheme 1, if the catalyst is fixed, in addition to copolymerization conditions, the initiator is the most important variable to determine the features of the resulting oligo(carbonate-ether) polyols. Many efforts have been spent on developing suitable initiators in last decades, among which oligo(carbonate-ether) polyols prepared using polypropylene glycol (PPG) as initiator have received considerable attention.16–20 Nevertheless, the high reaction temperature resulted in polyols with low carbonate unit (CU) content. Generally, polyols with molecular weights (Mn) below 2000 g mol−1 and CU contents of over 40% are still difficult to prepare, though PPG is a cheap initiator.21,22 Recently, we reported the tailor-made synthesis of oligo(carbonate-ether) diol using dicarboxylic acid as initiator, where the Mn was controllable in the range 1200–3000 g mol−1, and CU content was between 40% and 75%.23 Moreover, the induction period of the copolymerization could be significantly reduced from 425 min to 20 min at similar reaction conditions by simply replacing PPG with sebacic acid, suggesting the significant influence of the initiator on the copolymerization system. Many carboxylic acids initiators were then used to further investigate the effect of acidic initiator on the copolymerization.24,25 The manner of initiation of different acidic initiators is closely associated with their acidity (pKa1 value) in the initial copolymerization stage. For example, a weak organic acid (pKa1: 4.43–4.72) like sebacic acid only acts as the chain transfer agent, directly participating in the copolymerization of CO2 with PO, while a relatively strong organic acid (1.87 < pKa1 < 4.2) acts as the initiate-transfer agent which first initiates PO homopolymerization, following which the in situ formed polyols act as a new chain transfer agent in the early stages of copolymerization.
 | ||
| Scheme 1 Copolymerization of CO2 with PO catalyzed by Zn–Co–DMC catalyst in the presence of initiator. | ||
In addition to performing the role of the chain transfer agent or chain initiate-transfer agent, the cost of the initiator must be considered as well, because the initiator usually accounts for 10–20 wt% in the resulting polyols, especially for polyols with low molecular weight, since the initiator loading increases significantly with the decrease of the Mn of polyols. For example, sebacic acid has been confirmed as an effective initiator, in that it can assure Mn and CU content control in addition to high productivity in a short copolymerization time; however, the high price of sebacic acid may limit its industrial application. Oxalic acid is the cheapest saturated dicarboxylic acid,26 and its price is merely one sixth that of sebacic acid. We wondered whether oxalic acid can be used to prepare thermally stable oligo(carbonate-ether) diol. In a preliminary study, we found that the reaction time for complete PO conversion was as long as 255 min using oxalic acid as initiator (the reaction time for sebacic acid was only 40 min at similar conditions). This extension of reaction time will inevitably increase the cost of production. To accelerate the reaction, here we propose a novel preactivation approach based on understanding of the reaction mechanism, by homopolymerization of PO over oxalic acid, then the resulting oligo-ether-diol acts as chain transfer agent for the following copolymerization reaction, shortening the reaction time to 150 min.
Results and discussion
Immortal polymerization, discovered by Inoue in 1985,27 is an effective method to synthesize end-functional polymers with controllable Mn and narrow polydispersity index (PDI). The copolymerization of CO2 with PO over Zn–Co–DMC catalyst in the presence of an active proton containing initiator is one such immortal polymerization. However, when oxalic acid is used as the initiator, the copolymerization reaction needs 255 min for complete PO conversion. According to our previous report,25 oxalic acid, pKa1 (1.25) < 4.2, should act as initiate-transfer agent in the early stage of the copolymerization. Since the initiators usually affect the initial stage of copolymerization, clarification of the intermediates in this stage is vitally important to understand the long reaction time of the oxalic acid system. Fig. 1 shows the ESI-MS spectra of the reaction products at different reaction times, where the copolymerization was carried out at 80 °C and 4 MPa, using 0.32 g oxalic acid, 20 mg DMC and 10 ml PO. After 4 min of reaction, four species were identified from Fig. 1(a) (for detailed information of each m/z species see the bottom of each picture).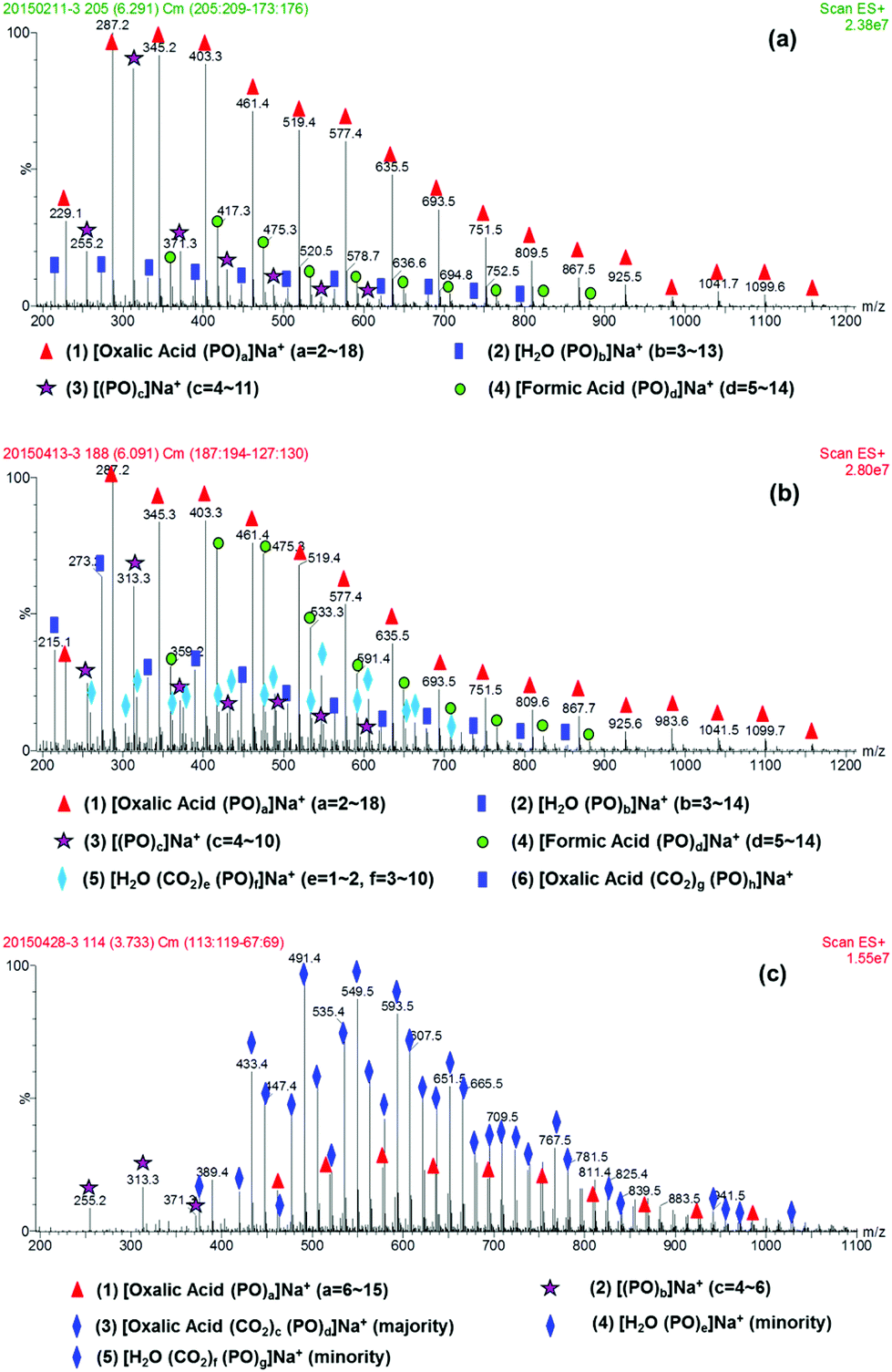 | ||
| Fig. 1 ESI-MS spectra of copolymerization products at different reaction time: (a) 4 min, (b) 30 min, (c) 180 min. The copolymerization reaction was carried out at 80 °C and 4 MPa using 0.32 g oxalic acid, 20 mg DMC and 10 ml PO. | ||
The major species (1) was assigned to be the oxalic acid initiating polyether, showing dihydroxyl end-groups and a single unit of the incorporated initiator. Species (2) with low relative abundance represented polyether diol prepared from the adventitious H2O present in the reaction system. Species (3) was a cyclic oligo-propylene oxide generated through a neutral loss of H2O from [H2O(PO)b]Na+ under ESI-MS test conditions.28 The species (4) was assigned to be the formic acid initiated polyether, which was observed with weak relative abundance. Considering the chemically unstable structure of oxalic acid,26 we could reasonably assume that the formic acid based polyether was generated through decarboxylation from the free carboxylic acid group present in the oxalic acid initiated polyether under ESI-MS testing. Thus, oxalic acid first initiated PO homopolymerization in the early stage of copolymerization, as evidenced by the exclusive formation of polyether.25 A clear assignment of oxalic acid based polycarbonate-ether was not feasible due to its overlap with H2O initiated polyether or polycarbonate-ether in the ESI MS spectrum. With extending reaction time, we observed an increasing H2O initiated polyether or polycarbonate-ether signal, whereas the relative abundance of oxalic acid based polyether declined (Fig. 1a, b and S1–6†). Since the water content remained constant in the reaction system, this implies that the above species is possibly attributed to oxalic acid based oligo(carbonate-ether) diol. Notably, the relative abundance of species, [oxalic acid (CO2)c(PO)d]Na+, increased significantly after 30 min and became dominant at 180 min (Fig. 1c, at which point the copolymerization mixture was mainly composed of oxalic acid based CO2 and PO copolymer). Therefore, we could conclude that the induction period of oxalic acid for copolymerization was about 30 min, while that for adventitious H2O was about 15 min (Fig. S2†), according to our previous definition.24 Interestingly, the maximum number of PO homopolymerization initiated by oxalic acid and H2O before 30 min was constant as 18 and 13 PO monomers, respectively, which was dependent on themselves rather than reaction time. Moreover, the thermal stability of the products increased with time, as evidenced by the disappearance of the species [formic acid (PO)d]Na+ at 180 min, in the ESI-MS spectrum (Fig. 1c).
In order to understand the chain propagation reaction, the apparent kinetics of the CO2/PO copolymerization in the presence of oxalic acid was investigated (Fig. 2).
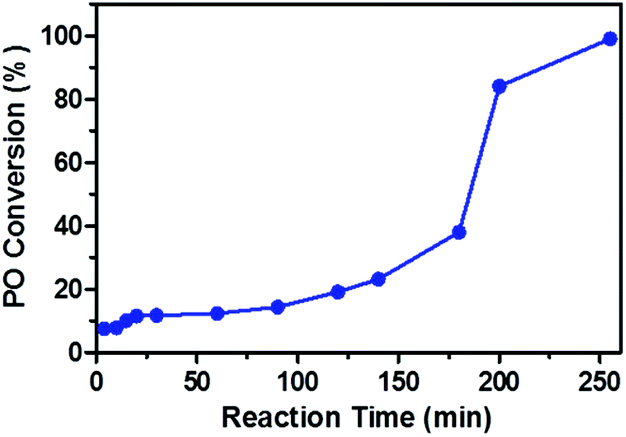 | ||
| Fig. 2 Plot of PO conversion versus time for the copolymerization of CO2 and PO carried out at 80 °C and 4 MPa, using 0.32 g oxalic acid, 20 mg DMC and 10 ml PO. | ||
Plots of PO conversion versus reaction time exhibited a sigmoid shape indicative of a slow initiation reaction.29 The PO conversion was only 20% at 120 min, and it increased to about 40% at 180 min, and rapidly increased to 85% at 200 min. This result suggested that the reaction time at which the PO conversion increased significantly corresponds to the time that the carboxylic acid group disappeared (180 min from Fig. 1c). Therefore, the low PO conversion or long reaction time might result from the existence of free carboxylic acid group in the reaction system.
FTIR spectroscopy is powerful for the detection carbonyl groups in the sample. Fig. 3 shows the FTIR spectra in the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching region for the copolymerization products at different reaction times. For the reaction product at 4 min (oxalic acid initiated polyether), two nearly equivalent absorption peaks at 1745 cm−1 and 1769 cm−1 characteristic of stretching modes of CO group were observed, suggesting almost complete reaction of the two carboxylic acid groups in oxalic acid, as proved by model compound diethyl oxalate.30 The relative intensity of the peaks at 1745 cm−1 to 1769 cm−1 increased with reaction time, due to the formation of CO2 based copolymer (νCO in carbonate segment was 1745 cm−1), which matched the induction period of H2O and oxalic acid (15 and 30 min, respectively). For the reaction product at 60 min, the absorption peak at 1769 cm−1 weakened and appeared as a shoulder peak, which was suppressed by the incorporation of CO2 in the copolymer. The increase of absorption intensity at 1745 cm−1 after 30 min further proved that species (2) in Fig. 1b and S2–6† was assigned to the oxalic acid initiated copolymer of CO2 and PO.
O stretching region for the copolymerization products at different reaction times. For the reaction product at 4 min (oxalic acid initiated polyether), two nearly equivalent absorption peaks at 1745 cm−1 and 1769 cm−1 characteristic of stretching modes of CO group were observed, suggesting almost complete reaction of the two carboxylic acid groups in oxalic acid, as proved by model compound diethyl oxalate.30 The relative intensity of the peaks at 1745 cm−1 to 1769 cm−1 increased with reaction time, due to the formation of CO2 based copolymer (νCO in carbonate segment was 1745 cm−1), which matched the induction period of H2O and oxalic acid (15 and 30 min, respectively). For the reaction product at 60 min, the absorption peak at 1769 cm−1 weakened and appeared as a shoulder peak, which was suppressed by the incorporation of CO2 in the copolymer. The increase of absorption intensity at 1745 cm−1 after 30 min further proved that species (2) in Fig. 1b and S2–6† was assigned to the oxalic acid initiated copolymer of CO2 and PO.
 | ||
| Fig. 3 FTIR spectra in the CO stretching region for the copolymerization products at different reaction time: (a) 4 min, (b) 15 min, (c) 30 min, (d) 60 min. The copolymerization was carried out at 80 °C and 4 MPa using 0.32 g oxalic acid, 20 mg DMC and 10 ml PO. | ||
As discussed above, the existence of a free carboxylic acid group might result in a low PO conversion or long reaction time. We wondered whether the reaction time can be shortened by elimination of the free carboxylic acid group in the reaction system. Considering the relatively strong acidic character of oxalic acid (pKa1 = 1.25, pKa2 = 4.14), the two, carboxylic acid groups can be almost completely converted by reaction with PO. Therefore, a novel preactivation approach was proposed to reduce the copolymerization time. At first, oxalic acid was converted to oligo-ether-diol by reaction with PO, late DMC catalyst was added, and CO2 was pressurized to initiate the copolymerization. ESI-MS spectroscopy was employed to characterize the resulting oligo-ether-diol. No formic acid based polyether was detected from the pure product of oxalic acid and PO at 80 °C for a short reaction time of 4 min (see ESI-MS spectrum of reaction product in Fig. S7†), indicating that the polyether was capped with two terminal hydroxyl groups. The microstructure of the oligo-ether-diol was further characterized by 1H NMR and COSY NMR spectra. The absorption peaks were assigned and the possible structure of oligo-ether-diol is shown in the inset of Fig. 4a. However, it should be noted that the absorption peak assigned to PO directly connected to oxalic acid overlapped with that of the carbonate group in oligo(carbonate-ether) diol, moreover, there are no characteristic protons present in the oxalic acid, which added difficulty for the precise calculation of CU content and Wpc of the final product.
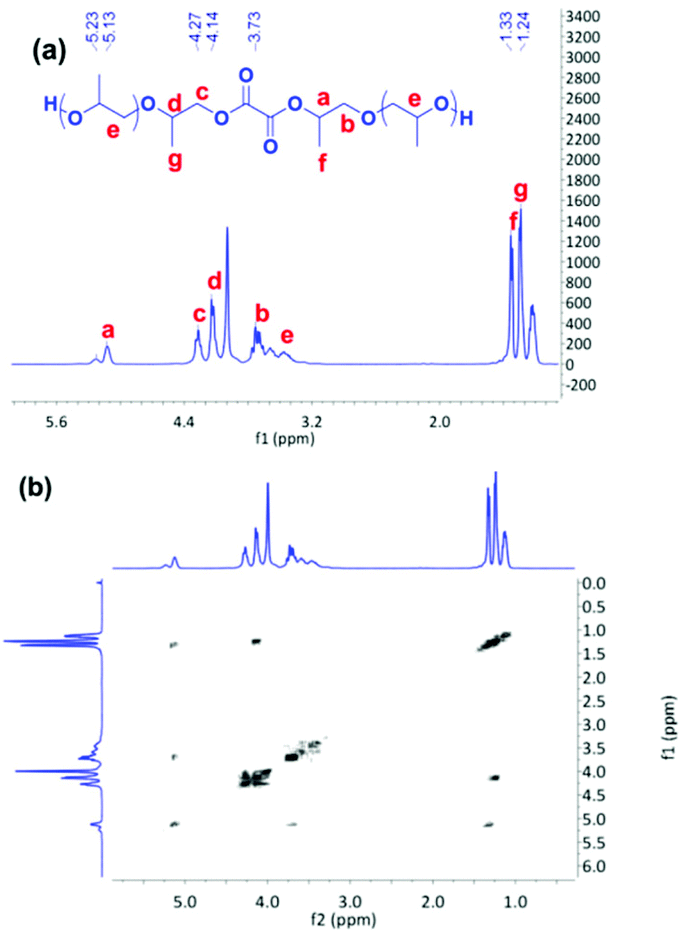 | ||
| Fig. 4 1H NMR (a) and COSY NMR (b) spectra of oligo-ether-diol (diol from Fig. S7†). | ||
The effect of the preactivation time on the copolymerization of CO2/PO is summarized in Table 1. All polymerizations showed reduced copolymerization time relative to the control experiment (255 min), and the copolymerization time decreased at first and then increased with extending preactivation time from 0 min to 120 min (entries 1–7 and 10). Notably, a minimum copolymerization time of 150 min was observed at the preactivation time of 15 min (Fig. 5).
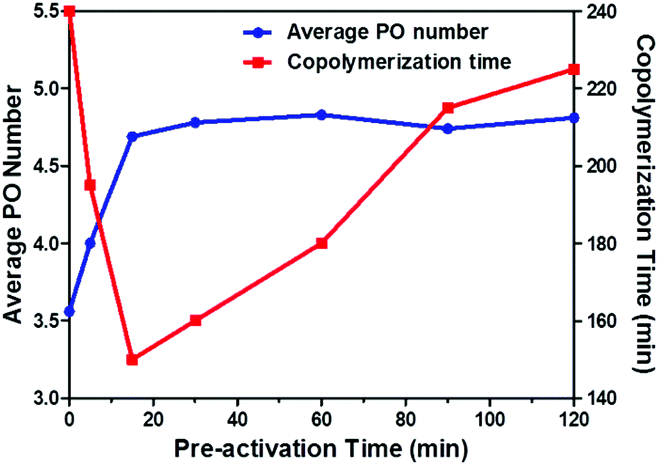 | ||
| Fig. 5 Typical plot of average PO number and copolymerization time versus time for the copolymerization of CO2 and PO carried out at 80 °C and 4 MPa, using 0.32 g oxalic acid, 20 mg DMC and10 ml PO. | ||
| Entry | Time-1b (min) |
![[N with combining macron]](https://www.rsc.org/images/entities/i_char_004e_0304.gif) PO
PO![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) c c |
Time-2d (min) | CUe (%) |
W
pce (%) |
M
nf (g mol−1) |
PDIf |
|---|---|---|---|---|---|---|---|
|
a The preactivation reaction between oxalic acid (0.32 g) and PO (10 ml) was carried out at 80 °C for different times, then 20 mg Zn–Co–DMC was added and the reaction system was pressurised to 4 MPa to initiate copolymerization.
b Time-1 refers to the preactivation time.
c
PO was average PO number calculated by 1H NMR, PO = (A3.5 − 2A5.12)/(3A5.12 + A4.14–4.27) + 2.
d Time-2 is the time needed for complete PO conversion after preactivation.
e Calculated by 1H NMR spectrum, CU = (A5.0 + A4.2 − 2.73A5.2 − 2A4.58)/(A5.0 + A4.2 − 2.73A5.2 − 2A4.58 + A3.5 + 3.73A5.2), Wpc = 102A1.5/[102(A5.0 + A4.2 − 2.73A5.2 − 2A4.58 + A1.5) + 58(A3.5 + 3.73A5.2)], see Fig. S8.
f Measured by GPC.
g 0.2 g oxalic acid and 15 mg DMC were used, other conditions were the same as in note [a].
h 0.4 g oxalic acid and 25 mg DMC were used, other conditions were the same as in note [a].
i Control experiment, without pre-activation process.
|
|||||||
| 1 | 0 | 3.56 | 240 | 40.8 | 7.2 | 5000 | 1.33 |
| 2 | 5 | 4.0 | 195 | 39.4 | 6.9 | 4800 | 1.29 |
| 3 | 15 | 4.69 | 150 | 41.4 | 7.6 | 4800 | 1.28 |
| 4 | 30 | 4.78 | 160 | 41.2 | 7.3 | 4800 | 1.28 |
| 5 | 60 | 4.83 | 180 | 39.9 | 6.7 | 4800 | 1.29 |
| 6 | 90 | 4.74 | 215 | 42.2 | 6.0 | 4900 | 1.29 |
| 7 | 120 | 4.81 | 225 | 42.4 | 6.5 | 4900 | 1.28 |
| 8g | 15 | 4.73 | 190 | 45.8 | 6.4 | 7400 | 1.34 |
| 9h | 15 | 4.72 | 200 | 38.7 | 7.3 | 3500 | 1.26 |
| 10i | — | — | 255 | 37.8 | 7.6 | 5200 | 1.30 |
The total reaction time (165 min) was still significantly shorter than 255 min, suggesting the feasibility of this preactivation approach, which is vitally important for industrial production. Moreover, the similar results (CU, Wpc, Mn, PDI of the diol) of the preactivation experiment and control experiment showed that the oxalic acid initiated oligoether diol participated in the copolymerization rather than existing as a blend in the final diol.
The average PO number (PO) of the preactivation product was an important parameter to observe the initiation character of oxalic acid. As shown in Fig. 5, PO of the preactivation product increased dramatically to 3.56 at 5 min preheating (corresponding to 0 min in Fig. 5), the PO remained constant at 4.75 PO after 15 min. The slight change of PO was in good agreement with the variation in PO conversion before 120 min (Fig. 2). Moreover, different oxalic acid loading displayed a negligible effect on PO (entires 4, 8 and 9). Generally, about 4.75 PO monomers were initiated by oxalic acid in the early stage of homopolymerization, which was independent of time and initiator loading. It was for this reason that the drop in molecular weight resulted in the decrease of CU content in the final oligo(carbonate-ether) diol.
As discussed above, it is difficult to calculate the CU content and Wpc of the final oligo(carbonate-ether) diol, due to the overlapping of absorption peak of PO directly connected to oxalic acid (δ = 4.1–4.2, 3.7 ppm) with that of the carbonate group in oligo(carbonate-ether) diol, therefore, these peak areas should be subtracted in the calculation to obtain accurate data. Fortunately, though there are no characteristic protons in the oxalic acid molecule, the molar ratio of α, β ring opening method was constant at 2.73/3 in PO directly attached by the initiator (Fig. S9†), and the peaks at 5.2 ppm could be integrated distinctly. Therefore, the peak areas at 4.1–4.2 and 3.7 ppm were equal to 2.73A5.2 and 2A5.2, respectively, which coincidently solved the problem of precisely calculating the CU, PO and Wpc of the final product. It should be noted that the peaks of the cyclic propylene carbonate (4.89 and 4.04 ppm) also overlapped with the peaks of the copolymer; all these peak areas should be considered in the calculation. Based on above analysis, CU, PO and Wpc of the final product can be calculated according to the eqn (1)–(3):
| CU = (A5.0 + A4.2 − 2.73A5.2 − 2A4.58)/(A5.0 + A4.2 − 2.73A5.2 − 2A4.58 + A3.5 + 3.73A5.2) | (1) |
| PO = (A3.5 − 2A5.12)/(3A5.12 + A4.14–4.27) + 2 | (2) |
| Wpc = 102A1.5/[102(A5.0 + A4.2 − 2.73A5.2 − 2A4.58 + A1.5) + 58(A3.5 + 3.73A5.2)] | (3) |
Next, we wished to demonstrate that the shortening of copolymerization time was due to the formation of polyether diol. After preactivation for 15 min, the copolymerization stopped even at the beginning (after only preheating for 5 min). The absorption peak characteristics of the carbonate unit appeared in the 1H NMR, proving the incorporation of CO2 (Fig. S10†). Therefore, the oxalic acid based polyether diol did act as chain transfer agent during the copolymerization. We can reasonably conclude that the complete conversion of oxalic acid or the disappearance of the carboxylic acid group in the initial stage was the key to shortening the reaction time.
Upon further consideration, oxalic acid is also sensitive to heat, therefore the question of thermal instability arose for the oxalic acid based diol due to the existence of two neighboring carboxylic acid groups. It was encouraging to find that almost the same starting gravimetric temperature (∼190 °C) of oxalic acid-based and sebacic acid-based oligo(carbonate-ether) diol (Mn = 5200 g mol−1) was observed from TGA (Fig. 6), which was higher than that of anhydrous oxalic acid (125 °C, part of which might be caused by sublimation), suggesting the formation of the highly stable oxalic acid based oligo(carbonate-ether) diol. Moreover, as shown in Fig. 6, even though the Mn of the oxalic acid diol changed from 5200 g mol−1 to 1200 g mol−1, the starting gravimetric temperature remained constant, further proving the thermal stability of the resulting oligo(carbonate-ether) diol.
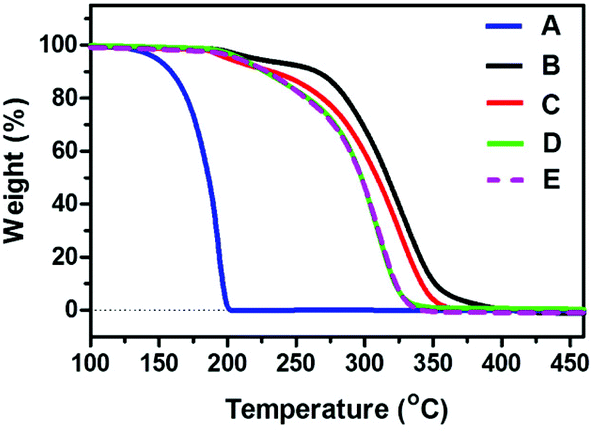 | ||
| Fig. 6 Thermogravimetric Analyzer (TGA) curves of (A) anhydrous oxalic acid; (B) sebacic acid based oligo(carbonate-ether) diol (Mn = 5200, CU = 46%, PDI = 1.25); oxalic acid based oligo(carbonate-ether) diol (C) (Mn = 5200, CU = 42%, PDI = 1.3); (D) (Mn = 3500, CU = 34%, PDI = 1.25); (E) (Mn = 1200, CU = 15%, PDI = 1.22). | ||
Experimental
Materials
Anhydrous oxalic acid was purchased from Alfa Aesar and dried for 48 h in vacuum at 50 °C prior to use. K3[Co(CN)6] was also provided by Alfa Aesar and recrystallized from deionized water prior to use. ZnCl2 and tert-butanol (t-BuOH) were analytical grade and used without further purification. PO was refluxed over calcium hydride and then distilled under an argon atmosphere. Carbon dioxide with purity over 99.99% was used as received.Zn–Co–DMC catalyst was prepared according to the previous report.6
Copolymerization
One-pot copolymerization: in a glove box free of oxygen and water, a calculated quantity of DMC catalyst, acidic initiator and PO were added to a 50 ml professional stainless-steel autoclave with magnetic stirring. The reaction vessel was pressurized with CO2 and the reaction was carried out at the determined conditions. After copolymerization, the autoclave was cooled to room temperature, and the CO2 pressure was released by opening the outlet valve. A small aliquot of the copolymerization mixture was taken out for 1H NMR spectroscopy.Pre-activation approach: in a glove box free of oxygen and water, a calculated quantity of acidic initiator and PO were added to a 50 ml professional stainless-steel autoclave with magnetic stirring. The reaction was carried out at the determined conditions. After pre-activation, the autoclave was cooled to room temperature, a small aliquot of the reaction mixture was taken out for 1H NMR spectroscopy. Then a calculated quantity of DMC catalyst was added to the autoclave and the vessel was pressurized with CO2 and stabilized for 5 min. After copolymerization, the autoclave was cooled to room temperature, and the CO2 pressure was released by opening the outlet valve. A small aliquot of the copolymerization mixture was taken out for 1H NMR spectroscopy.
Caution: The polymerization of PO is highly exothermic. Because of the possibility of runaway reactions, the reactor needs to be equipped with an appropriate pressure release system and operated in an explosion-proof safety box.
Measurements
Fourier transform infrared (FTIR) spectra were recorded by casting an acetone solution of the collected product onto a disk of KBr by a Bruker TENSOR-27 spectrometer with 30 scans per experiment at a resolution of 4 cm−1. 1H NMR, 13C NMR and COSY NMR spectra were recorded at room temperature on a Unity-500 NMR spectrometer using CDCl3 as solvent. The Mn and PDI of the oligo(carbonate-ether) diol were measured by gel permeation chromatography (GPC) at 35 °C using a polystyrene standard on a Waters 410 GPC instrument (tandem double columns: WAT044222, 7.8 × 300 mm, molecular weight range 500–30000 g mol−1; WAT044234, 7.8 × 300 mm, molecular weight range 100–5000 g mol−1, RID detector) with CH2Cl2 as eluent, where the flow rate was set at 1.0 ml min−1. Electrospray ionization mass spectrometry (ESI-MS) analyses were performed on Waters Quattro Premier XE mass spectrometer, using methanol/water (4:1) as solvent. Thermogravimetric analyzer (TGA) testing was carried out on a Perkin-Elmer Pyris 1 TGA thermal analyser from 40 °C to 500 °C under nitrogen protection at a heating rate of 10 °C min−1.
Conclusions
Oxalic acid initiated thermally stable oligo(carbonate-ether) diol was cheaply and quickly synthesized by the immortal copolymerization of CO2 and PO using Zn–Co–DMC catalyst via a novel preactivation approach. The existence of the carboxylic acid group of oxalic acid resulted in the prolongation of the copolymerization time. By using a preactivation approach, the reaction time could be significantly reduced from 255 min to 150 min, and the resulting oligo(carbonate-ether) diol showed almost the same structure to that prepared by the one-pot method. The studies revealed that about 4.75 PO monomers were initiated during preactivation, accounting for the CU content dropping with decreasing Mn. The studies also highlighted that the oligo-ether-diol formed by the preactivation approach almost immediately participated in the copolymerization of CO2 and PO, therefore shortening the reaction time. Thus, such a preactivation approach may be an interesting means to shorten the reaction time of polyols in strongly acidic initiator systems.Acknowledgements
The authors thank the National Natural Science Foundation of China (grant no. 51321062, 21134002, and 51273197) for financial support.Notes and references
- P. Krol, Prog. Mater. Sci., 2007, 52, 915–1015 CrossRef CAS.
- E. Delebecq, J. P. Pascault, B. Boutevin and F. Ganachaud, Chem. Rev., 2013, 113, 80–118 CrossRef CAS PubMed.
- J. Langanke, A. Wolf, J. Hofmann, K. Bohm, M. A. Subhani, T. E. Muller, W. Leitner and C. Gurtler, Green Chem., 2014, 16, 1865–1870 RSC.
- N. von der Assen and A. Bardow, Green Chem., 2014, 16, 3272–3280 RSC.
- I. Kim, M. J. Yi, K. J. Lee, D. W. Park, B. U. Kim and C. S. Ha, Catal. Today, 2006, 111, 292–296 CrossRef CAS.
- Z. Li, Y. Qin, X. Zhao, F. Wang, S. Zhang and X. Wang, Eur. Polym. J., 2011, 47, 2152–2157 CrossRef CAS.
- X. H. Zhang, Z. J. Hua, S. Chen, F. Liu, X. K. Sun and G. R. Qi, Appl. Catal., A, 2007, 325, 91–98 CrossRef CAS.
- J. Kuyper, P. W. Lednor and G. A. Pogany, US Pat, 1989/4826952, 1989 Search PubMed.
- G. W. Coates and D. R. Moore, Angew. Chem., Int. Ed., 2004, 43, 6618–6639 CrossRef CAS PubMed.
- S. Klaus, M. W. Lehenmeier, C. E. Anderson and B. Rieger, Coord. Chem. Rev., 2011, 255, 1460–1479 CrossRef CAS.
- X. B. Lu and D. J. Darensbourg, Chem. Soc. Rev., 2012, 41, 1462–1484 RSC.
- Y. S. Qin, X. F. Sheng, S. J. Liu, G. J. Ren, X. H. Wang and F. S. Wang, J. CO2 Util., 2014 DOI:10.1016/j.jcou.2014.10.003.
- K. Nakano, K. Kobayashi and K. Nozaki, J. Am. Chem. Soc., 2011, 133, 10720–10723 CrossRef CAS PubMed.
- B. Y. Liu, Y. H. Gao, X. Zhao, W. D. Yan and X. H. Wang, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 359–365 CrossRef CAS.
- M. R. Kember and C. K. Williams, J. Am. Chem. Soc., 2012, 134, 15676–15679 CrossRef CAS PubMed.
- C. Gürtler, S. Grasser and A. Wolf, US Pat, 2013/0211042A1, 2013 Search PubMed.
- C. Gürtler, J. Hofmann, T. E. Müller, A. Wolf, S. Grasser and B. Köhler, US Pat, 2013/0072602, 2013 Search PubMed.
- S. Grasser, C. Gürtler, J. Hofmann and A. Wolf, US Pat, 2012/0289732A1, 2012 Search PubMed.
- D. Mijolovic, M. Kummeter, M. Stoesser and S. Bauer, US Pat, 2010/0048935A1, 2010 Search PubMed.
- W. Hinz, E. M. Dexheimer, E. Bohres and G. H. Grosch, US Pat, 2004/6762278B2, 2004 Search PubMed.
- Y. G. Gao, Y. S. Qin, X. J. Zhao, F. S. Wang and X. H. Wang, J. Polym. Res., 2012, 19, 9 CrossRef.
- C. Gürtler, J. Hofmann, A. Wolf and S. Grasser, US Pat, 2013/0184432A1, 2013 Search PubMed.
- Y. G. Gao, L. Gu, Y. S. Qin, X. H. Wang and F. S. Wang, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 5177–5184 CrossRef CAS.
- S. J. Liu, Y. S. Qin, X. S. Chen, X. H. Wang and F. S. Wang, Polym. Chem., 2014, 5, 6171–6179 RSC.
- S. J. Liu, Y. Y. Miao, L. J. Qiao, Y. S. Qin, X. S. Chen, X. H. Wang and F. S. Wang, under revision, 2015.
- W. Riemenschneider and M. Tanifuji, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, 2000, DOI:10.1002/14356007.a18_247.
- S. Inoue, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 2861–2871 CrossRef CAS.
- X. H. Zhang, R. J. Wei, X. K. Sun, J. F. Zhang, B. Y. Du, Z. Q. Fan and G. R. Qi, Polymer, 2011, 52, 5494–5502 CrossRef CAS.
- Y. Zhu, C. Romain, V. Poirier and C. K. Williams, Macromolecules, 2015, 48, 2407–2416 CrossRef CAS.
- http://sdbs.db.aist.go.jp/sdbs/cgi-in/direct_frame_top.cgi .
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5py01338k |
| This journal is © The Royal Society of Chemistry 2016 |