
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Cucurbit[7]uril inclusion complexation as a supramolecular strategy for color stabilization of anthocyanin model compounds†
Barbara
Held‡
a,
Hao
Tang‡
 b,
Palani
Natarajan§
b,
Cassio Pacheco
da Silva
a,
Volnir
de Oliveira Silva
a,
Cornelia
Bohne
*b and
Frank H.
Quina
*b
b,
Palani
Natarajan§
b,
Cassio Pacheco
da Silva
a,
Volnir
de Oliveira Silva
a,
Cornelia
Bohne
*b and
Frank H.
Quina
*b
aInstituto de Química, Universidade de São Paulo, C.P. 26077, São Paulo 05513-970, Brasil
bDepartment of Chemistry, University of Victoria, PO Box 3065, Victoria, BC, Canada V8W 3V6. E-mail: cornelia.bohne@gmail.com; quina@usp.br
First published on 22nd April 2016
Abstract
Host–guest complexation with cucurbit[7]uril of anthocyanin model compounds in which acid–base equilibria are blocked resulted in essentially complete stabilization of their color. The color protection is a thermodynamic effect and establishes a strategy to stabilize these colored compounds at pH values of interest for practical applications.
Introduction
Anthocyanins are the natural plant pigments responsible for the majority of the red, blue and purple colors of fruits, flowers, and leaves.1 The basic chromophore of anthocyanins is the flavylium cation, whose color depends primarily on the substituents present on the chromophore and the local pH.2 Above about pH 3 in aqueous solution, however, the flavylium cation form (Fl+) of anthocyanins typically undergoes hydration to form the hemiacetal (B), followed by ring-opening tautomerization to give the Z-chalcone (CZ) and subsequent isomerization to the E-chalcone (CE). These equilibria are indicated in Scheme 1 for the 3′,4′,7-trimethoxyflavylium ion (B-TMF), together with the structure of the 7-methoxy-4-methylflavylium cation (MMF+), a compound that does not hydrate under the same conditions.3 | ||
| Scheme 1 Reactivity of B-TMF, and structures of CB[7] and MMF+. | ||
In nature, the color of anthocyanins can be stabilized to some extent by complexation with metal cations or colorless organic molecules, called co-pigments.4,5 Guest-host complexation is an alternative strategy that might potentially preserve the color of the flavylium cation chromophore more effectively than co-pigmentation by shielding the chromophore from contact with water. Achieving the stabilization of the colored flavylium cation is relevant to the ways in which flavylium ions, and by inference anthocyanins, can potentially be stabilized as photoprotectors.4,5 Guest-host complexes with hosts such as molecular clips6 and cucurbit[7]uril (CB[7])7–9 were shown to slow down the decoloration rate. In this context, cucurbiturils are particularly attractive as hosts since they have a high affinity for cationic species10–12 and can change the spectroscopic properties and reactivity of guest molecules that are included in their interior.13–17 CB[7] complexation of flavylium cations containing a hydroxyl group at the 7-positon resulted in only partial color stabilization because the decrease of the rate of the hydration reaction was offset by the deprotonation of the flavylium cation at higher pH.7–9 We report here that the flavylium cation form (Fl+) of B-TMF, in which the acid–base equilibria are blocked, indeed exhibits an impressive, essentially complete stabilization against hydration upon inclusion in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 guest-host complex with CB[7]. Kinetic experiments showed that this is a thermodynamic and not a kinetic effect.
:2 guest-host complex with CB[7]. Kinetic experiments showed that this is a thermodynamic and not a kinetic effect.
Experimental
MMF+ was synthesized as previously described.18 The modified synthesis for B-TMF19,20 and CB[7]21–23 and the titration of CB[7]24 are described in the ESI (Fig. S1 and S2 in the ESI† for B-TMF). Cobaltocenium hexafluorophosphate (Cob+ PF6−, Aldrich 98%), HCl (Anachemia, ACS reagent grade) and methanol (EMD Omnisol, spectrograde) were used as received. De-ionized water (Barnstead Nanopure System, ≥17.8 MΩ cm) was used for all aqueous solutions. B-TMF stock solutions (1.1 mM) were prepared either in acidified methanol (pH = 0.6) or by directly dissolving the solid in water containing 1 mM HCl. Stock solutions of CB[7] were prepared in 1 mM HCl aqueous solutions. Binding isotherm determinations and stopped-flow experiments were performed in the presence of 1 mM HCl. The initial concentrations for the stopped-flow experiments were 0.4 μM of B-TMF and between 0 and 12.6 μM of CB[7]. Separate solutions of B-TMF and CB[7] were mixed in the stopped-flow experiment in a 1:1 ratio and the final concentrations were one half of the initial ones. The B-TMF and MMF+ concentrations for the binding isotherm studies were 0.2 μM. The methods for the fitting of the binding isotherms are described in the ESI.†
The kinetic absorption experiments on the minutes time scale at pH values of 4.3 and higher were performed with 5.0 μM B-TMF. Two different experiments were performed: (i) B-TMF was injected into aqueous solutions without and with CB[7]. (ii) B-TMF was pre-incubated in water at pH 4.6 for 12 h. After this period the solution was injected with water (control) or aqueous CB[7] solutions. Spectra were collected at regular intervals or the kinetics were followed at 470 nm, where Fl+ absorbs.
The apparent hydration constant (pKap) was determined by adding B-TMF (18 μM) to solutions prepared at different pH values and measuring the absorption spectra 2 h and 24 h after the B-TMF addition.
Absorption spectra were measured with Cary 100 UV-Vis or Cary 50 Bio spectrometers, or a Hewlett Packard 8452A diode array spectrometer. Kinetic studies at 470 nm, where Fl+ absorbs, were performed on the Cary 100 or Cary 50 Bio spectrometers where multi-cell holders were used (6 to 18) and the absorption was measured sequentially at defined intervals.
Fluorescence measurements with B-TMF were performed with a PTI QM-2 fluorimeter with excitation and emission monochromator bandwidths of 5 nm. The samples were excited at 470 nm and the emission spectra were measured between 500 and 700 nm. The spectrum of a water sample was subtracted from the experimental fluorescence spectra to remove the Raman scattering peak of water at ca. 560 nm. The experiments were performed at 20 °C. Fluorescence measurements with MMF+ were measured with a Hitach F-4500 where the bandwidths for the excitation and emission monochromators were 5.0 nm. The samples were excited at 416 nm and the emission spectra were measured between 430 and 650 nm. No spectral subtraction was required in this case because the Raman scattering was negligible. The experiments were performed at 25 °C.
Stopped-flow experiments were performed with a SX20 system from Applied Photophysics. Samples were excited at 470 nm using a 2.3 nm bandwidth on the excitation monochromator, and the emission was detected at 90 degrees with respect to the excitation beam using a 515 nm cut-off filter. The two syringes, one containing B-TMF and the other with CB[7], were kept at 20 °C for 10 min before the start of mixing. The mixing ratio was 1:1. Each experiment corresponds to the average of 24 individual kinetic traces. The control experiments were the following: (i) mixing of two water solutions to determine the “zero” reading and (ii) the mixing of water with B-TMF to determine the fluorescence intensity of B-TMF in the absence of CB[7]. The kinetic data were analyzed using the Pro-Data Viewer software from Applied Photophysics. The quality of the fits was judged by the randomness of the residuals. All the kinetics fit well to a mono-exponential function.
Results and discussion
In the absence of CB[7] in aqueous solution at ca. pH 1, the dominant form of B-TMF is the Fl+ form, with a maximum absorption at 470 nm. Transfer to a solution of pH 4.3 results in spectral changes characteristic of the hydration and subsequent ring-opening tautomerization and isomerization reactions, since B, CZ and CE do not absorb above 400 nm (Fig. 1). From the pH dependence of the loss of the color of the Fl+ form, a value of pKap = 3.0 ± 0.3 was found for the negative logarithm of the apparent hydration constant (inset Fig. 1, see details in the ESI†), in good agreement with the value (3.1 ± 0.3) estimated from the correlations of Freitas et al.25 | ||
| Fig. 1 Absorption spectra for B-TMF (18 μM) in water at different pH values after a 24 h equilibration period. The pH value for the highest absorbance for the band centered at 470 nm was 0.98 followed by pH values of 1.32, 1.80, 2.50, 3.01, 3.44, 3.86, 4.01, 4.61, 4.99, 5.45 and 6.04. Inset: Dependence of log (xFl/(1 − xFl)) with pH for solutions equilibrated for 2 h (black) and 24 h (red). The solid lines correspond to the fit of the data to eqn (S12) in the ESI.† | ||
Addition of CB[7] led to the stabilization of Fl+ at pH 4.3 (Fig. 2) which was dependent on the CB[7] concentration. At a 24-fold excess the absorption spectra for B-TMF were the same right after the addition of B-TMF and after 7 h, while with a lower concentration of CB[7] some hydration occurred. The kinetics at 470 nm showed that in the presence a 2.4-fold excess of CB[7] over B-TMF, the rate of the hydration reaction of the Fl+ form was noticeably slower (Fig. 3A) and, in the presence of a 24-fold excess of CB[7], this reaction was essentially completely inhibited. The same effects were observed at pH 5.5 in absorbance and at pH 4.3 by monitoring the fluorescence of the Fl+ form of B-TMF (see Fig. S3 and S4 in the ESI†).
 | ||
| Fig. 2 Absorption spectra at pH 4.3 measured 7 h after the addition of B-TMF (5 μM) in the absence (black) and presence of 12 μM (blue) and 120 μM (red) CB[7]. The inset shows the spectra right after the addition of B-TMF. For 120 μM CB[7], the spectra are still unchanged 7 h after the addition of B-TMF. | ||
 | ||
| Fig. 3 Kinetics measured at 470 nm for B-TMF (5 μM) at pH 4.6. A: after addition of B-TMF to an aqueous solution without added CB[7] (black) or addition to aqueous solutions of B-TMF containing 12 (blue) or 120 μM CB[7] (red). B: after addition of water (black), or 12 (blue) or 470 μM CB[7] (red) to B-TMF pre-incubated for 12 h at pH 4.6. | ||
That this is a true thermodynamic rather than a mere kinetic stabilization of the flavylium cation form Fl+ of B-TMF was shown by initially incubating B-TMF (5 μM) at pH 4.6 for 12 h to obtain an equilibrium mixture containing predominantly the hydration product B plus CZ and CE. Addition of 12 μM or 120 μM CB[7] then resulted in a shift of the equilibrium toward Fl+ (Fig. 3B) at a rate and to an extent dependent on the concentration of added host (Fig. S5 in the ESI† for fluorescence measurements). It is important to note that the kinetics after incubation for 12 h are not the same as those for the direct addition of B-TMF to a solution of CB[7] where the effect of the slow conversion of CE to CZ is absent.
The binding kinetics of guests with cucurbiturils can be fast, with relaxation processes that occur in the sub-second to seconds time-domain.26–28 The binding kinetics were studied by stopped-flow, accompanying the increase in fluorescence due to the complexation of Fl+ upon mixing of a solution of B-TMF with a solution containing CB[7] (Fig. 4, top). The kinetics levelled off within 0.25 s showing the formation of the CB[7] complex. The formation of B, CZ and CE was not observed on this time scale. The experiments were run under conditions where the concentration of CB[7] was at least 10 times higher than the concentration of Fl+ and all kinetic traces fit well to first-order kinetics. Although both Fl+ and B were initially present in solution ([HCl] = 1 mM) at the time of mixing, the kinetic traces showed no initial off-set that would indicate a fast process occurring within the stopped-flow mixing time of 1 ms, which could be related to the binding of B. Therefore under the pseudo-first order conditions used, the concentration of B is not relevant for the analysis of the kinetic data.
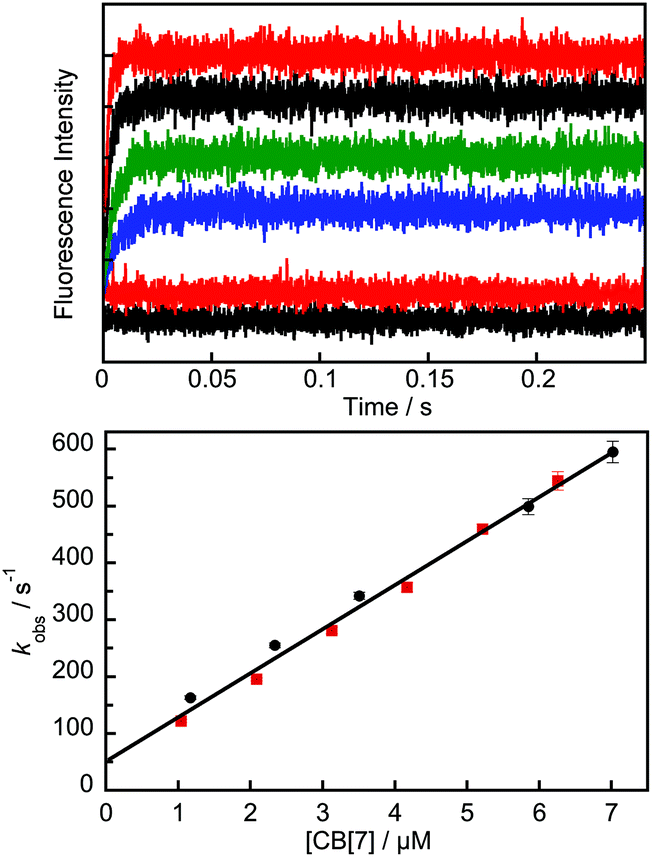 | ||
| Fig. 4 Top: Stopped-flow kinetics for the mixing of B-TMF (0.2 μM) with CB[7] (top to bottom: 6.3 μM (red), 4.2 (black), 2.1 (green), 1.0 (blue) and 0 (red)). The lowest trace (black) corresponds to the baseline intensity for water in the mixing cell. Bottom: Dependence of the observed rate constant with the concentration of CB[7] for two independent experiments (black and red symbols) fit simultaneously to eqn (1). Error bars not shown are smaller than the data points. | ||
The kinetics of Fl+ binding with CB[7] could not be followed at concentrations of CB[7] above 7 μM because the reaction rate exceeded the time-resolution of the equipment (rate constant >600 s−1). In the accessible time range, the observed first-order rate constants were a linear function (eqn (1)) of the CB[7] concentration (Fig. 4 bottom), providing values for the bimolecular association (k11+) and unimolecular dissociation (k11−) rate constants of (7.7 ± 0.2) × 107 M−1 s−1 and 50 ± 10 s−1. The ratio of these values leads to an equilibrium constant of (1.5 ± 0.3) × 106 M−1, which was assigned to the formation of the 1:1 complex between Fl+ and CB[7] (K11), because at the low CB[7] concentrations the formation of the 1:2 complex is negligible since K12 is much lower than K11 (see below). Two relaxation processes would have been observed if the rate constants for hydration/dehydration were of the same order as those for complex formation. The values of kh+ and kh− for B-TMF were estimated to be ca. 0.05 s−1 and 104 M−1 s−1, respectively, from the value of pKap and the values reported by Pina et al.2 for analogous compounds. Thus, at pH 3, kh−[H+] should be ca. 10 s−1, which is at least an order of magnitude smaller than the lowest value of kobs measured.
| kobs = k11+ [CB[7]] + k11− | (1) |
Because the fluorescence intensities of the Fl+ form of B-TMF and MMF+ increase upon addition of CB[7] (Fig. S6 in the ESI†), the changes in fluorescence were used to obtain the binding isotherms. The binding isotherm for B-TMF could not be fit with a model that considered the formation of only a 1:1 complex (Fig. 5). The intensity decrease observed for the calculated fit for a 1:1 stoichiometry at high CB[7] concentrations occurs because the fluorophore was diluted as CB[7] was added. This dilution was taken into account in the numerical fit. The binding isotherm did however fit nicely to a sequential 1:1 and 1:2 B-TMF:CB[7] binding stoichiometry. Because the emission efficiencies of the 1:1 and 1:2 complexes were similar, the equilibrium constants derived from the unrestrained fits had relatively large errors. For this reason, the value for K11 determined from the kinetic studies (1.5 × 106 M−1) was treated as a fixed parameter. This provided a fit with good residuals and an estimated K12 value of ≤3 × 104 M−1 (see ESI† for details), showing that the binding of the second CB[7] to Fl+@CB[7] is less efficient than the binding of the first CB[7]. For the objectives of the current work, an absolute value of K12 is not required; the crucial point being the demonstration that the higher order 1:2 complex is indeed formed at the higher CB[7] concentrations and is the key species in the protection of the flavylium cation of B-TMF from hydration. Likewise, fitting of the binding isotherms of MMF+ with CB[7] also required the assumption of a 1:2 sequential binding model with K11 = (9 ± 2) × 105 M−1 and K12 = (8 ± 5) × 105 M−1 (Fig. S7 in the ESI†). The fact that MMF+ does not hydrate under these conditions shows that the formation of the 1:2 complex is a feature of the flavylium cation framework for both MMF+ and Fl+. The slightly smaller K11 value and much larger K12 value for MMF+ compared to B-TMF+ are attributed to differences in steric hindrance caused by the 4-methyl substitution in MMF+vs. dimethoxy substitution in the B-ring of B-TMF. The formation of the 1:2 complex between flavylium derivatives and CB[7] in addition to the 1:1 complex was previously not reported.8,9,29,30 However, the position and hydrophobicity of the substituents were shown to affect the magnitude of the equilibrium constants for the 1:1 complex,29,30 to determine which ring is primarily incorporated into the CB[7] and the ability of CB[7] to shuttle between the two rings.29
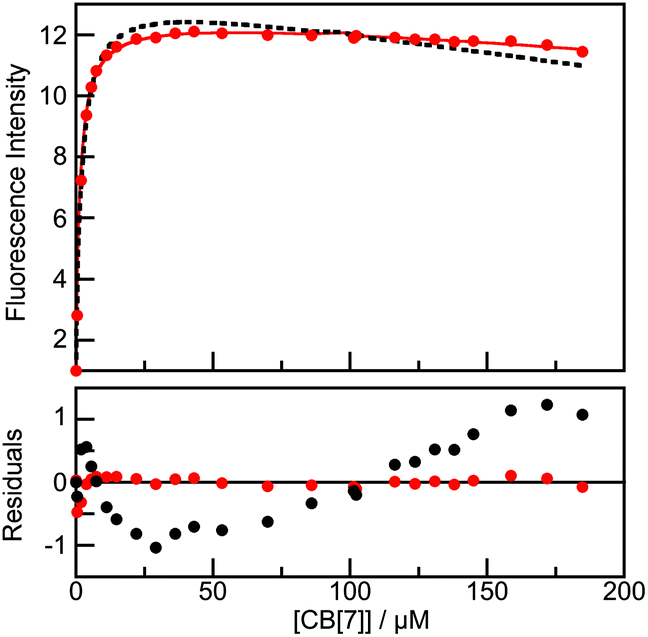 | ||
| Fig. 5 Top: Fit of the binding isotherm for B-TMF (0.2 μM) with CB[7] assuming a pKap of 3.0 to a 1:1 stoichiometry (black, dashed line) and to sequential 1:1 and 1:2 B-TMF:CB[7] stoichiometries (red, solid line). Bottom: Residuals between the data and calculated values for the fits assuming a 1:1 stoichiometry (black) and sequential 1:1 and 1:2 stoichiometries (red). | ||
The kinetic experiments show that the rate of formation of the 1:1 complex is faster than the hydration reaction (Scheme 2). The rate constant for association of the Fl+ form of B-TMF with CB[7] is ca. 4 times higher than that of berberine (Chart 1), a guest with a centrally located positive charge27 similar to Fl+, but one order of magnitude lower than that for the 2-naphthyl-1-ethylammonium cation,26 in which the positive charge is located at an extremity of the molecule. In contrast, the dissociation rate constants for Fl+ and the 2-naphthyl-1-ethylammonium cation26 were the same, while the value for the larger berberine molecule was ca. 60 times lower. The intermediate behavior observed for Fl+ suggests that it fits snuggly into the CB[7] cavity, implying that the larger 1-benzopyrylium moiety is the portion of the molecules that is preferentially included in the 1:1 complex. The inclusion of the remaining portion (the B ring) of the flavylium ion protruding from the 1:1 complex into the cavity of the second CB[7], together with steric and/or dipolar repulsion between the two juxtaposed CB[7] host molecules would nicely rationalize the weaker 1:2 binding.
 | ||
| Scheme 2 Representation of the competitive reactions of B-TMF in the aqueous phase and for incorporation in CB[7]. | ||
 | ||
| Chart 1 Structures of other cationic CB[7] guests and their association and dissociation rate constants with CB[7]. | ||
The selective binding of the Fl+ forms of these two flavylium cations to CB[7] is fully consistent with the known preference of CB[7] for cationic species over neutral ones,17,31,32 such as the hemiacetal B and the chalcones CZ and CE. At the higher CB[7] concentrations, where the Fl+ form is sequestered as the 1:2 complex, essentially complete protection of the Fl+ form of B-TMF against hydration is observed, even at pH values where hydration is predominant in water. At lower CB[7] concentrations, the observation of only partial protection can be ascribed to the competition between the dissociation of Fl+@CB[7] to yield free Fl+, which can undergo hydration, and the association of the Fl+@CB[7] with a second CB[7] forming an unreactive complex (Scheme 2).
Inhibition of the reactivity of CB[n] bound guests has been observed previously,33–38 mainly by protecting the guest from bimolecular reactions with reactants in water. Cucurbituril stabilization of the color of synthetic anthocyanin analogues against reaction with water at pHs of importance in technological applications, such as in the food industry, is a potential strategy with an advantage that cucurbiturils with different cavity sizes could be employed to encapsulate anthocyanidins or minimally glycosylated naturally-occurring anthocyanins of different sizes in the form of 1:2 complexes. The stabilization of the different isomers of anthocyanin derivatives is also of importance in the development of photoprotectors active in different wavelengths regions.
Conclusions
Complete stabilization of the color of a flavylium cation was achieved at moderate pH by complexation with CB[7]. Key to this stabilization is the use of a flavylium cation that does not have the hydroxyl substituent in the 7-position of the flavylium framework and the formation of a 1:2 guest-CB[7] complex. The formation of this higher order complex ensures that the hydration reaction is non-competitive with the formation of guest-host complex.
Acknowledgements
B. H. thanks FAPESP for a doctoral fellowship (2010/00383-9) and F. H. Q., C. P. S. and V. O. S. thank the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) for fellowship support. Funding from the CNPq, INCT-Catalysis and NAP-PhotoTech, the USP Research Consortium for Photochemical Technology is gratefully acknowledged. The authors at UVic thank the Natural Sciences and Engineering Council of Canada (RGPIN-121389-2012) for funding.Notes and references
- R. Brouillard, Anthocyanins as Food Colors, Academic Press, New York, 1982 Search PubMed.
- F. Pina, M. J. Melo, C. A. T. Laia, J. Parola and J. C. Lima, Chemistry and applications of flavylium compounds: a handful of colours, Chem. Soc. Rev., 2012, 41, 869–908 RSC.
- P. F. da Silva, J. C. Lima, A. A. Freitas, K. Shimizu, A. L. Maçanita and F. H. Quina, Charge-Transfer Complexation as a General Phenomenon in the Copigmentation of Anthocyanins, J. Phys. Chem. A, 2005, 109, 7329–7338 CrossRef PubMed.
- P. F. da Silva, L. Paulo, A. Barbafina, F. Elisei, F. H. Quina and A. L. Maçanita, Photoprotection and the Photophysics of Acylated Anthocyanins, Chem. – Eur. J., 2012, 18, 3736–3744 CrossRef PubMed.
- F. H. Quina, P. F. Moreira, C. Vautier-Giogo, D. Rettori, R. F. Rodrigues, A. A. Freitas, P. F. Silva and A. L. Maçanita, Photochemistry of anthocyanins and their biological role in plant tissues, Pure Appl. Chem., 2009, 81, 1687–1694 CrossRef CAS.
- R. Gomes, A. J. Parola, F. Bastkowski, J. Polkowska and F. G. Klärner, Host–Guest Interactions between Molecular Clips and Multistate Systems Based on Flavylium Salts, J. Am. Chem. Soc., 2009, 131, 8922–8938 CrossRef CAS PubMed.
- N. Basilio and F. Pina, Flavylium Network of Chemical Reactions in Confined Media: Modulation of 3′,4′,7-Trihydroxyflavilium Reactions by Host–Guest Interactions with Cucurbit[7]uril, ChemPhysChem, 2014, 15, 2295–2302 CrossRef CAS PubMed.
- N. Basílio, L. Cabrita and F. Pina, Mimicking Positive and Negative Copigmentation Effects in Anthocyanin Analogues by Host–Guest Interaction with Cucurbit[7]uril and β–Cyclodextrins, J. Agric. Food Chem., 2015, 63, 7624–7629 CrossRef PubMed.
- N. Basílio, C. A. T. Laia and F. Pina, Excited-State Proton Transfer in Confined Medium. 4–Methyl-7-hydroxyflavylium and β–Naphthol Incorporated in Cucurbit[7]uril, J. Phys. Chem. B, 2015, 119, 2749–2757 CrossRef PubMed.
- J. Lagona, P. Mukhopadhyay, S. Chakrabarti and L. Isaacs, The cucurbit[n]uril family, Angew. Chem., Int. Ed., 2005, 44, 4844–4870 CrossRef CAS PubMed.
- M. V. Rekharsky, T. Mori, C. Yang, Y. H. Ko, N. Selvapalam, H. Kim, D. Sobransingh, A. E. Kaifer, S. Liu, L. Isaacs, W. Chen, S. Moghaddam, M. K. Gilson, K. Kim and Y. Inoue, A synthetic host–guest system achieves avidin-biotin affinity by overcoming enthalpy–entropy compensation, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 20737–20742 CrossRef CAS PubMed.
- L. Cao, M. Sekutor, P. Y. Zavalij, K. Mlinaric-Majerski, R. Glaser and L. Isaacs, Cucurbit[7]uril·Guest Pair with an Attomolar Dissociation Constant, Angew. Chem., Int. Ed., 2014, 53, 988–993 CrossRef CAS PubMed.
- R. N. Dsouza, U. Pischel and W. M. Nau, Fluorescent Dyes and Their Supramolecular Host/Guest Complexes with Macrocycles in Aqueous Solution, Chem. Rev., 2011, 111, 7941–7980 CrossRef CAS PubMed.
- L. Isaacs, Stimuli Responsive Systems Constructed Using Cucurbit[n]uril-Type Molecular Containers, Acc. Chem. Res., 2014, 47, 2052–2062 CrossRef CAS PubMed.
- N. Vallavoju and J. Sivaguru, Supramolecular photocatalysis: combining confinement and non-covalent interactions to control light initiated reactions, Chem. Soc. Rev., 2014, 43, 4084–4101 RSC.
- G. Parvari, O. Reany and E. Keinan, Applicable Properties of Curcurbiturils, Isr. J. Chem., 2011, 51, 646–663 CrossRef CAS.
- E. Masson, X. Ling, R. Joseph, L. Kyeremeh-Mensah and X. Lu, Cucurbituril chemistry: a tale of supramolecular success, RSC Adv., 2012, 2, 1213–1247 RSC.
- J. C. Lima, I. Abreu, R. Brouillard and A. L. Maçanita, Kinetics of ultra-fast excited state proton transfer from 7-hydroxy-4-methylflavylium chloride to water, Chem. Phys. Lett., 1998, 298, 189–195 CrossRef CAS.
- A. Robertson and R. Robinson, CCXXV. Experiments on the Synthesis of Anthocyanins, J. Chem. Soc., 1926, 1713–1720 RSC.
- D. D. Pratt and R. Robinson, CLXXXVIII. A Synthesis of Pyrylium Salts of Anthocyanidin Type, J. Chem. Soc. Trans., 1922, 1577–1585 RSC.
- J. Kim, I.-S. Jung, S.-Y. Kim, E. Lee, J.-K. Kang, S. Sakamoto, K. Yamaguchi and K. Kim, New Cucurbituril Homologues: Syntheses, Isolation, Characterization, and X-ray Crystal Structures of Cucurbit[n]uril (n = 5, 7, and 8), J. Am. Chem. Soc., 2000, 122, 540–541 CrossRef CAS.
- A. Day, A. P. Arnold, R. J. Blanch and B. Snushall, Controlling Factors in the Synthesis of Cucurbituril and Its Homologues, J. Org. Chem., 2001, 66, 8094–8100 CrossRef CAS PubMed.
- C. Marquez, F. Huang and W. M. Nau, Cucurbiturils: Molecular Nanocapsules for Time-Resolved Fluorescence-Based Assays, IEEE Trans. Nanobiosci., 2004, 3, 39–45 CrossRef PubMed.
- S. Yi and A. E. Kaifer, Determination of the Purity of Cucurbit[n]uril (n = 7, 8) Host Samples, J. Org. Chem., 2011, 76, 10275–10278 CrossRef CAS PubMed.
- A. A. Freitas, L. G. Dias, A. L. Maçanita and F. H. Quina, Substituent effects on the pH-dependent multiequilibria of flavylium salt analogs of anthocyanins, J. Phys. Org. Chem., 2010, 24, 1201–1208 CrossRef.
- H. Tang, D. Fuentealba, Y. H. Ko, N. Selvapalam, K. Kim and C. Bohne, Guest Binding Dynamics with Cucurbit[7]uril in the Presence of Cations, J. Am. Chem. Soc., 2011, 133, 20623–20633 CrossRef CAS PubMed.
- Z. Miskolczy and L. Biczók, Kinetics and Thermodynamics of Berberine Inclusion in Cucurbit[7]uril, J. Phys. Chem. B, 2014, 118, 2499–2505 CrossRef CAS PubMed.
- S. S. Thomas and C. Bohne, Determination of the kinetics underlying the pKa shift for the 2-aminoanthracenium cation binding with cucurbit[7]uril, Faraday Discuss., 2015, 185, 381–398 RSC.
- N. Basilio, V. Petrov and F. Pina, Host–Guest Complexes of Flavylium Cations and Cucurbit[7]uril: The Influence of Flavylium Substituents on the Structure and Stability of the Complex, ChemPlusChem, 2015, 80, 1779–1785 CrossRef CAS.
- B. C. MacGillivray, PhD thesis, Host–Guest Chemistry between Cucurbit[7]uril and Cationic and Neutral Guests, Department of Chemistry, University of Queens, Kingston, Ontario, Canada, 2012 Search PubMed.
- W. L. Mock and N. Y. Shih, Structure and selectivity in host–guest complexes of cucurbituril, J. Org. Chem., 1986, 51, 4440–4446 CrossRef CAS.
- M. Shaikh, J. Mohanty, P. K. Singh, W. M. Nau and H. Pal, Complexation of acridine orange by cucurbit[7]uril and beta-cyclodextrin: photophysical effects and pKa shifts, Photochem. Photobiol. Sci., 2008, 7, 408–414 CAS.
- R. Wang, L. Yuan and D. H. Macartney, Inhibition of C(2)-H/D exchange of a bis(imidazolium) dication upon complexation with cucurbit[7]uril, Chem. Commun., 2006, 2908–2910 RSC.
- N. Basilio, L. Garcia-Rio, J. A. Moreira and M. Pessego, Supramolecular Catalysis by Cucurbit[7]uril and Cyclodextrins: Similarity and Differences, J. Org. Chem., 2010, 75, 848–855 CrossRef CAS PubMed.
- L. S. Berbeci, W. Wang and A. E. Kaifer, Drastically Decreased Reactivity of Thiols and Disulfides Complexed by Cucurbit[6]uril, Org. Lett., 2008, 10, 3721–3724 CrossRef PubMed.
- Z. Miskolczy, M. Megyesi, G. Tárkányi, E. Mizsei and L. Biczók, Inclusion complex formation of sanguinarine alkaloid with cucurbit[7]uril: inhibition of nucleophilic attack and photooxidation, Org. Biomol. Chem., 2011, 9, 1061–1070 CAS.
- Z. Miskolczy and L. Biczók, Photochromism in Cucurbit[8]uril Cavity: Inhibition of Hydrolysis and Modification of the Rate of Merocyanine Spiropyran Transformations, J. Phys. Chem. B, 2011, 115, 12577–12583 CrossRef CAS PubMed.
- H. Ren, Z. Huang, H. Yang, H. Xu and X. Zhang, Controlling the Reactivity of the Se-Se Bond by the Supramolecular Chemistry of Cucurbituril, ChemPhysChem, 2015, 16, 523–527 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthesis of B-TMF and CB[7], fitting methods, determination of the apparent hydration constant, stabilization of flavylium cation and binding isotherms. See DOI: 10.1039/c6pp00060f |
| ‡ These two authors contributed equally. |
| § Present address: Department of Chemistry & CAS, Panjab University, Chandigarh-160014, India. |
| This journal is © The Royal Society of Chemistry and Owner Societies 2016 |