
Application of spectroscopic and theoretical methods in the studies of photoisomerization and photophysical properties of the push–pull styryl-benzimidazole dyes†
B.
Jędrzejewska
*a,
B.
Ośmiałowski
a and
R.
Zaleśny
bc
aFaculty of Chemical Technology and Engineering, UTP University of Science and Technology, Seminaryjna 3, 85-326 Bydgoszcz, Poland. E-mail: beata@utp.edu.pl
bDepartment of Chemistry, Faculty of Natural Sciences, Matej Bel University, Tajovského 40, 974 01 Banská Bystrica, Slovak Republic
cFaculty of Chemistry, Wrocław University of Technology, Wyb. Wyspiańskiego 27, PL-50370 Wrocław, Poland
First published on 9th December 2015
Abstract
The synthesis and spectroscopic properties of a series of substituted 1,3-dimethyl-2-aminostyrylbenzimidazolium iodides are described and discussed. The products were identified by NMR, IR and UV-Vis spectroscopy and elemental analysis. Their electronic absorption and fluorescence band positions are affected by the character of the substituent and by the solvent polarity. The fluorescence decay of the dyes shows two lifetimes interpreted in terms of emission from two forms of the dye in the excited state. Moreover, the photochemical trans → cis isomerization is reported for these compounds. It occurs from the first excited singlet state of the trans isomer to the cis isomer following a trans-S0 → S1 excitation. The electron-donating character of the substituent in a styrene moiety is one of the crucial factors influencing the photoisomerization process. The structure of the cis isomer was established by 1H and 15N NMR. Experimental studies are supported by the results of quantum chemical calculations.
Introduction
It has become clear that the development of molecular and supramolecular electronics, light harvesting and photocatalysis would greatly benefit from the design and synthesis of organic, electroactive and photoactive materials.1 Organic molecules are already utilized in optoelectronic devices based on electroluminescence, in photoconductors, light emitting devices (LED), solid-state lasers,2,3 biochemical fluorescent technology and non-linear optics.4–6 Among organic functional materials, compounds possessing electron-donating (D) and electron-accepting (A) groups on opposite sides of a π-conjugated linker are very important.Generally, there are two kinds of D–A systems: molecular complexes with intermolecular charge transfer, which are usually encountered in bulk heterojunction solar cells for instance, and D–A systems joined by covalent bonds exhibiting the intramolecular charge transfer (ICT) within molecules.7 The photochemical and photophysical properties of the intramolecular D–A systems depend on the excited-state processes that occur after absorption of a photon (interaction with solvent molecules, rotation about single bonds, photoisomerisation etc.). After excitation, the generated excitons are stabilized and further separated because of the extended conjugation (π-conjugated spacer). In such molecules rapid electron transfer from D to A takes place.7 Thus, the properties of molecules of D–π-A type can be readily changed by modifying the donors or/and acceptors.1 Regarding the acceptor side, several heterocyclic moieties were described, i.e.: quinoline, dimethylindoline, benzoxazole, benzothiazole or benzimidazole.8,9 Among these the benzimidazole moiety is very useful in the synthesis of such molecules. This heterocycle plays an important role in, for instance, the medical field. The pharmacological activities of benzimidazole such as antiviral, antiulcer, antihypertension, and anticancer properties are well documented.7 Benzimidazole derivatives and their metal complexes10,11 have been extensively investigated e.g. due to their prominence in studies on the interaction of a transition metal complex with nucleic acids.12 They were also used in determining the mechanism of metal ion toxicity.13 Furthermore, the benzimidazole derivatives were applied as chromophores with high extinction coefficient, readily tunable absorption wavelength, and fluorophoric properties and they are also desirable as large planar synthetic building blocks in supramolecular chemistry.9
The important role of benzimidazoles in biomedical research, in organocatalysis, organometallic, and materials chemistry stems from two reasons related to their molecular structure. Firstly, imidazole is a precursor to N-heterocyclic carbenes.14 Secondly, the benzene ring provides a convenient scaffold for easy functionalization modifying the spatial and electronic characteristics of this moiety. This combination of a reactive carbene center with a modifiable backbone is responsible for increasing attention and the use of benzimidazoles and their N-heterocyclic derivatives.7,9–13,15
Essentially, styrylbenzimidazole dyes represent stilbene-like structures, with two aromatic moieties and a vinylic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond, offering the possibility of trans → cis isomerism. Thus, one should expect styrylbenzimidazole dyes to exist in trans and/or cis configurations as well as in two resonance structures16 that should be taken into account when analyzing their properties. It should be emphasized that a quinoid structure might also be considered as an ICT state. In principle the trans and quinoid structures can be treated as mesomeric resonance structures, with the positive charge localized on different nitrogen atoms, i.e. on the benzimidazole nitrogen in the case of the trans isomer and on the substituent's nitrogen atom (vide infra) in the quinoid form.17,18 Therefore, to gain an insight into the electronic properties of the ground and excited states of these dyes it is necessary to collect experimental spectroscopic data.
C double bond, offering the possibility of trans → cis isomerism. Thus, one should expect styrylbenzimidazole dyes to exist in trans and/or cis configurations as well as in two resonance structures16 that should be taken into account when analyzing their properties. It should be emphasized that a quinoid structure might also be considered as an ICT state. In principle the trans and quinoid structures can be treated as mesomeric resonance structures, with the positive charge localized on different nitrogen atoms, i.e. on the benzimidazole nitrogen in the case of the trans isomer and on the substituent's nitrogen atom (vide infra) in the quinoid form.17,18 Therefore, to gain an insight into the electronic properties of the ground and excited states of these dyes it is necessary to collect experimental spectroscopic data.
In this study, we describe the synthesis and perform an analysis of the structure–property relationship for hemicyanine dyes based on a benzimidazole skeleton. We also explore the essential aspects of the trans → cis photoisomerization for these dyes. It is worth mentioning that a similar photoisomerization was recently described19–21 also in combination with the supramolecular chemistry approach.
Experimental section
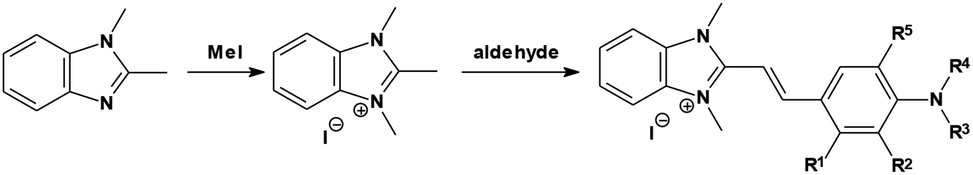
Synthetic procedure
In the present study we have followed the synthetic method (Scheme 1) described earlier.22–24 2-[4-(N,N-dialkylamino)-styryl]-1,3-dimethyl-benzimidazolium iodides were obtained by refluxing (4–6 hours) an appropriate amount of p-aminobenzaldehyde (0.01 mol) with 1,2,3-trimethylbenzimidazolim iodide (0.01 mol) in methanol (20 mL) in the presence of piperidine (a few drops). The precipitate formed after cooling down the reaction mixture was filtered out and recrystallized from ethanol or methanol. The purity of the dyes under study was checked by thin layer chromatography on aluminum oxide IB-F gel, eluting with a mixture of chloroform and ethanol (5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 v/v).
:2 v/v).
 | ||
| Scheme 1 General route for the styrylbenzimidazolium dye synthesis. | ||
Materials and methods
The following solvents, purchased from either Aldrich or Lancaster Chemical Co., were used for spectroscopic measurements: acetonitrile (MeCN), N,N-dimethylformamide (DMF), acetone (AcMe), 1-methyl-2-pyrrolidone (MP), tetrahydrofuran (THF), ethylene glycol dimethyl ether (EGDME), propionitrile (EtCN), toluene and benzene. Absorption and emission spectra were recorded at room temperature using a Shimadzu UV-Vis Multispec-1501 spectrophotometer and a Hitachi F-4500 spectrofluorometer, respectively. The concentration of the dye in solvents of different polarities was 1.0 × 10−5 M and 1.0 × 10−6 M for absorption and fluorescence measurements, respectively. The fluorescence quantum yields of the dyes were calculated using eqn (1). | (1) |
The fluorescence lifetimes were measured using an Edinburgh Instruments single-photon counting system (FLS920P Spectrometers). The apparatus utilizes a picosecond diode laser for the excitation generating pulses of about 55 ps at 375 nm. The dyes were studied at concentrations at which they exhibit similar absorbance at 375 nm (0.2–0.3 in the 10 mm cell). The fluorescence decays were fitted to two-exponential functions. The average lifetime, τav is calculated as
 | (2) |
The photochemical bleaching process was studied in a quartz cuvette with dimensions 4 × 1 × 1 cm. In order to ensure complete absorption of light, the cuvette was placed in a horizontal position and irradiated with diode-pumped solid state (DPSS) laser light (λEM = 408 nm; intensity 20 mW) (or xenon lamp equipped with monochromator, λEM = 280 nm) through the bottom wall (the optical path length = 4 cm). The solution was stirred during irradiation. The photobleaching quantum yield was determined using the following formula:
 | (3) |
The electronic structure calculations were performed using the Kohn–Sham formulation of the density functional theory using the linear response polarizable continuum model (LR-PCM26). For a recent comparative study of continuum solvation models we refer to the work of Chibani et al.27 Optimization of the ground state geometry was carried out using two exchange–correlation functionals: B3LYP and CAMB3LYP. Vertical excitation energies were computed employing the time dependent density functional theory. For all quantum-chemical calculations the 6-311++G(d,p) basis set was used. All electronic structure calculations were performed using the Gaussian 2009 (revision D01) program.28
Results and discussion
Molecular design and synthetic procedures
Nine hemicyanine dyes (Fig. 1) have been synthesized by the reaction (Knoevenagel condensation) of 1,2,3-trimethyl-benzimidazolium iodide with an appropriate amount of p-aminobenzaldehyde.22–24 It is fair to mention that an alternative method (solvent-free microwave irradiation) of the hemicyanine synthesis was described by Lan-Ying Wang et al.29,30 The 1H and 13C NMR chemical shift data are in agreement with the dyes structures. The two characteristic doublets related to vinyl protons in a trans conformation (3JH,H = 15–16 Hz) are localized roughly at 7 and 8 ppm. Detailed analytical and spectroscopic data for the novel styrylbenzimidazolium dyes are available in the ESI.†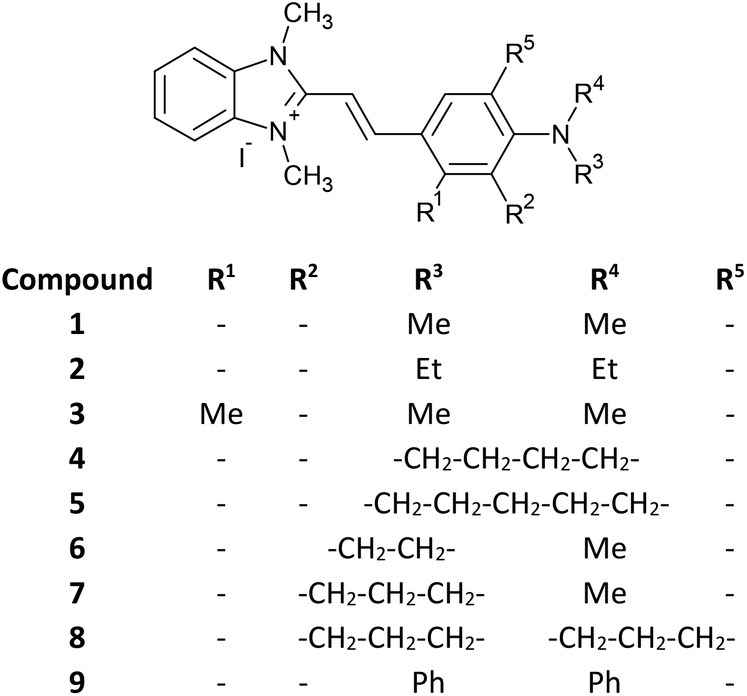 | ||
| Fig. 1 Dyes studied. | ||
UV-Vis spectroscopic properties
The spectroscopic data of dyes 1–9 in nine solvents of different polarities are collected in Table S1.† All hemicyanine dyes studied in this work show a characteristic absorption band with the maximum at the range 418–475 nm and a corresponding, clearly Stokes-shifted, emission band (λEX = 404 nm). The broad absorption bands correspond to the π → π* ICT transition involving the amino nitrogen and the cationic benzimidazolium moiety.18,22 Indeed, the results of time-dependent density functional calculations for compound 1 (cf. Table S2 in ESI†) reveal that the π → π* excitation to the lowest-energy state is dominated by the HOMO → LUMO transition. The shape of the two frontier orbitals for trans-1 and cis-1, shown in Fig. 2, confirms that the density reorganization takes place from dimethylamino to the benzimidazolium moiety. The short-wavelength bands are attributed to the π → π* transitions. As an example, the spectra of the dye are shown in Fig. 3. The molar absorption coefficients of the CT bands are in the range 31200–45600 M−1 cm−1. The high value of the molar absorption coefficient indicates an extensive conjugation of π-electrons, thus suggesting a planar structure of the dye molecule in its ground state. In fact this is confirmed by the results of calculations.
 | ||
| Fig. 2 Frontier orbitals involved in the lowest-energy excitation for trans-1 (top) and cis-1 (bottom) in DMF solvent. | ||
 | ||
| Fig. 3 Normalized electronic absorption spectra of selected hemicyanine dyes (2, 5, 7 and 8 in MeCN at 293 K). | ||
The selected bond lengths, as predicted by quantum chemistry calculations, for trans and cis forms are summarized in Table 1. The theoretical calculations show that the bond lengths of the π-spacer show a single–double–single bond character confirming the ethenic linkage character in the ground state. The values of bond angles between imidazole carbon and the central double bond (and between the central double bond and benzene carbon) are equal to 127.64° (125.65°) for trans and 132.66° (129.3°) for cis forms, respectively. The torsional angle of C–CHCH–C equals 179.57° for the trans form, thus confirming its planarity in the ground state. The corresponding angle for the cis isomer is −11.33°.
The absorption spectra of the studied compounds are affected by the type of dialkyl(aryl)amino group in the dye molecule. In all the solvents the maximum absorption positions (Table S1†) differ in some cases up to 40 nm and this indicates that the energy of electronic transition substantially depends on the structure of the electron-donating moiety. It can be seen that the replacement of the NMe2 group (dye 1) by the NEt2 one (dye 2) shifts the absorption band toward a longer wavelength by about 10 nm. Additionally, a bathochromic effect is observed when the methyl group is introduced in the ortho position to the central double bond (compound 3) or upon elimination of the free rotation of the dialkylamino group by the bridging (compounds 6, 7 and 8). The absorption spectra of the largely bridged compound (8) are the most shifted to lower energies. This is probably due to reduction of conformational flexibility in the ground state and it is connected with an increasingly rigidized molecular skeleton.8,17,34 On the other hand, the blue shift of the CT absorption band appears when the alkylamino group is twisted because the coplanar conformation decreases the probability of radiative transitions.
The UV-Vis absorption spectra recorded in solvents of different polarities show weak solvatochromism. Upon increasing the solvent polarity, the CT absorption band is slightly blue-shifted as for instance the change of the solvent from benzene to DMF in the case of 1 changes the maximum absorption wavelength from 438 nm to 424 nm. The results indicate that these molecules might possess fairly similar polarities between the excited state and the ground state and thus the energy gap is barely influenced by the increasing polarity of the solvent.35 In order to gain a deeper insight into the polarity changes upon photoexcitation to the lowest-energy π → π* excited state, we performed quantum chemistry calculations for the trans isomer of compound 1. Note that unlike dipole moment components in the ground and excited states, only their differences are translationally invariant for a charged system. The orientation of trans-1 for property calculations is shown in Fig. 4. The dipole moment change upon photoexcitation along the conjugation pathway, i.e. along the x direction, is an order of magnitude larger than that for the other directions. The value of Δμx for trans-1 is 8.6 D and 10.1 D in benzene and DMF, respectively. Thus, as already mentioned, one observes an increase of Δμ with increasing solvent polarity.
 | ||
| Fig. 4 Orientation of trans-1 in Cartesian coordinate system for the ground- and excited-state dipole moment calculations. | ||
As in the case of electronic absorption spectra, the structure of the electron donor group and solvent polarity also affect the position of the emission band. The rigidity and planarization of the amino group lead to the red shift in the fluorescence (see Table S1†). A solvent change from benzene to acetonitrile causes a bathochromic shift roughly by few nanometers for its fluorescence band, e.g. the maximum emission peak for 1 is at 541 nm in benzene and at 546 nm in MeCN. Only in the case of dye 9 a more pronounced effect is observed. Solvent change from benzene to acetonitrile causes a bathochromic shift in fluorescence maximum equal to 120 nm. In the same compound the absorption peak position shifts to the blue only by 10 nm. For N,N-diphenylamine-substituted dye (9) a clear red-shift of the fluorescence spectra with increasing polarity of the solvents indicates the occurrence of an intramolecular charge transfer process from the electron-donating triphenylamine moiety to the electron-accepting benzimidazole moiety. As a result the excited state energy decreased with increasing polarity of the solvents due to enhanced solvent-solute dipole–dipole interactions, and the energy gap between the ground state and excited state declined, which explains the sensitivity of the emission spectra of the compound toward solvent polarity.34,35
Generally, data collected in Table S1† reveal that the long wavelength absorption band undergoes a hypsochromic shift as the solvent polarity increases whereas there is a bathochromic shift of the fluorescence band, with an increase of the Stokes shift as the solvent polarity increases (Fig. S1†). On changing the solvent from nonpolar to polar, the change in the Stokes shift equals ca. 4400 cm−1 for 9 and ca. 1000 cm−1 for other dyes, respectively. The large value of the Stokes shift indicates that the excited-state geometry differs significantly from that of the ground state.36,37
Table S1† shows that the Stokes shifts of the studied dyes are large (>3000 cm−1). Presumably, this can be linked to the extensive ICT across the molecule, which is facilitated by the presence of a typical D–π-A structure. An increase in the Stokes shift with increasing solvent polarity indicates an increase in the dipole moment upon excitation.36,37
To gain a further insight into the emission process the fluorescence decay profiles were recorded by using the time correlated single-photon counting system. The samples were excited at 375 nm and the emissions were followed at 550 nm. The decay curves were fitted to two-exponential decay and the lifetimes were extracted from the measured decay curves by deconvolution of the instrument response function. The fluorescence decay profile of selected dyes is shown in Fig. 5. The results of lifetime measurements are summarized in Table 2.
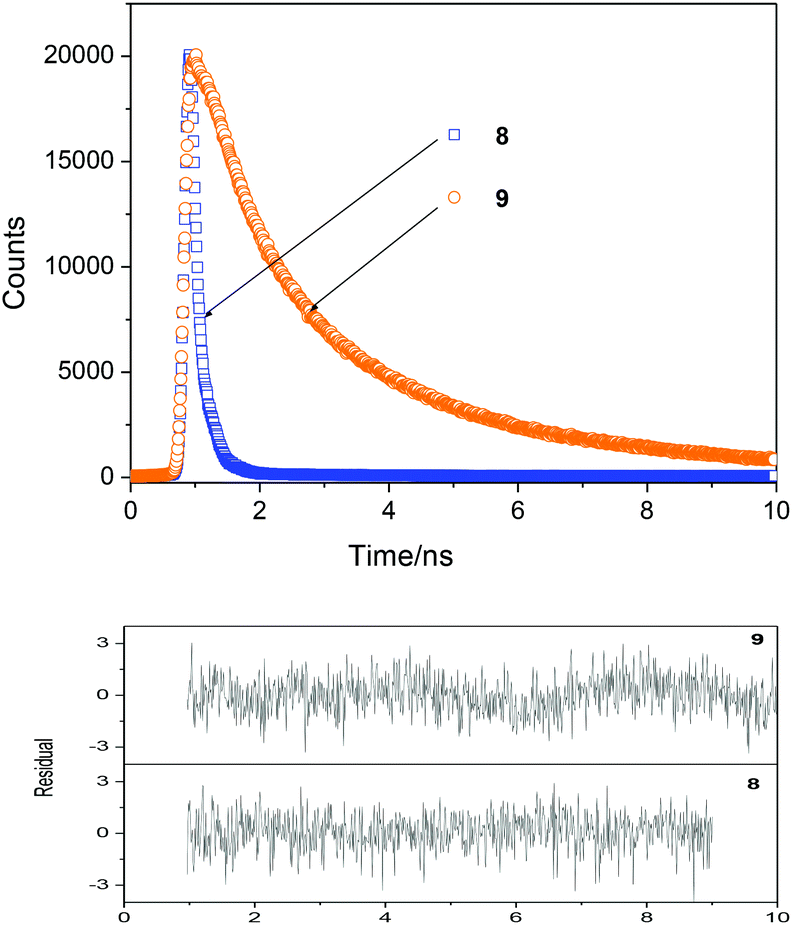 | ||
| Fig. 5 Fluorescence decays of 8 and 9 dyes (weighted residuals are shown in the bottom panels) in AcOEt–NMP mixture (10:1 v/v) (A ≈ 0.2–0.3 in the 10 mm cell, λEX = 375 nm, λEM = 550 nm, at r.t.). | ||
| Dye | Φ fl (%) | τ 1 (ps) | τ 2 (ps) | α 1 | α 2 | τ av (ps) | χ 2 |
|---|---|---|---|---|---|---|---|
| AcOEt – MP mixture (10:1 v/v) |
|||||||
| 1 | 0.81 | 65 | 1719 | 96.7 | 3.3 | 120 | 1.28 |
| 2 | 0.95 | 67 | 2694 | 81.3 | 18.7 | 558 | 1.06 |
| 3 | 0.38 | 53 | 2436 | 90.5 | 9.5 | 279 | 1.25 |
| 4 | 1.13 | 71 | 1411 | 92.2 | 7.8 | 176 | 1.22 |
| 5 | 1.32 | 79 | 1887 | 94.5 | 5.5 | 178 | 1.06 |
| 6 | 0.70 | 69 | 960 | 96.8 | 3.2 | 98 | 1.29 |
| 7 | 0.75 | 66 | 1194 | 98.8 | 1.2 | 79 | 1.35 |
| 8 | 1.22 | 80 | 1046 | 98.4 | 1.6 | 96 | 1.08 |
| 9 | 14.73 | 107 | 2372 | 69.5 | 30.5 | 798 | 1.10 |
| MeCN | |||||||
| 1 | 0.73 | 36 | 1040 | 99.8 | 0.2 | 38 | 1.81 |
| 2 | 0.95 | 38 | 1249 | 99.3 | 0.7 | 46 | 1.77 |
| 3 | 0.42 | 31 | 729 | 98.5 | 1.5 | 41 | 1.08 |
| 4 | 1.02 | 38 | 1421 | 99.2 | 0.8 | 49 | 1.83 |
| 5 | 1.14 | 41 | 872 | 99.9 | 0.1 | 41 | 1.02 |
| 6 | 0.66 | 35 | 1213 | 99.4 | 0.6 | 40 | 1.86 |
| 7 | 0.98 | 39 | 1105 | 99.6 | 0.4 | 43 | 1.52 |
| 8 | 0.80 | 37 | 1326 | 98.9 | 1.1 | 51 | 1.42 |
| 9 | 2.71 | 64 | 1131 | 93.7 | 6.3 | 131 | 1.94 |
The two-exponential character of the decays is consistent with the observations of Röcker et al.38 and Rettig et al.39 for similar compounds. From their work it is known that hemicyanine dyes exist as a mixture of rotamers. Rettig et al. investigation39 shows that single bonds adjacent to the ethylenic group lead to low lying twisted intramolecular charge transfer states with possibly emissive properties while the twist of the dimethylamino group leads to an energetically higher lying twisted intramolecular charge transfer state, unlikely to be populated thermally.
On the other hand, twisting around the central bond leads to an excited state considerably lower in energy than the planar one. This state possesses a nonemissive character because of the small energy gap between the ground and excited states.39 So, the emitting structures at room temperature are linked to conformations with close to planar geometries. In this approach, the dominant component in fluorescence decay of the studied dyes can be attributed to the emission from the trans isomer.18 The short lifetime (τ1) in this case reflects the fast reaction from the planar Franck–Condon state to a non-emissive partially twisted state (the so-called “phantom” state) which quickly leads to internal conversion to S0 as a mixture of trans + cis isomers. The long lifetime component may instead be assigned to the perpendicular quinoid (Q⊥) or ICT form, respectively.17,18,40–44 However, because of the small percentage of long-lived components only the main component of the fluorescence decay curve (above 90%) was selected for further analysis.
The radiative rate constants kr and the total nonradiative rate constants knr from the singlet-excited state of tested dyes were estimated from the fluorescence quantum yield (Φfl) and the main lifetime (τ1) values using well-known equations.36 Thus, the calculated rate constants are kr = (0.71–13.76) × 108 s−1 and knr = (0.8–3.21) × 1010 s−1 (Table 3). This estimation indicates that for the tested hemicyanines the non-radiative transition rate is at least two orders of magnitude faster than the radiative rate.
| Dye | k r (108 s−1) | k nr (1010 s−1) | k nr/kr | Φ bl | k (10−4 s−1) | |||
|---|---|---|---|---|---|---|---|---|
| A | B | A | B | A | B | |||
| 1 | 1.24 | 2.02 | 1.53 | 2.76 | 122.8 | 136.4 | 0.144 | 4.75 |
| 2 | 1.42 | 2.50 | 1.48 | 2.61 | 104.1 | 104.3 | 0.148 | 4.93 |
| 3 | 0.71 | 1.35 | 1.88 | 3.21 | 263.0 | 237.3 | 0.155 | 5.40 |
| 4 | 1.59 | 2.69 | 1.39 | 2.60 | 87.5 | 96.9 | 0.147 | 5.34 |
| 5 | 1.67 | 2.79 | 1.25 | 2.41 | 74.6 | 86.5 | 0.137 | 4.53 |
| 6 | 1.01 | 1.89 | 1.44 | 2.84 | 142.3 | 150.0 | 0.091 | 4.19 |
| 7 | 1.13 | 2.52 | 1.50 | 2.54 | 132.6 | 100.8 | 0.133 | 4.45 |
| 8 | 1.51 | 2.17 | 1.23 | 2.68 | 81.1 | 123.8 | 0.119 | 4.02 |
| 9 | 13.76 | 4.24 | 0.80 | 1.52 | 5.8 | 35.9 | 0.002 | 0.10 |
The results of spectroscopic and lifetime measurements indicate the existence of multiple species of the styrylbenzimidazolium dye in organic solvents. In the case of hemicyanine dyes, the presence of a positive charge at two different types of nitrogens in the molecule (two resonant forms) may cause that some specific solvents can stabilize these two structures to different extents. The degree of the charge transfer can influence the properties of dyes including the bond order and thus the bond rotational barrier.16,17,30,34–37,40,41
Trans → cis photoisomerization
Due to the presence of the carbon–carbon double bond, the hemicyanine dyes may undergo photoreactions typical of alkenes. The most characteristic reaction is the trans–cis isomerization. It is obvious that the photophysical properties of such molecules may be dependent on the trans–cis isomerism. Thus, it is crucial to gain an insight into the photophysics of these dyes.Irradiation with a laser beam (λ = 408 nm, 20 mW) of a solution of 1 prepared in the dark causes a progressive decrease in the absorbance of the long-wavelength band (λmax initially at 425 nm) with a concomitant increase in the absorbance intensity at ca. 280 nm until a stationary state is reached (Fig. 6 top). The sharp isosbestic point at ca. 370 nm is evidence for a transformation between two species, but not yet proof for a photoisomerization (any photodegradation could lead to a similar behavior).16 Once a photostationary state had been reached, we have changed the excitation wavelength to a region where an increase in absorption was observed and irradiated the sample at 280 nm (xenon lamp equipped with a monochromator). The results (Fig. 6 bottom) show a reversal trend: an increase of the main band (around 425 nm) and a decrease of absorption at ca. 280 nm.
 | ||
| Fig. 6 The dependence of UV-Vis absorption spectra of 1 in MeCN upon irradiation with DPSS laser beam (λ = 408 nm, 20 mW, top) and xenon lamp (λ = 280 nm, bottom). Arrows indicate the direction of changes with time of irradiation. | ||
In order to distinguish the substrate from the product formed after irradiation of the solution NMR analysis was performed. The spectra were recorded for (a) the initial solution prepared in the dark, (b) after heating in the dark and (c) after irradiation (Fig. S2†).
The analysis of 1H NMR spectra of 1 in DMSO-d6 indicates that the heating of solution (b) did not cause any changes in the signal position. There are well-separated pairs of doublets for two olefinic protons. The coupling constant 3JH,H = 16.4 Hz is characteristic for the trans configuration. After irradiation to a photostationary state and transition, the proton spectrum of the same solution shows considerable changes in chemical shifts. It becomes evident that for the new pair of doublets the coupling constant is equal to 12.0 Hz, which is in perfect agreement with the cis arrangement (the similar is observed in acetonitrile).45 It follows from the studies of Steiner et al.16 that in the cis configuration, as in cis-stilbene, two arene rings must be twisted from the coplanar orientation relative to the central double bond. Upon rotation of the aromatic parts of the molecule around the single bonds adjacent to the central double bond, Hd and He (Fig. 7) protons may come into the screening cone of the anisotropic ring current. This would result in different shielding in cis and trans isomers. Simultaneously, the aryl protons in the ortho position to the olefinic spacer (Hf) are located well within the shielding cone of the benzimidazole ring in the cis conformation, and thus experience a significant high-field shift. Such an effect is in fact observed.
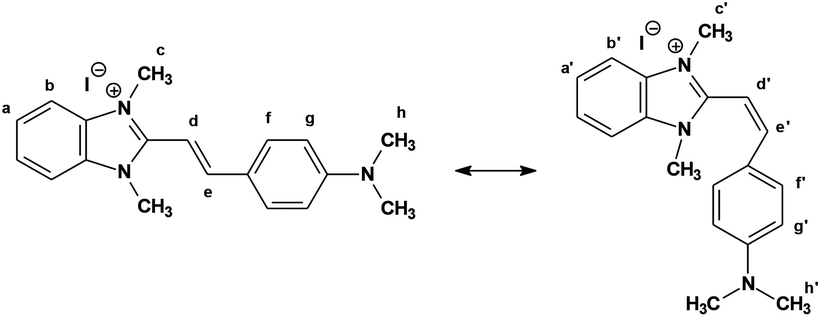 | ||
| Fig. 7 The trans and cis forms of the dye 1. | ||
The additional experiments were focused on the possibility of thermal isomerization of 1. The following steps were recorded (1H and 15N spectra) for (1) the parent sample (trans form), (2) the sample heated at 80 °C for at least 4 h, (3) the irradiated sample (visible light, r.t., time = 4 h), (4) the re-heated sample that was irradiated before (from step 3, in order to check if the cis-to-trans thermal isomerization takes place, 80 °C, 3 h). The results are summarized in Table 3, while the stacked spectra are collected in Fig. S2.†
Analysis of the NMR and electronic absorption spectra recorded under various conditions (data presented in the ESI†) indicates that the trans isomer is thermally stable but is partially converted to the cis-isomer under visible light irradiation. Based on the NMR spectra of 1 recorded for the photostationary state the trans/cis photoisomer ratio was calculated by integrating respective peaks corresponding to the vinylic doublets and/or the methyl groups in cis and trans forms. The re-heated sample (step 4, 3 h at 80 °C) did not exhibit any significant changes in the absorption and NMR spectra. However, the abundance of the trans form increased. Thus, a more precise study of the thermal back-reaction was performed. The 1H NMR spectra of a previously irradiated sample of 1 at r.t., recorded 16 h, 36 h, 39 h and 74 h after heating (see Fig. S3†) show that the peaks attributable to the cis isomer decrease in intensity. Signal integration suggests that almost all of the cis isomer reverts thermally to the trans isomer after 74 h at 373 K (a percent of the remainder cis form equals ca. 2.8%). This indicates that the cis-to-trans thermal interconversion is slow and inefficient and this is, most probably, due to the high value of cis → trans activation energy. Similarly to Mishra et al. studies,46 the photoisomerization of styrylbenzimidazolium iodides follow the non-adiabatic path and occur via the perpendicular state. However, in contrast to the neutral molecule, the trans and cis isomers of the tested dyes exist only in one confomeric form.
Further, calculations were based on the geometry of trans and cis forms. The shieldings of the nuclei in 1 were used to calculate the chemical shifts. It is known that the nitrogen atom is sensitive to the environment47 thus its chemical shift may be a method of choice for distinguishing between two isomeric forms. This is especially true for compounds, which exhibit intramolecular charge transfer. Such charge transfer is dependent on the geometry of the molecule and thus it can be directly linked to the cis–trans isomerism. The GIAO-based48,49 calculations yield various chemical shifts for imidazole and NMe2 group nitrogen atoms in trans and cis forms (see row c in Table 4). Table 5 collects the experimental and calculated 15N NMR data (with respect to MeNO2). The same table contains (in italic) the values of the 1H chemical shifts of methyl groups that gave a cross-peak in 1H,15N HMBC spectra. It is clearly seen that methyl protons are also sensitive to isomerism of 1.
| Step (see text) | 1H (trans)a | 15N (trans) | 1H (cis)b | 15N (cis) | % of trans formc |
|---|---|---|---|---|---|
| a Chemical shift of the vinylic protons (trans doublet with 3JH,H = 16.3 Hz). b Chemical shift of the vinylic proton (cis doublet with 3JH,H = 12.0 Hz). c Ratio based on integration of the vinylic doublets ([trans]/[trans] + [cis]). | |||||
| 1 | 7.196 | −230.7 | — | — | 100 |
| 7.746 | −317.2 | ||||
| 2 | 7.196 | −231.5 | — | — | 100 |
| 7.744 | −317.7 | ||||
| 3 | 7.197 | −230.7 | 6.400 | −225.8 | 12 |
| 7.742 | −317.2 | 7.525 | −319.7 | ||
| 4 | 7.197 | −231.0 | 6.399 | −226.0 | 19 |
| 7.742 | −318.0 | 7.525 | −320.5 | ||
| Atom (form) | Exp. | Calcd | Δ trans-to-cisa | Δ NMe2-to-imidazole N |
|---|---|---|---|---|
| a First and second row represent experimental and calculated (bold) values, respectively. | ||||
| NMe2 (trans) | −317.2 (NMe2, 3.04) | −332.7 | 2.5 | 86.5 |
| NMe2 (cis) | −319.7 (NMe2, 2.93) | −336.8 | 4.1 | 71.3 |
| Imidazole N (trans) | −230.7 (CH3, 4.10) | −261.4 | 4.9 | 93.9 |
| Imidazole N (cis) | −225.8 (CH3, 3.77) | −252.9 | 8.5 | 83.9 |
The differences between experimental and calculated chemical shifts may be related to the fact that under experimental conditions the averaged structure related to thermal motions is observed while in calculations only one conformation, i.e. the optimal one, is taken into account. Despite the differences in experimental conditions and model calculations, it is also worth emphasizing that (a) the trends in calculations are preserved with respect to the experimental data and (b) the difference between experimental and calculated values of 10 ppm (15N) is less than 3% error on the 15N scale. Moreover, in experiments the solvent–solute interactions exist and the I− anion is present while in calculations the solvent is treated as a continuum and only the organic cation is optimized. However, the calculations reflect the general trends observed in DMSO-d6 solution after irradiation (photoisomerization). In the case of trans conformation, the chemical shifts are lower for imidazole nitrogen atoms and higher for the NMe2 group. The same trend is found based on the results of computations. Finally, both the measured and calculated difference in chemical shifts for NMe2 and imidazole are larger for the cis conformation. This is in agreement with the fact that in the case of trans arrangement the charge transfer is more effective than in the case of cis form. This enables a higher degree of equalization of shielding the nuclei by electrons.
The above presented discussion indicates that the investigated styrylbenzimidazole molecules undergo a photoinduced trans-to-cis isomerization along the central carbon–carbon double bond. The photochemical conversion from the trans-form to the cis-form and vice versa is induced by absorbed light. The trans-form absorbs more light relative to the cis-form at 408 nm, while ultraviolet light (280 nm) is mainly absorbed by the cis-form.
Substitution effect and photoisomerization
The kinetics of the photoisomerization was studied by measuring the changes in the area under corresponding peaks in 1H NMR spectra as a function of irradiation time (see Fig. S4–S12 in the ESI†). The kinetic equation for the photochemical reaction takes the following form:| [trans] = [trans]0 exp(−kt) | (4) |
Since all signals in 1H NMR spectra for the trans and cis forms are well separated, small changes in the trans:cis ratio were readily determined. Fig. 8 compares the kinetic behavior for selected styrylbenzimidazole dyes containing various donor groups. The kinetic data fit well to a mono-exponential function, giving the rate constant of trans–cis photoisomerization (k) in DMSO-d6. The obtained data are compiled in Table 2.
 | ||
| Fig. 8 (a) Changes in abundance of trans form during white light irradiation as monitored by 1H NMR spectroscopy. (b) Kinetics of styrylbenzimidazole photoisomerization induced by irradiation. | ||
The substitution (the structure of the donor part of dyes) has a strong impact on the kinetics of the trans–cis reaction as seen in Fig. 8. An acceleration of the reaction is achieved by stronger electron-donating groups, while less electron-releasing groups slow down the reaction kinetics. This is in agreement with ICT and the quinoid-like structure, i.e. the central vinyl bond has a more singular character with increasing electron-donating ability of a substituent.
To quantify the substitution effect, besides the NMR data, the electronic absorption spectra were also analyzed. The obtained results allow us to calculate the photobleaching quantum yield (Table 2). The photobleaching quantum yields and rate constants of the photochemical process have the smallest values for 9, while the highest values are observed for compound 3. The dyes possessing the stiffened –NR2 group have much better photostability, whereas the compounds with alkyl groups which are linked to the N position are less stable. The dye with the methyl group in the meta position with respect to the –NR2 substituent is the least stable. The less electron-releasing group improves photostability compared with the dyes with a stronger electron-donating group which decreases photostability obviously.
The influence of the substitution by various amino groups affecting the rate of the photoisomerization was quantitatively described using the Hammett relationship with known ground-state parameters, σ.50Fig. 9 shows the Hammett correlations for the trans–cis process. Since in 9 some other processes are present (as mentioned above) manifested by much larger Stokes’ shifts especially in polar solvents (Table S1†) we performed a correlation with and without this substituent.
 | ||
| Fig. 9 Hammett correlation for the trans–cis photoisomerization in 1–8 (top) and 1–9 (bottom) (R2 correlation coefficient). | ||
The linear Hammett relationship for the investigated reaction shows that a pronounced structure–reactivity relationship exists for investigated compounds.
A quantitative correlation between the empirically derived Hammett constants and the reaction rates suggests that the type of amino substituent controls the photochemical behavior of the styrylbenzimidazole molecules. Such a correlation was earlier observed for other stilbene derivatives.51–53 The observed substituent effects indicate that the product formation during trans–cis isomerization of styrylbenzimidazole molecules occurs via a transition state (TS) with effective delocalization of a positive charge.54,55
The photostability of the hemicyanine dyes is strongly dependent on their chemical structure. The influence of substitution on the photochemical rate is also reflected in fluorescence spectroscopy. Since time-resolved fluorescence is a very sensitive technique to detect multiple emissive species,36,56 in the present study we carried out such measurements, recording the fluorescence decays of the dye 1 in MeCN solutions, as a function of the monitoring emission wavelength.
As indicated by the time-resolved emission spectra (TRES) constructed based on the emission wavelength dependent fluorescence decays, the fluorescence intensity decreases sharply for the initial time span. During this time, however, there is hardly any shift in the spectral position, as clearly indicated in Fig. 10. These observations are in accordance with our inference that the major contribution in the fluorescence decays is due to the excited trans-dye having a reasonably short fluorescence lifetime of ca. 0.04 ns in MeCN.
 | ||
| Fig. 10 (a) Time-resolved emission spectra (TRES) and (b) time-resolved area-normalized emission spectra (TRANES) of 1 in MeCN constructed from the decay transients obtained at different wavelengths. | ||
At the long time spans, there is a significant blue shift in the TRANES. This observation evidently indicates that the major emitting species at the latter time span have largely blue-shifted emission spectra in comparison with that of trans form. The clearly visible isoemissive point at 500 nm indicates that the emission comes from two species. The blue-shifted low intensity emission in the TRANES might be connected with the cis form or more probably come from a locally excited state due to deconjugation.
It is worth mentioning that the same effect was observed in steady-state fluorescence measurements as seen in Fig. 11. Due to difficulty in isolating the sterically hindered isomers we extended our fluorescence studies to include pure trans and a mixture of trans and cis isomers. The trans isomer is characterized by a strong component of the short lifetime τ1 (see Fig. S13 in the ESI†)) and fluorescence maximum at 545 nm in MeCN. The fluorescence spectrum of the trans–cis mixture in MeCN has two maxima, one at 545 nm and the second at 435 nm. The fluorescence intensity of the long-wavelength band decreases, but no shift is observed in that case. The emission decrease indicates the formation of a cis isomer after irradiation. In most cases, the cis isomers are observed to be either very weakly fluorescent or completely non-fluorescent in nature, mainly because of their fast nonradiative de-excitation rates.18,39,42–44,57,58 The fluorescence decay of a mixture of trans and cis isomers of 1 is also two-exponential in MeCN. A steady increase in the long-lived fluorescence lifetime τ2 with the increase in population of the cis isomer was observed (see Fig. S13 in the ESI†). These results suggest that the cis isomer converts to the same excited state as that of the trans.
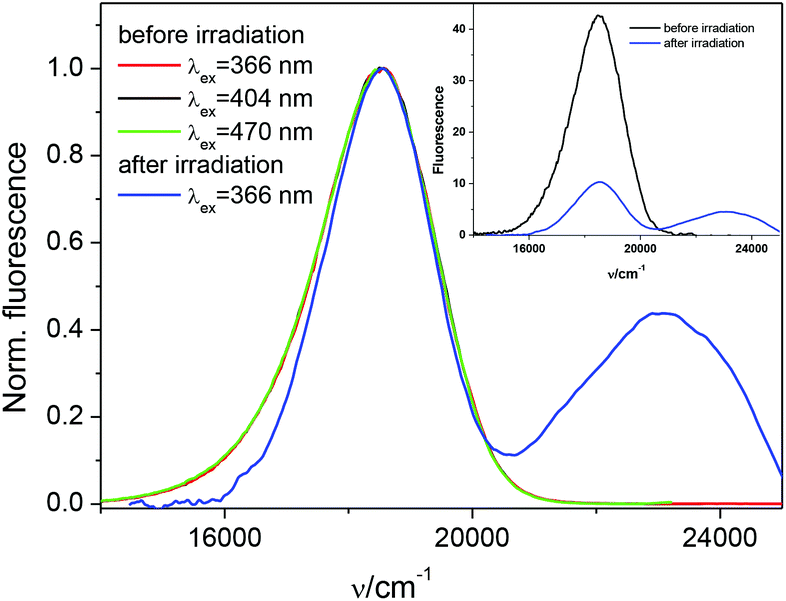 | ||
| Fig. 11 Steady-state fluorescence spectra before and after irradiation of diluted solution of 1 in MeCN. | ||
To sum up, trans-to-cis and cis-to-trans photoisomerization of styrylbenzimidazole molecules occurs through a common intermediate in the excited state, which is the transition state (TS). Presumably, the TS corresponds to a quinoid form with a twist of the two aromatic planes on either side of the double bond.18,34,37,38
Conclusions
The synthetic method described provides an attractive and environmentally friendly pathway to several useful hemicyanine dyes.Visible light promotes the conversion of the trans-isomer of styrylbenzimidazolium dyes to the cis-isomer that is confirmed by several instrumental methods and the regularity of properties with respect to the substituent constants. It was found that the stronger electron-releasing substituent has a more prominent effect on the observed photostability. The changes in the reaction rate of the trans–cis photoisomerization are successfully correlated with the Hammett constants.
Moreover, the hemicyanine dyes with an electron acceptor and an electron donor group on opposite sides of a π system exhibit an ICT character in solvents of variable polarity. The solvatochromism of dyes under study exhibited regular changes. We observed the blue-shifted absorption and red-shifted emission of the hemicyanine dyes with increasing polarity of solvent. The observed fluorescent behavior was ascribed to ICT upon excitation. An increase in the polarity of solvent decreases the activation barrier and enhances the process.
Acknowledgements
This work was supported by the Ministry of Science and Higher Education (MNiSW) (grant No 9/2014) and by PL-Grid Infrastructure. Robert Zaleśny is grateful to the project Mobilities – enhancing research, science and education at the Matej Bel University (ITMS code: 26110230082, under the Operational Program Education financed by the European Social Fund). The authors express their gratitude to Professor Marek Józefowicz for the assistance in the measurements and to Dr Miroslav Medved for reading the manuscript.References
- Q. Zhong, L. Chen, Y. Yang, Y. Cui, G. Qian and M. Wang, Synthesis and Spectroscopic Properties of Conjugated Triphenylaminosubstituted Chromophores, Dyes Pigm., 2008, 76, 677 CrossRef CAS.
- J. Ostrauskaite, H. R. Karickal, A. Leopold, D. Haarer and M. Thelakkat, Poly[bis(triphenylamine)ether]s with Low Glass Transition Temperatures as Photoconductors in Fact Photorefractive Systems, J. Mater. Chem., 2002, 12, 58 RSC.
- Y. Yasuda, T. Kamiyama and Y. Shirota, Ionic Conductivities of Low Molecular Weight Organic Gels and Their Application as Electrochromic Materials, Electrochim. Acta, 2000, 45, 1537 CrossRef CAS.
- J. P. Chen, H. Tanabe, X.-C. Li, T. Thoms, Y. Okamura and K. Ueno, Novel Organic Hole Transport Material with Very High Tg for Light-Emitting Diodes, Synth. Met., 2003, 132, 173 CrossRef CAS.
- I. Reva, L. Lapinski, N. Chattopadhyay and R. Fausto, Vibrational Spectrum and Molecular Structure of Triphenylamine Monomer: a Combined Matrix-Isolation FTIR and Theoretical Study, Phys. Chem. Chem. Phys., 2003, 5, 3844 RSC.
- S.-I. Kato, T. Matsumoto, T. Ishi, T. Thiemann, M. Shigeiwa, H. Gorohmaru, S. Maeda, Y. Yamashita and S. Mataka, Strongly Red-Fluorescent Novel Donor-π-Bridge-Acceptor-π-Bridge-Donor (D-π-A-π-D) Type 2,1,3-Benzothiadiazoles with Enhanced Two-Photon Absorption Cross-Sections, Chem. Commun., 2004, 2342–2343 RSC.
- Y.-T. Chang, S.-L. Hsu, G.-Y. Chen, M.-H. Su, T. A. Singh, E. W.-G. Diau and K.-H. Wei, Intramolecular Donor–Acceptor Regioregular Poly(3-hexylthiophene)s Presenting Octylphenanthrenyl-Imidazole Moieties Exhibit Enhanced Charge Transfer for Heterojunction Solar Cell Applications, Adv. Funct. Mater., 2008, 18, 2356 CrossRef CAS.
- L. Strekowski, in Topics in Heterocyclic Chemistry, ed. R. R. Gupta, Springer, Berlin, Heidelberg, 2008, vol. 14, p. 183 Search PubMed.
- R. Walia, M. Hedaitullah, S. F. Naaz, K. Iqbal and H. S. Lamba, Benzimidazole Derivatives – an Overview, Int. J. Res. Pharm. Chem., 2011, 1, 565 CAS.
- M. Boča, R. Boča, G. Kickelbick, W. Linert, I. Svoboda and H. Fuess, Novel Complexes of 2,6-bis(Benzthiazol-2-yl)pyridine, Inorg. Chim. Acta, 2002, 338, 36 CrossRef.
- H. Y. Shrivastava, M. Kanthimathi and B. U. Nair, Copper(II) Complex of a Tridentate Ligand: an Artificial Metalloprotease for Bovine Serum Albumin, Biochim. Biophys. Acta, 2002, 1573, 149 CrossRef CAS.
- V. G. Vaidyanathan and B. U. Nair, Synthesis, Characterization and DNA Binding Studies of a Ruthenium(II) Complex, J. Inorg. Biochem., 2002, 91, 405 CrossRef CAS PubMed.
- R. Vijayalakshmi, M. Kanthimathi, V. Subramanian and B. U. Nair, Interaction of DNA with [Cr(Schiff base)(H2O)2]ClO4, Biochim. Biophys. Acta, 2000, 1475, 157 CrossRef CAS.
- M. N. Hopkinson, C. Richter, M. Schedler and F. Glorius, An Overview of N-Heterocyclic Carbenes, Nature, 2014, 510, 485 CrossRef CAS PubMed.
- X. Han, H. Ma and Y. Wang, p-TsOH Catalyzed Synthesis of 2-Arylsubstituted Benzimidazoles, ARKIVOC, 2007, XIII, 150 Search PubMed.
- U. Steiner, M. H. Abdel-Kader, P. Fischer and H. E. A. Kramer, Photochemical cis/trans Isomerization of a Stilbazolium Betaine. A Protolytic/Photochemical Reaction Cycle, J. Am. Chem. Soc., 1978, 100, 3190 CrossRef CAS.
- B. Jędrzejewska, M. Pietrzak and J. Pączkowski, Solvent Effects on the Spectroscopic Properties of Styrylquinolinium Dyes Series, J. Fluoresc., 2010, 20, 73 CrossRef PubMed.
- A. Mishra, G. B. Behera, M. M. G. Krishna and N. Periasamy, Time-Resolved Fluorescence Studies of Aminostyryl Pyridinium Dyes in Organic Solvents and Surfactant solutions, J. Luminescence, 2001, 92, 175 CrossRef CAS.
- H. Jacob, S. Ulrich, U. Jung, S. Lemke, T. Rusch, C. Schutt, F. Petersen, T. Strunskus, O. Magnussen, R. Herges and F. Tuczek, Monitoring the Reversible Photoisomerization of an Azobenzene-functionalized Molecular Triazatriangulene Platform on Au(111) by IRRAS, Phys. Chem. Chem. Phys., 2014, 16, 22643 RSC.
- G. Vantomme and J.-M. Lehn, Reversible Adaptation to Photoinduced Shape Switching by Oligomer-Macrocycle Interconversion with Component Selection in a Three-State Constitutional Dynamic System, Chem. – Eur. J., 2014, 20, 16188 CrossRef CAS PubMed.
- M. Yamamura, K. Yamakawa, Y. Okazaki and T. Nabeshima, Coordination-Driven Macrocyclization for Locking of Photo- and Thermal cis-trans Isomerization of Azobenzene, Chem. – Eur. J., 2014, 20, 16258 CrossRef CAS PubMed.
- C. Cernatescu and E. Comanita, Benzazole Derivatives. IV. Reaction of 1,2,3-Trimethylbenzimidazolium Salts with Aromatic Aldehydes, J. Serb. Chem. Soc., 2005, 70, 1381 CrossRef CAS.
- B. Jędrzejewska, J. Kabatc and J. Pączkowski, 1,3-Bis[4-(p-aminostyryl)-pyridinyl]-propane Dibromide Derivatives: Synthesis and Spectroscopic Investigation, Dyes Pigm., 2007, 73, 361 CrossRef.
- B. Jędrzejewska and A. Rudnicki, The Synthesis and Spectroscopic Investigation of Dichromophoric Hemicyanine Dyes, Dyes Pigm., 2009, 80, 297 CrossRef.
- J. Olmsted III, Calorimetric Determinations of Absolute Fluorescence Quantum Yields, J. Phys. Chem., 1979, 83, 2581 CrossRef.
- R. Cammi and B. Mennucci, Linear Response Theory for the Polarizable Continuum Model, J. Chem. Phys., 1999, 110, 9877 CrossRef CAS.
- S. Chibani, S. Budzak, M. Medved, B. Mennucci and D. Jacquemin, Full cLR-PCM Calculations of the Solvatochromic Effects on Emission Energies, Phys. Chem. Chem. Phys., 2014, 16, 26024 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- L.-Y. Wang, X.-G. Zhang, Y.-P. Shi and Z.-X. Zhang, Microwave-Assisted Solvent-Free Synthesis of Some Hemicyanine Dyes, Dyes Pigm., 2004, 62, 21 CrossRef CAS.
- L. Wang, X. Zhang, F. Li and Z. Zhang, Microwave-Assisted Solvent-Free Synthesis of Some Styryl Dyes with Benzimidazole Nucleus, Synth. Commun., 2004, 34, 2245 CrossRef CAS.
- W. A. Schenk, T. Beucke, N. Burzlaff, M. Klüglein and M. Stemmler, The Coordination Chemistry of the CS Function. XX. Ruthenium Complexes of Thiocinnamaldehydes: Synthesis, Structure and [4 + 2]-Cycloaddition Reactions, Chem. – Eur. J., 2006, 12, 4821 CrossRef CAS PubMed.
- L. Hamdellou, O. Hernandez and J. Meinnel, 4-Dimethylamino-β-Nitrostyrene and 4-Dimethylamino-β-Ethyl-β-Nitrostyrene at 100 K, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2006, 62, o557 Search PubMed.
- X. H. Zhang, L. Y. Wang, G. H. Zhai, Z. Y. Wen and Z. X. Zhang, X-ray and DFT Studies of the Structure and Spectral Property of 2-[2-(4-Dimethylaminophenyl)ethenyl]-1-methyl-pyridinium Iodide, J. Mol. Struct., 2008, 881, 117 CrossRef CAS.
- H. Zhou, F. Zhou, P. Wu, Z. Zheng, Z. Yu, Y. Chen, Y. Tu, L. Kong, J. Wu and Y. Tian, Three New Five-Coordinated Mercury (II) Dyes: Structure and Enhanced Two-Photon Absorption, Dyes Pigm., 2011, 91, 237 CrossRef CAS.
- M. Kong, T. Wang, X. Tian, F. Wang, Y. Liu, Q. Zhang, H. Wang, H. Zhou, J. Wu and Y. Tian, Tunable Two-photon Absorption Near-infrared Materials Containing Different Electron-donors and a π-Bridge Center with Applications in Bioimaging in Live Cells, J. Mater. Chem. C, 2015, 3, 5580 RSC.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Plenum Press, New York, 2006 Search PubMed.
- M. Sauer, J. Hofkens and J. Enderlein, Handbook of Fluorescence Spectroscopy and Imaging, WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim, 2011 Search PubMed.
- C. Röcker, A. Heilemann and P. Fromherz, Time-Resolved Fluorescence of a Hemicyanine Dye: Dynamics of Rotamerism and Resolvation, J. Phys. Chem., 1996, 100, 12172 CrossRef.
- B. Strehmel, H. Seifert and W. Rettig, Photophysical Properties of Fluorescence Probes. 2. A Model of Multiple Fluorescence for Stilbazolium Dyes Studied by Global Analysis and Quantum Chemical Calculations, J. Phys. Chem. B, 1997, 101, 2232 CrossRef CAS.
- L. L. Zhou, Y. C. Jiang, W. G. Ju, X. H. Zhang and S. K. Wu, Emission Behavior of Non-Planar Intra-Molecular Conjugated Charge Transfer Compounds, Acta Phys. Chim. Sin., 2003, 19, 670 CAS.
- M. Cigáň, A. Gáplovský, I. Sigmundová, P. Zahradník, R. Dědic and M. Hromadová, Photostability of D-π-A Nonlinear Optical Chromophores Containing a Benzothiazolium Acceptor, J. Phys. Org. Chem., 2011, 24, 450 CrossRef.
- J. Kim and M. Lee, Excited-State Photophysics and Dynamics of a Hemicyanine Dye in AOT Reverse Micelles, J. Phys. Chem. A, 1999, 103, 3378 CrossRef CAS.
- M. Sczepan, W. Rettig, L. Y. Bricks, Y. L. Slominski and A. I. Tolmachev, Unsymmetric Cyanines: Chemical Rigidization and Photophysical Properties, J. Photochem. Photobiol., A, 1999, 124, 75 CrossRef CAS.
- X. Cao, R. W. Tolbert, J. L. McHale and W. D. Edwards, Theoretical Study of Solvent Effects on the Intramolecular Charge Transfer of a Hemicyanine Dye, J. Phys. Chem. A, 1998, 102, 2739 CrossRef CAS.
- R. M. Silverstein, F. X. Webster and D. Kiemle, Spectrometric Identification of Organic Compounds, Wiley, 2005 Search PubMed.
- A. Mishra, S. Chaterjee and G. Krishnamoorthy, Intramolecular Charge Transfer Emission of trans-2-[4′-(dimethylamino)styryl]benzimidazole: Effect of Solvent and pH, J. Photochem. Photobiol., A, 2013, 260, 50 CrossRef CAS.
- M. Barfield and P. Fagerness, Density Functional Theory/GIAO Studies of the 13C, 15N, and 1H NMR Chemical Shifts in Aminopyrimidines and Aminobenzenes: Relationships to Electron Densities and Amine Group Orientations, J. Am. Chem. Soc., 1997, 119, 8699 CrossRef CAS.
- R. Ditchfield, Self-Consistent Perturbation Theory of Diamagnetism. I. A Gauge-Invariant LCAO Method for N.M.R. Chemical Shifts, Mol. Phys., 1974, 27, 789 CrossRef CAS.
- K. Woliński, J. F. Hinton and P. Pulay, Efficient Implementation of the Gauge-Independent Atomic Orbital Method for NMR Chemical Shift Calculations, J. Am. Chem. Soc., 1990, 112, 8251 CrossRef.
- R. Gawinecki, E. Kolehmainen and R. Kauppinen, 1H and 13C NMR Studies of para-Substituted Benzaldoximes for Evaluation of the Electron Donor Properties of Substituted Amino Groups, J. Chem. Soc., Perkin Trans. 2, 1998, 25 RSC.
- T. Cordes, T. Schadendorf, B. Priewisch, K. Rück-Braun and W. Zinth, The Hammett Relationship and Reactions in the Excited Electronic State: Hemithioindigo Z/E-Photoisomerization, J. Phys. Chem. A, 2008, 112, 581 CrossRef CAS PubMed.
- V. Papper and G. I. Likhtenshtein, Substituted Stilbenes: A New View on Well-Known Systems: New Applications in Chemistry and Biophysics, J. Photochem. Photobiol., A, 2001, 140, 39 CrossRef CAS.
- V. Papper, D. Pines, G. Likhtenshtein and E. Pines, Photophysical Characterization of trans-4,4′-Disubstituted Stilbenes, J. Photochem. Photobiol., A, 1997, 111, 87 CrossRef CAS.
- M. B. Smith and J. March, March's Advanced Organic Chemistry, Wiley-Interscience, New York, 2001 Search PubMed.
- A. Williams, Free Energy Relationships in Organic and Bio-Organic Chemistry, The Royal Chemical Society, Cambridge, UK, 2003 Search PubMed.
- W. Becker, Advanced Time Correlated Single Photon Counting Technique, Springer, New York, 2005 Search PubMed.
- R. Lapouyade, K. Czeschka, W. Majenz, W. Rettig, E. Gilabert and C. Rulliere, Photophysics of Donor-Acceptor Substituted Stilbenes. A Time-Resolved Fluorescence Study Using Selectively Bridged Dimethylamino Cyano Model Compounds, J. Phys. Chem., 1992, 96, 9643 CrossRef CAS.
- E. Abraham, C. Grauby-Heywang, S. Selector and G. Jonusauskas, Characterization of Hemicyanine Langmuir-Blodgett Films by Picosecond Time-Resolved Fluorescence, J. Photochem. Photobiol., A, 2008, 93, 44 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Figures giving 1H and 13C NMR and IR spectra of dyes 1–9, HPLC chromatograms, computational details including the optimized geometries (Cartesians) of molecule 1, changes in 1H NMR spectra of trans isomers of compounds 1–9 upon irradiation in DMSO-d6 and fluorescence decays of dye 1 in acetonitrile before and after irradiation. See DOI: 10.1039/c5pp00361j |
| This journal is © The Royal Society of Chemistry and Owner Societies 2016 |