
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Stereoselective alkyne semihydrogenations with an air-stable copper(I) catalyst†
Niklas O.
Thiel
and
Johannes F.
Teichert
 *
*
Institut für Chemie, Technische Universität Berlin, Strasse des 17. Juni 115, 10623 Berlin, Germany. E-mail: johannes.teichert@chem.tu-berlin.de; Fax: +49 (0)30 314 28829; Tel: +49 (0)30 314 22791
First published on 20th October 2016
Abstract
An air-stable and preactivated copper(I) hydroxide/N-heteroyclic carbene (NHC) complex for alkyne semihydrogenations is reported. Next to an enhanced practicability of the process, the resulting alkenes are obtained with high Z-selectivities and no overreduction to the corresponding alkanes.
Introduction
Catalytic stereoselective alkyne semihydrogenations are powerful and atom-economic synthetic alternatives to olefination reactions.1,2 The resulting alkenes are valuable building blocks especially for diastereoselective follow-up reactions.3 For Z-selective alkyne semihydrogenations, the Lindlar catalyst4 has become the first choice, however, it suffers from E/Z-isomerisation processes and overreduction to the corresponding alkanes.2 While the latter leads to loss of the desired functionality, the former can be problematic with foresight to tedious separations and consecutive diastereoselective transformations.Hydrogenations catalysed by readily available first row transition metals are desirable from an economic point of view.5 Among them, homogeneous catalysts based on copper(I) have recently emerged as viable alternatives for Z-selective alkyne semihydrogenations.6–10 Key reactivity for these catalytic processes is the reported stereoselective insertion of alkynes into copper(I) hydride bonds.11–13 While most of the disclosed catalysts allow for good to excellent Z-stereoselectivity in alkyne semihydrogenations, all studied copper(I) complexes need to be prepared in situ as the active catalysts are unstable. This feat can be ascribed to the formation of a Cu–O-bond, which allows for H2 activation14 but at the same time renders the corresponding complexes sensitive to air and moisture. The need for preactivation hampers the practicability of the overall processes, as can be seen from the studied catalysts so far: a triphenylphosphine/copper(I) complex can be used under an H2 amosphere (5 bar) at elevated temperatures in combination with iso-propanol to transform alkynes into the corresponding Z-alkenes (Scheme 1a).6 This catalyst has to be activated with an alkoxide at elevated temperatures (100 °C) and is limited to mainly unfunctionalised substrates.15
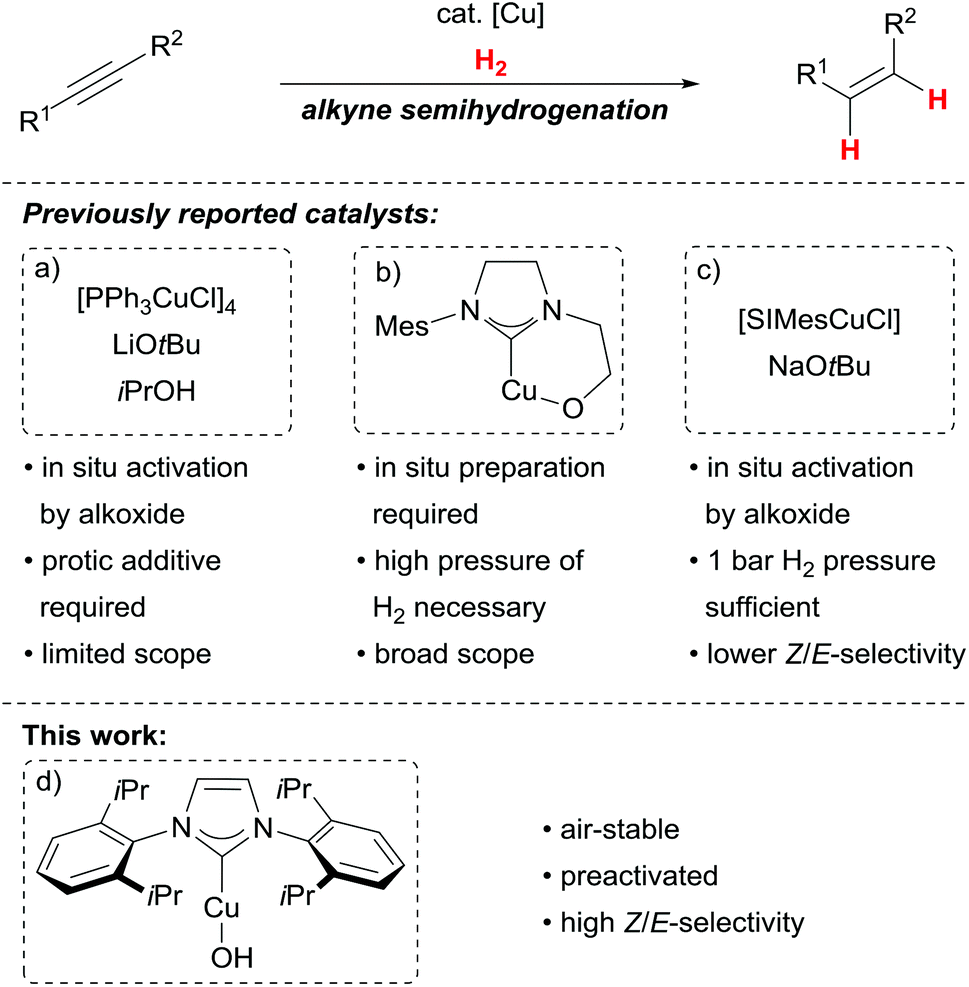 | ||
| Scheme 1 Approaches to Cu-catalysed alkyne semihydrogenations. | ||
We have introduced a highly stereoselective alkyne semihydrogenation based upon copper(I)/N-heterocylic carbene (NHC) complexes bearing an alkoxide tether (Scheme 1b).7 This system requires high H2 pressure (100 bar) and the catalyst needs to be generated in situ from sensitive mesitylcopper(I). More recently, an NHC/copper(I) complex has been reported which allows for alkyne semihydrogenations at 1 bar H2. The catalyst has to be generated in situ from a copper(I) chloride/NHC precursor with sodium tert-butanolate and shows somewhat reduced Z/E-selectivity (Scheme 1c).8
Herein, we report on the identification of a preactivated and air-stable NHC/copper(I) hydroxide complex, [IPrCuOH],16 for highly Z-selective alkyne semihydrogenations. The stability of the precatalyst allows for a more practical effectuation of the semihydrogenation without jeopardizing the stereoselectivity.17
Results and discussion
Optimisation of the alkyne semihydrogenation
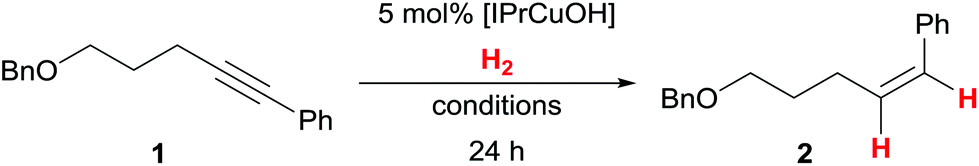
The alkyne semihydrogenation has been optimised using pentynol-derived internal alkyne 1 (Table 1): generally, E/Z-isomerisation processes and/or overreduction remained negligible. Under previously optimised reaction conditions (40 °C, 100 bar H2)7 as well as with slightly reduced H2 pressure of 80 bar, [IPrCuOH] shows complete conversion of the internal alkyne to the corresponding Z-styrene derivative 2 (Table 1, entries 1 and 2). Alkene 2 could be isolated with 93% yield. Lowering the pressure to 50 bar H2 led to incomplete conversion of 1 in THF (Table 1, entry 3). At these limiting conditions (40 °C, 50 bar H2), the alkyne semihydrogenation in DMF or toluene gave little turnover (25% and 26% respectively, Table 1, entries 4 and 5). During the investigation of the substrate scope (see below), we found that generally higher turnovers were obtained at higher H2 pressure. Additionally, for some compounds, more forcing conditions (100 bar H2, 60 °C) were required for full conversion (as in Table 1, entry 6). When lowering the H2 pressure to 1 bar (balloon) an alkyne semihydrogenation could not be observed (Table 1, entry 7). The sterically more demanding copper(I) hydroxide complex [IPr*CuOH]17b displayed lower activity under forcing conditions (Table 1, entry 8).18|
|
||
|---|---|---|
| Entry | Conditions | Conversionc |
a Reactions were carried out on 0.13 mmol scale.
b In all cases, the E/Z/alkane ratio was >99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1.
c Determined by 1H NMR and GC analysis.
d Isolated yield, 0.26 mmol scale. :1:1.
c Determined by 1H NMR and GC analysis.
d Isolated yield, 0.26 mmol scale.
|
||
| 1 | THF, 40 °C, 100 bar H2 | Full |
| 2 | THF, 40 °C, 80 bar H2 | Full (93%d) |
| 3 | THF, 40 °C, 50 bar H2 | 87% |
| 4 | DMF, 40 °C, 50 bar H2 | 25% |
| 5 | Toluene, 40 °C, 50 bar H2 | 26% |
| 6 | THF, 60 °C, 100 bar H2 | Full |
| 7 | THF, 60 °C, 1 bar H2 | n.d. |
| 8 | [IPr*CuOH] instead of [IPrCuOH], 60 °C, 100 bar H2 | 67% |
Substrate scope
With optimised reaction conditions in hand, we set out to investigate the substrate scope of the Z-selective alkyne semihydrogenation with [IPrCuOH] and found generally broad applicability, while Z-stereoselectivity remained high (Scheme 2). A variety of electron-rich and electron-poor aryl/alkyl-substituted alkynes 3a–3e based upon the pentynol-framework gave the corresponding Z-alkenes 4a–4e in high yields. Unlike our previously reported copper(I)/NHC complex (Scheme 1b),7 [IPrCuOH] does not fully tolerate ketone functional groups, which is showcased by partial overreduction of 4f to the benzylic alcohol (ratio ketone/benzylic alcohol = 88:12). We hypothesise that the presence of an intermediate alcohol(ate) disturbs the overall chemoselectivity, as in this case overreduction to the alkane was substantial (15%). This effect of additional alcohol(ate)s mirrors those of our previous study.7 In contrast, the tolerance of heterocycles differs from our earlier results: thiophene 4g, which was unreactive with the tethered catalyst,7 can now be obtained in good yield (94%): pyridine isomers 3h–3j, viable substrates with our earlier catalyst,7 give varying results in terms of overreduction and/or E/Z isomerisation. This displays a vulnerability of the copper(I) catalyst to strongly coordinating substrates. The present protocol is applicable to diaryl- and dialkylalkynes alike: tolane (3k) and its derivatives prove to be suitable precursors for Z-stilbenes 4k–4m under the semihydrogenation conditions. Notably, methoxy-substituted tolane 3l required a somewhat higher H2 pressure (100 bar). In a similar vein, dialkylalkynes 3n and 3o require slightly more forcing conditions (100 bar H2, 60 °C) to allow full conversion to the desired Z-alkenes 4n and 4o. The protected allylic alcohol 4p is available from the corresponding propargylic silyl ether in high yield and excellent stereoselectivity using elevated temperature and H2 pressure. This example marks one of the strong points of our catalytic process, as Z-allylic alcohols are important building blocks for diastereoselective follow-up reactions (see below for a synthetic elaboration of 4p). Finally, methyl-2-nonynoate (3q) shows excellent selectivity towards the corresponding Z-acrylate in our semihydrogenation protocol, albeit with low conversion (20%). Further investigations are needed to identify superior catalysts for this challenging, yet synthetically valuable semihydrogenation of propiolates.
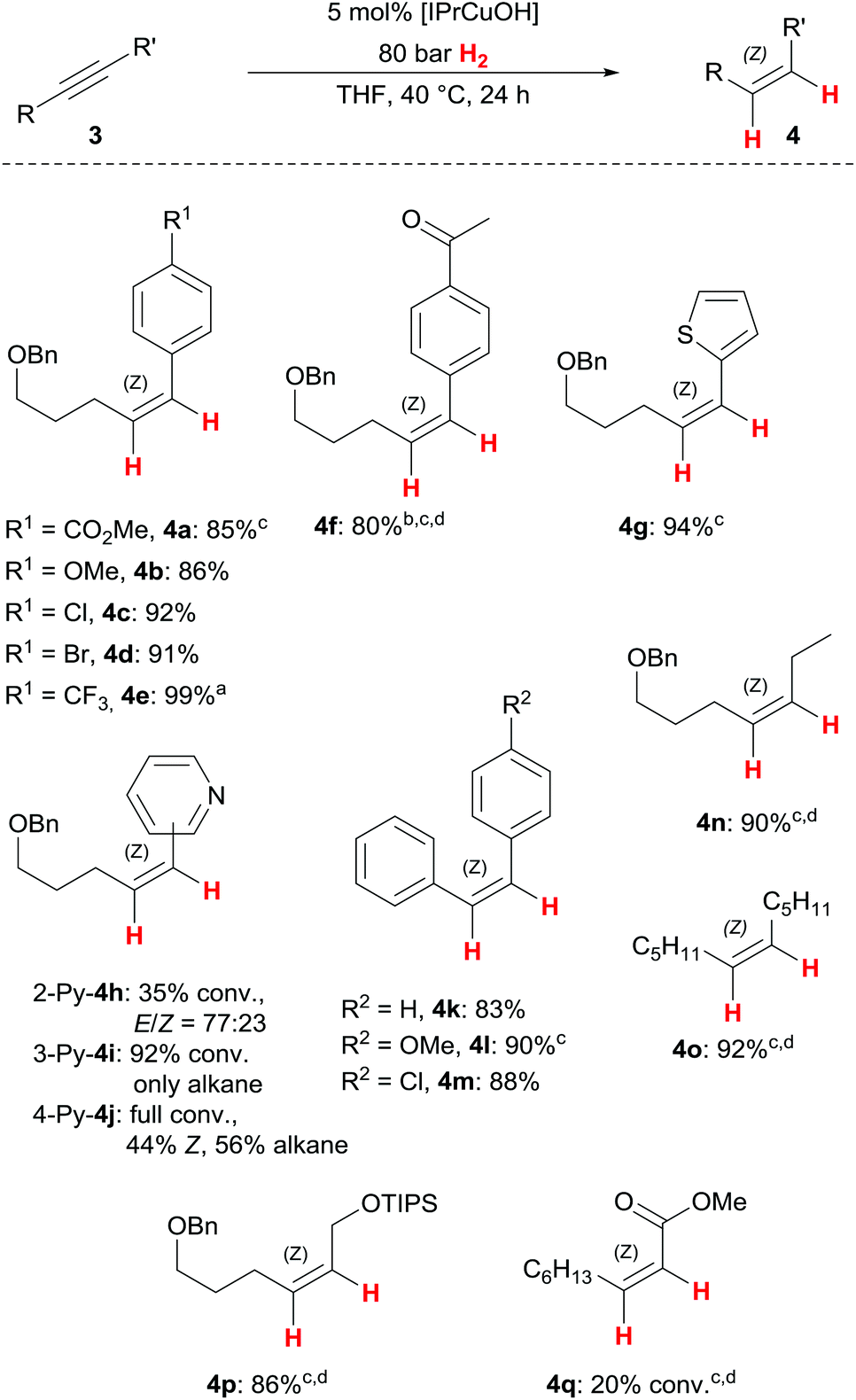 | ||
| Scheme 2 Cu-catalyzed alkyne semihydrogenation with [IPrCuOH], substrate scope. If not noted otherwise, the ratio Z/E/alkane is >99:1:1. aContains 5% alkane. bKetone/benzylic alcohol ratio 88:12, contains 15% alkane. Reaction was run for 48 h. c100 bar H2 employed. dReaction at 60 °C. | ||
When investigating diyne 5, selectively only the E,E-diene 6 was isolated with high yield (87%) with our catalyst (Scheme 3). This is of note, as this class of compounds has been reported to get reduced to a Z-monoalkene by an earlier copper(I) hydrogenation catalyst6 in alkyne semihydrogenation. To investigate the possible origin of this unexpected E-selectivity, we prepared E-enyne 7, a potential reaction intermediate in a stepwise diyne semihydrogenation from 5 towards 6. From this experiment, we isolated 96% of the E,Z-diene 8, representing only minor loss of stereochemical integrity of the primarily installed E-alkene. With this result, it seems reasonable to conclude that diynes such as 5 do not react step-wise as isolated triple bonds. A potential alkenylcopper(I) intermediate could equilibrate to a butatrienylcopper(I) intermediate19 (not shown), which accounts for the formation of the thermodynamically more preferred E,E-diene 6 from diyne 5.
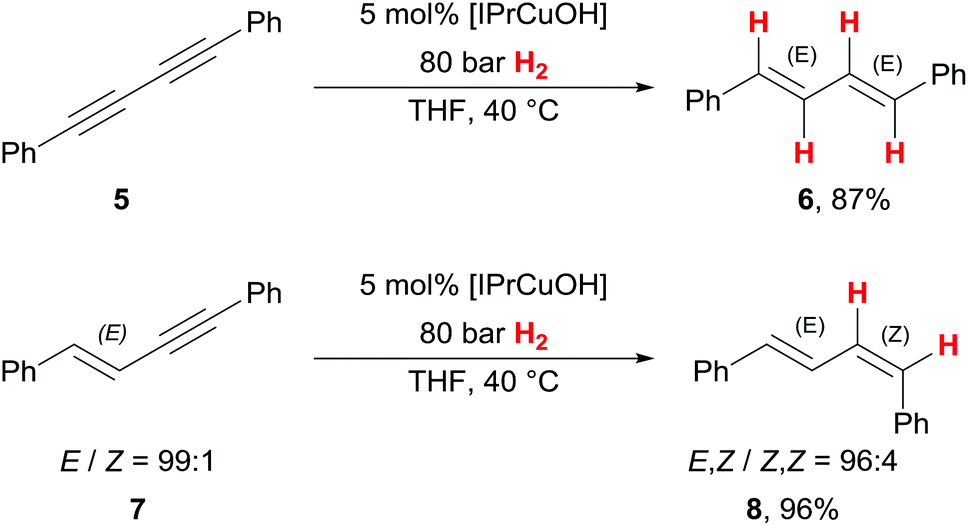 | ||
| Scheme 3 Cu-catalysed semihydrogenation of a diyne and an enyne. | ||
Follow-up chemistry of (Z)-allylic alcohols
Finally, to demonstrate the usefulness of the present highly Z-selective alkyne semihydrogenation, we further elaborated silyl ether 4p (Scheme 4): after silyl ether deprotection with TBAF to the allylic alcohol 9 (83%), subsequent oxidation with the Dess–Martin periodinane20 gave Z-acrolein-derivative 10 (88%). A subsequent Horner–Wadsworth–Emmons reaction was carried out to yield E,Z-sorbic acid derivative 11 in 86% yield. This approach underlines that a stereoselective alkyne semihydrogenation with [IPrCuOH] can serve as key step to generate alkene geometries with high selectivity.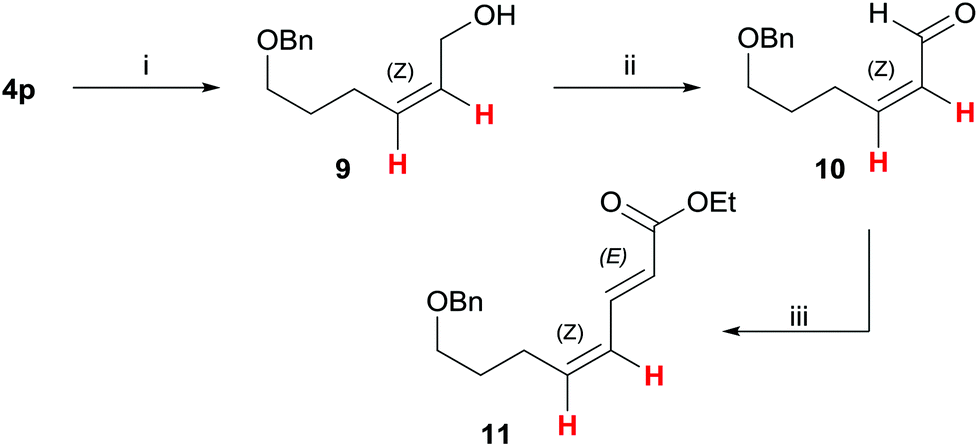 | ||
| Scheme 4 Derivatisation of allyl silyl ether 4p. Reaction conditions: (i) 1.1 equiv. TBAF, THF, rt, 3 h, 83%; (ii) 1.4 equiv. DMP, CH2Cl2, rt, 3 h, 88%; (iii) 1.5 equiv. triethyl phosphonoacetate, 1.5 equiv. NaH, THF, 0 °C to rt, 22 h, 86%, Z,E/Z,Z = >20:1. | ||
Conclusions
In summary, we have developed a highly Z-stereoselective alkyne semihydrogenation protocol, relying on an air-stable and preactivated NHC/copper(I) hydroxide complex, [IPrCuOH]. The practicability of this catalyst circumvents previous shortcomings of other copper(I) complexes as it does not require preactivation. A variety of products are accessible via this protocol in high yields and excellent Z-selectivities. The findings presented here could make a contribution towards widely applicable catalysis with easily accessible first-row transition metals.Experimental
All reactions were carried out in flame-dried glassware under a nitrogen atmosphere using standard Schlenk techniques. NMR spectra were recorded on AvanceII 400 MHz or AvanceIII 500 MHz or 700 MHz instruments (Bruker). Chemical shifts are reported in parts per million (ppm) and are referenced to the residual solvent resonance as the internal standard according to literature values.21 Data are reported as follows: chemical shift, multiplicity (br s = broad singlet, s = singlet, d = doublet, t = triplet, q = quartet, sept = septet, m = multiplet, mc = centrosymmetric multiplet), coupling constants (Hz), integration. All hydrogenation reactions were carried out in glass vials (50 × 14 mm, Schütt), equipped with a magnetic stir bar, a rubber septum pinched with a needle (0.90 × 50 mm, Braun) in autoclaves Berghof BR-100 or BR-300 equipped with heating blocks. For preparation and characterisation of the starting materials, see the ESI.†General procedure alkyne semihydrogenation
The reaction vessel was placed in a N2-purged autoclave under a counterflow of N2. The autoclave was purged with N2 (3 × 10 bar) and H2 (3 × 20 bar) before the appropriate H2 pressure was applied (pressure is given as initial pressure before heating). The heating block was pre-heated before the autoclave was placed inside. After the reported reaction time the autoclave was allowed to cool to rt and H2 was released. The autoclave was purged with N2 (3 × 10 bar) before the reaction vessel was taken out. The reaction mixture was filtered through a small plug of silica (1 mL tert-butyl methyl ether as eluent), and all volatiles were removed under reduced pressure. Reactions were subsequently analysed either by GC and/or NMR. The crude mixture was then subjected to purification as indicated with the appropriate substrates.(Z)-(5-(Benzyloxy)pent-1-en-1-yl)benzene (2)
Prepared from 1 (64 mg, 0.26 mmol, 1.0 equiv.) and [IPrCuOH] (6.0 mg, 13 μmol, 5.0 mol%) according to the general procedure. The reaction mixture was stirred under H2 atmosphere (80 bar) at 40 °C for 19 h. Purification by flash column chromatography on silica gel using cyclohexane/tert-butyl methyl ether 50:1 as eluent afforded 2 (60 mg, 0.24 mmol, 93%) as a colorless oil. Rf = 0.65 (cyclohexane/tert-butyl methyl ether 10:1). 1H NMR (500 MHz, CDCl3): δ = 1.76–1.84 (m, 2H), 2.48 (mc, 2H), 3.53 (t, J = 6.5 Hz, 2H), 4.50 (s, 2H), 5.69 (dt, J = 11.7 Hz, J = 7.4 Hz, 1H), 6.46 (mc, 1H), 7.22–7.26 (m, 1H), 7.28–7.36 (m, 9H) ppm; 13C NMR (126 MHz, CDCl3): δ = 25.4, 30.1, 69.9, 73.0, 126.6, 127.6, 127.7, 128.3, 128.5, 128.9, 129.5, 132.4, 137.8, 138.7 ppm; HRMS (APCI) calcd for C18H21O+ [(M + H)+]: Calculated 253.1587, found: 253.1582. The data is in accordance with literature.7
Methyl (Z)-4-(5-(benzyloxy)pent-1-en-1-yl)benzoate (4a)
Prepared from 3a (79 mg, 0.26 mmol, 1.0 equiv.) and [IPrCuOH] (6.0 mg, 13 μmol, 5 mol%) according to the general procedure. The reaction mixture was stirred under H2 atmosphere (100 bar) at 40 °C for 24 h. Purification by flash column chromatography on silica gel using cyclohexane/tert-butyl methyl ether 25:1 as eluent afforded 4a (67 mg, 0.22 mmol, 85%) as a colorless oil. Rf = 0.33 (cyclohexane/tert-butyl methyl ether 9:1); 1H NMR (500 MHz, CDCl3): δ = 1.79 (mc, 2H), 2.46 (mc, 2H), 3.51 (t, J = 6.4 Hz, 2H), 3.92 (s, 3H), 4.48 (s, 2H), 5.78 (dt, J = 11.7 Hz, J = 7.4 Hz, 1H), 6.46 (d, J = 11.7 Hz, 1H), 7.26–7.36 (m, 7H), 7.99 (mc, 2H) ppm; 13C NMR (126 MHz, CDCl3): δ = 25.6, 30.0, 52.1, 69.7, 73.1, 127.6, 127.7, 128.2, 128.5, 128.7, 128.8, 129.6, 134.6, 138.6, 142.4, 167.1 ppm; HRMS (APCI) for C20H23O3+ [(M + H)+]: Calculated 311.1642, found 311.1642. The data is in accordance with literature.7
(Z)-1-(5-(Benzyloxy)pent-1-en-1-yl)-4-methoxybenzene (4b)
Prepared from 3b (72 mg, 0.26 mmol, 1.0 equiv.) and [IPrCuOH] (6.0 mg, 13 μmol, 5 mol%) according to the general procedure. The reaction mixture was stirred under H2 atmosphere (80 bar) at 40 °C for 19 h. Purification by flash column chromatography on silica gel using cyclohexane/tert-butyl methyl ether 50:1 as eluent afforded 4b (62 mg, 0.22 mmol, 86%) as a colorless oil. Rf = 0.41 (cyclohexane/tert-butyl methyl ether 10:1); 1H NMR (500 MHz, CDCl3): δ = 1.79 (mc, 2H), 2.45 (mc, 2H), 3.52 (t, J = 6.4 Hz, 2H), 3.81 (s, 3H), 4.49 (s, 2H), 5.58 (dt, J = 11.7 Hz, J = 7.3 Hz, 1H), 6.38 (mc, 1H), 6.86 (mc, 2H), 7.24 (mc, 2H), 7.27–7.36 (m, 5H) ppm; 13C NMR (126 MHz, CDCl3): δ = 25.4, 30.2, 55.4, 69.9, 73.0, 113.7, 127.6, 127.7, 128.5, 128.9, 130.1, 130.5, 130.8, 138.8, 158.4 ppm; HRMS (EI) calcd for C19H22O2+ [(M)+]: 282.1614, found: 282.1610. The data is in accordance with literature.7
(Z)-1-(5-(Benzyloxy)pent-1-en-1-yl)-4-chlorobenzene (4c)
Prepared from 3c (71 mg, 0.25 mmol, 1.0 equiv.) and [IPrCuOH] (6.0 mg, 13 μmol, 5 mol%) according to the general procedure. The reaction mixture was stirred under H2 atmosphere (80 bar) at 40 °C for 19 h. Purification by flash column chromatography on silica gel using cyclohexane/tert-butyl methyl ether 50:1 as eluent afforded 4c (66 mg, 0.23 mmol, 92%) as a colorless oil. Rf = 0.59 (cyclohexane/tert-butyl methyl ether 10:1). 1H NMR (500 MHz, CDCl3): δ = 1.78 (mc, 2H), 2.42 (mc, 2H), 3.51 (t, J = 6.4 Hz, 2H), 4.48 (s, 2H), 5.69 (dt, J = 11.7 Hz, J = 7.4 Hz, 1H), 6.39 (d, J = 11.7 Hz, 1H), 7.22 (m, 2H), 7.27–7.30 (m, 5H), 7.33–7.36 (m, 2H) ppm; 13C NMR (126 MHz, CDCl3): δ = 25.4, 30.0, 69.7, 73.1, 127.6, 127.7, 128.3, 128.4, 128.5, 130.2, 132.4, 133.1, 136.2, 138.6 ppm; HRMS (EI) calcd for C18H19ClO+ [(M)+]: 286.1119, found: 286.1133; IR (ATR) ν = 2854 (m), 1490 (s), 1453 (m), 1362 (m), 1091 (s), 1013 (m), 840 (s), 734 (s), 696 (s) cm−1.
(Z)-1-(5-(Benzyloxy)pent-1-en-1-yl)-4-bromobenzene (4d)
Prepared from 3d (83 mg, 0.25 mmol, 1.0 equiv.) and [IPrCuOH] (6.0 mg, 13 μmol, 5 mol%) according to the general procedure. The reaction mixture was stirred under H2 atmosphere (80 bar) at 40 °C for 19 h. Purification by flash column chromatography on silica gel using cyclohexane/tert-butyl methyl ether 50:1 as eluent afforded 4d (77 mg, 0.23 mmol, 91%) as a colorless oil. Rf = 0.69 (cyclohexane/tert-butyl methyl ether 10:1); 1H NMR (500 MHz, CDCl3): δ = 1.77 (mc, 2H), 2.41 (mc, 2H), 3.50 (t, J = 6.4 Hz, 2H), 4.47 (s, 2H), 5.69 (dt, J = 11.7 Hz, J = 7.4 Hz, 1H), 6.36 (mc, 1H), 7.15 (mc, 2H), 7.27–7.30 (m, 3H), 7.33–7.36 (m, 2H), 7.43 (mc, 2H) ppm; 13C NMR (126 MHz, CDCl3): δ = 25.4, 30.0, 69.7, 73.1, 120.5, 127.6, 127.7, 128.4, 128.5, 130.5, 131.4, 133.2, 136.6, 138.6 ppm; HRMS (APCI) calcd for C18H20BrO+ [(M + H)+]: 331.0692, found: 331.0689; IR (ATR) ν = 2930 (m), 2856 (m), 1486 (s), 1453 (m), 1102 (s), 1071 (s), 1028 (m), 1009 (s), 837 (s), 734 (s), 696 (s) cm−1.
(Z)-1-(5-(Benzyloxy)pent-1-en-1-yl)-4-(trifluoromethyl)benzene (4e)
Prepared from 3e (82 mg, 0.26 mmol, 1.0 equiv.) and [IPrCuOH] (3.0 mg, 6.4 μmol, 2.5 mol%) according to the general procedure. The reaction mixture was stirred under H2 atmosphere (80 bar) at 40 °C for 19 h. Purification by flash column chromatography on silica gel using cyclohexane/tert-butyl methyl ether 50:1 as eluent afforded 4e (87 mg, 0.26 mmol, 99%) as a colorless oil, containing 5% of the corresponding alkane. Rf = 0.32 (cyclohexane/tert-butyl methyl ether 30:1); 1H NMR (500 MHz, CDCl3): δ = 1.79 (mc, 2H), 2.44 (mc, 2H), 3.51 (t, J = 6.3 Hz, 2H), 4.48 (s, 2H), 5.79 (dt, J = 11.7 Hz, J = 7.4 Hz, 1H), 6.46 (d, J = 11.7 Hz, 1H), 7.27–7.35 (m, 5H), 7.38 (mc, 2H), 7.56 (mc, 2H) ppm; 13C NMR (126 MHz, CDCl3): δ = 25.5, 30.0, 69.7, 73.1, 124.4 (q, J = 272 Hz), 125.2 (q, J = 3.7 Hz), 127.7, 127.8, 128.3, 128.5, 129.1, 134.6, 138.6, 141.3 ppm; 19F NMR (470 MHz, CDCl3): δ = −62.4 ppm; HRMS (EI) calcd for C12H12F3O+ [(M − Bn)+]: 229.0835, found: 229.0839; IR (ATR) ν = 2861 (w), 1616 (m), 1454 (m), 1323 (s), 1162 (s), 1112 (s), 1066 (s), 1016 (s), 852 (m), 744 (m), 697 (m) cm−1.
(Z)-1-(4-(5-(Benzyloxy)pent-1-en-1-yl)phenyl)ethan-1-one (4f)
Prepared from 3f (74 mg, 0.26 mmol, 1.0 equiv.) and [IPrCuOH] (6.0 mg, 13 μmol, 5 mol%) according to the general procedure. The reaction mixture was stirred under H2 atmosphere (100 bar) at 60 °C for 48 h. Purification by flash column chromatography on silica gel using cyclohexane/tert-butyl methyl ether 30:1 as eluent afforded 4f (59 mg, 0.20 mmol, 80%) as a colorless oil, containing 15% of the corresponding alkane. Rf = 0.39 (cyclohexane/tert-butyl methyl ether 4:1); 1H NMR (500 MHz, CDCl3): δ = 1.79 (mc, 2H), 2.46 (mc, 2H), 2.58 (s, 3H), 3.51 (t, J = 6.3 Hz, 2H), 4.47 (s, 2H), 5.79 (dt, J = 11.7 Hz, J = 7.4 Hz, 1H), 6.46 (d, J = 11.7 Hz, 1H), 7.25–7.38 (m, 5H), 7.37 (mc, 2H), 7.90 (mc, 2H) ppm; 13C NMR (126 MHz, CDCl3): δ = 25.6, 26.7, 30.0, 69.7, 73.1, 127.6, 127.7, 128.4, 128.5, 128.7, 129.0, 134.8, 135.3, 138.6, 142.6, 197.8 ppm; HRMS (EI) calcd for C20H22O2+ [(M)+]: 294.1614, found: 294.1621. The data is in accordance with literature.7
(Z)-2-(5-(Benzyloxy)pent-1-en-1-yl)thiophene (4g)
Prepared from 3g (69 mg, 0.27 mmol, 1.0 equiv.) and [IPrCuOH] (6.0 mg, 13 μmol, 5 mol%) according to the general procedure. The reaction mixture was stirred under H2 atmosphere (100 bar) at 40 °C for 19 h. Purification by flash column chromatography on silica gel using cyclohexane/tert-butyl methyl ether 60:1 as eluent afforded 4g (63 mg, 0.24 mmol, 94%) as a colorless oil. Rf = 0.68 (cyclohexane/tert-butyl methyl ether 10:1); 1H NMR (500 MHz, CDCl3): δ = 1.85 (mc, 2H), 2.53 (mc, 2H), 3.56 (t, J = 6.5 Hz, 2H), 4.52 (s, 2H), 5.59 (dt, J = 11.5 Hz, J = 7.3 Hz, 1H), 6.55 (mc, 1H), 6.99–7.02 (m, 2H), 7.25 (m, 1H), 7.27 (mc, 1H), 7.34 (mc, 4H) ppm; 13C NMR (126 MHz, CDCl3): δ = 26.1, 29.7, 70.0, 73.1, 122.3, 125.1, 126.9, 127.3, 127.6, 127.8, 128.5, 130.5, 138.8, 140.8 ppm; HRMS (APCI) calcd for C16H19OS+ [(M + H)+]: 259.1151, found: 259.1146; IR (ATR) ν = 2854 (w), 1452 (m), 1362 (m), 1102 (s), 1048 (m), 1027 (m), 851 (m), 826 (m), 734 (s), 693 (s) cm−1.
(Z)-1,2-Diphenylethene (4k)
Prepared from 3k (46 mg, 0.26 mmol, 1.0 equiv.) and [IPrCuOH] (6.0 mg, 13 μmol, 5 mol%) according to the general procedure. The reaction mixture was stirred under H2 atmosphere (80 bar) at 40 °C for 19 h. Purification by flash column chromatography on silica gel using cyclohexane as eluent afforded 4k (38 mg, 0.21 mmol, 83%) as a colorless oil. Rf = 0.58 (cyclohexane); 1H NMR (500 MHz, CD2Cl2): δ = 6.63 (s, 2H), 7.18–7.27 (m, 10H) ppm; 13C NMR (126 MHz, CD2Cl2): δ = 127.5, 128.6, 129.2, 130.6, 137.8 ppm; HRMS (EI) calcd for C14H12+ [(M)+]: 180.0934, found: 180.0933. The data is in accordance with literature.7(Z)-1-Methoxy-4-styrylbenzene (4l)
Prepared from 3l (53 mg, 0.26 mmol, 1.0 equiv.) and [IPrCuOH] (6.0 mg, 13 μmol, 5 mol%) according to the general procedure. The reaction mixture was stirred under H2 atmosphere (100 bar) at 40 °C for 24 h. Purification by flash column chromatography on silica gel using cyclohexane/tert-butyl methyl ether 100:1 as eluent afforded 4l (48 mg, 0.23 mmol, 90%) as a colorless oil. Rf = 0.64 (cyclohexane/tert-butyl methyl ether 10:1); 1H NMR (500 MHz, CD2Cl2): δ = 3.77 (s, 3H), 6.54 (mc, 2H), 6.75 (mc, 2H), 7.17–7.27 (m, 7H) ppm; 13C NMR (126 MHz, CD2Cl2): δ = 55.5, 113.9, 127.3, 128.6, 129.1, 129.2, 130.1, 130.2, 130.5, 138.1, 159.2 ppm; HRMS (APCI) calcd for C15H15O+ [(M + H)+]: 211.1117, found: 211.1124. The data is in accordance with literature.22
(Z)-1-Chloro-4-styrylbenzene (4m)
Prepared from 3m (54 mg, 0.26 mmol, 1.0 equiv.) and [IPrCuOH] (6.0 mg, 13 μmol, 5 mol%) according to the general procedure. The reaction mixture was stirred under H2 atmosphere (80 bar) at 40 °C for 24 h. Purification by flash column chromatography on silica gel using pentane as eluent afforded 4m (48 mg, 0.22 mmol, 88%) as a colorless oil. Rf = 0.67 (pentane); 1H NMR (500 MHz, CDCl3): δ = 6.54 (d, J = 12.2 Hz, 1H), 6.64 (d, J = 12.2 Hz, 1H), 7.17–7.20 (mc, 4H), 7.22–7.27 (m, 5H) ppm; 13C NMR (126 MHz, CDCl3): δ = 127.5, 128.5, 128.6, 128.9, 129.1, 130.4, 131.1, 132.9, 135.8, 137.0 ppm; HRMS (APCI) calcd for C14H11+ [(M − Cl−)+]: 179.0855, found: 179.0857. The data is in accordance with literature.22(Z)-((Hept-4-en-1-yloxy)methyl)benzene (4n)
Prepared from 3n (52 mg, 0.26 mmol, 1.0 equiv.) and [IPrCuOH] (6.0 mg, 13 μmol, 5.0 mol%) according to the general procedure. The reaction mixture was stirred under H2 atmosphere (100 bar) at 60 °C for 24 h. Purification via flash column chromatography on silica using cyclohexane/tert-butyl methyl ether 100:1 as eluent afforded 4n (47 mg, 0.23 mmol, 90%) as a colorless oil. Rf = 0.79 (cyclohexane/tert-butyl methyl ether 10:1); 1H NMR (700 MHz, CDCl3): δ = 0.96 (t, J = 7.5 Hz, 3H), 1.68 (mc, 2H), 2.05 (mc, 2H), 2.14 (mc, 2H), 3.49 (t, J = 6.5 Hz, 2H), 4.51 (s, 2H), 5.33 (dtt, J = 10.8 Hz, J = 7.2 Hz, J = 1.3 Hz, 1H), 5.39 (dtt, J = 10.8 Hz, J = 7.2 Hz, J = 1.3 Hz, 1H), 7.27–7.31 (m, 1H), 7.33–7.36 (m, 4H) ppm; 13C NMR (175 MHz, CDCl3): δ = 14.5, 20.6, 23.9, 29.9, 70.0, 73.1, 127.6, 127.8, 128.5, 128.5, 132.4, 138.8 ppm; HRMS (APCI) for C14H21O+ [(M + H)+]: Calculated 205.1587, found 205.1585. The data is in accordance with literature.7
(Z)-Dodec-6-ene (4o)
Prepared from 3o (52 mg, 0.26 mmol, 1.0 equiv.) and [IPrCuOH] (6.0 mg, 13 μmol, 5 mol%) according to the general procedure. The reaction mixture was stirred under H2 atmosphere (100 bar) at 60 °C for 24 h. Purification by flash column chromatography on silica gel using cyclohexane as eluent afforded 4o (48 mg, 0.24 mmol, 92%) as a colorless oil. 1H NMR (500 MHz, CDCl3): δ = 0.89 (t, J = 7.0 Hz, 6H), 1.28–1.36 (m, 12H), 1.99–2.04 (m, 4H), 5.36 (mc, 2H) ppm; 13C NMR (126 MHz, CDCl3): δ = 14.2, 22.7, 27.3, 29.6, 31.7, 130.1 ppm; HRMS (EI) calcd for [(M)+]: 168.1873, found: 168.1874. The data is in accordance with literature.7(Z)-((6-(Benzyloxy)hex-2-en-1-yl)oxy)triisopropylsilane (4p)
Prepared from 3p (92 mg, 0.26 mmol, 1.0 equiv.) and [IPrCuOH] (6.0 mg, 13 μmol, 5 mol%) according to the general procedure. The reaction mixture was stirred under H2 atmosphere (100 bar) at 60 °C for 24 h. Purification by flash column chromatography on silica gel using cyclohexane/tert-butyl methyl ether 50:1 as eluent afforded 4p (80 mg, 0.22 mmol, 86%) as a colorless oil. Rf = 0.68 (cyclohexane/tert-butyl methyl ether 10:1); 1H NMR (500 MHz, CDCl3): δ = 1.04–1.14 (m, 21H), 1.69 (mc, 2H), 2.15 (mc, 2H), 3.48 (t, J = 6.5 Hz, 2H), 4.31 (d, J = 6.0 Hz, 2H), 4.50 (s, 2H), 5.42 (dtt, J = 11.0 Hz, J = 7.4 Hz, J = 1.6 Hz, 1H), 5.58 (dtt, J = 11.0 Hz, J = 5.9 Hz, J = 1.5 Hz, 1H), 7.26–7.31 (m, 1H), 7.33–7.36 (m, 4H) ppm; 13C NMR (126 MHz, CDCl3): δ = 12.2, 18.2, 24.4, 29.7, 59.8, 69.8, 73.0, 127.6, 127.7, 128.5, 129.7, 130.7, 138.8 ppm; 29Si DEPT (99 MHz, J = 20 Hz, CDCl3): δ = 13.8 ppm; HRMS (EI) calcd for C19H31O2Si+ [(M − C3H7)+]: 319.2088, found: 319.2095; IR (ATR) ν = 2941 (m), 2864 (s), 1454 (m), 1362 (m), 1091 (s), 1068 (s), 881 (s), 804 (m), 732 (s), 676 (s), 657 (s) cm−1.
(1E,3E)-1,4-Diphenylbuta-1,3-diene (6)
Prepared from 5 (53 mg, 0.26 mmol, 1.0 equiv.) and [IPrCuOH] (6.0 mg, 13 μmol, 5 mol%) according to the general procedure. The reaction mixture was stirred under H2 atmosphere (80 bar) at 40 °C for 19 h. Purification by flash column chromatography on silica gel using cyclohexane as eluent afforded 6 (46 mg, 0.22 mmol, 87%) as a white crystalline solid. Rf = 0.35 (cyclohexane); 1H NMR (500 MHz, CD2Cl2): δ = 6.71 (mc, 2H), 7.00 (mc, 2H), 7.23–7.26 (m, 2H), 7.35 (mc, 4H), 7.46–7.47 (m, 4H) ppm; 13C NMR (126 MHz, CD2Cl2): δ = 126.8, 128.0, 129.1, 129.6, 133.2, 137.8 ppm; HRMS (EI) calcd for C16H14+ [(M)+]: 206.1090, found: 206.1093. The data is in accordance with literature.22(1Z,3E)-1,4-Diphenylbuta-1,3-diene (8)
Prepared from 7 (52 mg, 0.26 mmol, 1.0 equiv.) and [IPrCuOH] (6.0 mg, 13 μmol, 5.0 mol%) according to the general procedure. The reaction mixture was stirred under H2 atmosphere (80 bar) at 40 °C for 19 h. Purification by filtration through a plug of silica (3 cm) using cyclohexane as eluent afforded 8 (51 mg, 0.25 mmol, 96%) as a colorless oil. E,Z/Z,Z = 96:4 as judged by 1H NMR. Rf = 0.41 (cyclohexane); 1H NMR (500 MHz, CD2Cl2): δ = 6.45 (m, 1H), 6.55 (d, J = 11.5 Hz, 1H), 6.75 (d, J = 15.6 Hz, 1H), 7.23–7.43 (m, 11H) ppm. Spectrum contains <5% alkane-containing products, which were not further characterised; 13C NMR (126 MHz, CD2Cl2): δ = 125.6, 127.0, 127.5, 128.1, 128.8, 129.0, 129.5, 130.6, 130.7, 135.3, 137.8, 138.1 ppm; HRMS (EI) calcd for C16H14+ [(M)+]: 206.1090, found: 206.1091. The data is in accordance with literature.22
(Z)-6-(Benzyloxy)hex-2-en-1-ol (9)
In a flame-dried Schlenk tube (15 mL) equipped with a magnetic stirring bar 4p (0.70 g, 1.9 mmol, 1.0 equiv.) was dissolved in THF (4 mL). To this mixture, TBAF (1.0 M in THF, 2.1 mL, 2.1 mmol, 1.1 equiv.) was added dropwise. After 3 h the reaction was quenched by addition of H2O (10 mL). The aqueous phase was extracted with tert-butyl methyl ether (2 × 10 mL) and the combined organic layers were dried over Na2SO4. All volatiles were removed under reduced pressure to give the crude product. Purification via flash column chromatography on silica using cyclohexane/tert-butyl methyl ether 3:1 as eluent afforded 9 (0.33 g, 1.6 mmol, 83%) as a colorless oil. Rf = 0.35 (cyclohexane/tert-butyl methyl ether 1:1); 1H NMR (500 MHz, CDCl3): δ = 1.64 (br s, 1H), 1.70 (mc, 2H), 2.21 (mc, 2H), 3.49 (t, J = 6.3 Hz, 2H), 4.17 (d, J = 6.9 Hz, 2H), 4.50 (s, 2H), 5.50–5.56 (m, 1H), 5.63–5.68 (m, 1H), 7.27–7.31 (m, 1H), 7.32–7.37 (m, 4H) ppm; 13C NMR (126 MHz, CDCl3): δ = 24.0, 29.4, 58.5, 69.3, 73.0, 127.6, 127.7, 128.5, 129.4, 132.3, 138.5 ppm; HRMS (ESI) calcd for C13H19O2+ [(M + H)+]: 207.1380, found: 207.1383; IR (ATR) ν = 3354 (m), 2859 (m), 1453 (m), 1363 (m), 1098 (s), 1028 (s), 734 (s), 696 (s), 612 (m) cm−1.
(Z)-6-(Benzyloxy)hex-2-enal (10)
In a flame dried Schlenk tube (15 mL) equipped with a magnetic stirring bar 9 (0.10 g, 0.49 mmol, 1.0 equiv.) was dissolved in CH2Cl2 (0.15 M, 3.2 mL) and DMP (0.29 g, 0.68 mmol, 1.4 equiv.) was added. The mixture was stirred at rt for 3 h. The reaction mixture was filtered and concentrated under reduced pressure. Purification via flash column chromatography on silica using cyclohexane/tert-butyl methyl ether 4:1 as eluent afforded 10 (87 mg, 0.43 mmol, 88%) as a colorless oil. Rf = 0.41 (cyclohexane/tert-butyl methyl ether 3:1); 1H NMR (500 MHz, CDCl3): δ = 1.82 (mc, 2H), 2.73 (mc, 2H), 3.52 (t, J = 6.1 Hz, 2H), 4.49 (s, 2H), 5.97 (ddt, J = 11.2 Hz, J = 8.1 Hz, J = 1.5 Hz, 1H), 6.62 (dt, J = 11.2 Hz, J = 8.2 Hz, 1H), 7.27–7.37 (m, 5H), 10.08 (d, J = 8.1 Hz, 1H) ppm; 13C NMR (126 MHz, CDCl3): δ = 24.9, 29.3, 68.9, 73.2, 127.8, 128.6, 130.7, 138.3, 152.5, 191.1 ppm; HRMS (EI) calcd for C13H16O2+ [(M)+]: 204.1145, found: 204.1144; IR (ATR) ν = 2858 (m), 1676 (s), 1452 (m), 1363 (m), 1099 (s), 1027 (m), 736 (s), 697 (s), 608 (m) cm−1.
Ethyl (2E,4Z)-8-(benzyloxy)octa-2,4-dienoate (11)
To a cooled (0 °C) suspension of NaH (60 wt% in mineral oil, 44 mg, 1.1 mmol, 1.5 equiv.) in THF (0.15 M, 5 mL) was added triethyl phosphonoacetate (0.24 g, 1.1 mmol, 1.5 equiv.) dropwise. The resulting mixture is stirred at 0 °C for 30 min. Compound 10 (0.15 mg, 0.72 mmol, 1.0 equiv.) was added to this mixture. After completion of the addition the resulting mixture was warmed to rt and stirred for 22 h. The reaction was quenched by addition of sat. aq. NH4Cl (4 mL). The aqueous phase was extracted with tert-butyl methyl ether (2 × 5 mL) and the combined organic layers were washed with brine (15 mL) and dried over Na2SO4. All volatiles were removed under reduced pressure. Purification via flash column chromatography on silica using cyclohexane/tert-butyl methyl ether 20:1 as eluent afforded 11 (0.17 g, 0.62 mmol, 86%) as a colorless oil. Rf = 0.56 (cyclohexane/tert-butyl methyl ether 4:1); 1H NMR (500 MHz, CDCl3): δ = 1.29 (t, J = 7.1 Hz, 3H), 1.74 (mc, 2H), 2.42 (mc, 2H), 3.49 (t, J = 6.4 Hz, 2H), 4.21 (q, J = 7.1 Hz, 2H), 4.50 (s, 2H), 5.85 (dt, J = 11.2 Hz, J = 7.9 Hz, 1H), 5.88 (d, J = 15.3 Hz, 1H), 6.14 (mc, 1H), 7.27–7.30 (m, 1H), 7.33–7.36 (m, 4H), 7.62 (ddd, J = 15.2 Hz, J = 11.2 Hz, 1H) ppm; 13C NMR (126 MHz, CDCl3): δ = 14.4, 25.1, 29.5, 60.4, 69.5, 73.2, 121.7, 127.1, 127.7, 127.8, 128.5, 138.6, 139.4, 140.7, 167.3 ppm; HRMS (EI) calcd for C17H22O3+ [(M)+]: 274.1564, found: 274.1571; IR (ATR) ν = 2856 (m), 1709 (s), 1634 (s), 1605 (m), 1304 (m), 1266 (s), 1166 (s), 1098 (s), 961 (s), 995 (m), 872 (m), 735 (s), 696 (s) cm−1.
Acknowledgements
This research was supported by the Fonds der chemischen Industrie (Liebig-Stipendium for J. F. T.) and the Daimler-und-Benz-Foundation (Postdoctoral Scholarship for J. F. T.). We kindly thank Prof. Dr Martin Oestreich (TU Berlin) for generous support.Notes and references
- (a) M. Crespo-Quesada, F. Cárdenas-Lizana, A.-L. Dessimoz and L. Kiwi-Minsker, ACS Catal., 2012, 2, 1773–1786 CrossRef CAS; (b) A. Molnar, A. Sarkany and M. Varga, J. Mol. Catal. A: Chem., 2001, 173, 185–221 CrossRef CAS.
- C. Oger, L. Balas, T. Durand and J.-M. Galano, Chem. Rev., 2013, 113, 1313–1350 CrossRef CAS PubMed.
- W.-Y. Siau, Y. Zhang and Y. Zhao, Top. Curr. Chem., 2012, 327, 33–58 CrossRef CAS PubMed.
- H. Lindlar, Helv. Chim. Acta, 1952, 35, 446–450 CrossRef CAS.
- R. M. Bullock, Science, 2013, 342, 1054–1055 CrossRef CAS PubMed.
- K. Semba, R. Kameyama and Y. Nakao, Synlett, 2015, 318–322 CrossRef CAS.
- F. Pape, N. O. Thiel and J. F. Teichert, Chem. – Eur. J., 2015, 21, 15934–15938 CrossRef CAS PubMed.
- T. Wakamatsu, K. Nagao, H. Ohmiya and M. Sawamura, Organometallics, 2016, 35, 1354–1357 CrossRef CAS.
- For alkyne semihydrogenation with stoichiometric use of copper(I) hydride complexes, see: J. F. Daeuble, C. McGettigan and J. M. Stryker, Tetrahedron Lett., 1990, 31, 2397–2400 CrossRef CAS.
- For catalytic alkyne semireductions using silanes, see: (a) A. M. Whittaker and G. Lalic, Org. Lett., 2013, 15, 1112–1115 CrossRef CAS PubMed; (b) K. Semba, T. Fujihara, T. Xu, J. Terao and Y. Tsuji, Adv. Synth. Catal., 2012, 354, 1542–1550 CrossRef CAS.
- N. P. Mankad, D. S. Laitar and J. P. Sadighi, Organometallics, 2004, 23, 3369–3371 CrossRef CAS.
- See also: A. J. Jordan, C. M. Wyss, J. Bacsa and J. P. Sadighi, Organometallics, 2016, 35, 613–616 CrossRef CAS.
- (a) A. J. Jordan, G. Lalic and J. P. Sadighi, Chem. Rev., 2016, 116, 8318–8372 CrossRef CAS PubMed; (b) C. Deutsch, N. Krause and B. H. Lipshutz, Chem. Rev., 2008, 108, 2916–2927 CrossRef CAS PubMed; (c) S. Rendler and M. Oestreich, Angew. Chem., Int. Ed., 2007, 46, 498–504 CrossRef CAS PubMed.
- G. V. Goeden and K. G. Caulton, J. Am. Chem. Soc., 1981, 103, 7354–7355 CrossRef CAS.
- It should be noted that this procedure represents a formal hydrogenation, as the proton equivalent stems from the iPrOH additive.
- G. C. Fortman, A. M. Z. Slawin and S. P. Nolan, Organometallics, 2010, 29, 3966–3972 CrossRef CAS.
- For the use of [IPrCuOH] as catalyst in related carboxylation reactions, see: (a) I. I. F. Boogaerts, G. C. Fortman, M. R. L. Furst, C. S. J. Cazin and S. P. Nolan, Angew. Chem., Int. Ed., 2010, 49, 8674–8677 CrossRef CAS PubMed; (b) O. Santoro, F. Lazreg, Y. Minenkov, L. Cavallo and C. S. J. Cazin, Dalton Trans., 2015, 44, 18138–18144 RSC.
- It should be noted that other copper(I) hydroxide complexes bearing NHC ligands are elusive at the moment, which hampers further investigation of catalysts.
- For the equilibration of related (sp3)-based propargylcopper(I) complexes, see: (a) J. Canisius, T. A. Mobley, S. Berger and N. Krause, Chem. – Eur. J., 2001, 7, 2671–2675 CrossRef CAS PubMed; (b) N. Yoshikai, T. Yamashita and E. Nakamura, Chem. – Asian J., 2006, 1, 322–330 CrossRef CAS PubMed.
- D. B. Dess and J. C. Martin, J. Org. Chem., 1983, 48, 4155–4156 CrossRef CAS.
- (a) G. R. Fulmer, A. J. M. Miller, N. H. Sherden, H. E. Gottlieb, A. Nudelman, B. M. Stoltz, J. E. Bercaw and K. I. Goldberg, Organometallics, 2010, 29, 2176–2179 CrossRef CAS; (b) H. E. Gottlieb, V. Kotlyar and A. Nudelman, J. Org. Chem., 1997, 62, 7512–7515 CrossRef CAS PubMed.
- D.-J. Dong, H.-H. Li and S.-K. Tian, J. Am. Chem. Soc., 2010, 132, 5018–5020 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Preparation and characterisation data as well as 1H and 13C NMR spectra of all compounds. See DOI: 10.1039/c6ob02271e |
| This journal is © The Royal Society of Chemistry 2016 |