
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCarbohydrate receptors combining both a macrocyclic building block and flexible side arms as recognition units: binding properties of compounds with CH2OH groups as side arms†
Felix
Amrhein
,
Jan
Lippe
and
Monika
Mazik
*
Institut für Organische Chemie, Technische Universität Bergakademie Freiberg, Leipziger Strasse 29, 09599 Freiberg, Germany. E-mail: monika.mazik@chemie.tu-freiberg.de; Fax: +03731393170; Tel: +03731392389
First published on 12th October 2016
Abstract
New representatives of compounds combining both a macrocyclic building block and two flexible side arms as recognition units were prepared and their binding properties toward selected carbohydrates were evaluated. The aim of this study was to examine the effects of the replacement of the heterocycle-bearing side arms by smaller units, such as hydroxy groups, on the binding capability. The design of this type of receptor was inspired by the participation of the side chain hydroxy group of serine and threonine in the biorecognition of carbohydrates. Such structural modifications enable the recognition of structure–activity relationships, which are of high importance in the development of carbohydrate receptors with predictable binding strength and selectivity.
Introduction
Inspired by the binding motifs observed in the crystal structure of the 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 receptor–glucopyranoside complex formed by an acyclic trimethylbenzene-based receptor bearing three heterocyclic recognition groups,1 we have designed a new type of effective artificial carbohydrate receptor consisting of a macrocyclic building block and two flexible side arms (see Fig. 1).2,3 Through the use of different types of bridges (units Y in Fig. 1) and side arms (units X) as building blocks, we succeeded in developing various receptors with interesting binding properties (see structures 1–11 in Fig. 2). The desired ability to form effective and selective 1:1 complexes with selected carbohydrates,2,3 instead of 2:1 receptor–sugar complexes as in the case of the acyclic receptor,1 could be achieved with the new receptor architecture, as shown exemplarily for compound 4 and octyl-β-D-glucoside in Fig. 3. Compounds bearing aminopyridine or -pyrimidine groups as flexible side arms and pyrrole-/benzene-based bridges (1–5) were shown to be more effective receptors than their analogues consisting of pyridine- or hydroxybenzene-based bridges (compounds 8–11, see Fig. 4). The considerable lower affinity of compounds 8–11 is mainly a consequence of the participation of their bridge units in intramolecular hydrogen bonds (particularly drastic reduction of the binding affinity was observed for compounds consisting of hydroxybenzene units). Furthermore, the involvement of the flexible side arms in intramolecular interactions, as in the case of compounds incorporating 8-hydroxyquinoline-based side arms (compounds 6 and 7),3 also results in a reduction of the binding affinity (in comparison to their analogues bearing aminopyridine and aminopyrimidine groups).
:1 receptor–glucopyranoside complex formed by an acyclic trimethylbenzene-based receptor bearing three heterocyclic recognition groups,1 we have designed a new type of effective artificial carbohydrate receptor consisting of a macrocyclic building block and two flexible side arms (see Fig. 1).2,3 Through the use of different types of bridges (units Y in Fig. 1) and side arms (units X) as building blocks, we succeeded in developing various receptors with interesting binding properties (see structures 1–11 in Fig. 2). The desired ability to form effective and selective 1:1 complexes with selected carbohydrates,2,3 instead of 2:1 receptor–sugar complexes as in the case of the acyclic receptor,1 could be achieved with the new receptor architecture, as shown exemplarily for compound 4 and octyl-β-D-glucoside in Fig. 3. Compounds bearing aminopyridine or -pyrimidine groups as flexible side arms and pyrrole-/benzene-based bridges (1–5) were shown to be more effective receptors than their analogues consisting of pyridine- or hydroxybenzene-based bridges (compounds 8–11, see Fig. 4). The considerable lower affinity of compounds 8–11 is mainly a consequence of the participation of their bridge units in intramolecular hydrogen bonds (particularly drastic reduction of the binding affinity was observed for compounds consisting of hydroxybenzene units). Furthermore, the involvement of the flexible side arms in intramolecular interactions, as in the case of compounds incorporating 8-hydroxyquinoline-based side arms (compounds 6 and 7),3 also results in a reduction of the binding affinity (in comparison to their analogues bearing aminopyridine and aminopyrimidine groups).
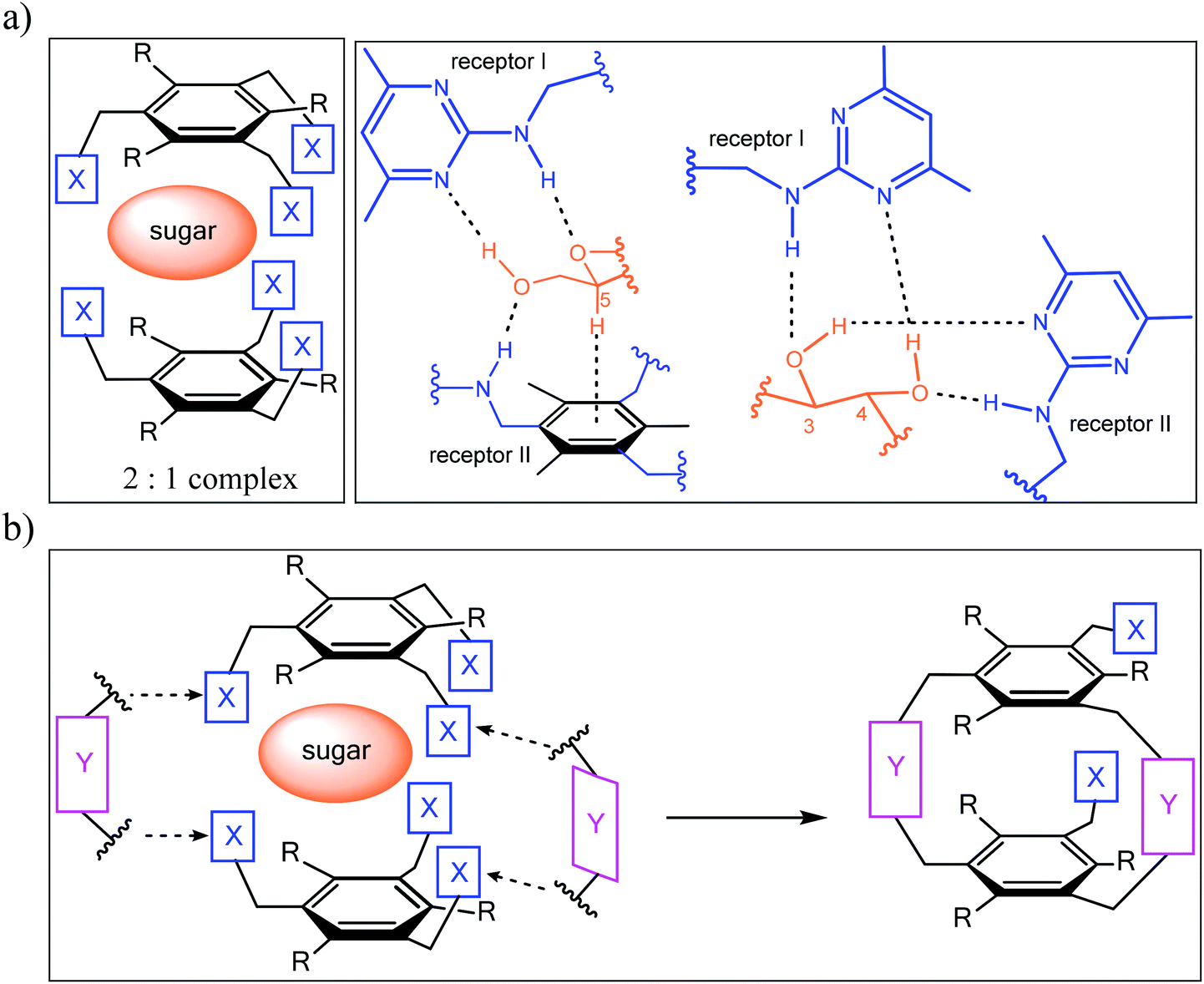 | ||
| Fig. 1 (a) Examples of hydrogen bonding and CH–π interactions in the crystalline 2:1 complex between an acyclic trimethylbenzene-based receptor (X = 2-amino-4,6-dimethylpyrimidine, R = Me) and octyl β-D-glucoside. (b) Schematic illustration of the design principle2,3 of carbohydrate receptors combining both a macrocyclic building block (units Y) and flexible side arms (units X). | ||
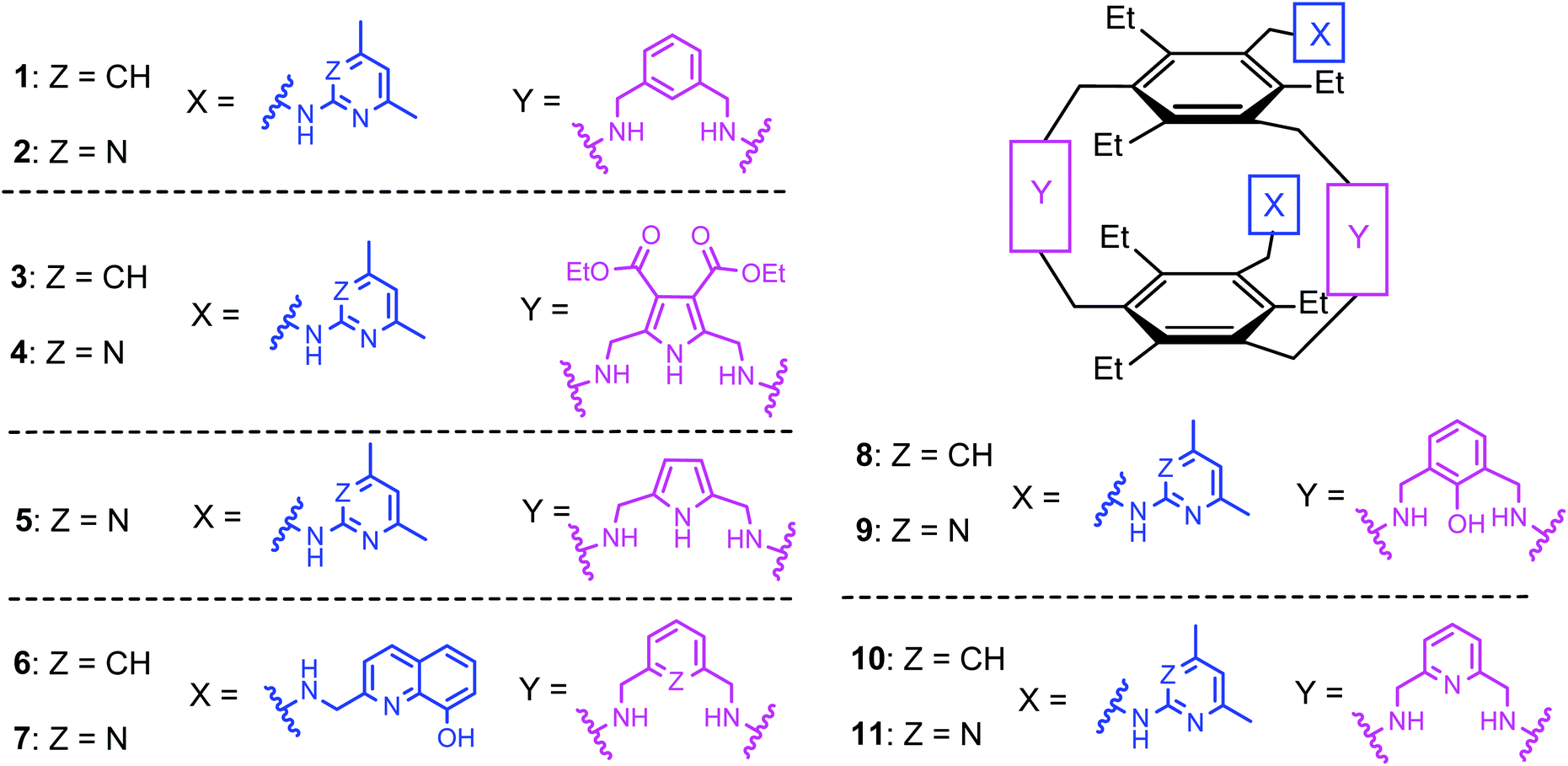 | ||
| Fig. 2 Structures of the previously described macrocyclic compounds 1–11 containing two flexible side arms as recognition units. | ||
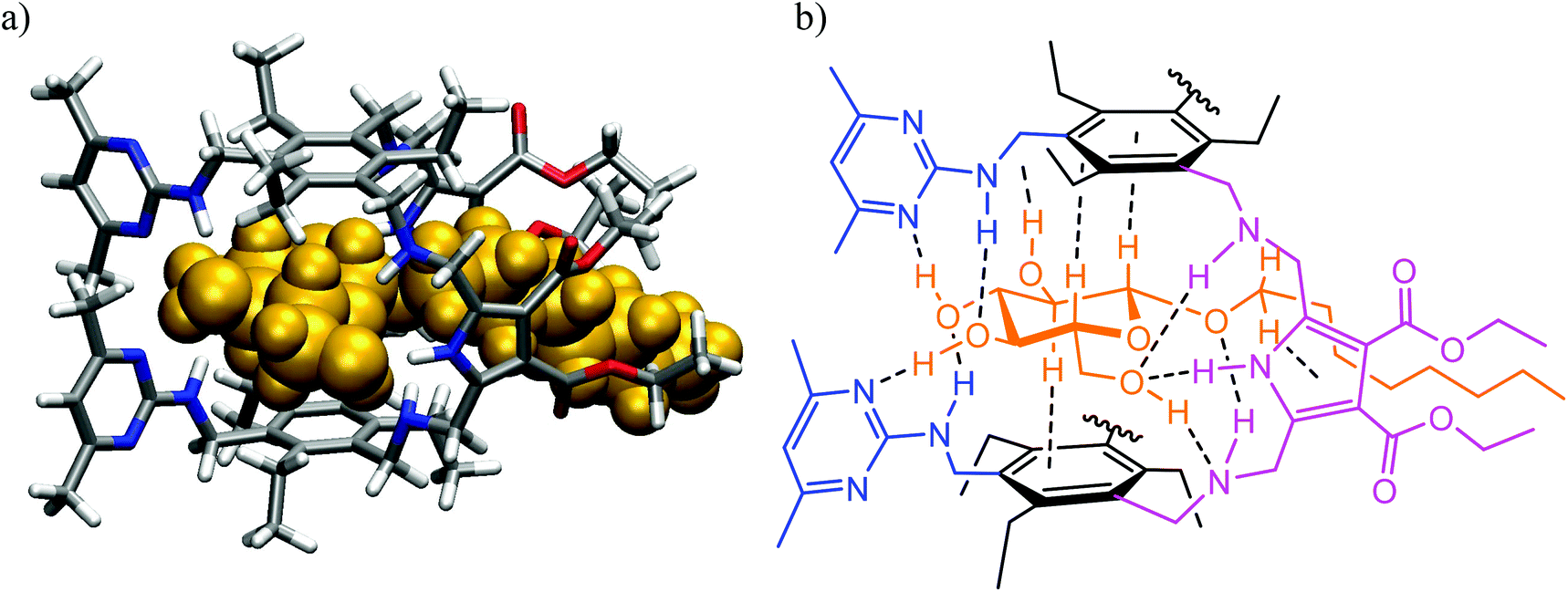 | ||
| Fig. 3 (a) Energy-minimized structure of the 1:1 complex formed between the receptor 4 and octyl β-D-glucoside (MacroModel V.8.5, OPLS 2001 force field, MCMM, 50000 steps). Color code: receptor N, blue; receptor O, red; receptor C, gray; the sugar molecule is highlighted in orange. (b) Schematic representation of the binding motifs in the complex (shown are interactions indicated by molecular modelling and confirmed by NMR spectroscopy).3 | ||
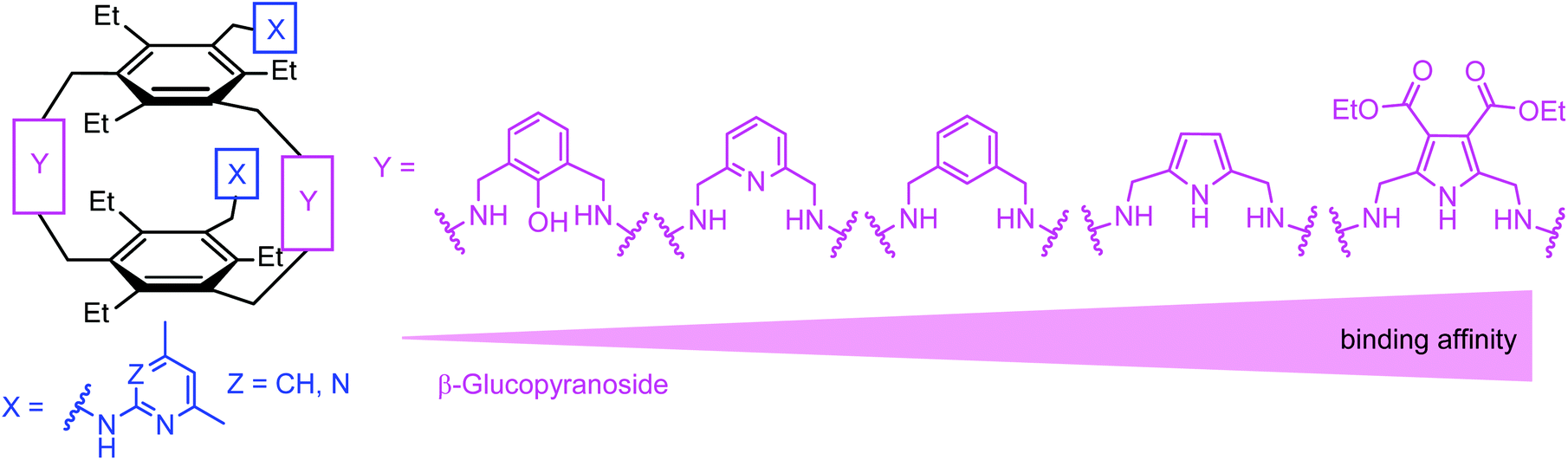 | ||
| Fig. 4 Schematic illustration of the influence of the bridge type (units Y) on the binding strength toward β-glucopyranoside in the case of macrocyclic compounds bearing two aminopyridine or -pyrimidine groups as flexible side-arms (units X). | ||
Our previous studies clearly showed the enormous potential of the designed receptor architecture for versatile structure variations through the combination of different types of bridges and side arms. In addition, the replacement of the two triethylbenzene units by other aromatic cores, having the ability to participate in CH⋯π interactions4–6 with the sugar CH units, provides further possibilities for structural modifications. Such versatile possibilities for structural changes enable the identification of structure–activity relationships, which are of particular importance for the design of artificial receptors with predictable binding strength and selectivity. It should be stressed at this point that the development of carbohydrate receptors with exact predictable binding properties still represents a great challenge.7
We were interested to see how the replacement of the heterocycle-bearing side arms by smaller units will affect the binding properties of the new compounds. The participation of the side chain hydroxy group of serine and threonine in the biorecognition of carbohydrates,8–10 as shown exemplarily in Fig. 5, has inspired us to design receptors bearing hydroxy groups11–14 as recognition sites. The aim of the present study was the synthesis and the evaluation of the binding properties of compounds 12–14 incorporating hydroxymethyl groups as flexible side arms (Fig. 6). Based on the results of our previous studies, we have selected benzene- and pyrrole-based subunits as bridges, since these building blocks led to the construction of particularly powerful carbohydrate receptors.
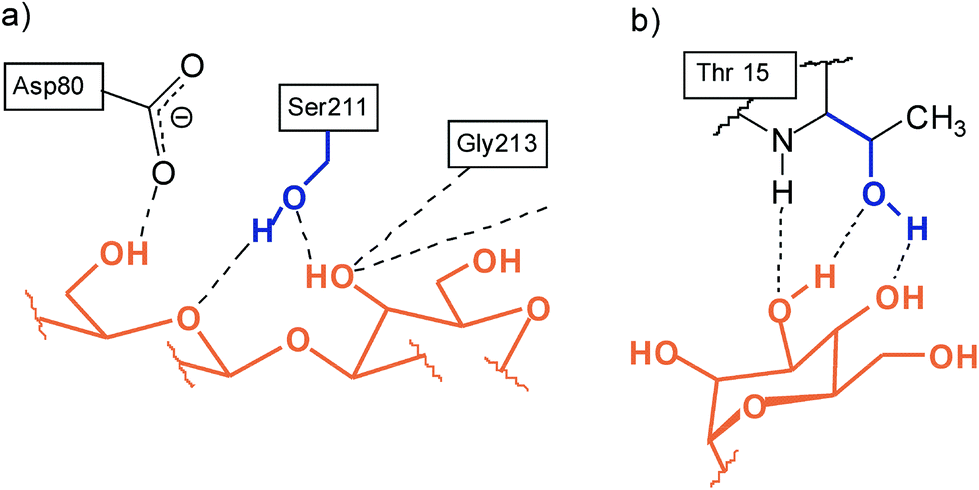 | ||
| Fig. 5 Examples for the participation of the side chain hydroxy group of serine and threonine in biorecognition of carbohydrates: (a) Galβ3GlcNAc in the combining site of peanut agglutinin;8,10a (b) neutral hydrogen bonds in the complex between Manα6(Manα3)Man and concanavaline A.9,10a | ||
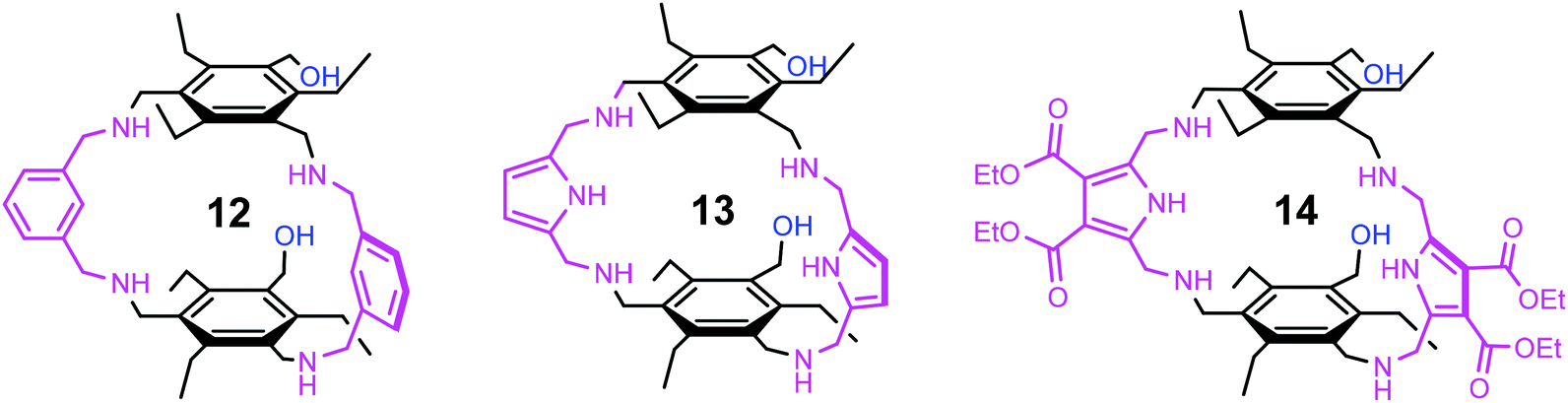 | ||
| Fig. 6 Structures of the macrocyclic compounds 12–14 bearing two hydroxymethyl-groups as flexible side arms. | ||
Compared to the earlier compounds 1–5, consisting of two aminopyridine- or aminopyrimidine-based side arms, which possess the ability to form more extensive interactions with the sugar substrate than the hydroxy groups of 12–14 (see Fig. 7a), a reduction in the binding capability of the new compounds was anticipated. In view of the potential of the pyrrole-based bridges15 to create more favourable intermolecular interactions than the benzene-bearing units (see Fig. 7b), compounds 13 and 14 were expected to be more powerful receptors than their analogue 12. Studies with compounds 12–14 confirmed these assessments and revealed again that the combination of a macrocyclic building block and two flexible side arms represents a particular valuable strategy for the construction of carbohydrate receptors with tunable binding properties.
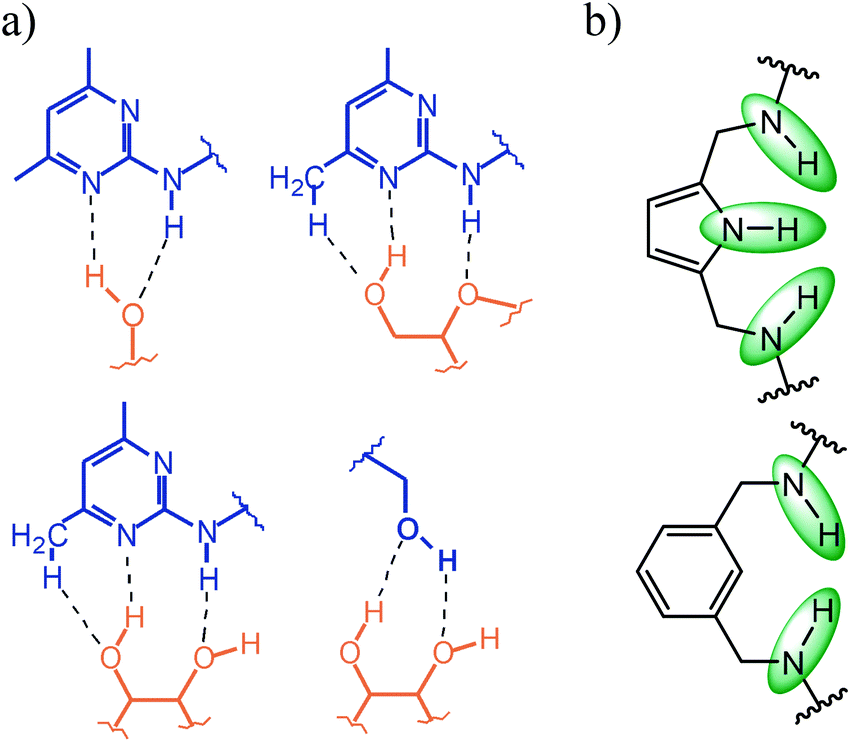 | ||
| Fig. 7 (a) Examples of noncovalent interactions, which can be formed by 2-amino-4,6-dimethylpyridine and the hydroxy group with carbohydrates; (b) hydrogen bonding sites (see green marking) of pyrrole- and benzene-based bridges (units Y in Fig. 1). | ||
Results and discussion
Synthesis
Compounds 12–14 were prepared on the basis of 3,5-bis(aminomethyl)-1-hydroxymethyl-2,4,6-triethylbenzene (18), the preparation of which started from 1,3,5-tris(bromomethyl)-2,4,6-triethylbenzene (15), as shown in Scheme 1. The reaction of 15 with potassium phthalimide (16) in water-containing dimethyl sulfoxide provided 1-hydroxymethyl-3,5-bis(phthalimidomethyl)-2,4,6-triethylbenzene (17) and 1,3,5-tris(phthalimidomethyl)-2,4,6-triethylbenzene, which could be separated out from each other by means of flash chromatography. The best yield of compound 17 (43%) could be achieved by using three equivalents of potassium phthalimide and 7%–10% water in dimethyl sulfoxide. The decrease in water content to 2–3% caused a reduction in the yield of 17. The use of only two equivalents of potassium phthalimide significantly decreased the yield of 17 to around 10%.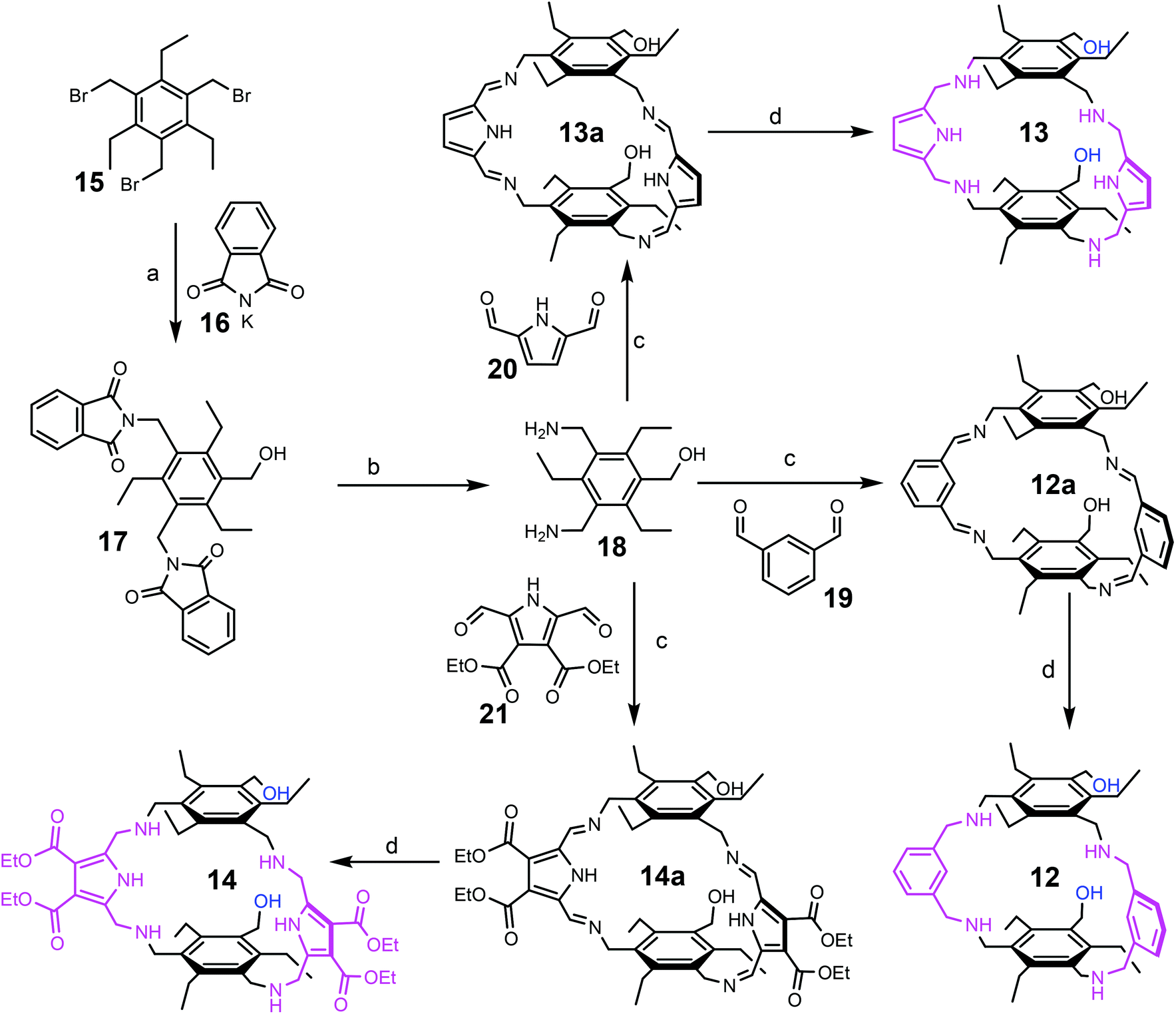 | ||
| Scheme 1 Reaction conditions: (a) potassium phthalimide (16), DMSO, 7–10% H2O; 43% of 17, (b) N2H4, EtOH/toluene; 91% of 18, (c) EtOH or ETOH/CH2Cl2, AcOH (catalytic amount), (d) NaBH4, MeOH; then H2O; 86% of 12, 83% of 13 and 41% of 14 (overall yields for step c and d). | ||
Compound 17 was converted into 3,5-bis(aminomethyl)-1-hydroxymethyl-2,4,6-triethylbenzene (18) by treatment with hydrazine hydrate. The condensation of 18 with the corresponding carbaldehyde, such as benzene-1,3-dicarbaldehyde (19), 1H-pyrrole-2,5-dicarbaldehyde (20) or diethyl 2,5-diformyl-1H-pyrrole-3,4-dicarboxylate (21), provided the corresponding imines (compounds 12a, 13a and 14a), which were further reduced with sodium borohydride to give the target compounds 12–14. The imines 12a and 13a were isolated via filtration and after their reduction the products 12 and 13 could be isolated in 86% and 83% yield, respectively. Due to the good solubility of the imine 14a, this imine was directly reduced without isolation. Product 14 was purified by column chromatography on silica gel using a chloroform/methanol mixture as the eluant and could be isolated in 41% yield. 1H-Pyrrole-2,5-dicarbaldehyde (20) and diethyl 2,5-diformyl-1H-pyrrole-3,4-dicarboxylate (21), used for the synthesis of compounds 13 and 14, respectively, were prepared according to known procedures.16,17
Binding studies
To compare the binding properties of the new compounds with those of the previously studied receptors, methyl β-D-glucoside (22) and methyl α-D-glucoside (23) were employed for binding studies in two-phase systems (extraction of the corresponding sugar from water into the organic phase), whereas octyl β-D-glucoside (24) was selected as a substrate for the binding studies in homogeneous media (1H NMR spectroscopic titrations and microcalorimetric titrations). Furthermore, ROESY studies were carried out and provided structural information for the receptor–sugar complexes.Extraction experiments: phase transfer of sugars from aqueous into organic solvents
Similarly to the case of the previously studied compounds 1–5, the extraction of methyl β-D-glucoside (22) and methyl α-D-glucoside (23) from aqueous into chloroform solution revealed a binding preference for β-glucoside 22, i.e. for a substrate with all-equatorial substitution pattern (similar to the receptors reported by Davis et al.18). Compared to the pyrimidine-bearing analogue 5, compound 13 showed a decreased affinity for β-glucoside 22 and was able to extract only 0.44 equiv. of the sugar (versus 0.74 equiv. for 5). It should however be noted that similar extractability was observed in the case of compounds 1 and 2 (0.40 and 0.50 equiv. of 22 for 1 and 2, respectively), consisting of pyridine- or pyrimidine-bearing side arms and benzene-based bridges. Thus, depending on the used combination of the side arms and bridges (X/Y combination), the extractabilities of β-glucoside by compounds 1, 2, 4, 5 and 13 decrease in the following sequence (see Table 1): aminopyrimidine/pyrrole > aminopyrimidine/benzene > hydroxy group/pyrrole ≥ aminopyridine/benzene.| Receptor | Combination of the building blocks X/Yc | β-Glucoside 22 | α-Glucoside 23 | β-Galactoside |
|---|---|---|---|---|
| a Values in mole equivalents with respect to the receptor; the 1H NMR signals of the corresponding sugar were integrated with respect to the receptor's signals to provide the sugar–receptor ratio (control experiments were performed in the absence of the receptor). b Results from ref. 2 and 3. c X = side arm unit, Y = bridge unit (pyrrole- or benzene-based, see Fig. 1). d Not determined. e Overlapping of sugar and receptor signals prevented the estimation of their ratio. | ||||
| 4 | Aminopyrimidine/pyrrole(diester) | 0.81 | 0.45 | 0.50 |
| 5 | Aminopyrimidine/pyrrole | 0.74 | 0.38 | 0.58 |
| 2 | Aminopyrimidine/benzene | 0.50 | n.d.d | 0.40 |
| 1 | Aminopyridine/benzene | 0.40 | 0.15 | 0.36 |
| 13 | Hydroxy group/pyrrole | 0.44 | 0.17 | n.d.d,e |
1H NMR spectroscopic titration and microcalorimetric titrations
The 1H NMR spectroscopic titrations with octyl β-D-glucoside (24) were carried out by adding increasing amounts of the sugar to a CDCl3 solution of the corresponding receptor 12–14 (according to the protocol described in the ESI†). The complexation between the binding partners was evidenced by several changes in the NMR spectra (for examples, see Fig. 8). In the case of compounds 13 and 14, bearing pyrrole-based bridges, the sequential additions of β-glucoside 24 caused successive replacement of the receptor signals by a new set of signals, as shown in Fig. 8a and b. Such spectral changes were also observed for the powerful receptors 2–52,3 and are consistent with complex formation, in which exchange between bound and unbound forms is slow on the NMR time-scale. The estimation of the binding constants directly from the relative ratios of the free and bound receptor provided indication of strong binding (K11 > 100000 M−1); however, significant errors did not allow the accurate determination of the binding constants on the basis of the 1H NMR data. For the previously tested compounds 2–5 the binding constants were determined on the basis of fluorescence titrations and found to be in the range of 210000–390000 M−1 in CDCl3,2,3 this technique could not be used for compounds 13 and 14.
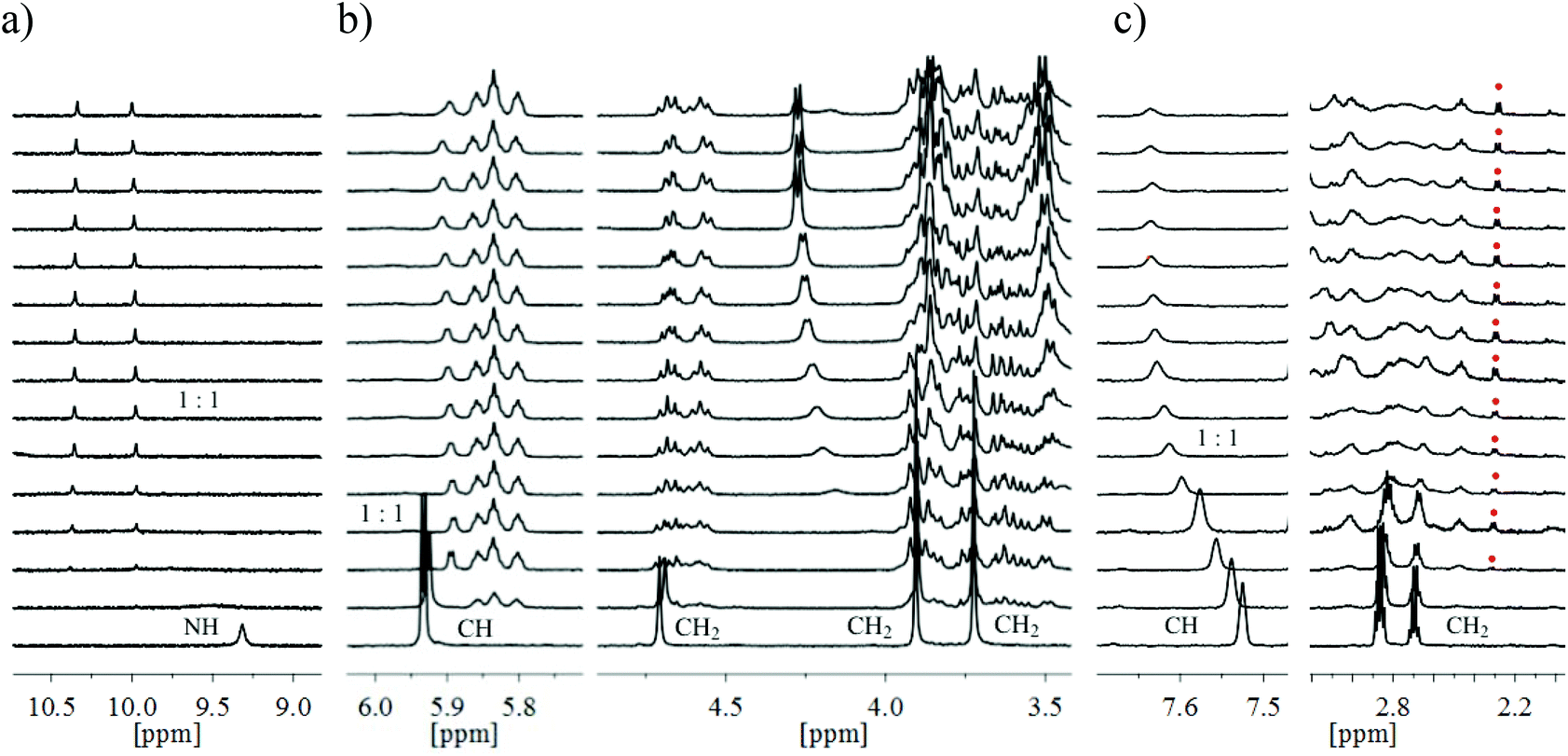 | ||
| Fig. 8 Partial 1H NMR (CDCl3, 500 MHz) spectra of receptors 14 (a), 13 (b) and 12 (c) after the addition of 0.00–3.50 equiv. of β-glucoside 24; [receptor] = 1.0 or 1.1 mM. (a, b) Complex formation with slow equilibration on the NMR time-scale, (c) simultaneous presence of fast- and slow-exchanging (red mark) complexes. Shown are the chemical shifts of the following signals of 12–14: pyrrole NH of 14, pyrrole CH and CH2 (side arms and bridge units) of 13 as well as benzene CH (bridge) and CH2 (ethyl groups) of 12. | ||
The spectra of compound 12, bearing benzene-based bridges, upon addition of β-glucoside 24 showed both significant movements of the receptor signals and the appearance of a new set of signals, as exemplarily shown in Fig. 8c. These results suggest complex formation with both fast and slow equilibration on the NMR time-scale (see also below, Fig. 11). Such a simultaneous presence of fast- and slow-exchanging complexes prevented quantitative calculation of the binding constants. It should, however, be noted that the analysis of the complexation-induced shifts for the predominant fast-exchanging complex provided clear indication for relatively weak interactions between 12 and 24 (K11 of about 8000 M−1).
The binding properties of compounds 12 and 13 were further analysed by isothermal titration calorimetry (ITC). The microcalorimetric titrations were carried out in CDCl3 and water-containing CDCl3 at 25 °C by adding increasing amounts of the sugar 24 to a solution of the corresponding receptor (according to the protocol described in the ESI†). Experimental parameters, such as concentration, temperature and calorimeter type (see the ESI†), were strictly maintained to be constant to allow the accurate comparison of the calorimetric data. The binding constants were determined from three independent microcalorimetric titrations and in all cases the best fit of the titration data was obtained with the 1:1 receptor–sugar binding model.
The experiments showed that 12 and 13 form enthalpy driven complexes with sugar 24 not only in dry CDCl3 but also in water-containing CDCl3 (0.035 and 0.07 mol L−1 H2O in CDCl3). The enthalpic driving force of all the investigated binding processes is partially compensated by a negative ΔS. As already indicated by the results of the NMR titrations, compound 13 was found to be a much more effective receptor for 24 than compound 12. The binding constants in dry CDCl3 were determined to be approximately 200000 M−1 and 7900 M−1 for 13·24 and 12·24, respectively (see Table 2 and Fig. 9). When the two bridge units are varied from benzene- (compound 12) to pyrrole-based units (compound 13), the increased gain in binding free enthalpy −ΔG (−22.2 kJ mol−1 for 12·24 compared with −30.2 kJ mol−1 for 13·24) originates from a more favourable enthalpic term (ΔH ≈ −78 kJ mol−1 for 12·24 compared with ΔH ≈ −87 kJ mol−1 for 13·24).
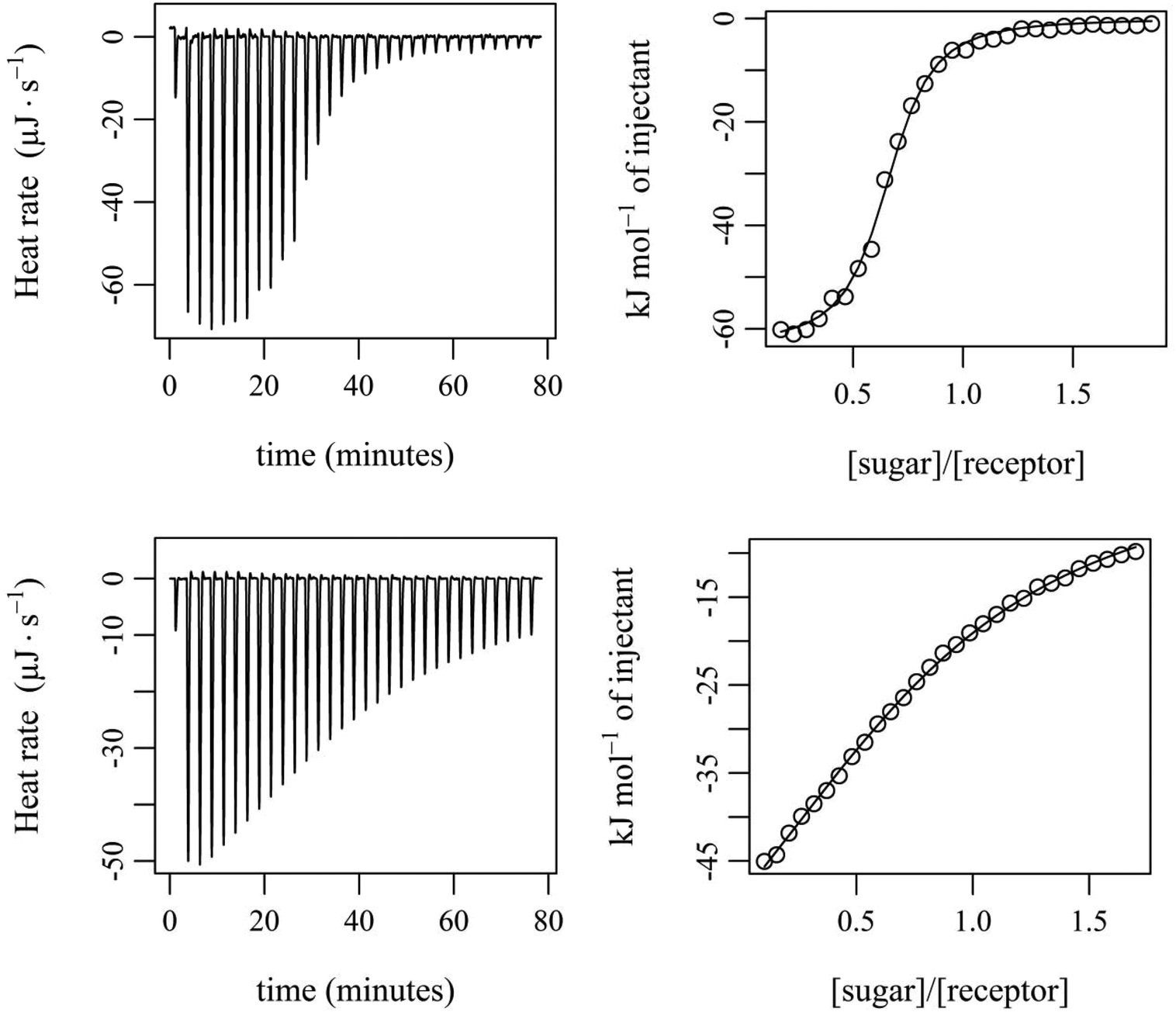 | ||
| Fig. 9 (a) ITC thermogram (left) and titration curve-fitting by independent model (right) of 13 with 24 in dry CDCl3. Titration mode: addition of β-glucoside (csyringe = 3.03 mM) into 24 (ccell = 0.31 mM) at 298 K in 30 steps. (b) ITC thermogram (left) and titration curve-fitting by independent model (right) of 12 with 24 in CDCl3. Titration mode: addition of β-glucoside (csyringe = 3.01 mM) into 12 (ccell = 0.31 mM) at 298 K in 30 steps. | ||
| Compound (solvent) | lgK11 (K11 [M−1]) |
ΔG [kJ mol−1] | ΔH [kJ mol−1] | TΔS [kJ mol−1] | ΔS [J mol−1 K−1] |
|---|---|---|---|---|---|
| a Used concentrations: [receptor] = 0.3 mM, [sugar] = 3 mM. b The errors listed are the standard deviations for a minimum of three replicate ITC titrations. | |||||
| 13 (dry CDCl3) | 5.29 ± 0.02 (195000 ± 9000) |
−30.2 ± 0.2 | −87.3 ± 0.9 | −57.1 | −192 |
| 13 (0.035 mol L−1 H2O) | 4.94 ± 0.01 (87100 ± 2000) |
−28.2 ± 0.1 | −56.8 ± 1.2 | −28.6 | −96 |
| 13 (0.07 mol L−1 H2O) | 4.75 ± 0.04 (56200 ± 5000) |
−27.1 ± 0.5 | −36.8 ± 0.7 | −9.7 | −33 |
| 12 (dry CDCl3) | 3.90 ± 0.02 (7900 ± 400) | −22.2 ± 0.2 | −78.6 ± 0.3 | −56.2 | −190 |
| 12 (0.07 mol L−1 H2O) | 4.06 ± 0.02 (11500 ± 500) |
−23.1 ± 0.1 | −54.6 ± 1.0 | −31.5 | −105 |
In the case of compound 13 the addition of water led to a significant decrease in binding strength and a less favourable complexation enthalpy (see Table 2), but a more favourable entropy term. The partial enthalpy–entropy compensation is also responsible for the relatively small variation in the free energy observed for the binding processes in the presence of 0.035 and 0.07 mol L−1 H2O in CDCl3.
In contrast to 13, compound 12 was found to exhibit a slightly higher binding constant in water-containing CDCl3 compared to that in dry solvent. The drop in the enthalpic driving force for binding is compensated by an increase in entropy in the water-containing solvent (resulting mostly from the desolvation process).
An increase in binding affinity in the presence of water in organic solvents was already observed for other carbohydrate receptors.1,19 The authors pointed out that the synthetic systems have the tendency to adopt the strategy widely used by nature and that the addition of water increases pyranoside binding “by filling in the gaps between the receptor and ligand”.19
The microcalorimetric titrations of 12 and 13 with sugar 24 revealed the expected more favourable binding properties of 13 compared to 12, confirmed the tendencies suggested by the results of the NMR titrations, clearly showed the strong influence of the nature of the bridge units on the binding strength and provided interesting information about the driving forces of the binding processes in dry and water-containing solvents.20
It is worth noting that the analysis of calorimetrically determined thermodynamic parameters for more than 60 protein–carbohydrate associations21 revealed that the enthalpy of binding is more negative than or equal to the free energy of binding and showed strong enthalpy–entropy compensation for associations in aqueous solution. Interestingly, experiments with compounds 12 and 13 in CDCl3 and water-containing CDCl3 also show similar tendency and revealed that the enthalpy of binding is more negative than the free energy of binding.
Exemplary ROESY and molecular modelling studies
Structural aspects of binding were also analysed in detail on the basis of ROESY studies (schematic representations of some short contacts between β-glucoside 24 and receptor 13 or 14 indicated by ROESY spectra are given in the ESI†). The ROESY experiments suggested the binding modes, which were indicated by molecular modelling calculations, and gave detailed structures for the receptor–sugar complexes, as shown exemplarily for 14·24 in Fig. 10. As observed for the previously studied receptor–sugar complexes,3 all OH groups of the bound sugar are involved in the formation of hydrogen bonds with 13 and 14. Furthermore, both sides of the pyranose ring are involved in CH⋯π interactions with the two central triethylbenzene rings of the receptor molecule, so that the sugar is fully encapsulated in the receptor cavity. It should be noted at this point that in the complexes of sugar binding proteins often one or two aromatic groups stack on the sugar ring.10 Interactions of the octyl chain of 24 with the pyrrole rings of 13 and 14 were also indicated by the performed studies. In the case of 12, the formation of complexes with different orientation of the sugar molecule in the cavity of 12 was indicated and is schematic illustrated in Fig. 11.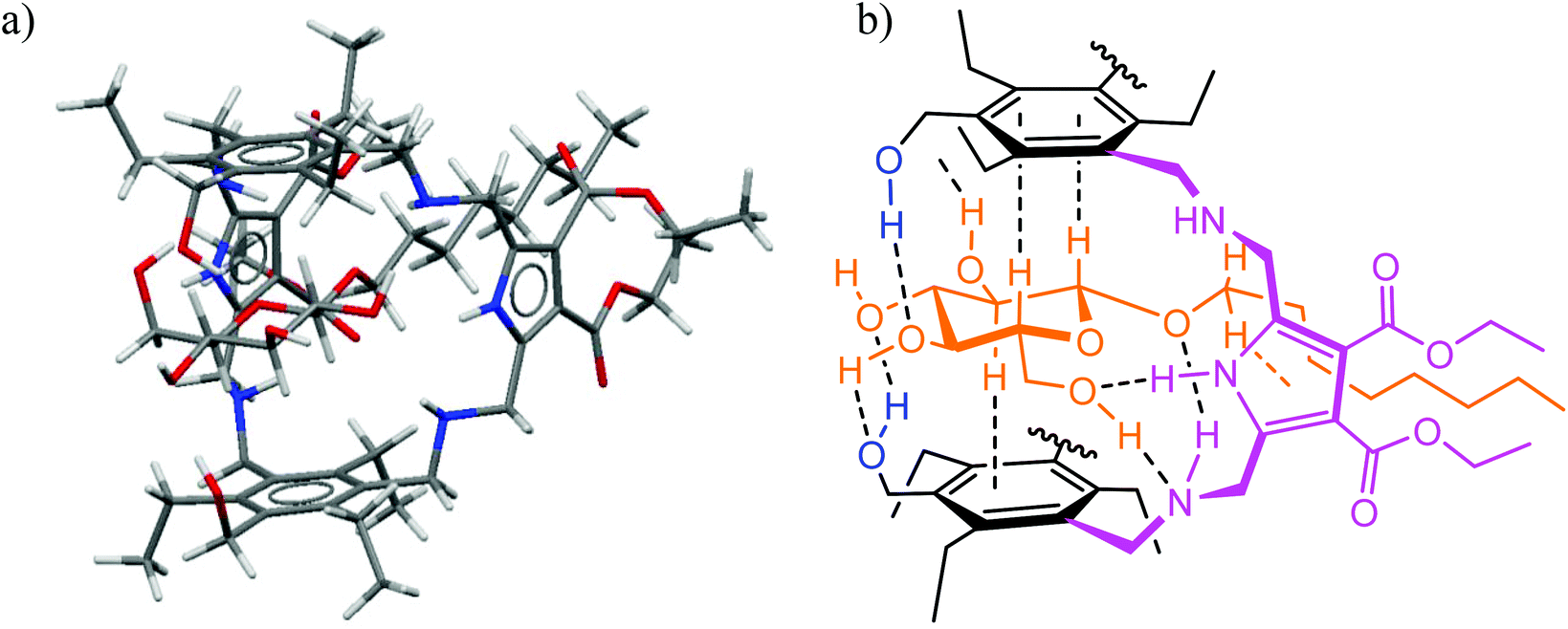 | ||
| Fig. 10 (a) Energy-minimized structure of the 1:1 complex formed between receptor 14 and octyl β-glucoside 24 (MacroModel V.8.5, OPLS 2001 force field, MCMM, 50000 steps). Color code: N, blue; O, red; C, gray. (b) Schematic representation of selected binding motifs indicated by molecular modelling and NMR spectroscopy for 14·24. | ||
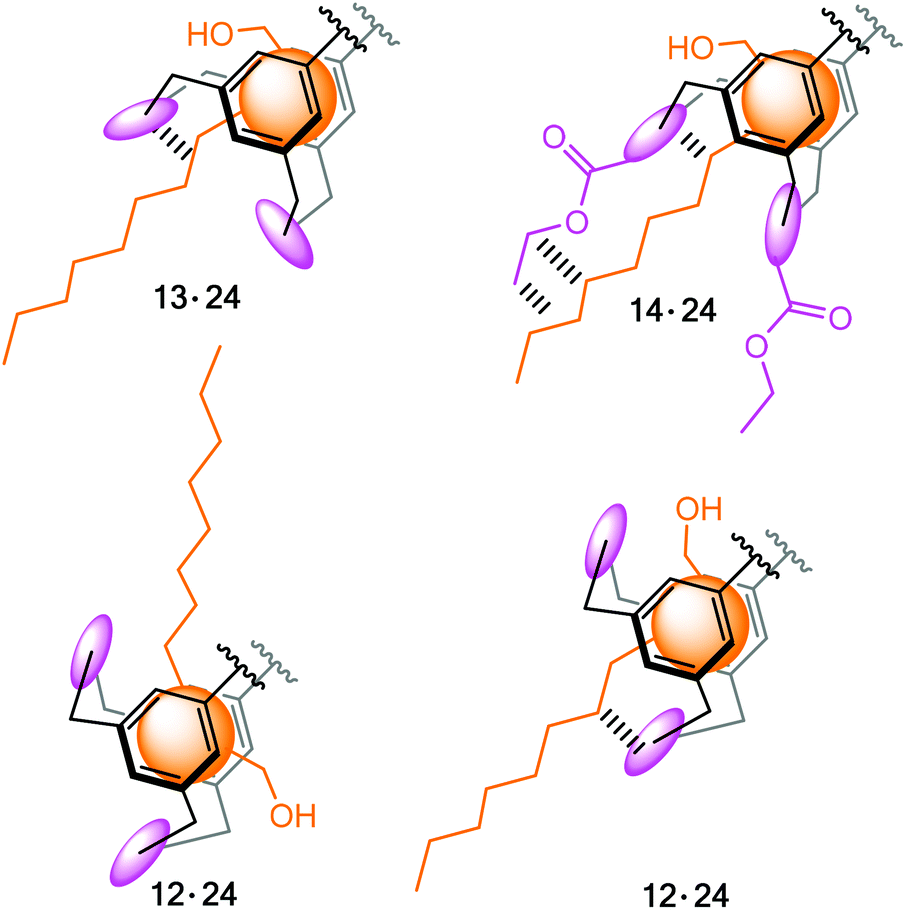 | ||
| Fig. 11 Schematic representation of the orientation of octyl β-glucoside 24 in the receptor cavity: complexes 12·24, 13·24 and 14·24 (bridge units of the receptors 12–14 are marked in magenta, the sugar molecule is highlighted in orange). | ||
Conclusion
Our previous studies with compounds consisting of both a macrocyclic building block and two flexible side arms revealed the high potential of this receptor architecture for versatile structural modifications, which allow the recognition of important structure–activity relationships. We were now interested to see how the replacement of the heterocycle-bearing side arms, used as building blocks for the construction of the earlier receptors,2,3 by hydroxy groups affects the binding capability of the new compounds. The use of hydroxy groups as recognition sites was inspired by the participation of the side chain hydroxy group of serine and threonine in the biorecognition of carbohydrates. New representatives of macrocyclic compounds bearing two hydroxymethyl groups as side arms were prepared and their binding properties toward selected carbohydrates were evaluated.Compound 12, containing benzene-based bridges, was prepared as an analogue of the earlier developed carbohydrate receptors 1 and 2, consisting of aminopyridine- or aminopyrimidine-bearing side arms. Compounds 13 and 14, incorporating pyrrole-based bridges, were prepared as analogues of the powerful receptors 3–5. Binding properties of the new compounds were evaluated on the basis of experiments in two-phase systems, such as phase transfer of sugars from aqueous into organic solvents, and in homogeneous organic media by performing 1H NMR spectroscopic titrations, ROESY experiments and microcalorimetric titrations.
Although the replacement of aminopyridine- or aminopyrimidine-bearing side arms by hydroxy groups caused a decrease in the binding affinity, the hydroxymethyl groups used in combination with suitable bridge units were revealed to be valuable building blocks for carbohydrate receptors of this type. For instance, the combination of hydroxymethyl groups with pyrrole-bearing bridges (compounds 13) provides a powerful receptor, having the ability to complex the tested β-glucoside with an association constant K11 of about 200000 M−1 in CDCl3. Molecular modelling calculations indicated the formation of the expected hydrogen bonds and CH–π interactions between the binding partners, the existence of which was also suggested by ROESY experiments. The replacement of the pyrrole-bearing bridges by benzene-based units significantly weakened binding by a factor of about 20. Such binding behaviour of 12 is mainly a consequence of the less favourable hydrogen bonding capabilities of the benzene-based bridges and may also be a result of the tendency of 12 to form more intramolecular interactions22 (OH⋯O, NH⋯O and NH⋯π) than 13, as indicated by molecular modeling calculations (see Fig. S6 and S7†). The microcalorimetric titrations revealed that the tested compounds form enthalpy driven complexes with sugar 24 in dry CDCl3 and in a water-containing solvent and that the enthalpic driving force is partially compensated by a negative ΔS.
The present results and the previously observed binding preferences indicate that the combination of two heterocycle- or hydroxyl-bearing side arms with pyrrole-based bridges represents a particularly suitable architecture for the recognition of β-glucoside. The comparison of the binding properties of 1–5 and 12–14 revealed that for the different combinations of the side arms and bridges (combinations of X and Y units), the affinity of the considered compounds increases in the order shown in Fig. 12.
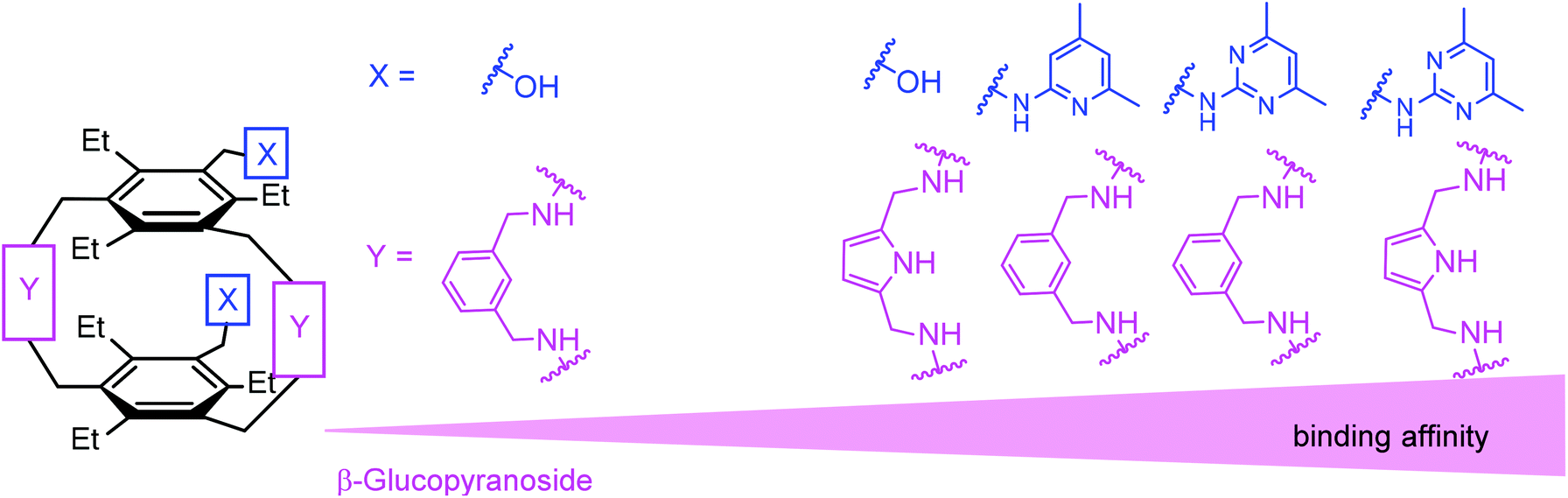 | ||
| Fig. 12 Schematic illustration of the influence of the used bridge/side arm combination (Y/X combination) on the binding strength toward β-glucopyranoside in the case of compounds consisting of both a macrocyclic building block and flexible side-arms. | ||
The performed studies show once again that the binding properties of the investigated type of receptor can be usefully modified by relatively simple structural variations. Further structural modifications, with which we hope to recognise characteristic structural features that are able to affect the binding properties in a predictable23 way, are currently in progress.
Experimental section
Analytical TLC was carried out on silica gel 60 F254 plates; column chromatography was performed on silica gel. Melting points are uncorrected. The tested monosaccharides 22–24 are commercially available. Descriptions of the binding studies in two-phase systems (extraction experiments), 1H NMR spectroscopic titrations and microcalorimetric titrations are given in the ESI.†General procedure for the synthesis of compounds 12 and 13
To a solution of 3,5-bis(aminomethyl)-1-hydroxymethyl-2,4,6-triethylbenzene (18) in dry EtOH (10 mL) the corresponding aldehyde (19 or 20) and one drop of acetic acid were added and the resulting mixture was stirred for 7 h (in the case of 12) or 4 h (in the case of 13) at 45 °C. After the mixture was cooled to room temperature, the precipitate (imine 12a or 13a) was filtered off, washed with small amounts of EtOH and suspended in dry MeOH (4 ml) or MeOH/CH2Cl2 (2:1 v/v, 6 mL; in the case of 13). To this solution NaBH4 (5 to 10 equiv.) was slowly added and the mixture was stirred at room temperature for 4 h. Then the solvent was evaporated, the residue was suspended in a mixture of H2O/CHCl3 (9:1 v/v, 10 mL) and the suspension was stirred again for another 12 h. Afterwards, the suspension was extracted with CHCl3 and the combined organic layers were washed with H2O and dried over MgSO4. The solvent was evaporated and the residue was dried under vacuum.
:1 v/v, 10 mL) and the suspension was stirred again for another 14 h. Afterwards, the suspension was extracted with CHCl3, the combined organic layers were dried over Na2SO4 and the solvent was removed under reduced pressure. The crude product was purified by column chromatography [silica gel, CHCl3/CH3OH (10:1 v/v) + 1% NH3 in MeOH (7 N)]. Product 3 was obtained as a slightly yellowish solid.
Yield: 41% (118 mg, 0.12 mmol). Mp 119–121 °C. 1H NMR (500 MHz, CDCl3): δ = 1.14 (m, 18H), 1.35 (t, J = 7.1 Hz, 12H), 2.74 (q, J = 7.3 Hz, 8H), 2.89 (q, J = 7.3 Hz, 4H), 3.72 (s, 8H), 4.04 (s, 8H), 4.29 (q, J = 7.1 Hz, 8H), 4.62 (s, 4H), 9.43 (br.s, 2H). 13C NMR (125 MHz, CDCl3): δ = 14.40, 16.52, 17.00, 22.31, 22.55, 45.96, 47.48, 58.61, 60.23, 112.10, 133.66, 134.78, 135.14, 142.39, 143.66, 165.07. HRMS (ESI) calcd for C54H79N6O10: 971.585219 [M + H]+, found 971.585277.
:1–2:1 (v/v))]. Product 17 was obtained as a white solid.
Yield: 43% (2.47 g, 4.84 mmol). Mp 129–131 °C. 1H NMR (500 MHz, CDCl3): δ = 0.99 (t, J = 7.6 Hz, 3H), 1.12 (t, J = 7.5 Hz, 6H), 2.97 (q, J = 7.5 Hz, 4H), 3.17 (q, J = 7.6 Hz, 2H), 4.75 (d, J = 5.2 Hz, 2H), 4.95 (s, 4H), 7.76–7.65 (m, 4H), 7.87–7.76 (m, 4H). 13C NMR (125 MHz, CDCl3): δ = 15.86, 16.35, 22.92, 23.44, 37.43, 59.05, 123.25, 129.58, 131.98, 134.00, 134.73, 144.70, 145.92, 168.28. MS (ESI) calcd for C31H30N2O5Na: 533.20, found 533.20 [M + Na]+. Elemental analysis, calcd for C31H30N2O5: C 72.92%, H 5.92%, N 5.49%, O 15.67%; found C 72.49%, H 6.24%, N 5.22%.
:1 v/v, 30 mL) and refluxed with hydrazine hydrate (64%, 0.18 mL) for 16 h. After cooling down to room temperature, the reaction mixture with the precipitate was stirred and a solution of 40% aq. KOH was added until the solid was completely dissolved. Then the organic layer was separated, dried over MgSO4 and the solvent was removed under vacuum. 18 was obtained as a white solid.
Yield: 91% (201 mg, 0.80 mmol). Mp 108–110 °C. 1H NMR (500 MHz, CDCl3): δ = 1.24 (m, 9H), 2.83 (q, J = 7.5 Hz, 2H), 2.87 (q, J = 7.5 Hz, 4H), 3.88 (s, 4H), 4.74 (s, 2H). 13C NMR [125 MHz, CDCl3/CD3OD 50:1 (v/v)]: δ = 16.83, 22.61, 22.85, 38.99, 58.31, 135.06, 136.37, 141.51, 142.17. MS (ESI) calcd for C15H26N2O: 250.20, found 251.15 [M + H]+, 273.15 [M + Na]+, 289.14 [M + K]+, 501.41 [2M + H]+.
Acknowledgements
Financial support of the Deutsche Forschungsgemeinschaft is gratefully acknowledged.References
- M. Mazik, H. Cavga and P. G. Jones, J. Am. Chem. Soc., 2005, 127, 9045–9052 CrossRef CAS PubMed.
- J. Lippe and M. Mazik, J. Org. Chem., 2013, 78, 9013 CrossRef CAS PubMed.
- J. Lippe and M. Mazik, J. Org. Chem., 2015, 80, 1427 CrossRef CAS PubMed.
- For discussions on the importance of carbohydrate–aromatic interactions, see: (a) J. L. Asensio, A. Arda, F. J. Caňada and J. Jiménez-Barbero, Acc. Chem. Res., 2013, 46, 946–954 CrossRef CAS PubMed; (b) S. Tsuzuki, T. Uchimaru and M. Mikami, J. Phys. Chem. B, 2009, 113, 5617–5621 CrossRef CAS PubMed; (c) G. Terraneo, D. Potenza, A. Canales, J. Jiménez-Barbero, K. K. Baldridge and A. Bernardi, J. Am. Chem. Soc., 2007, 129, 2890–2900 CrossRef CAS PubMed; (d) M. I. Chávez, C. Andreu, P. Vidal, N. Aboitiz, F. Freire, P. Groves, J. L. Asensio, G. Asensio, M. Muraki, F. J. Caňada and J. Jiménez-Barbero, Chem. – Eur. J., 2005, 11, 7060–7074 CrossRef PubMed; (e) J. Screen, E. C. Stanca-Kaposta, D. P. Gamblin, B. Liu, N. A. Macleod, L. C. Snoek, B. G. Davis and J. P. Simons, Angew. Chem., Int. Ed., 2007, 46, 3644–3648 CrossRef CAS PubMed; (f) S. H. Kiehna, Z. R. Laughrey and M. L. Waters, Chem. Commun., 2007, 4026–4028 RSC.
- For examples of CH–π interactions in the crystal structures of complexes formed between artificial receptors and carbohydrates, see ref. 1.
- (a) For a discussion on CH–π interactions, see: M. Nishio, Phys. Chem. Chem. Phys., 2011, 13, 13873–13900 RSC; (b) For a discussion on the importance of aromatic rings in chemical and biological recognition, see: L. M. Salonen, M. Ellermann and F. Diederich, Angew. Chem., Int. Ed., 2011, 50, 4808–4812 CrossRef CAS PubMed.
- For reviews on carbohydrate recognition with artificial receptors using noncovalent interactions, see: (a) A. P. Davis and T. D. James, in Functional Synthetic Receptors, ed. T. Schrader and A. D. Hamilton, Wiley-VCH, Weinheim, Germany, 2005, pp. 45–109 Search PubMed; (b) A. P. Davis and R. S. Wareham, Angew. Chem., Int. Ed., 1999, 38, 2978–2996 CrossRef; (c) D. B. Walker, G. Joshi and A. P. Davis, Cell. Mol. Life Sci., 2009, 66, 3177–3191 CrossRef CAS PubMed; (d) M. Mazik, Chem. Soc. Rev., 2009, 38, 935–956 RSC; (e) S. Jin, Y. Cheng, S. Reid, M. Li and B. Wang, Med. Res. Rev., 2010, 30, 171–257 CAS; (f) A. P. Davis, Org. Biomol. Chem., 2009, 7, 3629–3638 RSC; (g) S. Kubik, Angew. Chem., Int. Ed., 2009, 48, 1722–1725 CrossRef CAS PubMed; (h) M. Mazik, ChemBioChem, 2008, 9, 1015–1017 CrossRef CAS PubMed; (i) M. Mazik, RSC Adv., 2012, 2, 2630–2642 RSC.
- P. Adhikari, K. Bachhawat-Sikder, C. J. Thomas, R. Ravishankar, A. Arockia Jeyaprakash, V. Sharma, M. Vijayan and A. Surolia, J. Biol. Chem., 2001, 276, 40734–40739 CrossRef CAS PubMed.
- J. H. Naismith and R. A. Field, J. Biol. Chem., 1996, 271, 972–976 CrossRef CAS PubMed.
- For examples, see: (a) H. Lis and N. Sharon, Lectins, Kluwer Academic Publishers, Dordrecht, The Netherlands, 2003 Search PubMed; (b) H. Lis and N. Sharon, Chem. Rev., 1998, 98, 637–674 CrossRef CAS PubMed; (c) F. A. Quiocho, Pure Appl. Chem., 1989, 61, 1293–1306 CrossRef CAS; (d) W. I. Weiss and K. Drickamer, Annu. Rev. Biochem., 1996, 65, 441–473 CrossRef PubMed; (e) H. J. Gabius, The Sugar Code – Fundamentals of Glycoscience, Wiley-Blackwell, 2009 Search PubMed; (f) H.-J. Gabius, S. André, J. Jiménez-Barbero, A. Romero and D. Solis, Trends Biochem. Sci., 2011, 36, 298–313 CrossRef CAS PubMed.
- For examples of acyclic receptors bearing hydroxy groups, which we have previously studied, see: (a) M. Mazik and M. Kuschel, Eur. J. Org. Chem., 2008, 1517–1526 CrossRef CAS; (b) M. Mazik and A. König, Eur. J. Org. Chem., 2007, 3271–3276 CrossRef CAS; (c) For examples of other acyclic receptors studied by our group, see ref. 12–14.
- For examples of benzene-based receptors bearing phenanthroline, naphthyridine, quinoline, pyridine, pyridinium, quinolinium, alkylamino or oxime groups, see: (a) C. Geffert and M. Mazik, J. Org. Chem., 2013, 78, 292–300 CrossRef CAS PubMed; (b) M. Mazik and C. Geffert, Org. Biomol. Chem., 2011, 9, 2319–2326 RSC; (c) M. Mazik and C. Sonnenberg, J. Org. Chem., 2010, 75, 6416–6423 CrossRef CAS PubMed; (d) M. Mazik, A. Hartmann and P. G. Jones, Chem. – Eur. J., 2009, 15, 9147–9159 CrossRef CAS PubMed; (e) M. Mazik and A. Hartmann, J. Org. Chem., 2008, 73, 7444–7450 CrossRef CAS PubMed; (f) M. Mazik and H. Cavga, Eur. J. Org. Chem., 2007, 3633–3363 CrossRef CAS; (g) M. Mazik and H. Cavga, J. Org. Chem., 2006, 71, 2957–2963 CrossRef CAS PubMed; (h) M. Mazik, M. Kuschel and W. Sicking, Org. Lett., 2006, 8, 855–858 CrossRef CAS PubMed; (i) M. Mazik and W. Sicking, Tetrahedron Lett., 2004, 45, 3117–3121 CrossRef CAS; (j) M. Mazik and W. Sicking, Chem. – Eur. J., 2001, 7, 664–670 CrossRef CAS PubMed; (k) M. Mazik, H. Bandmann and W. Sicking, Angew. Chem., Int. Ed., 2000, 39, 551–554 CrossRef CAS; (l) M. Mazik, W. Radunz and R. Boese, J. Org. Chem., 2004, 69, 7448–7462 CrossRef CAS PubMed; (m) M. Mazik, W. Radunz and W. Sicking, Org. Lett., 2002, 4, 4579–4582 CrossRef CAS PubMed; (n) M. Mazik and A. C. Buthe, J. Org. Chem., 2007, 72, 8319–8326 CrossRef CAS PubMed; (o) M. Mazik and A. C. Buthe, Org. Biomol. Chem., 2008, 6, 1558–1568 RSC.
- For examples of carbohydrate receptors containing imidazole, benzimidazole, indole, pyrrole, pyrazole or pyrimidine groups and 2,4,6-triethyl-, trimethyl- or trimethoxybenzene scaffold, see: (a) J.-R. Rosien, W. Seichter and M. Mazik, Org. Biomol. Chem., 2013, 11, 6569–6579 RSC; (b) M. Mazik and M. Kuschel, Chem. – Eur. J., 2008, 14, 2405–2419 CrossRef CAS PubMed; (c) M. Mazik and A. Hartmann, Beilstein J. Org. Chem., 2010, 6(9) DOI:10.3762/bjoc.6.9; (d) C. Sonnenberg, A. Hartmann and M. Mazik, Nat. Prod. Commun., 2012, 7, 321–326 CAS; (e) M. Mazik and H. Cavga, J. Org. Chem., 2007, 72, 831–838 CrossRef CAS PubMed; (f) J. Lippe, W. Seichter and M. Mazik, Org. Biomol. Chem., 2015, 13, 11622 RSC; (g) N. Koch and M. Mazik, Synthesis, 2016, 48 Search PubMed.
- For examples of carbohydrate receptors consisting of a biphenyl-, diphenylmethane- or dimesitylmethane-based scaffold, see ref. 11b and: (a) M. Mazik and A. König, J. Org. Chem., 2006, 71, 7854–7857 CrossRef CAS PubMed; (b) M. Mazik and A. C. Buthe, Org. Biomol. Chem., 2009, 7, 2063–2071 RSC; (c) N. Koch, J.-R. Rosien and M. Mazik, Tetrahedron, 2014, 70, 8758–8767 CrossRef CAS.
- For examples of other macrocyclic carbohydrate receptors bearing pyrrole-based bridges, see: (a) G. Joshi and A. P. Davis, Org. Biomol. Chem., 2012, 10, 5760–5763 RSC; (b) O. Francesconi, M. Gentili, C. Nativi, A. Ardá, F. J. Cañada, J. Jimenez-Barbero and S. Roelens, Chem. – Eur. J., 2014, 20, 6081–6091 CrossRef CAS PubMed.
- G. Sudhakar, V. D. Kadam, S. Bayya, G. Pranitha and B. Jagadeesh, Org. Lett., 2011, 13, 5452–5455 CrossRef CAS PubMed and references therein.
- D. T. Gryko and M. Galęzowski, Org. Lett., 2005, 7, 1749–1752 CrossRef CAS PubMed.
- (a) C. Ke, H. Destecroix, M. P. Crump and A. P. Davis, Nat. Chem., 2012, 4, 718–723 CrossRef CAS PubMed; (b) N. P. Barwell and A. P. Davis, J. Org. Chem., 2011, 76, 6548–6557 CrossRef CAS PubMed; (c) E. Klein, M. P. Crump and A. P. Davis, Angew. Chem., Int. Ed., 2005, 44, 298–302 CrossRef CAS PubMed; (d) E. Klein, Y. Ferrand, E. K. Auty and A. P. Davis, Chem. Commun., 2007, 2390–2392 RSC; (e) Y. Ferrand, M. P. Crump and A. P. Davis, Science, 2007, 318, 619–622 CrossRef CAS PubMed; (f) Y. Ferrand, E. Klein, N. P. Barwell, M. P. Crump, J. Jiménez-Barbero, C. Vicent, G.-J. Boons, S. Ingale and A. P. Davis, Angew. Chem., Int. Ed., 2009, 48, 1775–1779 CrossRef CAS PubMed.
- (a) R. P. Bonar-Law and J. K. M. Sanders, J. Am. Chem. Soc., 1995, 117, 259–271 CrossRef CAS; (b) T. Mizutami, T. Kurahashi, T. Murakami, N. Matsumi and H. Ogoshi, J. Am. Chem. Soc., 1997, 119, 8991–9001 CrossRef.
- (a) For a recent discussion on experimental binding energies in supramolecular complexes, see: F. Biedermann and H.-J. Schneider, Chem. Rev., 2016, 116, 5216–5300 CrossRef CAS PubMed; (b) For a recent discussion on dispersive interactions in solution complexes, see: H.-J. Schneider, Acc. Chem. Res., 2015, 48, 1815–1822 CrossRef CAS PubMed.
- E. J. Toone, Curr. Opin. Struct. Biol., 1994, 4, 719–728 CrossRef CAS.
- For an analysis of the influence of intramolecular hydrogen bonds on the binding abilities of artificial receptors, see: B. Dolenský, R. Konvalinka, M. Jakubek and V. Král, J. Mol. Struct., 2013, 1035, 124–128 CrossRef.
- For a discussion on selectivity in supramolecular host–guest complexes, see: H.-J. Schneider and A. Yatsimirsky, Chem. Soc. Rev., 2008, 37, 263–277 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Descriptions of the binding studies (extraction experiments, 1H NMR spectroscopic titrations and microcalorimetric titrations). Further examples of molecular modelling calculations (Fig. S1–S3). Schematic representations of some short contacts between β-glucoside 24 and receptor 13 or 14 indicated by ROESY spectra (Fig. S4). Copies of the 1H and 13C NMR spectra of the new compounds 12–14, 17 and 18 (Fig. S5–S14). See DOI: 10.1039/c6ob01682k |
| This journal is © The Royal Society of Chemistry 2016 |