
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A trifunctional linker suitable for conducting three orthogonal click chemistries in one pot†
Astrid-Caroline
Knall
 *,
Manuel
Hollauf
,
Robert
Saf
and
Christian
Slugovc
*,
Manuel
Hollauf
,
Robert
Saf
and
Christian
Slugovc
Institute for Chemistry and Technology of Materials, NAWI Graz, Graz University of Technology, Stremayrgasse 9, 8010 Graz, Austria. E-mail: a.knall@tugraz.at
First published on 20th October 2016
Abstract
The feasibility of a one pot approach for conducting mutually orthogonal thiol-Michael addition, copper catalyzed azide–alkyne and inverse electron demand Diels–Alder click chemistry on a tri-functional substrate was demonstrated.
Click chemistry has become the catchphrase whenever conjugation of different molecules to each other is desired. Molecular biologists, and material and polymer chemists have rapidly picked up this type of chemistry and adapted it to match their needs in life sciences, polymer science and materials science.1 In many cases, these adaptions did not fulfil the well-established criteria for click chemistry,2 nevertheless, the aim – joining two different molecules in an efficient way – has been achieved. This is especially true for macromolecular chemistry, where the challenge is to be able to perform click chemistry with polymers, e.g. preparing block copolymers by using click-chemistry to link two chemically different macromolecular segments.3
In this context, a particularly intriguing target is to increase the complexity of the macromolecular architecture by being able to combine multiple click reactions to link three or more polymer chains together. This task has been previously accomplished by using a protecting group strategy and/or different catalysts but using the same click-reaction. Conceptually, it is more efficient and elegant to use orthogonal click reactions avoiding the use of these aids so that in an ideal setup,4 multiple conjugation reactions could be carried out in a single experiment by simultaneous addition of the reagents. Since this is a very desirable concept, orthogonal click chemistry has therefore been the objective of several theoretical5,6 as well as experimental studies.7–12 The most recent approaches aim at designing molecular scaffolds which allow multiple conjugations while avoiding the use of protecting groups.13,14 Triple-conjugated products could be obtained following a sequential approach. For a one-pot strategy, a yield of triple-click product of 28% after reverse-phase HPLC was obtained, which suggests interactions between the different reagents applied.14
In terms of click-chemistries, undoubtedly the copper(I)-catalysed azide–alkyne (CuAAC)15,16 and, in a more limited way, the thiol-Michael click reaction17 are the most used implementations. One recent addition to the click chemistry toolbox,18 which has emerged as an especially useful tool in polymer science and materials science, are inverse electron demand Diels–Alder (iEDDA) reactions, in which electron-rich dienophiles and electron-deficient dienes (e.g. 1,2,4,5-tetrazines) undergo a [4 + 2]-cycloaddition with subsequent elimination and oxidation steps resulting in pyridazines.19,20 A unique feature of iEDDA is that both reaction partners can be modified to influence the reaction rate.21–23 As opposed to CuAAC click chemistry, no catalysts are needed.24
Herein, we wish to report on advancing the scope of orthogonal click chemistry by performing three click reactions, namely CuAAC, thiol-Michael and iEDDA at a single substrate containing complementary functionalities for every click-reaction type in an one pot approach in high yield and purity.
First, an appropriate substrate was designed based on the following requirements: (a) straightforward preparation, (b) storage stability and, (c) most importantly, three different points of inherent reactivity with preference of participating in one of the three envisaged click reactions. With storage stability in mind (since e.g. a combination of a strained alkene (needed as reactant for iEDDA) and an azide as well as a thiol will most probably result in self reaction), the combination of a strained alkene (for iEDDA), an alkyne (for CuAAC) and an electron-deficient olefin (for thiol-Michael) in one molecule was envisaged. Azides, thiols and tetrazines, on the other hand, can be introduced quite straightforwardly to yield semitelechelic polymer chains. The synthesis of the corresponding scaffold 3 (see Scheme 1) was accomplished by ring opening of endo-5-norbornene-2,3-dicarboxylic anhydride (1) with propargyl alcohol followed by subsequent esterification of the free carboxylic acid group with 1-(2-hydroxyethyl)-1H-pyrrole-2,5-dione25 in overall 67% yield.
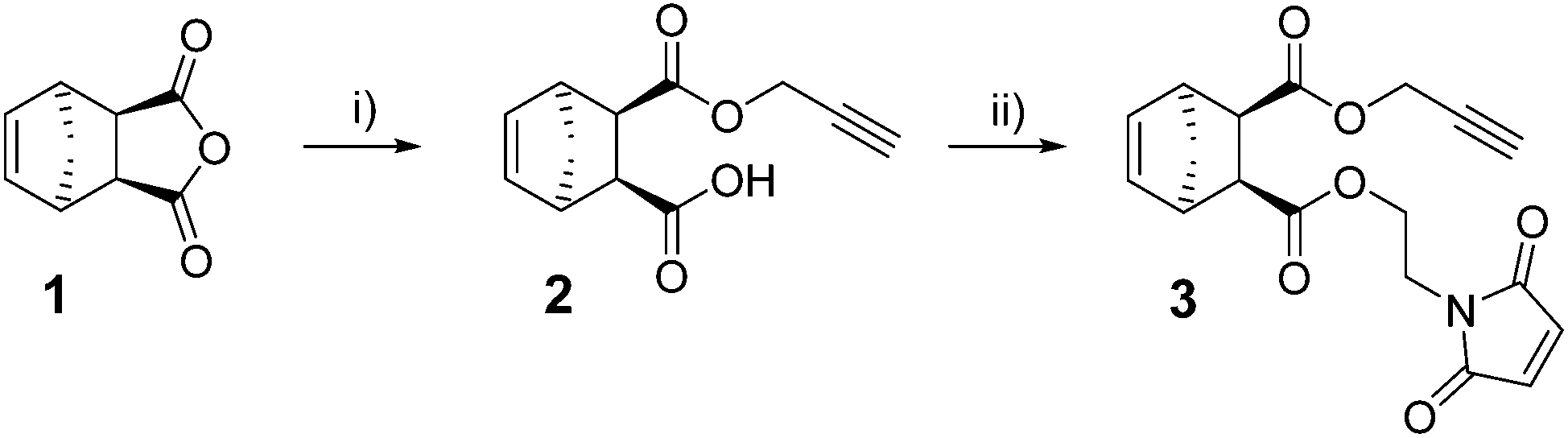 | ||
| Scheme 1 Synthesis of endo-3. (i) 1 (1 equiv.), propargyl alcohol (2 equiv.), triethylamine (Et3N, cat.), dichloromethane, reflux, 48 h, yield: 87%; (ii) 2 (1 equiv.), oxalyl chloride (1.2 equiv.), DMF (cat.), 3.5 h; then dichloromethane, 1-(2-hydroxyethyl)-1H-pyrrole-2,5-dione (1 equiv.), Et3N (1.5 equiv.), yield: 67%. | ||
Compound 3 was characterized by 1H and 13C-NMR spectroscopies and electron impact mass spectrometry. The stereochemistry was further confirmed by differential nuclear Overhauser effect measurements (see ESI†). It is worth noting that starting from exo-1 led to partial stereoinversion in the second step resulting in about 27% trans-configured by-product for exo-3 (see ESI†).
In order to learn about possible cross-selectivities with the other reactive centers, 3 was subjected to each individual click reaction (Scheme 2), using a slight excess of each reagent (1.1 equiv.). After 12 h reaction time, a simple work up (extraction with dichloromethane, drying and evaporation) was carried out. Finally, the product distribution was analysed by 1H-NMR spectroscopy (Fig. 1) and matrix-assisted laser desorption/ionization time of flight mass spectrometry (MALDI-TOF-MS, Table 1).
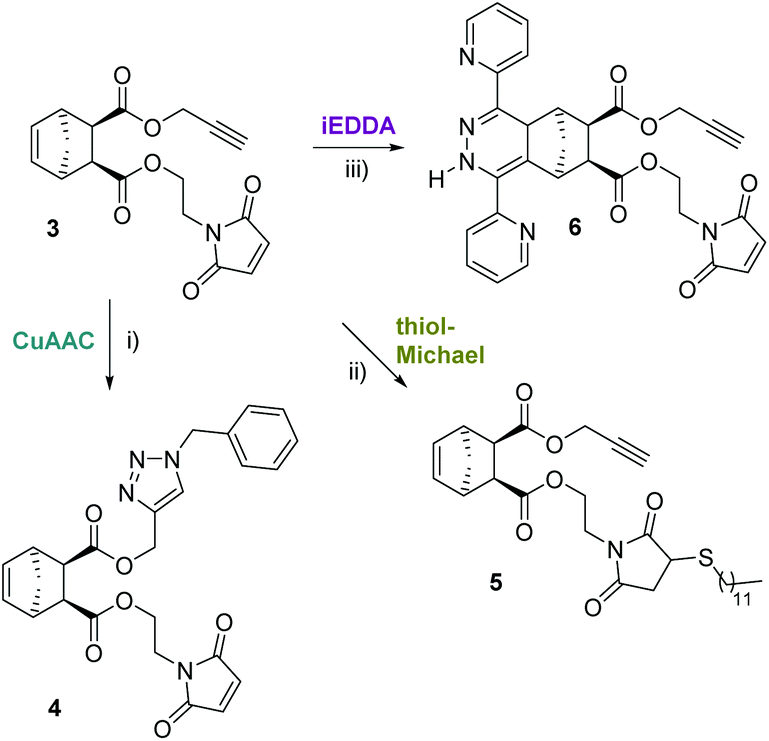 | ||
| Scheme 2 Individual “click” reactions. (i) Benzyl azide (1.1 equiv., stock solution in DMF), copper(II)sulfate, sodium ascorbate, dichloromethane/water 1/1, r.t., 12 h; (ii) dodecanethiol (1.1 equiv.), Et3N (0.1 equiv.), dichloromethane, r.t., 12 h; (iii) 3,6-di(pyridyl)tetrazine (1.1 equiv.), methanol/water, 25 °C, r.t., 12 h. | ||
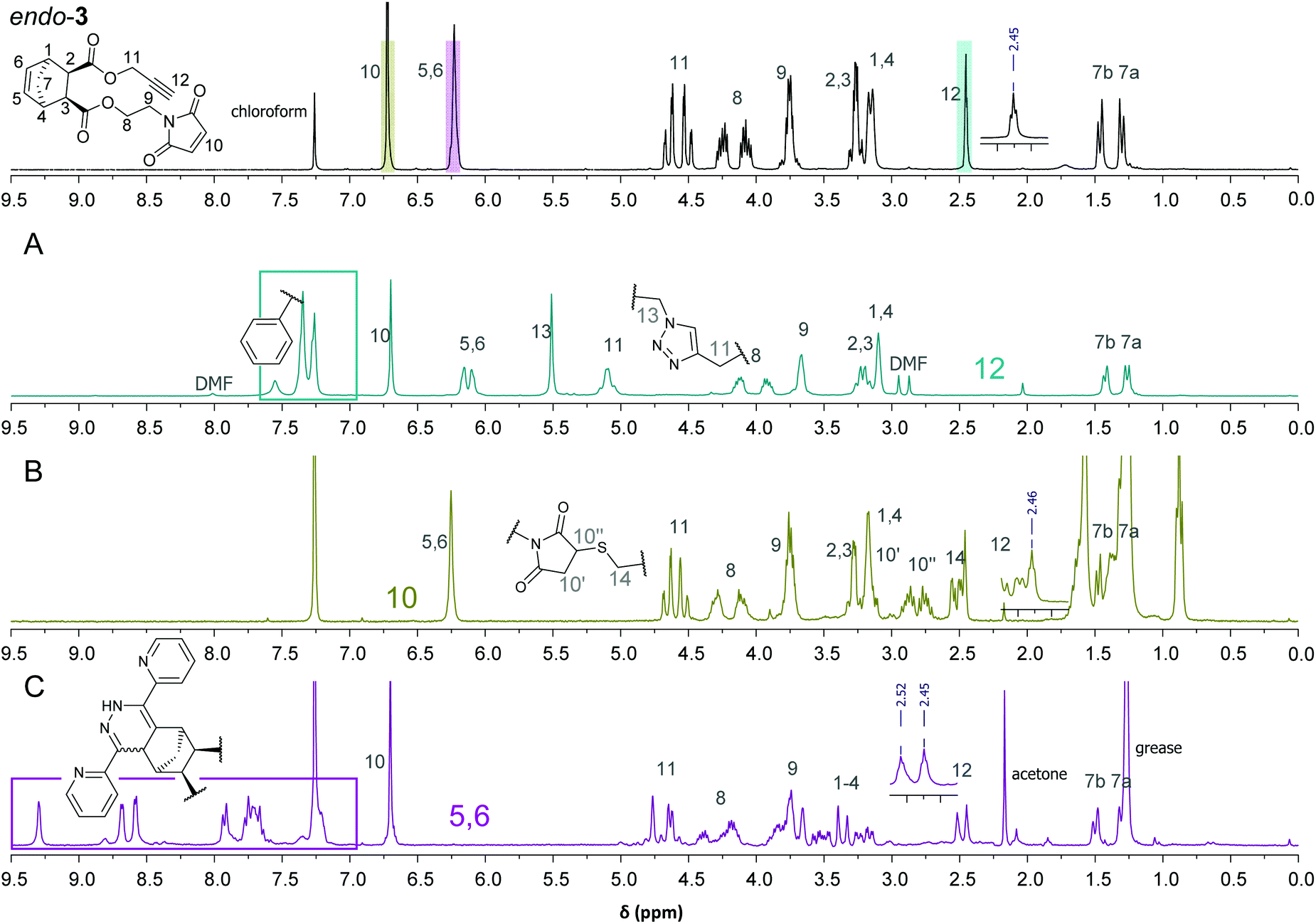 | ||
| Fig. 1 1H-NMR spectra of 3 and the individual “click” reaction products after extraction with DCM/H2O (A: CuAAC; B: thiol-Michael; C: iEDDA) and peak assignment. | ||
| Sample | Conditions | Functionalities consumed [%] | Conversion to side product [%] | Unreacted funct. remaining [%] |
|---|---|---|---|---|
| a Second thiol–en/thiol–yne reaction leading to a “double-click” product). b Partial conversion to pyridazine ([M − 2H] + H) detected. c Thiol–ene and iEDDA without azide–alkyne click product. d 95% isolated yield. | ||||
| A | CuAAC | 4 >99% | ∼8% | <1% |
| B | Thiol–ene | 5 >99% | ∼15%b | <1% |
| C | iEDDA | 6 >99% | <1% | <1%a |
| D | One pot | 7 <10% | >90% | |
| E | Seq. one pot | 7 >99%d | <1% | <1%a |
Fig. 1 (top) shows the 1H-NMR spectrum of 3 comprising the characteristic signals of the three clickable functionalities (C–H of the alkyne (H12, triplet at 2.45 ppm), C–H of the norbornene double bond (H5,6, two overlapping peaks at 6.23 ppm) and C–H of the maleimide double bond, (H10, singlet at 6.72 ppm)).
The 1H-NMR spectra of the individual “click” reactions (labelled A, B and C) were evaluated according to the method of Collins et al. treating the remaining functional groups as additives.26 The signals of H7b, H8 and/or H11 were used, as appropriate, for referencing. In the 1H-NMR spectrum of the product of the CuAAC reaction (4) the alkyne signal has entirely vanished (Fig. 1A). In addition, a new peak corresponding to the newly formed CH2-triazole group (H13) is observed at 5.51 ppm. Another indication for a successful azide–alkyne “click” reaction is a downfield shift of H11 (from 4.48–4.68 ppm to 5.09 ppm). The two H5,6 protons are shifted slightly upfield and give individual signals due to the bigger difference in their chemical environment. The other protons belonging to the norbornene skeleton and the maleimide arm are barely shifted or changed in shape. In addition, the according molecular mass was detected confirming the formation of the desired product. Using ligand-free conditions (by simply combining the azide and alkyne reactants in a biphasic water/dichloromethane mixture),27 an additional peak [632 m/z] was observed in addition to the desired main product [499 m/z], suggesting a double azide addition, which normally should only take place at elevated temperatures.10 However, in aqueous systems, strain-promoted azide addition to norbornenes has been reported.28
Treating 3 with dodecanethiol as the only reagent (Fig. 1B) resulted in preferred addition to the maleimide double bond (5) as indicated by the absence of the H10 peak at 6.70 ppm. Furthermore, new signals at 2.48–2.55 (H14, thioether), 2.67–2.93 (H′10) and 3.09–3.15 (H′′10 which is overlapped by H1,4) indicate the formation of a maleimide thioether. Compared to the integrals for H8 and H11, the integral for H5,6 is lower than expected which suggests a thiol–ene reaction involving the norbornene double bond. This is supported by an according peak in the MALDI-TOF MS spectrum [770 m/z]. A formation of 25% of the corresponding side product can be estimated which is in line with the integral of the CH3 group of the dodecyl residue. Another possibility would be a thiol–yne reaction with the terminal alkyne, which cannot be quantified due to the overlap of A and the alkyl-CH2-S signal of the newly formed thioether. MALDI-TOF MS corroborated the formation of a double thiol addition product.
iEDDA was conducted by adding 1.1 equiv. di(pyridyl)tetrazine to 3 in the absence of other reagents and resulted in the formation of the corresponding (dihydro)pyridazines [552 m/z]. Since the dihydropyridazine product 6 can be formed in endo- and exo-configuration as well as in two different orientations, in addition to being oxidised to the corresponding pyridazine, a product mixture is observed in the proton NMR spectrum (Fig. 1C) and in the MALDI-TOF MS spectrum. For example, the peak for the terminal alkyne (H12) is still visible but split into two signals, both with the characteristic triplet shape. The signal for H10 is unchanged, which indicates selective consumption of the norbornene double bond, whereas protons H1–4 are shifted downfield due to the influence of the attached dihydropyridazine. About 90% of the product is present in the dihydropyridazine form which can be estimated from the broad peak at 9.30 ppm corresponding to the pyridazine-NH. This is in line with the MALDI-TOF MS results where peaks for masses for both pyridazine and dihydropyridazine products could be found. However, the norbornene double bond was selectively consumed. The reaction was performed in different solvent mixtures with the expected result that while the reaction proceeded smoothly in a water/methanol mixture (within 8 hours, all of 3 was consumed), it took 48 h to be completed in dichloromethane as solvent. Interestingly, no significant difference in reactivity between endo-3, the exo analogue exo-3 and ±endo,exo-5-norbornene-2,3-dicarboxylic acid dimethylester (which was used as a reference) was observed when iEDDA reactions were performed under the same conditions. This is due to the overall rate-reducing effect of electron-withdrawing substituents on norbornenes in combination with steric shielding of the norbornene double bond as previously observed by us and others.21,23
According to these results, iEDDA can be considered the most selective of all three click reactions. Since in all cases the targeted functionality was preferably consumed, a one pot-triple click approach (D) was used adopting the preferred conditions for the azide–alkyne click reaction (biphasic solvent mixture). All three reactions took place at the preferred sites, which can be confirmed by the absence of double thiol–ene or double azide click products. However, the targeted “triple-click” product 7 was only formed with a yield of less than 10% (according to NMR, Fig. S12†), and only a weak signal could be detected by MALDI-TOF MS (Fig. S13†).
The iEDDA and thiol–ene reactions both proceeded quantitatively (as indicated by the complete absence of H5,6 and H10), but the signal for H12 is still present in the 1H-NMR spectrum of D, split into two individual triplets at 2.43 and 2.51 ppm, similar to what was previously observed for the individual iEDDA reaction.
Furthermore, another weak signal showed up in the mass spectrum at 1303.57 m/z (calcd 1303.53), which can be assigned to an alkyne–alkyne coupling product (potentially catalysed by Cu(II)).10,29 Accordingly, CuAAC and iEDDA, in spite of involving orthogonally reactive groups, cannot be performed simultaneously in this particular setup. We assume that the added Cu(II) is most probably forming a coordination compound with di(pyridyl)tetrazine or the formed di(pyridyl)(dihydro)pyridazines. Indeed, a dark blue precipitate was formed immediately after adding copper(II) sulphate to the reaction. Thus, the Cu(II) species in the reaction mixture are no longer reduced to Cu(I) which could explain why the CuAAC reaction does not take place with satisfactory yield.
Therefore, a sequential one pot approach was pursued. Ideally, the iEDDA reaction (as the most selective of all three click reactions used herein) should be performed prior to the others. However, due to the aforementioned deactivation of the copper catalyst, the CuAAC reaction had to be performed first. After 1 h, full conversion was detected by thin layer chromatography and di(pyridyl)tetrazine was added. After another 2 h, the CuAAC product 3 was entirely consumed and dodecanethiol was added to the reaction mixture. The resulting reaction mixture was stirred overnight and worked up by diluting the aqueous and organic layer followed by extraction. Fig. 2 shows a MALDI-TOF MS of the crude product 7.
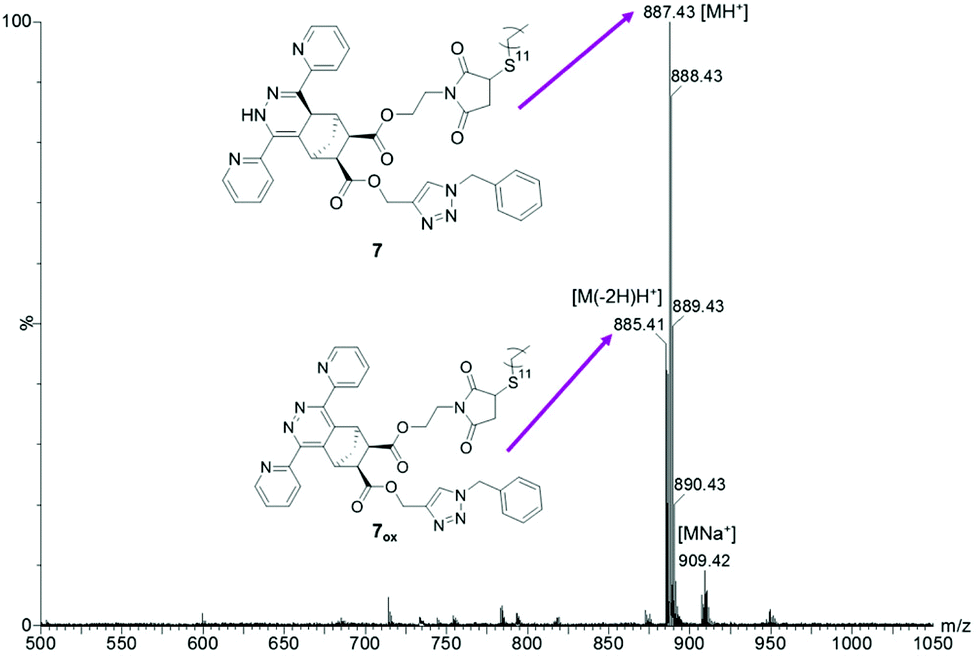 | ||
| Fig. 2 MALDI-TOF MS spectrum of endo-3 treated with benzyl azide (CuAAC), di(pyridine-2-yl)tetrazine and dodecanethiol in a sequential “one pot, triple-click” approach (Sample E). | ||
As opposed to the simultaneous approach, also the alkyne triple bond has been entirely consumed in the sequential approach due to avoiding deactivation of the copper catalyst by coordination which is proven by the absence of the H12 peak around 2.44 ppm and no evidence of cross-reactions or alkyne–alkyne coupling was found (i.e. addition of two azides or two tetrazines).
On 3 the triple-click approach can be considered successful since it is possible to perform all three reactions without using any protecting groups, different catalysts or purification steps in between. By selecting appropriate conditions and performing the “click” reactions in the correct order, all functional groups could be selectively addressed leading to the desired product in good conversion and purity. This makes 3 and similar building blocks suitable core-units for structures requiring multiple conjugation reactions, such as miktoarm terpolymers.
Financial support from the Austrian Science Fund (FWF): [T 578-N19] is gratefully acknowledged. We thank Petra Kaschnitz for performing the differential nuclear Overhauser experiments.
Notes and references
- A. S. Goldmann, M. Glassner, A. J. Inglis and C. Barner-Kowollik, Macromol. Rapid Commun., 2013, 34, 810 CrossRef CAS PubMed.
- C. Barner-Kowollik, F. E. Du Prez, P. Espeel, C. J. Hawker, T. Junkers, H. Schlaad and W. Van Camp, Angew. Chem., Int. Ed., 2011, 50, 60 CrossRef CAS PubMed.
- P. Espeel and F. E. Du Prez, Macromolecules, 2015, 48, 2 CrossRef CAS.
- C.-H. Wong and S. C. Zimmermann, Chem. Commun., 2013, 49, 1679 RSC.
- M. K. Narayanam, Y. Liang, K. N. Houk and J. M. Murphy, Chem. Sci., 2016, 7, 1257 RSC.
- Y. Liang, J. L. Mackey, S. A. Lopez, F. Liu and K. N. Houk, J. Am. Chem. Soc., 2012, 134, 17904 CrossRef CAS PubMed.
- A. Meyer, J.-J. Vasseur and F. Morvan, Eur. J. Org. Chem., 2013, 465 CrossRef CAS.
- J. M. Spruell, M. Wolffs, F. A. Leibfarth, B. C. Stahl, J. Heo, L. A. Connal, J. Hu and C. J. Hawker, J. Am. Chem. Soc., 2011, 133, 16698 CrossRef CAS PubMed.
- C. F. Hansell and R. K. O'Reilly, ACS Macro Lett., 2012, 1, 896 CrossRef CAS.
- R. J. Williams, I. A. Barker, R. K. O'Reilly and A. P. Dove, ACS Macro Lett., 2012, 1, 1285 CrossRef CAS.
- J. D. Thomas, H. Cui, P. J. North, T. Hofer, C. Rader and T. R. Burke, Bioconjugate Chem., 2012, 23, 2007 CrossRef CAS PubMed.
- D. M. Beal and L. H. Jones, Angew. Chem., Int. Ed., 2012, 51, 6320 CrossRef CAS PubMed.
- G. Clavé, H. Volland, M. Flaender, D. Gasparutto, A. Romieu and P.-Y. Renard, Org. Biomol. Chem., 2010, 8, 4329 Search PubMed.
- G. Viault, S. Dautrey, N. Maindron, J. Hardouin, P.-Y. Renard and A. Romieu, Org. Biomol. Chem., 2013, 11, 2693 CAS.
- J. E. Hein and V. V. Fokin, Chem. Soc. Rev., 2010, 39, 1302–1315 RSC.
- E. Haldón, M. C. Nicasio and P. J. Pérez, Org. Biomol. Chem., 2015, 13, 9528 Search PubMed.
- D. P. Nair, M. Podgórski, S. Chatani, T. Gong, W. Xi, C. R. Fenoli and C. N. Bowman, Chem. Mater., 2014, 26, 724 CrossRef CAS.
- R. K. Iha, K. L. Wooley, A. M. Nyström, D. J. Burke, M. J. Kade and C. J. Hawker, Chem. Rev., 2009, 109, 5620 CrossRef CAS PubMed.
- A.-C. Knall and C. Slugovc, Chem. Soc. Rev., 2013, 42, 5131 RSC.
- A.-C. Knall, S. Kovačič, M. Hollauf, D. Reishofer, R. Saf and C. Slugovc, Chem. Commun., 2013, 66, 7325 RSC.
- M. Vrabel, P. Kölle, K. M. Brunner, M. J. Gattner, V. López-Carrillo, R. de Vivie-Riedle and T. Carell, Chem. – Eur. J., 2013, 19, 13309 CrossRef CAS PubMed.
- D. Wang, W. Chen, Y. Zheng, C. Dai, K. Wang, B. Ke and B. Wang, Org. Biomol. Chem., 2014, 12, 3950 CAS.
- A.-C. Knall, M. Hollauf and C. Slugovc, Tetrahedron Lett., 2013, 55, 4763 CrossRef PubMed.
- N. K. Devaraj and R. Weissleder, Acc. Chem. Res., 2011, 44, 816 CrossRef CAS PubMed.
- M. Schaefer, N. Hanik and A. F. M. Kilbinger, Macromolecules, 2012, 45, 6807 CrossRef CAS.
- K. D. Collins and F. Glorius, Nat. Chem., 2013, 5, 597 CrossRef CAS PubMed.
- B.-Y. Lee, S. R. Park, H. B. Jeon and K. S. Kim, Tetrahedron Lett., 2006, 47, 5105 CrossRef CAS.
- J. W. Wijnen, R. A. Steiner and J. B. F. N. Engberts, Tetrahedron Lett., 1995, 36, 5389 CrossRef CAS.
- X. Jia, K. Yin, C. Li, J. Li and H. Bian, Green Chem., 2011, 13, 2175 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures and additional NMR and mass spectra. See DOI: 10.1039/c6ob02182d |
| This journal is © The Royal Society of Chemistry 2016 |