
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Stereospecific prenylation of tryptophan by a cyanobacterial post-translational modification enzyme†
Masahiro
Okada
*,
Tomotoshi
Sugita
,
Kohei
Akita
,
Yu
Nakashima
,
Tian
Tian
,
Chang
Li
,
Takahiro
Mori
and
Ikuro
Abe
*
Graduate School of Pharmaceutical Sciences, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo, 113-0033, Japan. E-mail: abei@mol.f.u-tokyo.ac.jp
First published on 15th September 2016
Abstract
Prenylation is a key post-translational reaction to increase the structural diversity and bioactivity of peptides and proteins. Until now, only one post-translational modification enzyme, ComQ, has been identified to mediate the prenylation of a tryptophan residue in ribosomally synthesized peptides. Here, we report the in vitro characterization of KgpF, a novel prenyltransferase which transfers dimethylallyl moieties to tryptophan residues during kawaguchipeptin A biosynthesis. The stereospecific prenylation by KgpF was determined by a combination of in vitro dimethylallylation of Fmoc-tryptophan by KgpF and chemical synthesis of dimethylallylated Fmoc-tryptophan diastereomers. KgpF modified the tryptophan derivative with a dimethylallyl group at the 3 position of its indole ring, resulting in the formation of a tricyclic structure with the same scaffold as prenylation by ComQ, but with the opposite stereochemistry.
Introduction
Numerous indole alkaloids are synthesized through tryptophan intermediates prenylated at each possible position of the indole ring.1–4 The prenylations of tryptophan contribute to the structural diversity and bioactivities of natural products. Indeed, several aromatic prenyltransferases have been identified as prenyl tryptophan synthases, and they are members of the ABBA prenyltransferase family that share a common structural motif known as the ABBA fold.5 In contrast, relatively few modification enzymes are known to mediate C–C bond formation for ribosomally synthesized and post-translationally modified peptides (RiPPs), even though new classes of post-translational modification enzymes have been reported in recent years.6,7 We first demonstrated post-translational prenylation of a tryptophan residue in a quorum sensing pheromone, ComX pheromone, produced by Bacillus subtilis and related bacilli.8 The tryptophan residue of ComX is modified by ComQ with either a geranyl or a farnesyl group at the gamma position, resulting in the formation of a tricyclic structure, including a newly formed pyrrolidine ring (Fig. 1).9–11 ComQ shows moderate similarity to isoprenyldiphosphate synthase, but no similarity to the ABBA family of aromatic prenyltransferases. Notably, in order to confirm the stereospecific prenylation, the chemical synthesis of the ComX pheromone and its diastereomers was required because the stereochemistry of the modified tryptophan residue could not be determined by NMR analyses of the ComX pheromone, such as NOESY and ROESY measurements.9–12 | ||
| Fig. 1 Chemical structure of the modified tryptophan residues. | ||
Another RiPP containing prenylated tryptophan residues, kawaguchipeptin A (1), was identified from the cyanobacterium Microcystis aeruginosa NIES-88 (Fig. 2).13 Kawaguchipeptin A (1), together with kawaguchipeptin B, are members of the cyanobactin family (cyclic RiPPs produced by cyanobacteria).14 Kawaguchipeptins A (1) and B are cyclic undecapeptides with very similar amino acid sequences. However, in contrast to kawaguchipeptin B, which possesses only L-amino acid residues (cyclic-[WLNGDNNWSTP]), kawaguchipeptin A (1) contains one D-leucine and two dimethylallylated tryptophan residues.13,14 Notably, it was suggested that the two tryptophan residues in 1 are modified with a dimethylallyl group at the C-3 position of their indole rings, resulting in the same scaffold as that of the ComX pheromone, but in the opposite configuration (Fig. 1). However, the stereoconfiguration of C-3 in 1 has not been assigned unambiguously, because the stereochemistry of 1 was determined only by comparing the coupling constants of NMR spectra with those of similar synthetic prenylated tryptophan compounds.13 Here, we report the stereoconfiguration of the modified tryptophan residues, solved by using a combination of in vitro dimethylallylation of protected tryptophan derivatives and the chemical synthesis of the dimethylallylated Fmoc-tryptophan, Fmoc-Trp*(β-Pre) (2b), according to our strategy for the structure determination of ComX pheromones.9–12
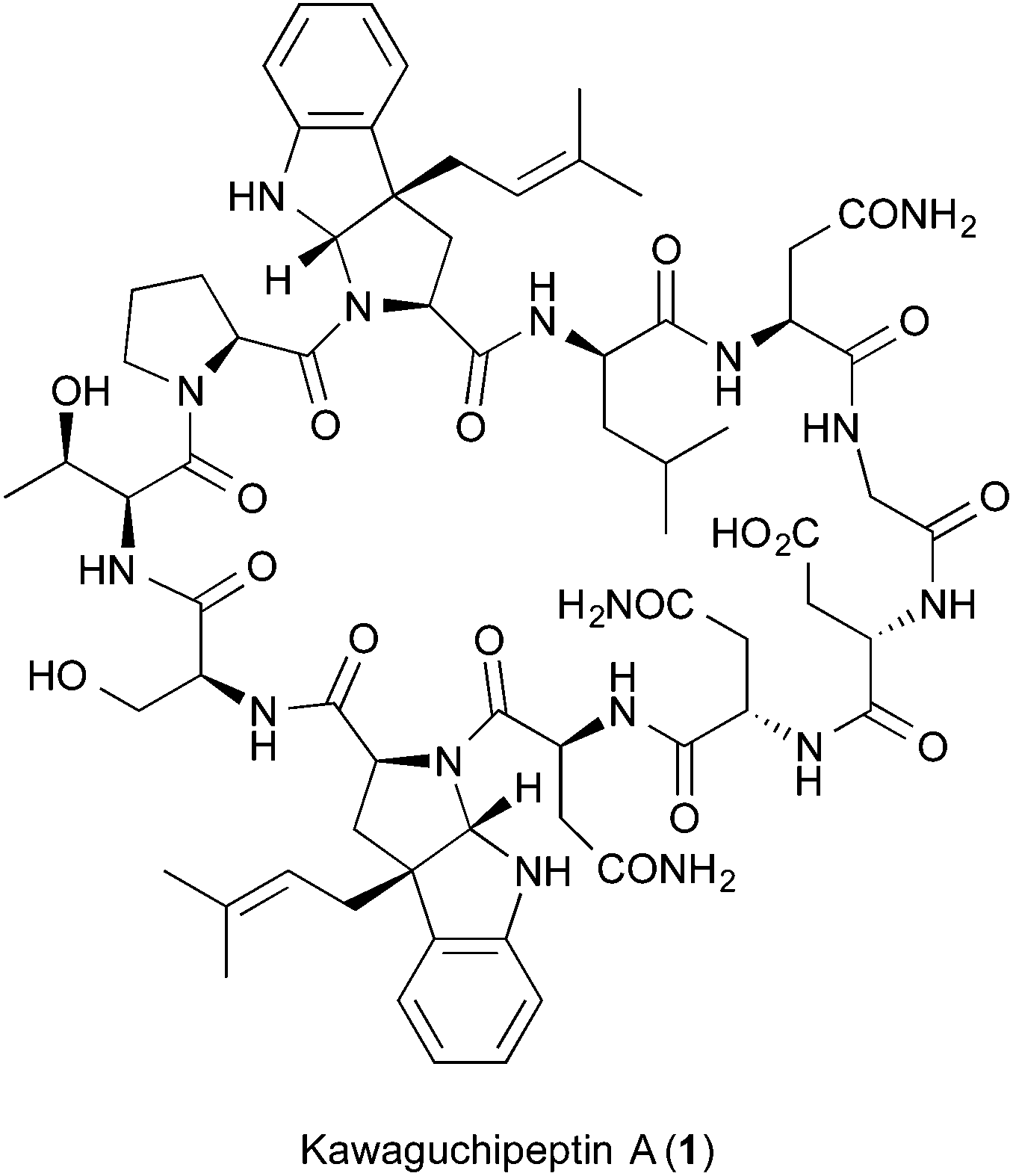 | ||
| Fig. 2 Chemical structure of kawaguchipeptin A (1). | ||
Results and discussion
In order to investigate the biosynthetic enzymes responsible for 1, we first performed genome sequencing of the kawaguchipeptin A producing strain, M. aeruginosa NIES-88. As a result, we identified approximately 7 kb of a gene cluster flanked by two genes encoding two proteases at each end, which is an indicator of a cyanobactin biosynthetic gene cluster (Fig. S1†). A short peptide possessing the amino acid sequence of kawaguchipeptins [WLNGDNNWSTP] was encoded in this region. In addition, an ABBA prenyltransferase was encoded and located adjacent to the gene in this region. These results suggested that the gene cluster is responsible for kawaguchipeptin biosynthesis, and the ABBA prenyltransferase, KgpF, mediates the prenylation of the tryptophan residues as well as other prenyltransferases for tyrosine, serine, or threonine residues reported previously in the biosynthesis of cyanobactins.15,16 Very recently, Parajuli et al. have independently identified the kawaguchipeptin gene cluster, named kgpA–G, in M. aeruginosa NIES-88, and demonstrated its involvement in kawaguchipeptin production in vivo.17 Although they also reported the in vitro prenylation of several cyclic or linear peptides with dimethylallyldiphosphate (DMAPP) by KgpF, the chemical structures of the modified tryptophan residues were never determined, much less the absolute configurations.17 To confirm the stereospecific prenylation of the tryptophan residues in 1 by KgpF, we investigated the in vitro prenylation of several protected tryptophan derivatives and precursor peptides. To our delight, we found that KgpF can prenylate Fmoc-tryptophan; however, prenylation was not observed at all when tripeptides [PWL] or [NWS], tryptophan, Boc-tryptophan, tryptophan methyl esters, Fmoc-Trp-OCH3 or Trp-OCH3, were used as substrates. In addition, Fmoc-tryptophan could not be modified by KgpF when GPP or FPP was used as an extender (Table 1).| Substrate | Extender | Product |
|---|---|---|
| a Others have independently confirmed the same result.16 | ||
| Pro-Trp-Leu | DMAPP | Not detected |
| Asn-Trp-Ser | DMAPP | Not detected |
| Trp | DMAPP | Not detected |
| Trp-OCH3 | DMAPP | Not detected |
| Boc-Trp | DMAPP | Not detecteda |
| Fmoc-Trp | DMAPP | 2b |
| Fmoc-Trp-OCH3 | DMAPP | Not detected |
| Fmoc-Trp | GPP | Not detected |
| Fmoc-Trp | FPP | Not detected |
To accurately confirm the stereoconfiguration of the prenylation product, we synthesized Fmoc-protected C-3 dimethylallylated L-tryptophan diastereomers, Fmoc-Trp*(α-Pre) (2a) and Fmoc-Trp*(β-Pre) (2b), according to the synthesis of the ComX pheromone and its diastereomers (Scheme 1).9–12,18 Dimethylallylation of the tricyclic compound 3 was accomplished by treating it with NaH and 3,3-dimethylallyl bromide to give predominantly the C-dimethylallylated compounds 4a and 4b, as an inseparable mixture of diastereomers with a ratio of 1.0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.6 (4a:4b, corresponding to R:S) in a high yield. Reductive cleavage of the benzoyl group of 4a and 4b was chemoselectively achieved by diisobutylaluminium hydride (DIBAL), although the reason for this highly selective reduction in the presence of the methyl ester was unknown. The diastereomers were separated by silica gel column chromatography to give analytically pure compounds 5a and 5b, and the stereochemistry was definitively determined by NOE analysis of 5a (Fig. S4†). However, due to the instability of 5a, the mixture was used in the next step reaction without purification. After Fmoc protection of 5a and 5b to give 6a and 6b, respectively, chemoselective reduction of the imines in 6a and 6b was achieved by lithium borohydride to give 7a and 7b, respectively, and then deprotection of Fmoc afforded the diastereomixture of 8a and 8b. The diastereomers were carefully separated by silica gel column chromatography to give the optically pure esters 8a and 8b. Although it was rather difficult to accurately determine the stereochemistry of 8a (or 8b) by NOE, except for the cis annulation, the small-scale synthesis of 8a (or 8b) using pure 5a (or 5b) as a reactant has made it possible for us to identify the absolute configurations of 8a and 8b. Finally, hydrolysis of ester 8a (or 8b), and successive Fmoc protection afforded the desired compound 2a (or 2b). The chemical structures were scarcely determined by the NMR analyses of 2a and 2b, because the 1H-NMR spectra of 2a and 2b were observed as 1:1 equilibrium mixtures between N-conformers, respectively (Fig. S14 and S15†). Therefore, due to the unambiguous confirmation of the synthesis of 2a (or 2b) without racemization, the small-scale synthesis of 8a (or 8b) using pure 2a (or 2b) via methyl esterification and Fmoc deprotection was carried out (data not shown).
:0.6 (4a:4b, corresponding to R:S) in a high yield. Reductive cleavage of the benzoyl group of 4a and 4b was chemoselectively achieved by diisobutylaluminium hydride (DIBAL), although the reason for this highly selective reduction in the presence of the methyl ester was unknown. The diastereomers were separated by silica gel column chromatography to give analytically pure compounds 5a and 5b, and the stereochemistry was definitively determined by NOE analysis of 5a (Fig. S4†). However, due to the instability of 5a, the mixture was used in the next step reaction without purification. After Fmoc protection of 5a and 5b to give 6a and 6b, respectively, chemoselective reduction of the imines in 6a and 6b was achieved by lithium borohydride to give 7a and 7b, respectively, and then deprotection of Fmoc afforded the diastereomixture of 8a and 8b. The diastereomers were carefully separated by silica gel column chromatography to give the optically pure esters 8a and 8b. Although it was rather difficult to accurately determine the stereochemistry of 8a (or 8b) by NOE, except for the cis annulation, the small-scale synthesis of 8a (or 8b) using pure 5a (or 5b) as a reactant has made it possible for us to identify the absolute configurations of 8a and 8b. Finally, hydrolysis of ester 8a (or 8b), and successive Fmoc protection afforded the desired compound 2a (or 2b). The chemical structures were scarcely determined by the NMR analyses of 2a and 2b, because the 1H-NMR spectra of 2a and 2b were observed as 1:1 equilibrium mixtures between N-conformers, respectively (Fig. S14 and S15†). Therefore, due to the unambiguous confirmation of the synthesis of 2a (or 2b) without racemization, the small-scale synthesis of 8a (or 8b) using pure 2a (or 2b) via methyl esterification and Fmoc deprotection was carried out (data not shown).
 | ||
| Scheme 1 Synthesis of Fmoc-protected prenylated tryptophans. | ||
The stereochemistry of the enzyme reaction was determined by comparing the retention times and mass fragmentations obtained from LC-MS with those of the optically pure synthetic standards 2a and 2b. The enzymatic product was consistent with 2b, but not with 2a, in LC-MS analysis (Fig. 3 and S16†). These results clearly demonstrated that KgpF modified Fmoc-tryptophan with a dimethylallyl group at the 3 position of its indole ring, resulting in the formation of a tricyclic structure with the same scaffold as ComX pheromones, but with the opposite stereochemistry.
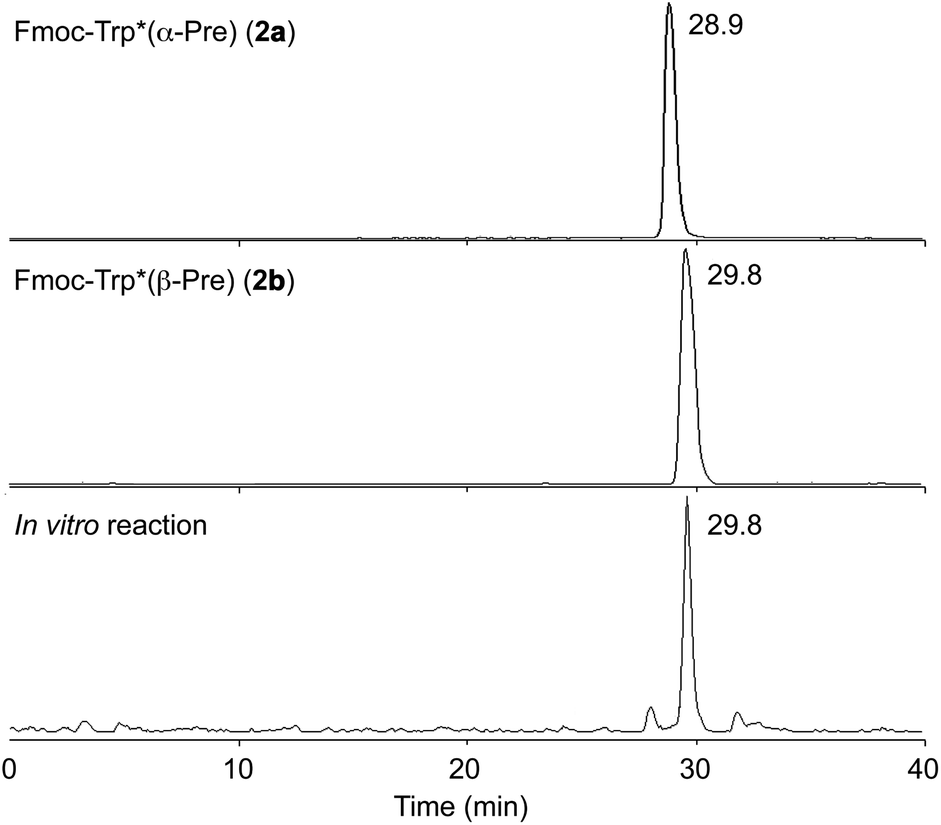 | ||
| Fig. 3 LC-MS analysis of in vitro reaction of Fmoc-Trp with KgpF. Extracted ion chromatograms (positive) of synthetic 2a (top), synthetic 2b (middle), and the in vitro reaction product (bottom) at m/z 495. | ||
In contrast to typical post-translational modifications, a specific amino acid sequence adjacent to the core peptide for directing KgpF is unlikely to be required. In addition, a specific amino acid motif in the core peptide is unlikely to be required for prenylation by KgpF, because there is no similarity between the sequences surrounding the two tryptophan residues prenylated by KgpF in kawaguchipeptin B. However, a recent paper reports that KgpF only modified the internal tryptophan residue of the two tryptophan residues in a linear undecapeptide [WLNGDNNWSTP], which possesses the same sequence as kawaguchipeptins from the N-terminal tryptophan, to the C-terminal proline.17 Considering the results together with our findings, N-terminal tryptophan residues and tryptophan derivatives with a free amino group are not recognized as a substrate by KgpF. More importantly, only one amino acid residue, Fmoc-tryptophan, was prenylated with DMAPP by KgpF. The substrate specificity of KgpF is similar to an ABBA prenyltransferase, LynF, for the biosynthesis of the cyanobactin, prenylagaramide B.16 LynF mediated the prenylation of not only a tyrosine residue of the original cyclic peptide, but also N-terminus protected tyrosine in the in vitro reaction.16 In a structural model of KgpF using the structures of a cyanobactin synthase, PatF (PDB ID codes 4BG2), and a dimethylallyl tryptophan synthase (PDB ID codes 3I4X) from Aspergillus fumigatus,19,20 the N-terminal Fmoc group of Fmoc-tryptophan was bound with aromatic amino acid residues, Tyr69 and Trp137 in KgpF (Fig. 4). It was suggested that these residues are important for its ability to bind Fmoc-tryptophan (Scheme 2). To obtain more detailed information about this enzyme, crystal structural studies of KgpF are needed in addition to in vitro prenylation studies.
 | ||
| Fig. 4 A structural model of KgpF generated by using SWISS MODEL. The N-terminal Fmoc group of Fmoc-tryptophan was bound with aromatic amino acid residues in KgpF. The two aromatic amino acid residues, Tyr69 and Trp137, are highlighted in yellow. | ||
 | ||
| Scheme 2 Plausible mechanism for the synthesis of 2b by KgpF. | ||
Since the indole side chain of a tryptophan residue is an efficient nucleophile for C–C bond formation, most patterns of tryptophan posttranslational modifications involve C–C bond formation, even though there are fewer classes of posttranslational modifications for tryptophan than for other amino acid residues.9–11,13,21,22 Among the modification enzymes, KgpF accepted even Fmoc-tryptophan as a substrate and mediated regioselective and stereoselective dimethylallylation of Fmoc-tryptophan. Therefore, KgpF may exhibit relaxed substrate specificity toward diverse tryptophan residues in peptides, which in turn would expand the chemical diversity of peptides with prenylated tryptophan among the residues. KgpF may have great potential as a promising enzyme for the synthesis of prenylated tryptophan derivatives and peptide libraries containing prenylated tryptophan residues.
Conclusion
In this study, we analyzed the in vitro prenylation of tryptophan derivatives by KgpF. The stereospecific prenylation by KgpF was determined using a combination of in vitro dimethylallylation of Fmoc-tryptophan and chemical synthesis of dimethylallylated Fmoc-tryptophan diastereomers. KgpF modified the tryptophan derivative with a dimethylallyl group at the 3 position of its indole ring, resulting in the formation of a tricyclic structure with the same scaffold as ComX pheromones, but with the opposite stereochemistry. Since KgpF exhibited relaxed substrate specificity, KgpF may attract attention as a target for enzyme engineering for the synthesis of prenylated tryptophan derivatives.Experimental
Genomic analysis of Microcystis aeruginosa NIES-88
M. aeruginosa NIES-88 was obtained from the National Institute for Environmental Studies (Japan) collection. The M. aeruginosa NIES-88 genomic DNA was isolated according to the previously published method.23 Sequencing of the genomic DNA was performed by an Ion PGM sequencer, with a total number of 618551 sequence reads (ca. 300 bp). These sequences were assembled de novo into contigs by using the Genious assembler (Biomatters), with the default medium sensitivity. Putative protein-coding sequences were determined by combining the prediction results from the FramePlot and Glimmer 3.02 programs into one large contig (7.4 kbp).24,25 The domain organizations were assessed by using BLASTP. The biosynthetic gene cluster of kawaguchipeptins is shown in Fig. S1.†Heterologous expression and purification of KgpF
The KgpF gene was amplified from the genomic DNA of M. aeruginosa NIES-88 with a set of primers. After purification, the gene was ligated into the pET-22b(+) vector digested with SacI and XhoI by using an In-Fusion HD Cloning Kit. The constructed plasmid was transformed into the Escherichia coli BL21(DE3) strain for heterologous overexpression. When the transformant was grown at 37 °C in Luria-Bertani medium with ampicillin at an OD600 of 0.6–0.8, the culture was cooled at 20 °C, and then IPTG (final conc. 1.0 mM) was added and incubated for 12 h at 20 °C. The cells were collected and resuspended in 100 mM Tris-HCl buffer (pH 7.5) containing 400 mM NaCl, 5 mM imidazole, and 10% (v/v) glycerol, and the cells were disrupted by ultrasonication. After centrifugation at 9000g for 20 min, the supernatant was loaded onto a Ni+-NTA-agarose column. After washing the resin with the buffer containing 15 mM imidazole, KgpF with a C-terminal His tag was eluted with the buffer containing 300 mM imidazole. The eluate was filtered through a 10 kDa MWCO filter to give KgpF solution (500 μL at 44 g L−1). The protein solution for the in vitro reaction was prepared before use. Forward primer; GGAGATTAACTGATGCCGGATATAATTGATATACC, reverse primer; GGTGGTGGTGCTCGAAGTTGGGAGAATCCCACC.In vitro assay
To a 50 mM HEPES buffer (pH 7.5) containing 5 mM MgCl2, 5 mM ZnCl2, dimethylallyl diphosphate solution (final conc. 300 μM), substrate solution (final conc. 300 μM), and KgpF solution (final conc. 200 μM) were added, and the total volume was adjusted to 100 μL with the buffer solution. After the reaction mixture was incubated for 12 h at 37 °C, it was quenched and neutralized with 100 μL of acetonitrile. After centrifugation at 12000g for 5 min, the supernatant was filtered through a membrane filter (0.45 μm) to prepare the sample for LC-MS analysis.
LC-MS analysis
LC-MS analysis was performed with a JMS-T100LP (JEOL) ESI-MS system and a 1100 series HPLC system (Agilent) with a X-select CSH C18 (Waters) column (2.1 × 150 mm). Each sample (10 μL) was injected into the LC column and eluted with a linear gradient of 58–95% CH3CN/H2O containing 0.5% acetic acid for 40 min at a flow rate of 0.1 mL min−1. The enlarged extracted ion chromatograms and mass spectrum are shown in Fig. S17.†Homology modeling
The structural model of KgpF was generated by the SWISS-MODEL web server (swissmodel.expasy.org) using the crystal structure of PatF (PDB ID; 4BG2), which belongs to the ABBA family of prenyltransferases,19 and dimethylallyl tryptophan synthase from Aspergillus fumigatus (PDB ID; 3I4X).20Acknowledgements
This work was supported in part by a Grant-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology, Japan (JSPS KAKENHI Grant Number JP15H01836, JP16H06443, and JP24688011) and the Takeda Science Foundation (to M. O.).References
- X. Yu and S.-M. Li, Methods Enzymol., 2012, 516, 259 CAS.
- T. Walsh, ACS Chem. Biol., 2014, 9, 2718 CrossRef PubMed.
- M. E. Tanner, Nat. Prod. Rep., 2015, 32, 88 RSC.
- J. A. McIntosh, M. S. Donia and E. W. Schmidt, Nat. Prod. Rep., 2009, 26, 537 RSC.
- M. Tello, T. Kuzuyama, L. Heide, J. P. Noel and S. B. Richard, Cell. Mol. Life Sci., 2008, 65, 1459 CrossRef CAS PubMed.
- L. M. Alkhalaf and K. S. Ryan, Chem. Biol., 2015, 22, 317 CrossRef CAS PubMed.
- P. G. Arnison, M. J. Bibb, G. Bierbaum, A. A. Bowers, T. S. Bugni, G. Bulaj, J. A. Camarero, D. J. Campopiano, G. L. Challis, J. Clardy, P. D. Cotter, D. J. Craik, M. Dawson, E. Dittmann, S. Donadio, P. C. Dorrestein, K. D. Entian, M. A. Fischbach, J. S. Garavelli, U. Goransson, C. W. Gruber, D. H. Haft, T. K. Hemscheidt, C. Hertweck, C. Hill, A. R. Horswill, M. Jaspars, W. L. Kelly, J. P. Klinman, O. P. Kuipers, A. J. Link, W. Liu, M. A. Marahiel, D. A. Mitchell, G. N. Moll, B. S. Moore, R. Muller, S. K. Nair, I. F. Nes, G. E. Norris, B. M. Olivera, H. Onaka, M. L. Patchett, J. Piel, M. J. Reaney, S. Rebuffat, R. P. Ross, H. G. Sahl, E. W. Schmidt, M. E. Selsted, K. Severinov, B. Shen, K. Sivonen, L. Smith, T. Stein, R. D. Sussmuth, J. R. Tagg, G. L. Tang, A. W. Truman, J. C. Vederas, C. T. Walsh, J. D. Walton, S. C. Wenzel, J. M. Willey and W. A. van der Donk, Nat. Prod. Rep., 2013, 30, 108 RSC.
- R. Magnuson, I. Solomon and A. D. Grossman, Cell, 1994, 77, 207 CrossRef CAS PubMed.
- M. Okada, I. Sato, S. J. Cho, H. Iwata, T. Nishio, D. Dubnau and Y. Sakagami, Nat. Chem. Biol., 2005, 1, 23 CrossRef CAS PubMed.
- M. Okada, H. Yamaguchi, I. Sato, F. Tsuji, J. Qi, D. Dubnau and Y. Sakagami, Biosci. Biotechnol., Biochem., 2007, 71, 1807 CrossRef CAS PubMed.
- M. Okada, H. Yamaguchi, I. Sato, F. Tsuji, D. Dubnau and Y. Sakagami, Biosci. Biotechnol., Biochem., 2008, 72, 914 CrossRef CAS PubMed.
- M. Okada, I. Sato, S. J. Cho, D. Dubnau and Y. Sakagami, Tetrahedron, 2006, 62, 8907 CrossRef CAS.
- K. Ishida, H. Matsuda, M. Murakami and K. Yamaguchi, Tetrahedron, 1996, 52, 9025 CrossRef CAS.
- K. Ishida, H. Matsuda, M. Murakami and K. Yamaguchi, J. Nat. Prod., 1997, 60, 724 CrossRef CAS PubMed.
- M. S. Donia, J. Ravel and E. W. Schmidt, Nat. Chem. Biol., 2008, 4, 341 CrossRef CAS PubMed.
- J. A. McIntosh, M. S. Donia, S. K. Nair and E. W. Schmidt, J. Am. Chem. Soc., 2011, 133, 13698 CrossRef CAS PubMed.
- A. Parajuli, D. H. Kwak, L. Dalponte, N. Leikoski, T. Galica, U. Umeobika, L. Trembleau, A. Bent, K. Sivonen, M. Wahlsten, M. Wang, E. Rizzi, G. De Bellis, J. Naismith, J. Jaspars, X. Liu, W. Houssen and D. P. Fewer, Angew. Chem., Int. Ed., 2016, 55, 3596 CrossRef CAS PubMed.
- F. Tsuji, K. Kobayashi, M. Okada, H. Yamaguchi, M. Ojika and Y. Sakagami, Bioorg. Med. Chem. Lett., 2011, 21, 4041 CrossRef CAS PubMed.
- A. F. Bent, K. Jesko, W. E. Houssen, M. C. M. Smith, M. Jaspars and J. H. Naismith, Acta Crystallogr., Sect. F: Struct. Biol. Commun., 2013, 69, 618 CAS.
- U. Metzger, C. Schall, G. Zocher, I. Unsöld, E. Stec, S.-M. Li, L. Heidea and T. Stehle, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 14309 CrossRef CAS PubMed.
- W. S. McIntire, D. E. Wemmer, D. E. A. Chistoserdov and M. E. Lindstrom, Science, 1991, 252, 817 CrossRef CAS PubMed.
- J. Hofsteenge, D. R. Müller, T. de Beer, A. Löffler, W. J. Richter and J. F. G. Vliegenthart, Biochemistry, 1994, 33, 13524 CrossRef CAS PubMed.
- N. Morin, T. Vallaeys, L. Hendrickx, L. Natalie and A. Wilmotte, J. Microbiol. Methods, 2010, 80, 148 CrossRef CAS PubMed.
- J. Ishikawa and K. Hotta, FEMS Microbiol. Lett., 1999, 174, 251 CrossRef CAS PubMed.
- A. L. Delcher, D. Harmon, S. Kasif, O. White and S. L. Salzberg, Nucleic Acids Res., 1999, 27, 4636 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ob01759b |
| This journal is © The Royal Society of Chemistry 2016 |