
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Intramolecular thermal stepwise [2 + 2] cycloadditions: investigation of a stereoselective synthesis of [n.2.0]-bicyclolactones†
Adam
Throup
,
Laurence H.
Patterson
and
Helen M.
Sheldrake
 *
*
Institute of Cancer Therapeutics, University of Bradford, Bradford, BD7 1DP, UK. E-mail: h.sheldrake@bradford.ac.uk
First published on 20th September 2016
Abstract
Fused cyclobutanes are found in a range of natural products and formation of these motifs in a straightforward and easy manner represents an interesting synthetic challenge. To this end we investigated an intramolecular variant of the thermal enamine [2 + 2] cyclisation, developing a diastereoselective intramolecular enamine [2 + 2] cyclisation furnishing δ lactone and lactam fused cyclobutenes in good yield and excellent diastereoselectivity.
Introduction
Cyclobutanes and cyclobutenes still provide a synthetic challenge1–3 particularly when fused to larger ring systems, a motif often found in natural products (Fig. 1).4–8 The photochemical [2 + 2] reactions traditionally used to access 4-membered rings can be problematic and unpredictable with a range of different products being produced; including dimers, regioisomers, and diastereomers.9 Several alternate methodologies have been developed to control the reaction outcome; for example, using photosensitising groups, or tuning the electronics of the system by using enol ethers and enones10 in combination (for a thorough review of recent developments in photochemical [2 + 2] cyclisations see Poplata et al.11). Brannock has described a thermal enamine [2 + 2] cyclisation12 between an enamine and an electron deficient Michael acceptor alkene, which controls the regiochemistry of cyclisation and prevents homo-dimerization. The diastereoselectivity of this cyclisation is controlled by the reversibility of the Michael addition and subsequent cyclisation leads to the thermodynamic trans substituted cyclobutanes as the major product.13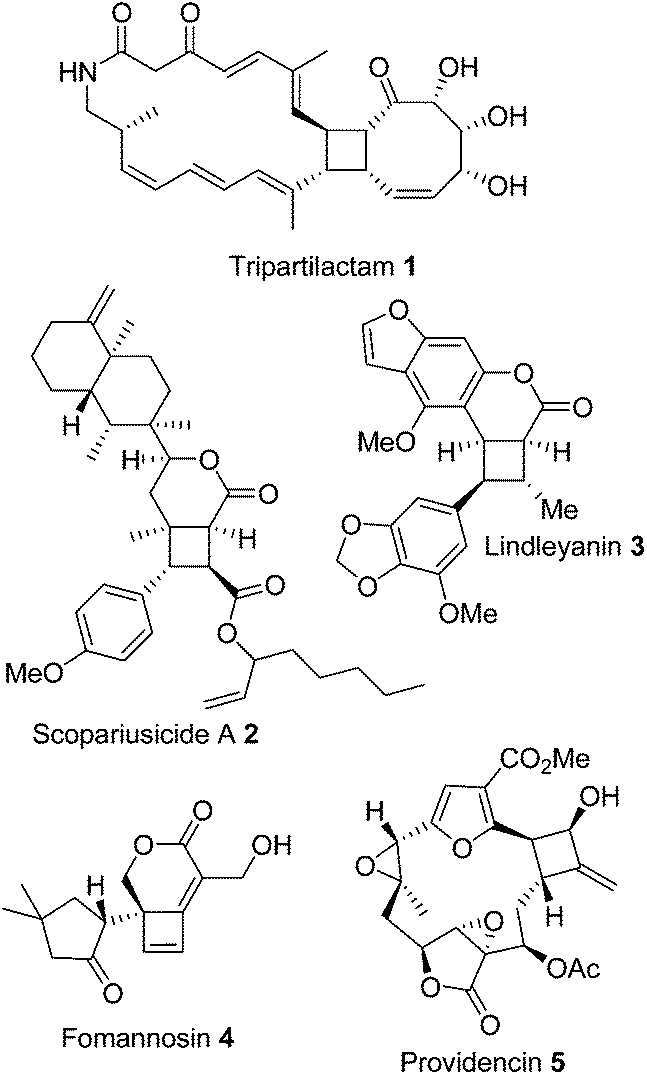 | ||
| Fig. 1 Examples of cyclobutane-containing complex natural product structures. | ||
Intramolecular variants of the photochemical [2 + 2] cyclisation have been developed and used in synthesis, but enamine [2 + 2] cyclisation has been underexploited to date and its full scope has not been explored. Intramolecular cycloaddition of ketenes or ketene-iminium salts with alkenes provides a useful route to [n.2.0] fused-cyclobutanones,14–18 however ketene cycloadditions provide a limited pattern of substituents on the cyclobutane ring and do not allow easy access to cyclobutenes. The development of an intramolecular enamine [2 + 2] cyclisation gives a complementary albeit contrasting route to functionalised fused cyclobutanes and cyclobutenes. The recent identification of the cyclobutene moiety as a suitable electrophile for targeted covalent modification of proteins19 suggests this functionality will have increasing importance in medicinal chemistry. The development of straightforward procedures for the synthesis of 4-membered rings leading to predictable product structures is therefore an important challenge.
Results and discussion
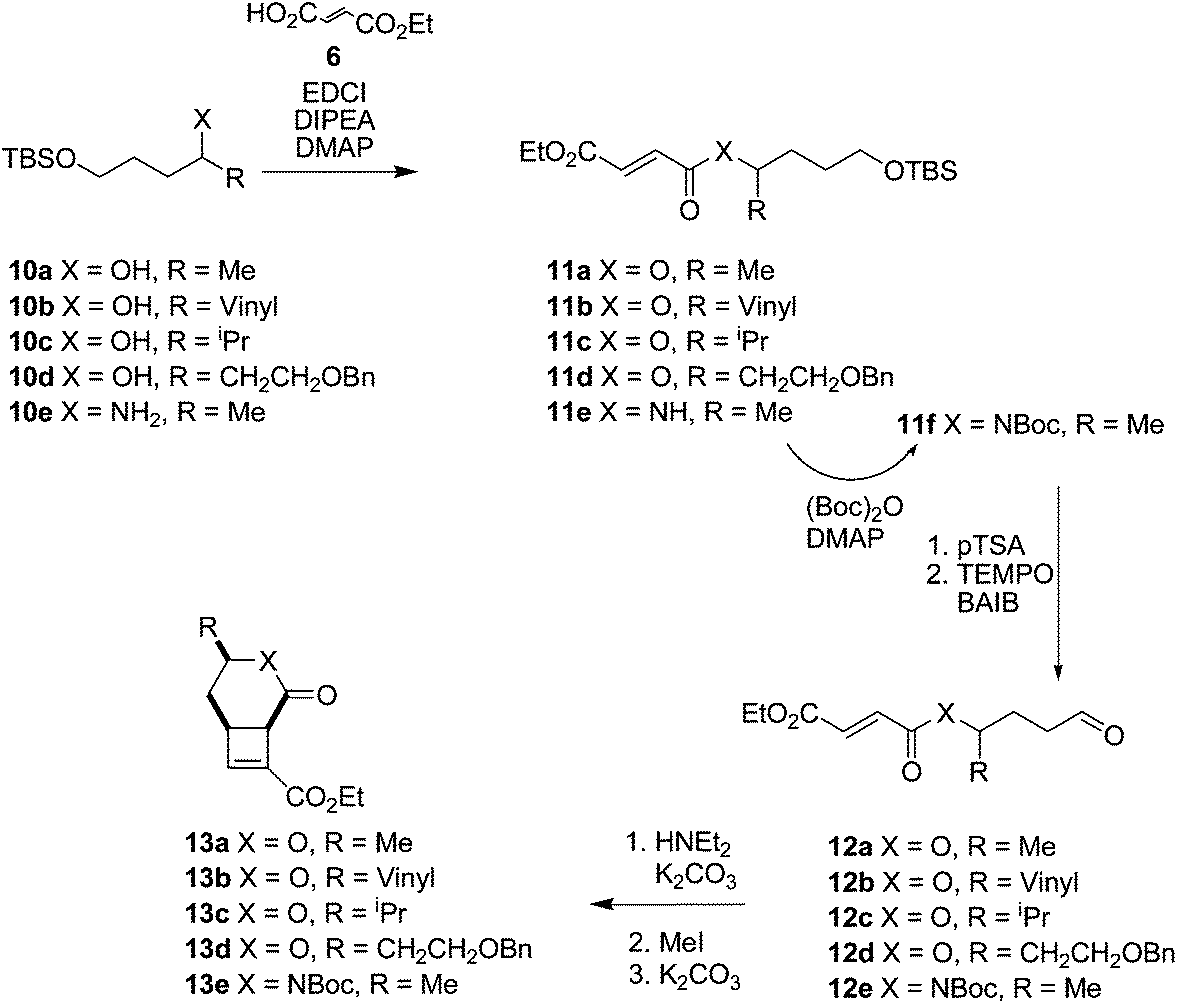
To investigate the range of ring sizes accessible by the reaction, aldehyde tethered fumarates 7a–e were synthesised from the corresponding diols (Table 1). The cyclisation precursors were treated with diethylamine in the presence of K2CO3 in acetonitrile to yield the intermediate cyclobutanes, which proved unstable and prone to hydrolysis so were not isolated. Treatment of the crude reaction mixture with MeI to give quaternary ammonium salts followed by Hoffmann elimination yielded the stable cis-fused cyclobutene (9b). The relative configuration of the ring junction was confirmed by nOe spectroscopy (see ESI†).|
|
|||
|---|---|---|---|
| Entry | Precursor | Product | Yield |
| 1 | 7a | 9a | 0 |
| 2 | 7b | 9b | 43% |
| 3 | 7c | 9c | 0 |
| 4 | 7d | 9d | 0 |
| 5 | 7e | 9e | 20% |
| 7 | 7f | 9f | 35% |
| 8 | 7g | 9g | 25% |
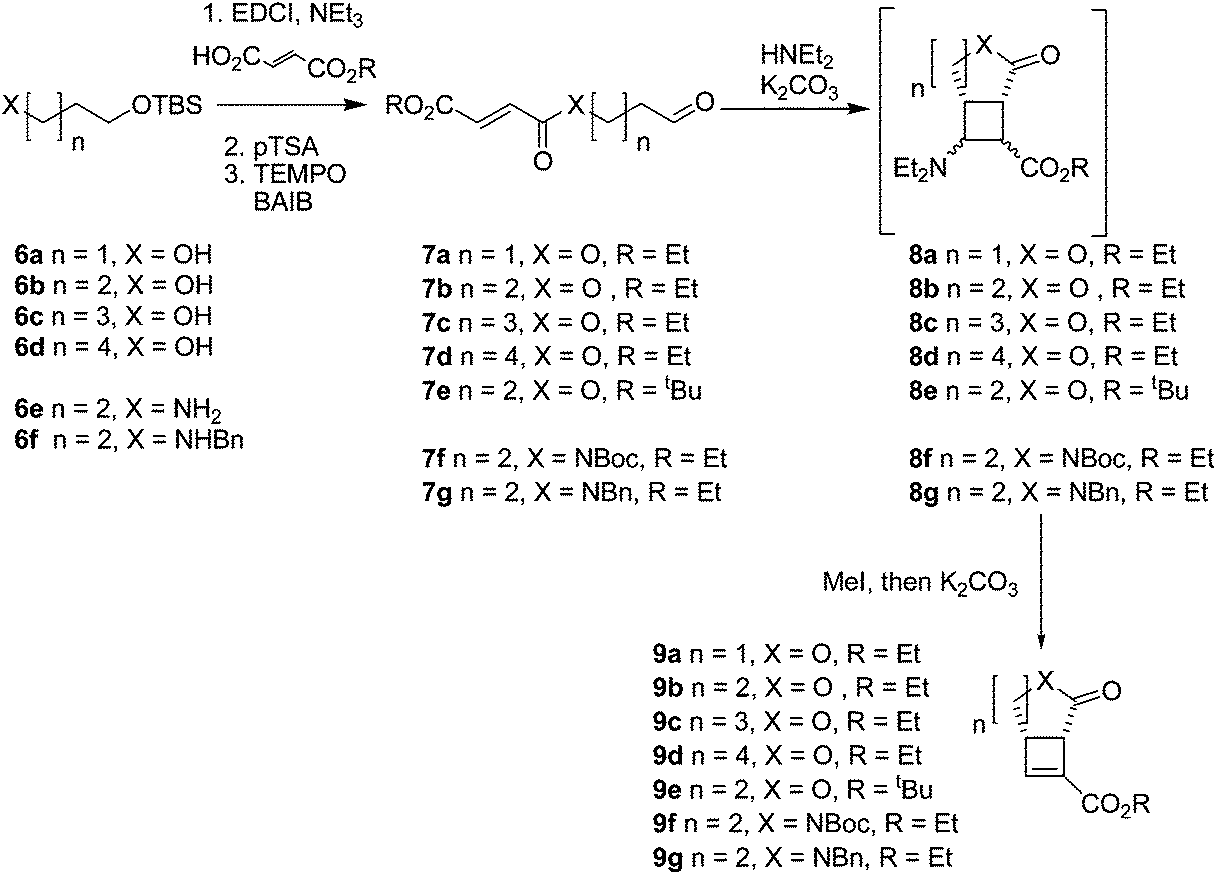
Only the 6,4 fused lactones 9b and 9e were isolated. The other length tethers failed to cyclise; in each case most of the aldehyde was consumed but the desired compounds (9a, 9c, 9d) were not isolable and the products 9 or corresponding intermediates (8a, 8c, 8d) were not observable by 1H NMR of the crude reaction mixtures. This indicated failure of the cyclisation step.
Cyclisation of 7a to form a 5,4 fused bicycle was probably prevented by the tether being too short to allow a favourable orientation of orbital overlap as the two reacting group approach one another; a conformation where the two halves of the molecule are close enough to allow cyclisation would place the π-system of the fumarate and aldehyde-derived enamine to be perpendicular to one another. Furthermore, Ghosh et al.20 have shown that a similar ester tethered system did not undergo photochemical cyclisation since the preferred conformation (7a S-cisFig. 2) holds the two ends of the chain at a distance from one another. Longer tethers (7c,d) will be affected by the entropic issues well-known for reducing efficiency of macrocycle formation. In this case, even the S-trans configuration does not bring the two ends of the molecule into sufficiently close proximity to allow a reaction. Small amounts of starting aldehyde were recovered from some reactions, along with complex mixtures of products also containing aldehyde groups (starting material was generally not recovered from successful reactions).
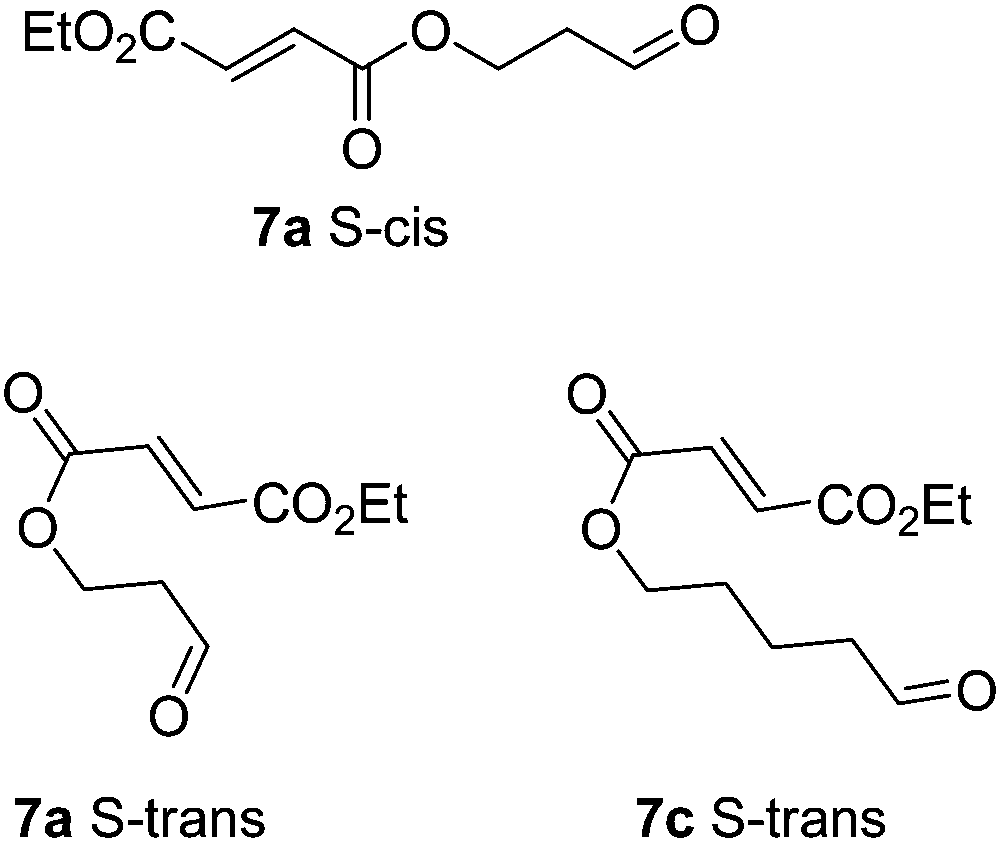 | ||
| Fig. 2 Non-reacting (S-cis) and reacting (S-trans) conformations of ester tethered cyclisation substrates. | ||
Attempts to optimise the reaction conditions were limited (Table 2) as it has already been established that this type of reaction tolerates a limited range of reagents and conditions; acetonitrile is required to stabilise the zwitterionic intermediates,12,13 (we have found that related reactions attempted in THF failed) and the presence of K2CO3 is required for cyclobutane formation.13 Decreasing the concentration of the reaction in an attempt to prevent polymerisation did not improve the yield of isolable cyclobutene products, which suggests polymerisation is not the major side reaction taking place. 7a may decompose in the presence of base by elimination of the fumarate group to yield acrolein. Michael addition of diethylamine to the starting material would reversibly prevent productive reaction of any substrate. Diethylamine was the only amine investigated which gave a successful reaction. Cyclic amines (entries 13–15) caused complete decomposition of the reaction mixture; (R)-N,α-dimethylbenzylamine did not cause cyclobutane formation, or react with the fumarate group in 7b. Chiral amine auxiliaries cannot be used to generate homochiral cyclobutanes by this method.
| Entry | Precursor | Concentration | Amine | Yield |
|---|---|---|---|---|
| a 10% aldehyde recovered. b 16% aldehyde recovered. c 35% aldehyde recovered. | ||||
| 1 | 7a | 0.045 M | Et2NH (1 eq.) | 0 |
| 2 | 7a | 0.045 M | Et2NH (2 eq.) | 0 |
| 3 | 7b | 0.050 M | Et2NH (1 eq.) | 29 (9b) |
| 4 | 7b | 0.051 M | Et2NH (2 eq.) | 37 (9b) |
| 5 | 7b | 0.066 M | Et2NH (2 eq.) | 43 (9b) |
| 6 | 7b | 0.075 M | Et2NH (2 eq.) | 41 (9b) |
| 7 | 7b | 0.034 M | Et2NH (10 eq.) | 28 (9b) |
| 8 | 7e | 0.045 M | Et2NH (2 eq.) | 20 (9e) |
| 9 | 7e | 0.097 M | Et2NH (2 eq.) | 16 (9e) |
| 10 | 7c | 0.050 M | Et2NH (2 eq.) | 0a |
| 11 | 7d | 0.032 M | Et2NH (2 eq.) | 0b |
| 12 | 7d | 0.024 M | Et2NH (2 eq.) | 0c |
| 13 | 7b | 0.050 M | Pyrrolidine (2 eq.) | 0 |
| 14 | 7b | 0.050 M | Morpholine (2 eq.) | 0 |
| 15 | 7b | 0.080 M | (R)-2-Methylpyrrolidine (2 eq.) | 0 |
| 16 | 7b | 0.080 M | (R)-N,α-Dimethylbenzylamine (2 eq.) | 0 |
Increasing the steric bulk in the substrate by switching from ethyl to tbutyl ester (7e) resulted in reduced yield of 9e after increased reaction time (5 days compared to the standard 2 days for intramolecular reaction of ethyl esters).
After the successful preparation of the lactone fused cyclobutene, synthesis of lactam fused cyclobutenes was attempted. Lactam 6,4 fused cyclobutanes have previously been accessed by photochemistry,21–23 but the majority of work involves quinolone-based structures. Lactam 5,4 fused cyclobutanes are accessible through a wider range of methods, frequently relying on prior preparation of a 1,2-cyclobutane dicarboxylic acid or photocyclisation of a maleimide.24–27 Here, amide tethered aldehydes were synthesised containing a Boc protected 7f, and benzyl protected 7g amide (the corresponding unprotected aldehyde was not accessible due to side reactions during oxidation). When subjected to the cyclisation conditions both the Boc and benzyl protected amides cyclised forming lactam fused cyclobutenes 9f,g, although in lower yields than their ester linked counterparts. The method therefore complements photochemical cyclisations, which tolerate the presence of an amide NH group.
Diastereoselective intramolecular enamine [2 + 2] cyclisations
We hypothesised that the presence of a side chain within the tether would control the way the molecule would fold into a reactive conformation thus giving diastereocontrol. Using enantiopure starting materials would therefore allow the enantiospecific synthesis of fused cyclobutenes.Cyclisation substrates 12a–e were prepared from a range of alcohols 10a–d and amine 10e by coupling with ethyl hydrogen fumarate, deprotection and oxidation to afford the required aldehydes. Cyclisation/eliminations proceeded in good to moderate yield over the 3 steps; all reactions yielded a single diastereomer (>95% de) (Table 3). This was shown to be the all cis compound by nOe spectroscopy; crosspeaks were observed between H-1 and H-6 indicating the expected cis-configuration at the ring junction, and between H-4 and H-6/H-1 indicating H-4 is on the same face of the molecule as the protons at the ring junction (e.g.Fig. 3).
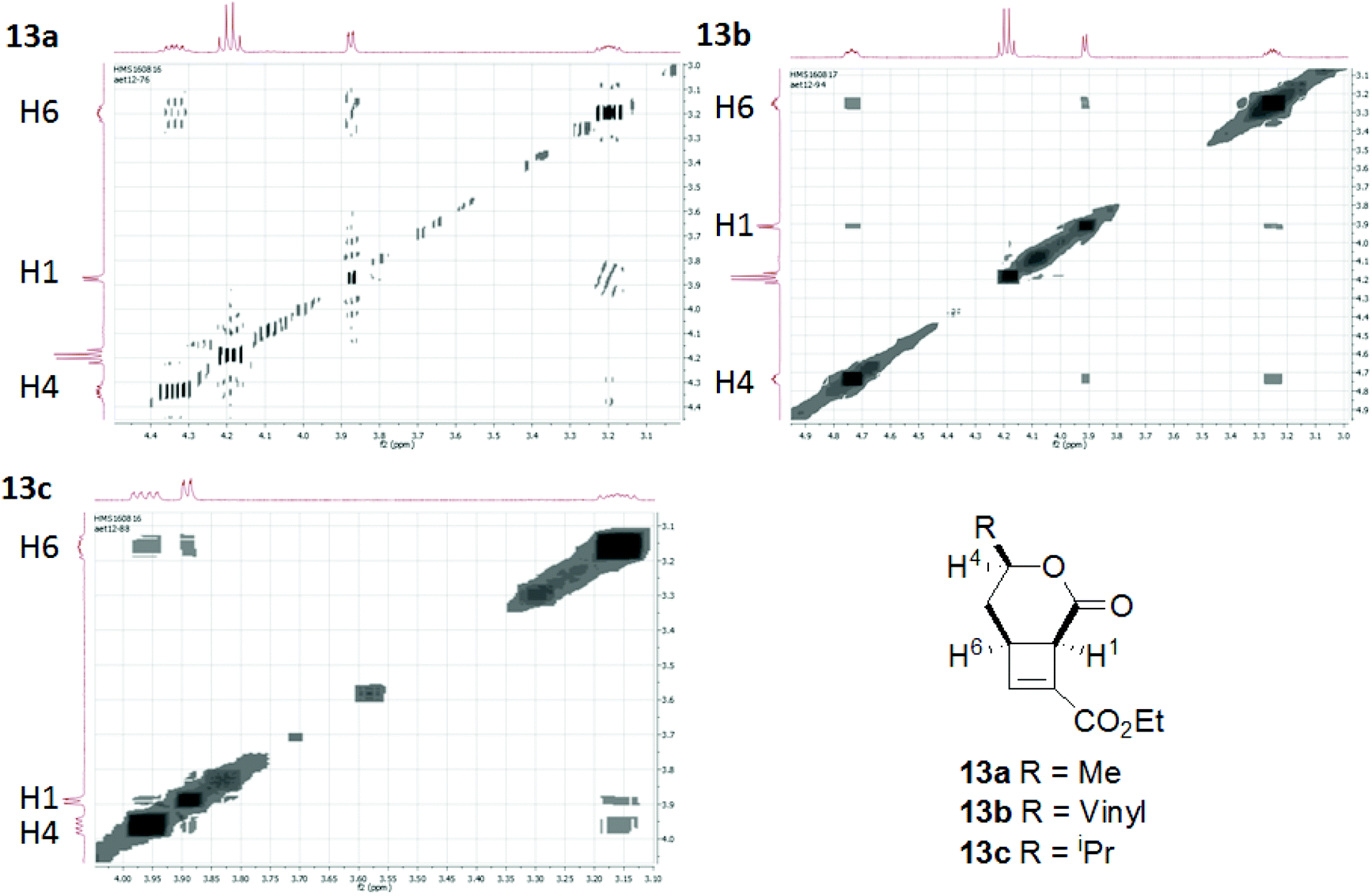 | ||
| Fig. 3 nOe correlations H1–H4–H6 of cyclobutenes 13a–c. | ||
|
|
||||
|---|---|---|---|---|
| Entry | Precursor | Product | Yield | de |
| 1 | 12a | 13a | 54% | >95% |
| 2 | 12b | 13b | 48% | >95% |
| 3 | 12c | 13c | 47% | >95% |
| 4 | 12d | 13d | 60% | >95% |
| 5 | 12e | 13e | 30% | >95% |
The excellent diastereoselectivity, and the cis arrangement of groups around the 6-membered ring, is hypothesised to arise through a chair-like transition state 14 (Fig. 4) where the side chain ‘R’ in a pseudoequatorial position is favoured. This conformation allows efficient overlap of the two π systems to facilitate cyclisation. The alternative conformation 15 places the π systems near perpendicular to one another, making efficient overlap to form a cis-fused bicycle not possible.
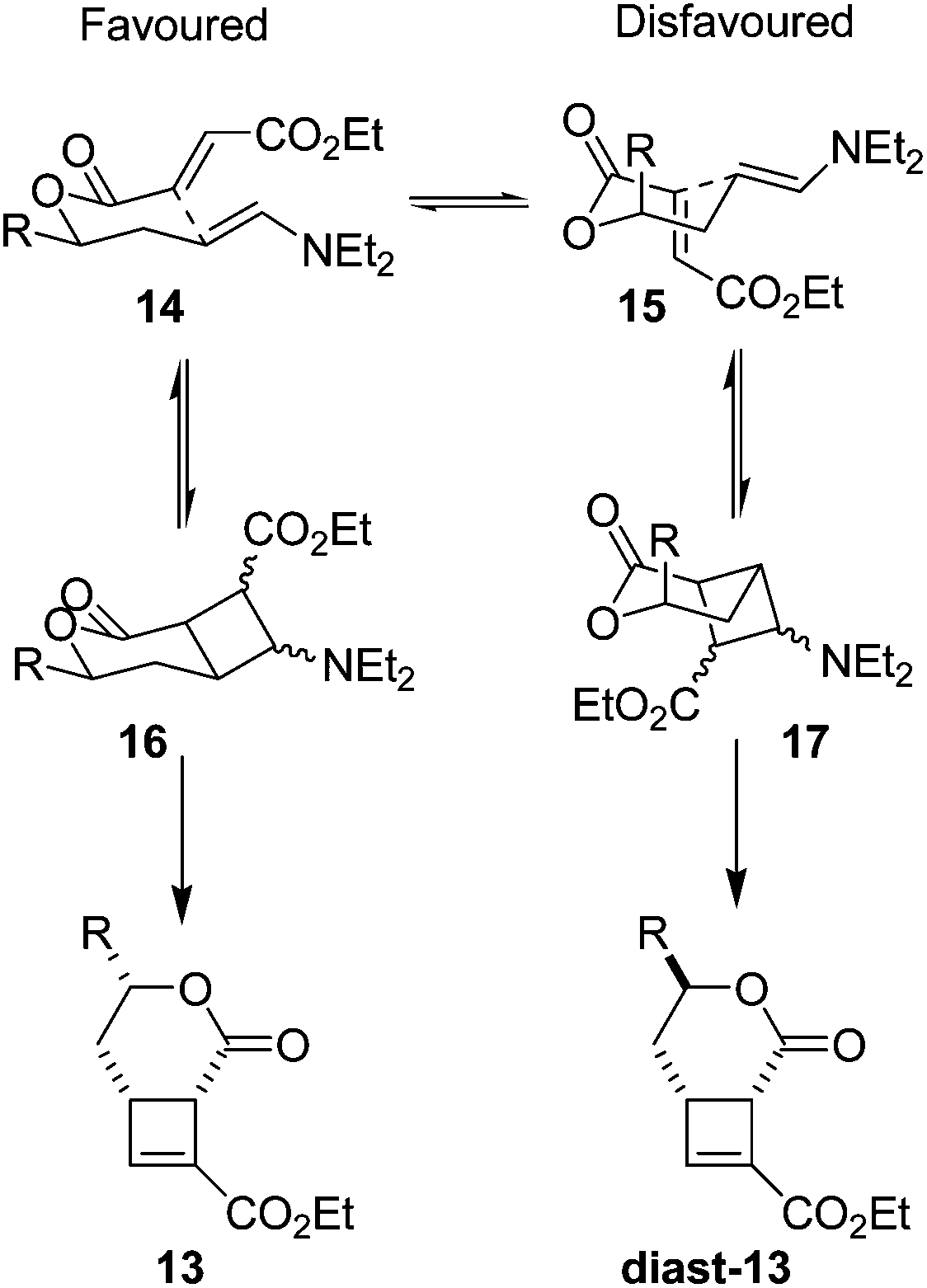 | ||
| Fig. 4 Proposed transition states for intramolecular cycloaddition. Pseudoequatorial arrangement of all sidechains during cyclisation leads to the observed product 13. | ||
The yields of cyclobutenes 13 were generally higher than the yields of cyclobutenes 9 obtained from cyclising unsubstituted precursors, presumably due to the presence of the sidechain reducing the conformational flexibility of the substrate and favouring a reactive conformation. However, the increased yield did not generalise to the formation of larger ring systems (Fig. 5). Compound 19 (an isomer of 13d) was of interest since both 19 and 13b are suitable starting points for synthesis of the highly substituted cyclobutane ring system found in providencin 5. Compound 21, which is similar to known intermediates such as 20,28 could be obtained from either 19 or 13b by rhodium-catalysed addition29 of furan to the cyclobutene and further elaboration of the sidechains. Subjecting aldehyde 18 to the cyclisation conditions for up to 7 days returned unreacted starting material along with small amounts of decomposition products resulting from aldol condensations of 18.
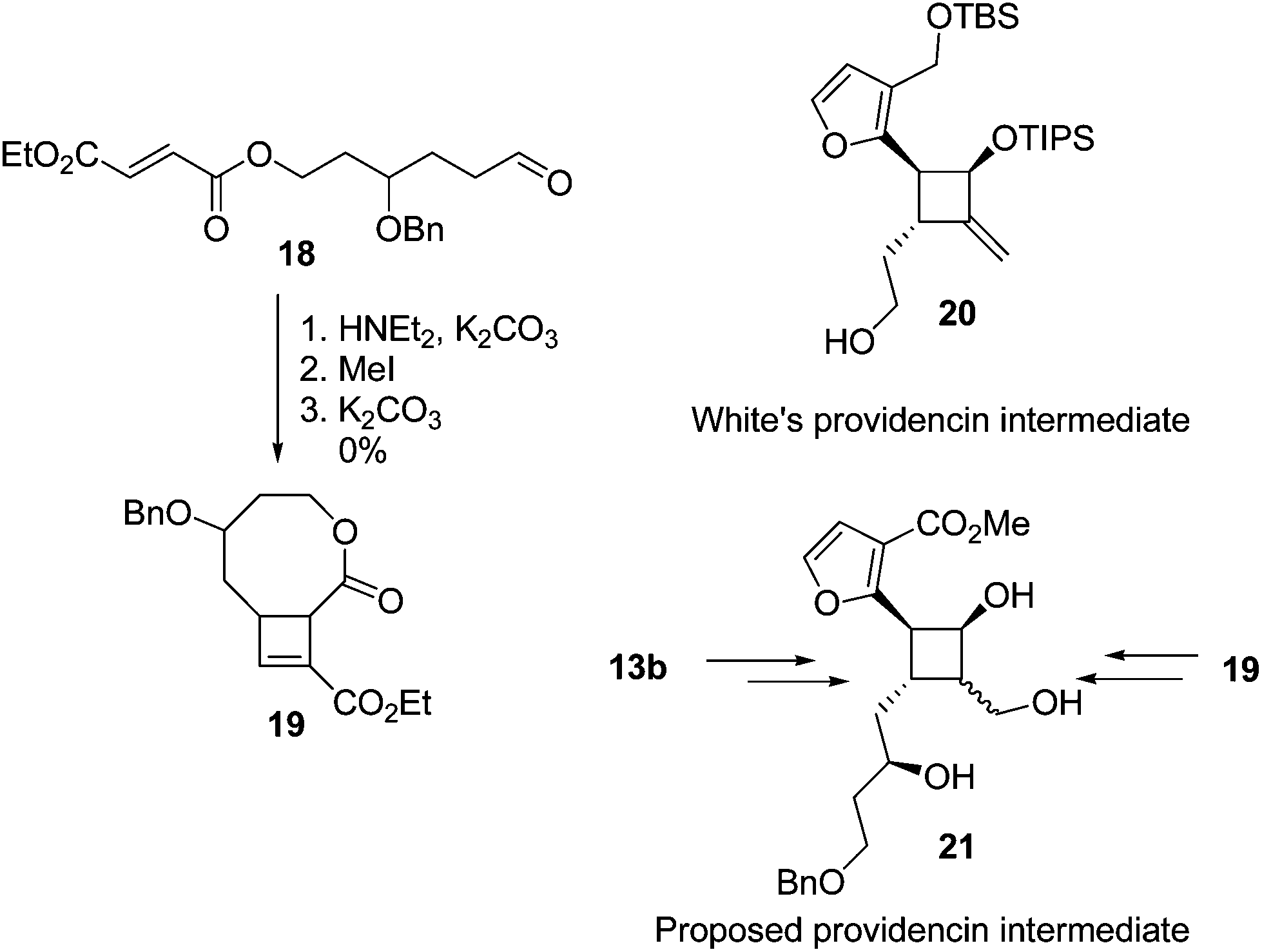 | ||
| Fig. 5 Attempted intramolecular enamine [2 + 2] cyclisation of a longer functionalised tether. | ||
In conclusion, we have demonstrated that an intramolecular variant of the enamine [2 + 2] cyclisation allows straightforward access to 6,4 fused bicyclic systems in a diastereoselective manner. This methodology provides a new means to access functionalized fused cyclobutenes bearing sidechains that allow further synthetic elaboration. Applications of the methodology in drug discovery, and to the total synthesis of the biologically active natural product providencin, are in progress.
This work was funded by Yorkshire Cancer Research (Award reference number B002-PhD). We thank the EPSRC UK National Mass Spectrometry Facility at Swansea University for measurement of HRMS data.
Example experimental procedure
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, EtOAc:PE) to give the title compound 13a as a clear oil (45 mg, 0.21 mmol, 54% yield); Rf 0.22 (1:1, EtOAc:PE); IR 2984.7 cm−1 (CH), 1710.8 cm−1 (C
:1, EtOAc:PE) to give the title compound 13a as a clear oil (45 mg, 0.21 mmol, 54% yield); Rf 0.22 (1:1, EtOAc:PE); IR 2984.7 cm−1 (CH), 1710.8 cm−1 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O); 1H NMR (400 MHz, CDCl3) δ 6.81 (s, 1H, H7), 4.40–4.28 (m, 1H, H4), 4.19 (q, J = 7.1 Hz, 2H, OCH2CH3), 3.88 (d, J = 4.8 Hz, 1H, H1), 3.27–3.14 (m, 1H, H6), 2.16 (ddd, J = 14.2, 7.8, 1.3 Hz, 1H, H5), 1.43 (app dt, J = 14.2, 10.7 Hz, 1H, H5′), 1.32 (d, J = 6.3 Hz, 3H, CH3), 1.25 (t, J = 7.1 Hz, 3H, OCH2CH3); 13C NMR (101 MHz, CDCl3) δ 169.9 (C), 161.2 (C), 148.7 (CH), 136.9 (C), 76.2 (CH), 60.8 (CH2), 42.2 (CH), 37.8 (CH), 35.4 (CH2), 21.0 (CH3), 14.2 (CH3); m/z (ES+) 233.1([M + Na]+, 100%) 211.1 ([M + H]+, 10%); HRMS found ([M + H]+) 211.0964. C11H15O4 req 211.0965; LCMS rt 2.71 minutes, m/z (ES+) 211.2 ([M + H]+, 100%), >95%.
O); 1H NMR (400 MHz, CDCl3) δ 6.81 (s, 1H, H7), 4.40–4.28 (m, 1H, H4), 4.19 (q, J = 7.1 Hz, 2H, OCH2CH3), 3.88 (d, J = 4.8 Hz, 1H, H1), 3.27–3.14 (m, 1H, H6), 2.16 (ddd, J = 14.2, 7.8, 1.3 Hz, 1H, H5), 1.43 (app dt, J = 14.2, 10.7 Hz, 1H, H5′), 1.32 (d, J = 6.3 Hz, 3H, CH3), 1.25 (t, J = 7.1 Hz, 3H, OCH2CH3); 13C NMR (101 MHz, CDCl3) δ 169.9 (C), 161.2 (C), 148.7 (CH), 136.9 (C), 76.2 (CH), 60.8 (CH2), 42.2 (CH), 37.8 (CH), 35.4 (CH2), 21.0 (CH3), 14.2 (CH3); m/z (ES+) 233.1([M + Na]+, 100%) 211.1 ([M + H]+, 10%); HRMS found ([M + H]+) 211.0964. C11H15O4 req 211.0965; LCMS rt 2.71 minutes, m/z (ES+) 211.2 ([M + H]+, 100%), >95%.
References
- E. Lee-Ruff and G. Miadenova, Chem. Rev., 2003, 103, 1449–1483 CrossRef CAS PubMed.
- R. Liu, M. Zhang, T. P. Wyche, G. N. Winston-McPherson, T. S. Bugni and W. Tang, Angew. Chem., Int. Ed., 2012, 51, 7503–7506 CrossRef CAS PubMed.
- Y. Xu, M. L. Conner and M. K. Brown, Angew. Chem., Int. Ed., 2015, 54, 11918–11928 CrossRef CAS PubMed.
- V. M. Dembitsky, J. Nat. Med., 2008, 62, 1–33 CrossRef CAS PubMed.
- A. Sergeiko, V. V. Poroikov, L. O. Hanus and V. M. Dembitsky, Open Med. Chem. J., 2008, 2, 26–37 CrossRef CAS PubMed.
- S.-H. Park, K. Moon, H.-S. Bang, S.-H. Kim, D.-G. Kim, K.-B. Oh, J. Shin and D.-C. Oh, Org. Lett., 2012, 14, 1258–1261 CrossRef CAS PubMed.
- M. Zhou, X.-R. Li, J.-W. Tang, Y. Liu, X.-N. Li, B. Wu, H.-B. Qin, X. Du, L.-M. Li, W.-G. Wang, J.-X. Pu and H.-D. Sun, Org. Lett., 2015, 17, 6062–6065 CrossRef CAS PubMed.
- J.-J. Tan, C.-H. Tan, Y.-Q. Wang, S.-H. Jiang and D.-Y. Zhu, Helv. Chim. Acta, 2006, 89, 117–121 CrossRef CAS.
- E. Lee-Ruff, in Patai's Chemistry of Functional Groups, John Wiley & Sons, Ltd, 2009 Search PubMed.
- P. Margaretha, Helv. Chim. Acta, 2014, 97, 1027–1035 CrossRef CAS.
- S. Poplata, A. Tröster, Y.-Q. Zou and T. Bach, Chem. Rev., 2016, 116, 9748–9815 CrossRef CAS PubMed.
- K. C. Brannock, A. Bell, R. D. Burpitt and C. A. Kelly, J. Org. Chem., 1964, 29, 801–812 CrossRef CAS.
- H. M. Sheldrake, T. W. Wallace and C. P. Wilson, Org. Lett., 2005, 7, 4233–4236 CrossRef CAS PubMed.
- I. Marko, B. Ronsmans, A.-M. Hesbain-Frisque, S. Dumas and L. Ghosez, J. Am. Chem. Soc., 1985, 107, 2192–2194 CrossRef CAS.
- B. B. Snider, R. A. H. F. Hui and Y. S. Kulkarni, J. Am. Chem. Soc., 1885, 107, 2194–2196 CrossRef.
- W. T. Brady, Y.-S. F. Giang, L. Weng and M. M. Dad, J. Org. Chem., 1987, 52, 2216–2220 CrossRef CAS.
- B. B. Snider, E. Ron and B. W. Burbaum, J. Org. Chem., 1987, 52, 5413–5419 CrossRef CAS.
- M. Lachia, H. C. Wolf and A. De Mesmaeker, Bioorg. Med. Chem. Lett., 2014, 24, 2123–2128 CrossRef CAS PubMed.
- M. E. Flanagan, J. A. Abramite, D. P. Anderson, A. Aulabaugh, U. P. Dahal, A. M. Gilbert, C. Li, J. Montgomery, S. R. Oppenheimer, T. Ryder, B. P. Schuff, D. P. Uccello, G. S. Walker, Y. Wu, M. F. Brown, J. M. Chen, M. M. Hayward, M. C. Noe, R. S. Obach, L. Philippe, V. Shanmugasundaram, M. J. Shapiro, J. Starr, J. Stroh and Y. Che, J. Med. Chem., 2014, 57, 10072–10079 CrossRef CAS PubMed.
- J. Panda, S. Ghosh and S. Ghosh, ARKIVOC, 2001, 146–153 CAS.
- P. Selig and T. Bach, J. Org. Chem., 2006, 71, 5662–5673 CrossRef CAS PubMed.
- E. Sato, Y. Ikeda and Y. Kanaoka, Liebigs Ann. Chem., 1989, 1989, 781–788 CrossRef.
- F. Yagishita, T. Mino, T. Fujita and M. Sakamoto, Org. Lett., 2012, 14, 2638–2641 CrossRef CAS PubMed.
- E. Kumarasamy, R. Raghunathan, S. Jockusch, A. Ugrinov and J. Sivaguru, J. Am. Chem. Soc., 2014, 136, 8729–8737 CrossRef CAS PubMed.
- H. M. Deutsch, L. T. Gelbaum, M. McLaughlin, T. J. Fleischmann, L. L. Earnhart, R. D. Haugwitz and L. H. Zalkow, J. Med. Chem., 1986, 29, 2164–2170 CrossRef CAS PubMed.
- M. Abou-Gharbia, U. R. Patel, M. B. Webb, J. A. Moyer, T. H. Andree and E. A. Muth, J. Med. Chem., 1988, 31, 1382–1392 CrossRef CAS PubMed.
- L. M. Rice and C. H. Grogan, J. Org. Chem., 1957, 22, 1100–1103 CrossRef CAS.
- J. D. White and S. Jana, J. Org. Chem., 2014, 79, 700–710 CrossRef CAS PubMed.
- R. Liu, M. Zhang, T. P. Wyche, G. N. Winston-McPherson, T. S. Bugni and W. Tang, Angew. Chem., Int. Ed., 2012, 51, 7503–7506 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ob01661h |
| This journal is © The Royal Society of Chemistry 2016 |