
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Biasing hydrogen bond donating host systems towards chemical warfare agent recognition†
Jennifer R.
Hiscock
 ab,
Neil J.
Wells
a,
Jayne A.
Ede
c,
Philip A.
Gale
*a and
Mark R.
Sambrook
*c
ab,
Neil J.
Wells
a,
Jayne A.
Ede
c,
Philip A.
Gale
*a and
Mark R.
Sambrook
*c
aChemistry, University of Southampton, Southampton, SO17 1BJ, UK. E-mail: philip.gale@soton.ac.uk; Fax: +44 (0)23 80596805; Tel: +44 (0)23 80593332
bSchool of Physical Sciences, University of Kent, Kent, CT2 7NZ, UK
cCBR Division, Dstl Porton Down, Salisbury, SP4 0JQ, UK. E-mail: msambrook@dstl.gov.uk; Tel: +44 (0)1980 614301
First published on 20th September 2016
Abstract
A series of neutral ditopic and negatively charged, monotopic host molecules have been evaluated for their ability to bind chloride and dihydrogen phosphate anions, and neutral organophosphorus species dimethyl methylphosphonate (DMMP), pinacolyl methylphosphonate (PMP) and the chemical warfare agent (CWA) pinacolyl methylphosphonofluoridate (GD, soman) in organic solvent via hydrogen bonding. Urea, thiourea and boronic acid groups are shown to bind anions and neutral guests through the formation of hydrogen bonds, with the urea and thiourea groups typically exhibiting higher affinity interactions. The introduction of a negative charge on the host structure is shown to decrease anion affinity, whilst still allowing for high stability host-GD complex formation. Importantly, the affinity of the host for the neutral CWA GD is greater than for anionic guests, thus demonstrating the potential for selectivity reversal based on charge repulsion.
Introduction
The highly toxic G-series organophosphorus (OP) nerve agents have been known since the 1940s, and their development, toxicology and physico-chemical properties have been well-documented since that time.1–4 Terrorist attacks in Matsumoto and Tokyo in the mid-1990s, and recent, well-publicised, events in Syria, have highlighted the long-standing threat posed by these materials.5–8Supramolecular approaches to the mitigation of the hazards posed by chemical warfare agents (CWAs) as a whole have been recently reviewed.9 Examples include the proposed use of functionalised cyclodextrins as medical countermeasures for G-agent poisoning,10,11 and the use of cavitand and basket-like structures for nerve agent detection.12,13 A number of researchers have also investigated the interactions of G-series CWAs, and their simulants, with transition or lanthanide metal complexes.14,15
Considering the commonality of phosphonyl and/or phosphoryl bonds to G- and V-series CWAs, and the ability of these groups to act as hydrogen bond acceptors, hydrogen bonding is an attractive means by which to mediate complex formation. In 2012 we published the first example of the recognition of an OP CWA, specifically O-pinacolyl methylphosphonfluoridate or soman (hereafter referred to as GD), by a designed, low molecular weight host molecule in which complex formation was driven by multiple hydrogen bonding interactions between NH donors and the phosphonyl P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O acceptor.16 Since then, we have exploited hydrogen bonding interactions in the development of responsive supramolecular gels17–20 and in receptors capable of accelerating the hydrolysis of GD and simulants.21,22
O acceptor.16 Since then, we have exploited hydrogen bonding interactions in the development of responsive supramolecular gels17–20 and in receptors capable of accelerating the hydrolysis of GD and simulants.21,22
We have been interested in extending our understanding of hydrogen bonding systems for OP CWA recognition, and in particular producing systems that will exhibit selective binding of neutral organophosphorus compounds over anionic phosphates. Given the oxobasicity and negative charge held by phosphates such as dihydrogen phosphate anions (H2PO4−), selectivity trends usually favour anionic guests over neutral species in hydrogen bonding complexes. This may present challenges in, for example, detection of G-agent targets in the presence of breakdown products/impurities, or in the poisoning of supramolecular catalysts based upon hydrogen bonding. Reversing this trend is challenging, but advantageous, so we proposed the use of negatively charged hosts to achieve this through decreasing the host–anion complex affinity due to electrostatic repulsion whilst still allowing for neutral guest recognition via hydrogen bonding (Fig. 1).
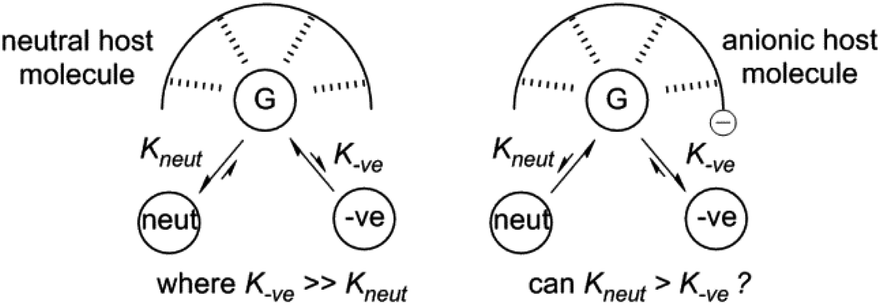 | ||
| Fig. 1 Can preference for neutral guest recognition by hydrogen bonds be favoured over charged guest binding through the use of electrostatic repulsion? G = guest binding site; neut = neutral guests species; −ve = anionic guest species. | ||
In an effort to further explore hydrogen bond recognition of OP CWAs and to determine the feasibility of charge repulsion as a route to reversing selectivity trends, a number of neutral ditopic or negatively charged monotopic receptors were synthesised that contain either a urea or thiourea hydrogen bond donating functionality and either a neutral boronic acid or a tetravalent, anionic, boron group for the recognition of anionic and neutral OP guest species, including GD.
Results and discussion
Receptor design
The design of two generic host structures is illustrated in Fig. 2. Both systems incorporate a central urea, or thiourea, motif that will present two hydrogen bond donors for guest recognition. A second functionality comprises either a boronic acid B(OH)2 or a negatively charged BFx− group. The former therefore yields a neutral, ditopic hydrogen bond-donor host, and the latter a negatively charged system to investigate the use of electrostatic repulsion to modify selectivity trends. | ||
| Fig. 2 Proposed receptor design. | ||
These receptor structures are closely related to a series of compounds reported by Hughes and Smith,23 in which an internal polarisation effect is exploited to drive enhanced binding of carboxylate guests in DMSO solution. In their difluoroboronate systems, two ‘limiting cases’ were proposed, with that represented by structure b in Fig. 3 confirmed by experimental studies.
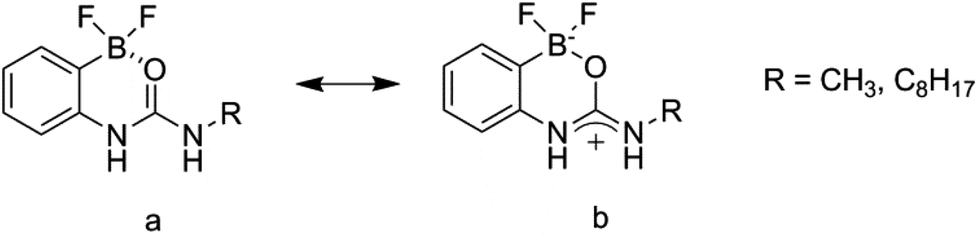 | ||
| Fig. 3 Internally polarised urea groups for anion guest binding as reported by Hughes and Smith.23 | ||
Receptor synthesis
It was found that some of the designed structures (2, 6 and 8; Fig. 4) underwent a similar cyclisation process to that reported by Hughes and Smith,23 and therefore yielded non-ideal structures for our own study. Contrastingly, a number retained a linear structure with intact urea/thiourea and boron functionalities (1, 3, 4, 5, and 7; Fig. 4), and these are the only receptors that can present appropriate, bidentate binding sites for guest recognition. | ||
| Fig. 4 Structures of receptors 1–8 and phosphonate guest species. | ||
To synthesise the receptors an isocyanate/isothiocyanate was added to a stirring solution of an (aminophenyl)boronic acid in anhydrous DMF. This mixture was then added to water which gave the crude product as a cream/white solid. The pure products were obtained by trituration or recrystallization/precipitation from ethyl acetate/methanol/hexane mixtures as appropriate giving receptors 1, 2, 5 and 6 in 36%, 55%, 74% and 95% yields, respectively. Receptors 3, 4, 7 and 8 were obtained by the reaction of receptors 1, 2, 5 and 6 with tetrabutylammonium (TBA) HF2 in a methanol/acetonitrile mixture. Receptor 4 was isolated directly from the supernatant mixture as a white crystalline solid in a 65% yield. Crude solids containing receptors 3, 7 and 8 were obtained by the addition of the reaction mixtures to water. The pure product was then obtained by trituration or recrystallization/precipitation from ethyl acetate/methanol/hexane mixtures as appropriate. This gave receptors 3, 7 and 8 as cream/white solids in 68%, 81% and 77% yields, respectively. The general synthetic scheme is outlined in Fig. 5 (see ESI† for full details).
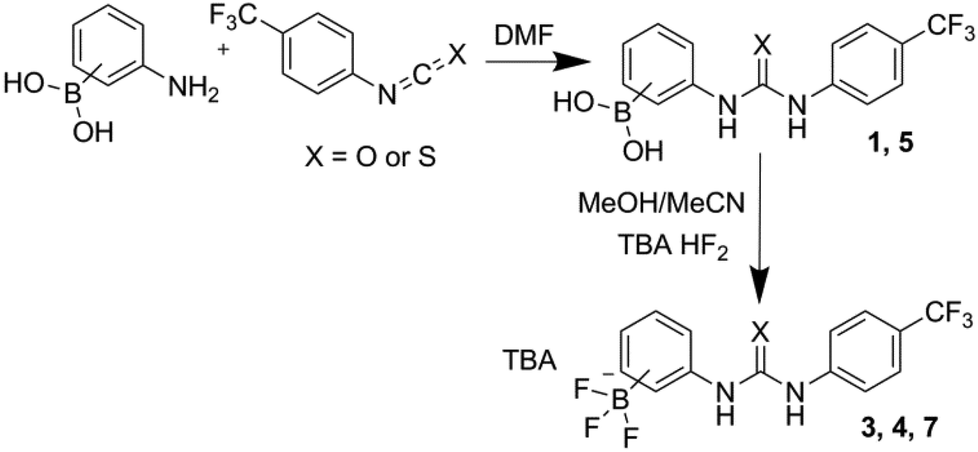 | ||
| Fig. 5 General reaction scheme for the synthesis of uncyclised compounds 1, 3, 4, 5 and 7. | ||
The structure of receptor 2 was identified by both 1H NMR and ROESY NMR techniques. The through-space correlations showed interactions between the boronic acid hydrogen and the boron substituted aromatic ring but not the CF3 substituted ring which would have been the case if this receptor had cyclised forming a B–N bond rather than a B–O bond as with receptors 6 and 8. The structure of receptor 8 was confirmed by 1H/19F HMBC. In this experiment a through-bond correlation was found between the fluorine atoms attached to the boron centre and both aromatic rings.
Solid state structures
The structure of receptor 6 was confirmed by single crystal X-ray diffraction, with the resulting structure shown in Fig. 6, and further supported by multinuclear NMR studies as detailed in the ESI.†‡ As with receptor 8, receptor 6 was found to cyclise through the formation of a B–N bond rather than a B–S bond.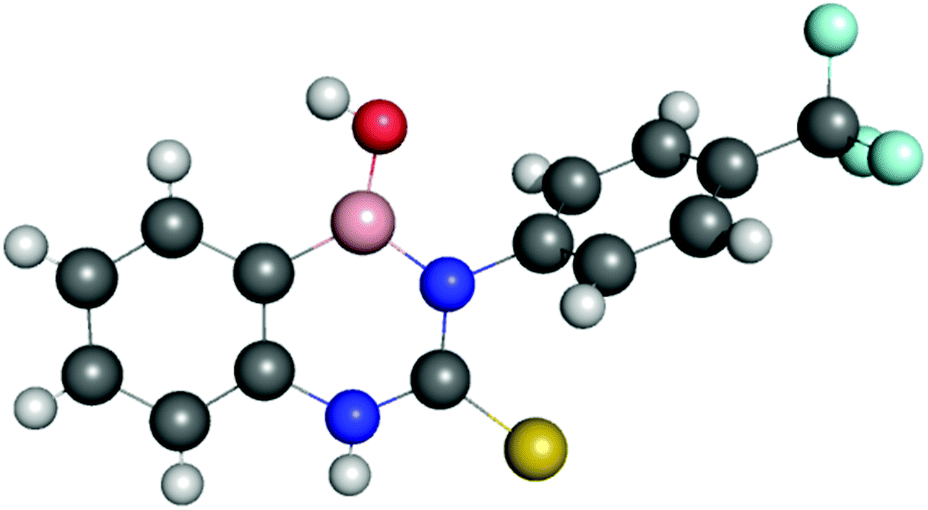 | ||
| Fig. 6 X-ray crystal structure confirming the presence of a B–N bond in the structure of receptor 6. A water molecule and a second receptor molecule have been omitted for clarity. Nitrogen atoms blue, hydrogen atoms white, carbon atoms grey, oxygen atoms red, boron atoms pink, sulfur atoms yellow and fluoride atoms turquoise. Crystals were obtained by slow evaporation from ethyl acetate. | ||
The structure of receptor 4 was confirmed by single crystal X-ray diffraction techniques as shown in Fig. 7, and clearly reveals the proposed linear receptor compound. The receptor was found to form dimers in the solid-state through the formation of intermolecular hydrogen bonds with the N–H functionality acting at the hydrogen bond donating group and the F of the R–BF3− functionality acting as the hydrogen bond accepting functionality. The most favourable hydrogen-bonds were found to have bond lengths of N⋯F 2.836(3)–2.854(3) Å, with bond angles of N–H⋯F 156(8)–160(8)°. The formation of N–H⋯F–B hydrogen bond contacts is assumed to be very low affinity and is not present in the solution phase.
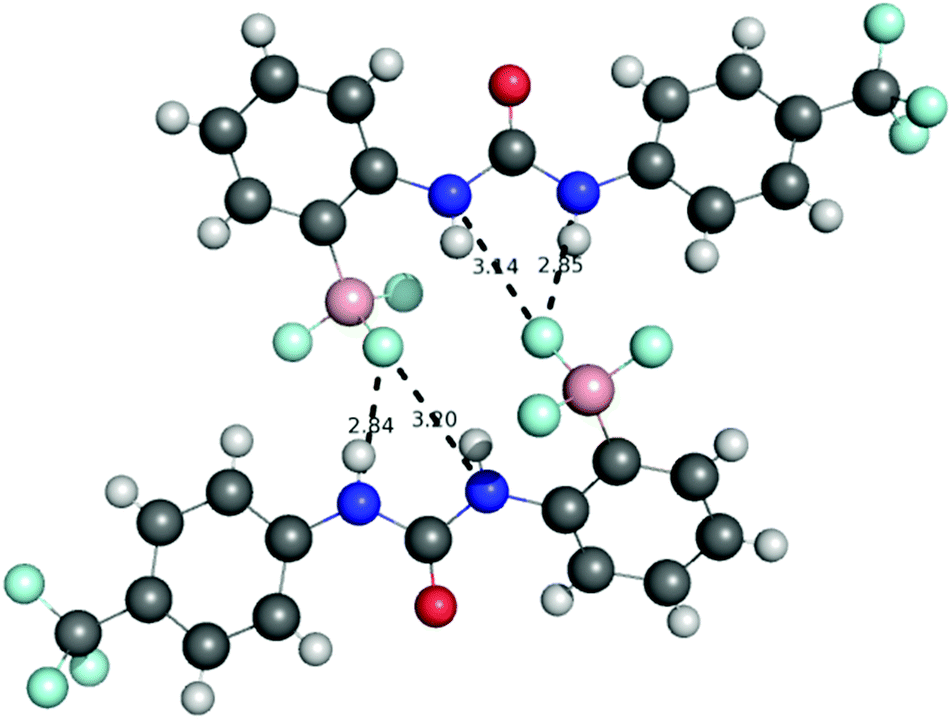 | ||
| Fig. 7 X-ray crystal structure of receptor 4 which was found to form hydrogen bonded dimers with hydrogen bonds formed between the urea NHs and R–BF3− functionality. The TBA counter-cations have been omitted for clarity. Nitrogen atoms blue, hydrogen atoms white, carbon atoms grey, oxygen atoms red, boron atoms pink, sulfur atoms yellow and fluoride atoms turquoise. Crystals were obtained by slow evaporation from acetonitrile in the presence of one equivalent of PMP. | ||
This solid-state structure also allows us to make a comparison between related receptor structures. The CO bond length is in the range 1.220–1.228 Å (Fig. 8), which is intermediate between that of the cyclised receptor reported by Smith and Hughes23 and a bis-nitrophenyl urea receptor reported by Etter et al.24 Similarly, comparison of the C–N bond lengths of the urea moiety reveals lengths of 1.367–1.369 Å and 1.382–1.388 Å for receptor 4, both of which are longer than the analogous lengths in the cyclised structure reported by Hughes and Smith.
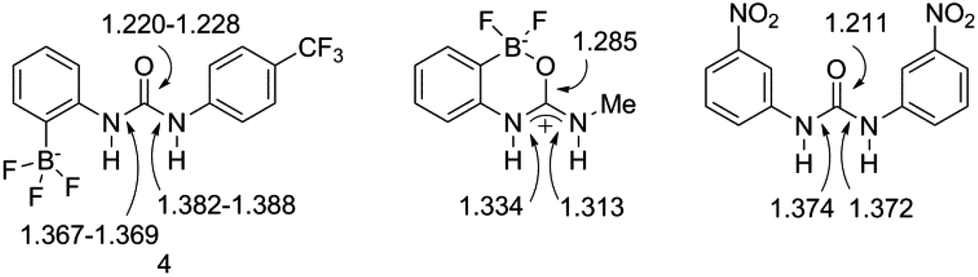 | ||
| Fig. 8 Comparison of bond lengths (Å) for receptor 4 to those in closely related receptors reported by Hughes and Smith,23 and by Etter et al.,24 as determined from solid-state structures. | ||
Anion recognition: Cl− and H2PO4−
Receptors 1–8 were first investigated with regard to their ability to bind anionic species, which are known to engage effectively in hydrogen bonding interactions with urea/thiourea25,26 and boronic acid functionalities.27 Chloride was selected as an initial guest to evaluate the behaviour of the ditopic hosts and the effects of a charged host on the formation of solution complexes. Dihydrogen phosphate (H2PO4−) presents the possibility of a bidentate hydrogen bond acceptor with a tetrahedral geometry, and was therefore also assessed as a potential guest. The results of these studies are outlined in Table 1.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and 1:2 H:G models where appropriate using the software package EQNMR.28 All errors <10% with the exception of 7 + H2PO4 where error is estimated at ±32%
:1 and 1:2 H:G models where appropriate using the software package EQNMR.28 All errors <10% with the exception of 7 + H2PO4 where error is estimated at ±32%
Receptors 1 and 5 have two potential anion binding sites – the urea or thiourea and the boronic acid. The addition chloride, as its tetrabutylammonium (TBA) salts, to a MeCN-d3 solutions of either receptor resulted in downfield chemical shifts of both the urea NH and boronic OH proton resonances, indicative of the formation of hydrogen bonding interactions. In the presence of approximately 2 molar equivalents of Cl− anions, the NH and B–OH resonances of receptor 1 were found to be perturbed by 2.49 and 1.15 ppm, respectively, with similar values obtained for receptor 5.
Quantitative 1H NMR titration experiments were conducted in MeCN-d3 and the shifts of the NH and OH proton resonances of receptors 1 and 5 followed. Titration curves in both cases provide evidence for the formation of both 1:1 and 1:2 host:guest complexes, as is expected due to the presence of two binding sites in the host molecule. For receptor 1, fitting of the NMR data obtained by monitoring the amide NH proton environments yielded association constants of K11 = 5540 M−1 and K12 = 270 M−1 using the software package EQNMR.28 Analysis of the boronic acid OH group protons yielded association constants of K11 = 2470 M−1 and K12 = 360 M−1. For receptor 5 data fitting of the thiourea NH data yielded K11 = 1360 M−1 and K12 = 98 M−1, and using the B(OH)2 data K11 = 1480 M−1 and K12 = 190 M−1. It should be noted that in order to construct binding curves from the titration data both (thio)urea proton environments were monitored, although in most cases at least one environment would be obscured by other resonances during the course of the experiment (typically at high guest equivalents). In those cases, a single environment was used to facilitate binding constant determination, whilst the second environment still provides confirmatory evidence (usually at lower molar equivalents) of the binding event. The (thio)urea protons used in the analysis of each host–guest pair are noted in the ESI.†
For both ditopic receptors the 1:1 complex is formed with a high affinity, and the binding of the second chloride anion is, unsurprisingly, much less favourable. Although the two K11 values obtained for receptor 1 through analysis of the NH and OH shift perturbations do possess a significant discrepancy it should be noted that they are both indicative of the formation of a high affinity complex and both yield close agreement for the K12 complex affinity. The analogous association constants for receptor 5 and both sets of K12 constants are in much closer agreement. Examination of the binding curves reveals much greater relative perturbations at lower molar equivalents of guest for the urea protons, and by 1 molar equivalent the majority of the host complex has been formed (Fig. 9a). Contrastingly, the S-shaped binding isotherm for the boronic acids plateaus at much higher molar equivalents (Fig. 9b). This, combined with the known affinity of urea groups for anions, allows us to postulate that the first high affinity (K11) binding site is the urea group, and a second, much weaker complex (K12), is formed at the boronic acid site (Fig. 10). This is further supported by the data for receptors 3 and 7 (vide infra).
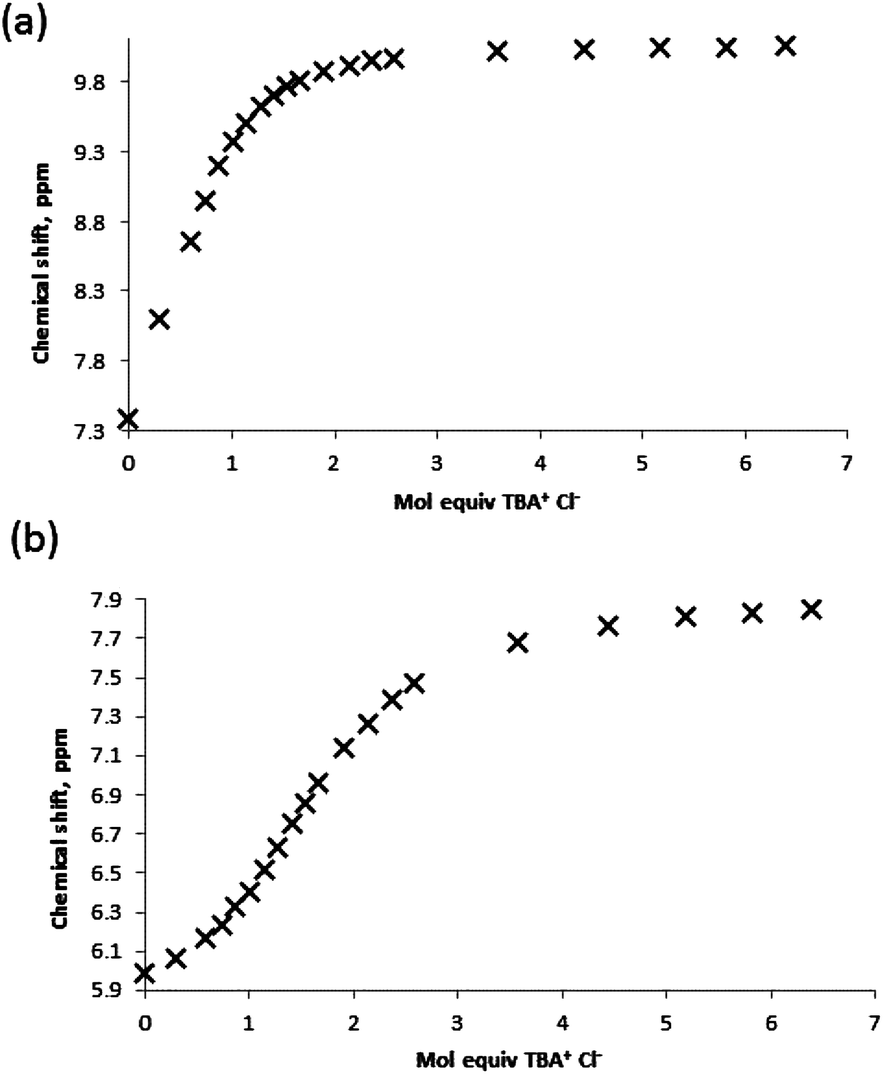 | ||
| Fig. 9 Binding curves obtained by titration of receptor 1 with TBA Cl guest in MeCN-d3 monitoring (a) urea NH protons and (b) boronic acid B(OH)2 protons. | ||
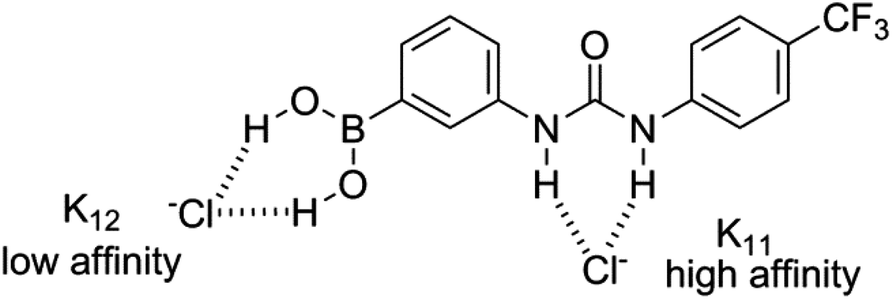 | ||
| Fig. 10 Proposed hydrogen bond-donating anion recognition sites for ditopic receptor 1, as illustrated for Cl− recognition. | ||
Upon addition of H2PO4−, again as a tetrabutylammonium salt, to solutions of receptors 1 and 5, and also to receptors 2 and 6 which also possess B–OH groups, a reaction was observed to occur between the receptor and the putative guest. In the 11B NMR a shift of the boron resonance is observed from ≈30 ppm, indicating a trivalent boron centre, to ≈5 ppm, indicating a tetravalent boron centre (see ESI†). There is also a new phosphorus signal identified at ≈−5 ppm in the 31P NMR spectrum indicating the presence of a new phosphorus containing species (see ESI†). It may be expected that integration of the two environments could be conducted to yield an estimate of the association, however, in this case overlap of the resonances was too great to allow for deconvolution and an association constant could not be determined. These results indicate that the arylboronic acid group forms a Lewis-type tetrahedral adduct with H2PO4− anions (Fig. 11), and is in agreement with the report of Martínez-Aguirre and Yatsimirsky.27
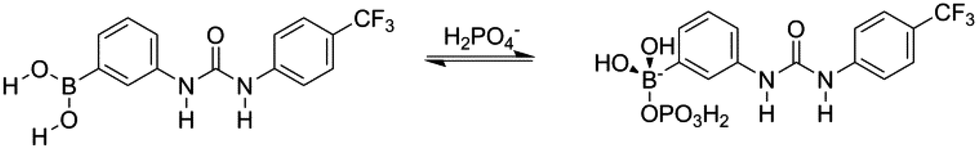 | ||
| Fig. 11 Proposed Lewis acidic behaviour of boronic acid groups in the presence of H2PO4−, as illustrated for receptor 1. | ||
Receptors 3, 4 and 7 possess only a single convergent binding site for guest recognition, and all three have pendent, negatively charged BF3− groups. Addition of chloride to solutions of the receptors resulted in downfield chemical shift perturbations in the urea/thiourea NH proton resonances. Analysis of titration curves generated from 1H NMR experiments yielded association constants K11 of 1050, <5 and 358 for 3, 4 and 7, respectively. Receptor affinities for H2PO4− were also determined for all three receptors, with Kassoc values of 351, <5 and 270 M−1 for 3, 4 and 7, respectively.
Some comparisons can be made between the two charged receptors 3 and 7 and the neutral, ditopic receptors 1 and 5. Our experiments indicate a marked decrease in the affinity of the urea NH binding site for Cl− in the charged receptors, with K11 values of 1051 vs. 5543 (or 2466) M−1 for 3 and 1, respectively, and K11 of 358 vs. 1364 M−1 for 7 and 5, respectively. In the case of H2PO4− comparisons cannot be made due the reactions observed between the guest and any receptors containing B(OH)x (x = 1, 2) groups.
Receptor 4, for which the neutral analogue was not available, did not have an appreciable affinity for either Cl− or H2PO4− anions. Although a binding curve could be generated from titration experiments with Cl−, it remained near linear up to the addition of 5–6 molar equivalents and represents a very low affinity estimated as <10 M−1. Nevertheless, significant perturbations in the chemical shift of the NH protons give us confidence in assigning the formation of a complex. It is unclear, however, whether the decrease in affinity is a result of electrostatic repulsion by the BF3− group as it is in closer proximity to the urea NH groups, or if it results from steric repulsion through partial blocking of the binding site.
Receptors 2, 6 and 8 differ from the others in that none of them present convergent hydrogen bonding sites for guest recognition; as such, they cannot be defined as host molecules. Affinities of 2 and 8 for Cl− are particularly weak with K11 values estimated at <10 M−1. For receptor 6, a higher affinity of 150 M−1 was determined by following the boronic OH protons. In comparison to receptor 2, the increased acidity of the NH proton due to the presence of a thioarea may account for this increased affinity. Conversely, receptor 8, in comparison to 6, carries a negative charge, which may decrease anion affinity through electrostatic repulsion. Similarly, H2PO4− was not observed to form complexes with receptor 8.
Neutral guest recognition: DMMP, PMP and GD
Potential recognition of three neutral OP guests, DMMP, PMP and the CWA GD, was studied in MeCN-d3 solution by quantitative NMR titration methods. These were selected based upon the common use of DMMP as an OP CWA simulant, and the fact that PMP is the hydrolysis product of GD and therefore shares many structural similarities.None of the receptors in the study were found to form a complex with DMMP to any significant extent; small perturbations in chemical shift that were not sufficient to generate a binding isotherm provided evidence of a very low affinity interaction. The association constant of the formation of these complexes is estimated to be <5 M−1 in all cases (Table 2).
:1 model using the software package EQNMR.28 Error is <10% with the exception of 3 + GD and 5 + PMP where errors are estimated at ±16% and ±32%, respectively. The value for 7 + GD is an indicative estimate obtained by fitting a partial data set and an estimated error of ±250 M−1 is indicated (see main text)
The guest species PMP was also found to interact very weakly with the receptors. Data fitting was more successful than with DMMP for some PMP–receptor pairs, although reactions were observed with receptors 3, 4 and 7. Association constants K11 were determined as <50 M−1 for PMP with receptors 2 and 6. For ditopic receptor 5 there was clear evidence of binding at both the urea and boronic acid binding sites, with K11 = 338 M−1 and K12 ≤ 10 M−1. The higher affinity complex is assigned to the formation of N–H⋯OP hydrogen bonds, analogous to the case of Cl− binding, and the second PMP guest molecule is bound at the boronic B(OH)2 site.
Receptor 8 was found to preferentially bind PMP over DMMP and, perhaps more surprisingly, H2PO4−. The selectivity for the binding of PMP over DHP and DMMP is postulated to be a result of the ability of this guest species to act as both a hydrogen bond donor and acceptor, due the presence of the acidic OH functionality. A proposed binding mode illustrating this is shown in Fig. 12, and would also be applicable to the complex formed between PMP and receptor 6.
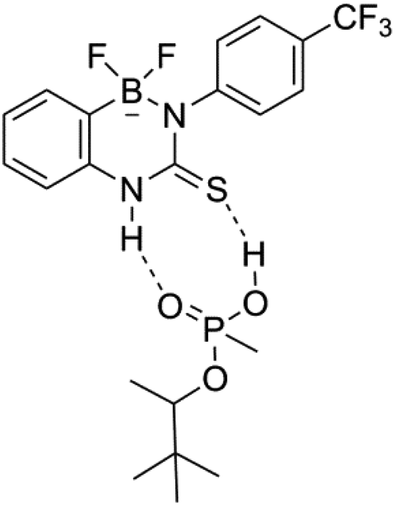 | ||
| Fig. 12 Possible binding motif for receptor 8 with PMP. | ||
A series of GD titrations were conducted with a selected set of the receptors, specifically 1, 3, 4, 5, 7 and 8. Analysis was not conducted with receptors 2 and 6, given the similarity of the structures to receptor 8 and in order to minimise experiments conducted with highly toxic GD samples.
For receptors 1 and 5 downfield chemical shift perturbations of both the urea NH and boronic acid OH proton environments were observed in the presence of GD. Monitoring the amide NH protons generated a binding curve that was not amenable to data refinement to generate an association constant, but was still highly indicative of the presence of receptor–GD complex formation. Following an initial, large (relative) downfield chemical shift perturbation upon addition of 0.2 molar equivalents of GD, the magnitude of the perturbation continued to increase until approximately 2–2.5 molar equivalent of GD was present. At this point, the change in chemical shift relative to the starting host began to decrease (Fig. 13a). Such behaviour is often attributed to the formation of higher-order lower-affinity complex formation, and as such association constants cannot be determined.16
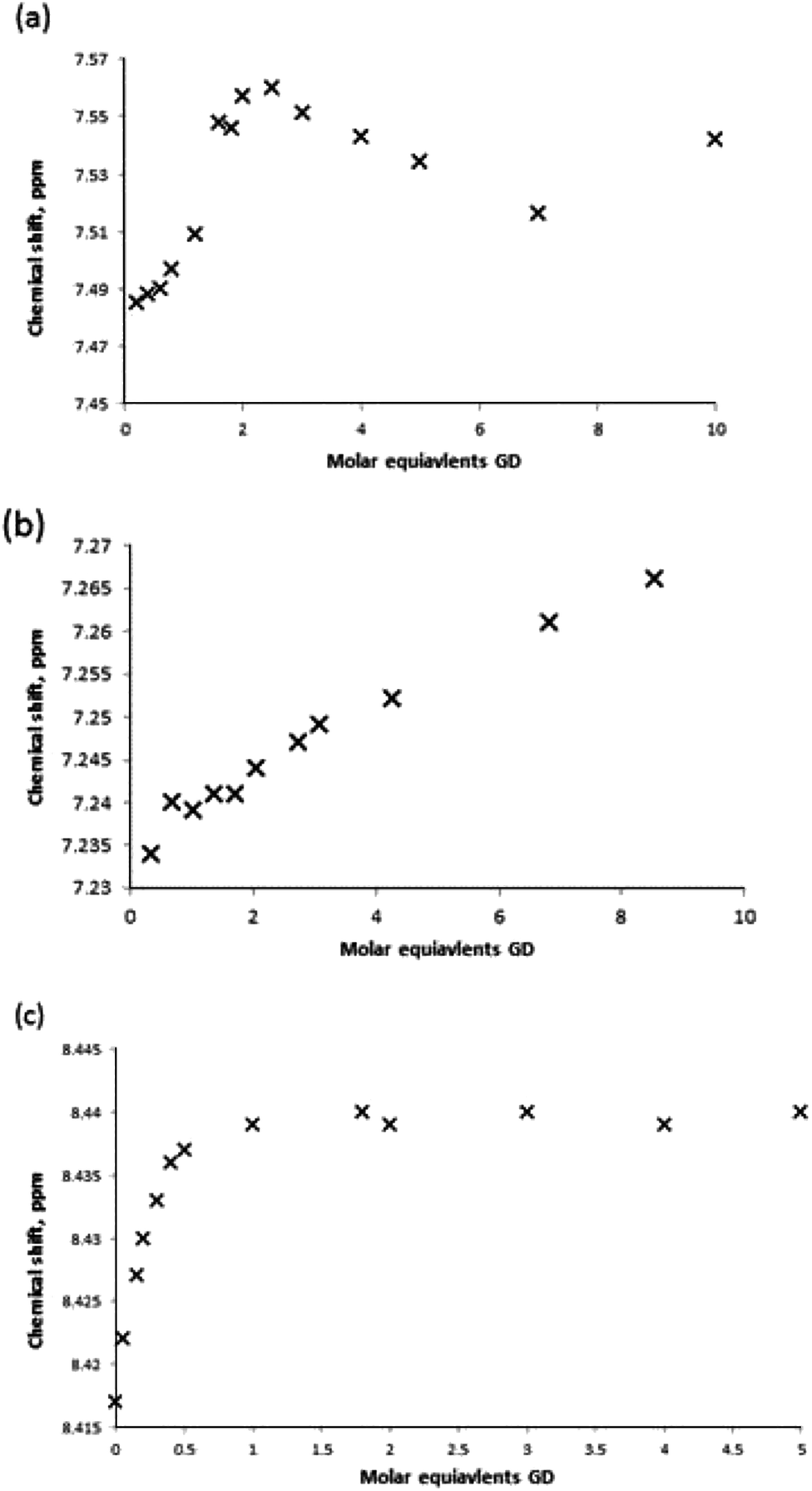 | ||
| Fig. 13 Binding curve generated by monitoring the urea NH protons of receptor (a) 1, (b) 3 and (c) 7 in the presence of increasing molar equivalents of GD in MeCN-d3. | ||
Monitoring the boronic acid OH protons also revealed strong evidence for GD recognition. Downfield shift perturbations were accompanied by rapid broadening of the proton resonance, resulting in the loss of the ability to monitor this environment by around 2–2.5 molar equivalents of GD present. 31P NMR of a sample of receptor 5 with 5 molar equivalents of GD revealed that the GD remained intact; confirming that loss of the B–OH resonance is due to complex formation and not the generation of reaction products.
The results of titration experiments with receptors 3 and 7 provided a number of points for consideration. Using standard NMR titration methods binding curves were generated for both receptors and that for receptor 3 was suitable for data fitting (Fig. 13b) and an association constant determined as Kassoc = 108 M−1. However, in both cases it was noted that there was a substantial relative increase in chemical shift in the region 0.2–0.4 molar equivalents, and that in the case of host 7 it was particularly pronounced and indicated the possibility of significantly underestimating the affinity. Therefore, additional NMR titration data was gathered over the very low guest concentration range for receptor 7, and a partial data set over the range 0–5 molar equivalents is shown in Fig. 13c (also see ESI†). This generated a titration curve for which fitting to a 1:1 binding model was partially successful and indicated a surprisingly high affinity with Kassoc = 1500 ± 250 M−1. It should be noted that the data for compound 7 in particular was challenging to fit, and the association constant should be taken as an estimate. Errors were estimated by continually modifying the fit model through changing of the input free host chemical shift (±0.002 ppm increments) and through the addition/removal of further data points at higher molar equivalents of guest (>1 mol equiv.). Acceptable fits were obtained using a data range that approximated 10–90% complex formation; data fitting became unacceptable by visual inspection where modifications to input data led to Kassoc estimates that differed by more than 250 from 1500 M−1, thus leading to the error estimate. This allows us to propose the general complex structure shown in Fig. 14, in which binding occurs through the formation of NH⋯OP hydrogen bond interactions.
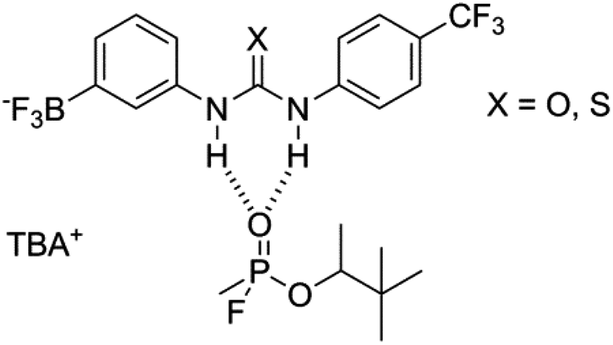 | ||
| Fig. 14 Proposed complexation of GD with receptors 3 (X = O) and 7 (X = S), where TBA = tetrabutylammonium. | ||
Titrations of receptor 4 with GD did not reveal strong evidence for complex formation, although an initial perturbation in the host urea NH proton of greater separation from the BF3− signal was observed. The formation of a very weak complex cannot be ruled out, although the lack of a binding curve amenable to data fitting and the lack of perturbations in the urea NH proton neighbouring the aryl-BF3− leads us to estimate an association constant close to zero. In this case, the position of the BF3− group may hinder complex formation through either (a) intramolecular B–F⋯H–N hydrogen bonding, or (b) steric blocking of the binding site unsurprisingly, receptor 8 did not demonstrate the ability to bind GD, and this likely arises from the presence of a single H-bond donor group.
Conclusions
A series of ditopic and anionic host molecules have been designed and synthesised. The recognition of chloride anion guests by urea NH and boronic acid B(OH)2 hydrogen bond donors has been demonstrated in ditopic host molecules, and association constants determined. The behaviour of these systems towards dihydrogen phosphate anions is dominated by their Lewis acidity, in agreement with the results reported by others.27 For the three receptors that possess pendant BF3− groups (3, 4 and 7), anion recognition at (thio)urea groups appears to be hindered, and we presume this is a result of coulombic repulsion. The binding of neutral OP compounds by both NH and B–OH hydrogen bond donors has been demonstrated. The commonly-used nerve agent simulant DMMP does not act as an effective hydrogen bond acceptor, with any host–guest complexes formed possessing low stabilities. Interestingly, this corresponds well with the findings of our recent studies on responsive supramolecular gels, in which DMMP was noticeably less effective at perturbing gel structures than GD.19 Interactions of host molecules with PMP lead to the formation of reaction products, or low affinity complexes, albeit more stable than the corresponding host–DMMP pairings. The CWA GD has been shown to interact with both NH and B–OH groups, although association constants for ditopic receptors could not be determined. A high affinity complex was observed with the negatively charged host 7, and it is comparably more stable than the corresponding complexes with Cl− and H2PO4− anions. Although we were unable to obtain complex affinities for GD with our neutral receptors, the results with anionic hosts indicate the possibility of using supramolecular interactions to significantly modify selectivity trends.Acknowledgements
JRH and PAG would like to thank EPSRC (EP/K503770/1) and Dstl (UK) for funding as well as the analytical departments at the University of Southampton for their assistance in obtaining mass spectrometry and single crystal X-ray diffraction data. M. R. S. and J. E. would like to thank Dr James Jones (Dstl) for assistance with NMR. M. R. S. would like to thank Professor Michael Hynes for his assistance and invaluable advice on data fitting. This article includes material subject to © Crown copyright (2015), Dstl.Notes and references
- K. Kim, O. G. Tsay, D. A. Atwood and D. G. Churchill, Chem. Rev., 2011, 111, 5345–5403 CrossRef CAS PubMed.
- R. M. Black and J. Harrison, The Chemistry of Organophosphorus Compounds, ed. F. R. Hartley, Wiley, Chichester (UK), 1996, vol. 4 Search PubMed.
- Y. C. Yang, J. A. Baker and R. J. Ward, Chem. Rev., 1992, 92, 1729–1743 CrossRef CAS.
- A. Grabowski, A. Richard and M. M. Blum, Decontamination of Warfare Agents, ed. A. Richard and M. M. Blum, Wiley, 2008, pp. 55–66 Search PubMed.
- H. Okudera, H. Morita, T. Iwashita, T. Shibata, T. Otagiri, S. Koayashi and N. Yanagisawa, Am. J. Emerg. Med., 1997, 15, 527–528 CrossRef CAS PubMed.
- R. Danzig, M. Sageman, T. Leighton, L. Hough, H. Yuki, R. Kotani and Z. M. Hosford, Shinrikyo – Aum Insights into How Terrorists Develop Biological and Chemical Weapons, Center for a New American Security, http://www.cnas.org.
- Anonymous, Chem. Eng. News, 2013, 91, 20 Search PubMed.
- Anonymous, Science, 2013, 341, 1324 Search PubMed.
- M. R. Sambrook and S. Notman, Chem. Soc. Rev., 2013, 42, 9251–9267 RSC.
- B. Desire and S. Saint-Andre, Fundam. Appl. Toxicol., 1986, 7, 646–657 CrossRef CAS PubMed.
- M. Zengerle, F. Braundhuber, C. Schneider, F. Worek, G. Reiter and S. Kubik, Beilstein J. Org. Chem., 2011, 7, 1205–1214 CrossRef PubMed.
- S. M. Daly, M. Grassi, D. K. Shenoy, F. Ugozzoli and E. J. Dalcanale, Mater. Chem., 2007, 17, 1809–1818 RSC.
- Y. Ruan, H. A. Taha, R. J. Yoder, V. Maslak, C. M. Hadad and J. D. Badjić, J. Phys. Chem. B, 2013, 117, 3240–3249 CrossRef CAS PubMed.
- K. Kim, I. Kim, N. Maiti, S. J. Kwon, D. Bucella, O. A. Egorova, Y. S. Lee, J. Kwak and D. G. Churchill, Polyhedron, 2009, 28, 2418–2430 CrossRef CAS.
- G. H. Dennison, M. R. Sambrook and M. R. Johnston, RSC Adv., 2014, 4, 55524–55528 RSC.
- M. R. Sambrook, J. R. Hiscock, A. Cook, A. C. Green, I. Holden, J. C. Vincent and P. A. Gale, Chem. Commun., 2012, 48, 5605–5607 RSC.
- J. R. Hiscock, F. Piana, M. R. Sambrook, N. J. Wells, A. J. Clark, J. C. Vincent, N. Busschaert, R. C. D. Brown and P. A. Gale, Chem. Commun., 2013, 49, 9119–9121 RSC.
- J. R. Hiscock, I. L. Kirby, J. Herniman, G. J. Langley, A. J. Clark and P. A. Gale, RSC Adv., 2014, 4, 45517–45521 RSC.
- J. R. Hiscock, M. R. Sambrook, J. A. Ede, N. J. Wells and P. A. Gale, J. Mater. Chem. A, 2015, 3, 1230–1234 CAS.
- F. Piana, M. Facciotti, G. Pileio, J. R. Hiscock, W. Van Rossom, R. C. D. Brown and P. A. Gale, RSC Adv., 2015, 5, 12287–12292 RSC.
- J. R. Hiscock, M. R. Sambrook, P. B. Cranwell, P. Watts, J. C. Vincent, D. J. Xuereb, N. J. Wells, R. Raja and P. A. Gale, Chem. Commun., 2014, 50, 6217–6220 RSC.
- A. Barba-Bon, A. M. Costero, M. Parra, M. S. Gil, R. Martinez-Manez, F. Sancenon, P. A. Gale and J. R. Hiscock, Chem. – Eur. J., 2013, 19, 1586–1590 CrossRef CAS PubMed.
- M. P. Hughes and B. D. Smith, J. Org. Chem., 1997, 62, 4492–4499 CrossRef CAS PubMed.
- M. C. Etter, Z. Urbañczyk-Lipkowska, M. Z. Ebrahimi and T. W. Panunto, J. Am. Chem. Soc., 1990, 112, 8415–8426 CrossRef CAS.
- V. Amendola, L. Fabbrizzi and L. Mosca, Chem. Soc. Rev., 2010, 39, 3889–3915 RSC.
- L. S. Evans, P. A. Gale, M. E. Light and R. Quesada, New J. Chem., 2006, 30, 1019–1025 RSC.
- M. A. Martínez-Aguirre and A. K. Yatsimirsky, J. Org. Chem., 2015, 80, 4985–4993 CrossRef PubMed.
- M. J. J. Hynes, J. Chem. Soc., Dalton Trans., 1993, 311–312 RSC.
- S. J. Coles and P. A. Gale, Chem. Sci., 2012, 3, 683–689 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthesis methods and characterisation, additional NMR, crystallographic and MS data, and titration data fitting. CCDC 1430064 and 1430065. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ob01210h |
| ‡ X-ray data were collected on a Rigaku AFC 12 diffractometer mounted on Rigaku FR-E+ Super Bright Ultra High Flux rotating anode CCD diffractometer equipped with VariMax very high flux (VHF) optics and Saturn 724+ CCD detector.29 Crystal data for receptor 6. CCDC 1430064, C28H22B2F6N4O3S2 (M = 662.23): triclinic, space group P Crystal data for receptor 4. CCDC 1430065, C30H46BF6N3O (M = 589.51): monoclinic, space group P21/n (no. 14), a = 17.561(6) Å, b = 16.385(5) Å, c = 23.157(8) Å, β = 103.246(5)°, V = 6486(4) Å3, Z = 8, T = 100(2) K, μ(MoKα) = 0.096 mm−1, Dcalc = 1.207 g mm−3, 63 |
![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (no. 2),
(no. 2), | This journal is © The Royal Society of Chemistry 2016 |