
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Recent advances in the synthesis of indolizines and their π-expanded analogues
Bartłomiej
Sadowski
,
Jan
Klajn
and
Daniel T.
Gryko
*
Institute of Organic Chemistry of the Polish Academy of Sciences, 01-224 Warsaw, Poland. E-mail: dtgryko@icho.edu.pl; Fax: +48 22 6326681; Tel: +48 22 3433063
First published on 28th June 2016
Abstract
Indolizine (pyrrolo[1,2-a]pyridine) is one of the five isomers of indole and it serves as a precursor for widespread indolizidine alkaloids. The straightforward synthesis of indolizines based on classical methodologies such as Scholtz or Chichibabin reactions has overshadowed numerous new strategies that have been revealed especially within the last ten years. The desire to achieve substitution patterns which were hard to build sparked the discovery of completely new pathways, e.g. transition metal-catalyzed reactions and approaches based on oxidative coupling. In this review, selected strategies toward indolizines published since 2005 are briefly summarized, commented upon, compared, and illustrated. The literature discussed here involves reactions based on either pyridine or pyrrole scaffolds, as well as selected methodologies leading to π-expanded indolizines.
 Bartłomiej Sadowski | Bartłomiej Sadowski was born in Nowy Dwór Mazowiecki, Poland, in 1990. In 2014, he graduated from the Warsaw University of Technology with a Masters in Engineering. He is currently undertaking a PhD at the Institute of Organic Chemistry of the Polish Academy of Sciences. His research interests focus on the synthesis and spectroscopy of advanced functional dyes with special emphasis on coumarins, pyrrole[3,2-b]pyrroles, as well as cross-conjugated chromophores. |
 Jan Klajn | Jan Klajn graduated from the Department of Chemistry, University of Wrocław under the supervision of Prof. Latos-Grażyński. Subsequently he joined Prof. Gryko's group in August 2008. He defended his PhD-thesis under his supervision at the Institute of Organic Chemistry, Polish Academy of Sciences in 2013. Currently, he works at the Department of Chemistry, University of Wrocław as a post-doctoral fellow under the guidance of Dr Pawlicki. His research interests focus on the chemistry of indolizines as well as on electron-rich functional dyes. |
 Daniel T. Gryko | Daniel T. Gryko obtained his PhD from the Institute of Organic Chemistry of the Polish Academy of Sciences in 1997 under the supervision of J. Jurczak. After post-doctoral studies with J. Lindsey at the North Carolina State University (1998–2000), he began his independent career in Poland. He became Full Professor in 2008. His current research interests are focused on the synthesis of corroles, π-expanded porphyrinoids and other functional dyes as well as on two-photon absorption, artificial photosynthesis, excited-state intramolecular proton transfer and fluorescence imaging. |
1. Introduction
Nitrogen-containing heterocycles play a key role in both medicinal and materials chemistry. Numerous drugs, dyes, and high-performance materials are inherently heterocyclic.1 The methods for their synthesis are continuously improving. One of the most important N-fused heterocyclic skeletons is indolizine (1) (Fig. 1), which is composed of two condensed rings: a pyrrole-type five-membered ring and a pyridine-type six-membered ring.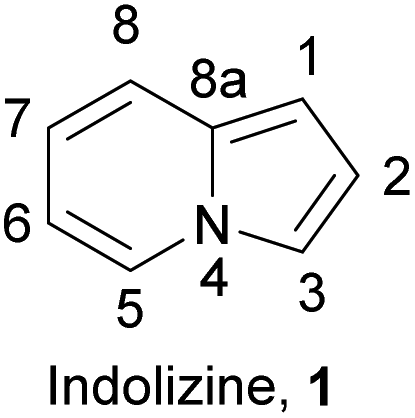 | ||
| Fig. 1 Structure and numbering system of indolizine skeleton. | ||
The partially or more often wholly hydrogenated derivatives of indolizine can be frequently found as backbones in many bioactive natural products such as swainsonine 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 2a or cryptowoline 3 (Fig. 2).2b Moreover, their synthetic derivatives have been found to exhibit a variety of biological activities, including as a potential inhibitor of vascular endothelial growth factor (VEGF) and for neuropilin-1 (NRP1) synergy (4),3a as agents with antibacterial, antiviral, anti-inflammatory properties,3b–d as well as phosphodiesterase V inhibitors (5),3e histamine H3 receptor antagonists,3f or as selective chemical probes for the bromodomains BAZ2A and BAZ2B.3g Indolizine derivatives have also been investigated for their suitability as dyes for dye sensitized solar cells (DSSC) (6)4a or organic light emitting devices (OLEDs),4b,c owing to their appreciable photophysical properties.
2a or cryptowoline 3 (Fig. 2).2b Moreover, their synthetic derivatives have been found to exhibit a variety of biological activities, including as a potential inhibitor of vascular endothelial growth factor (VEGF) and for neuropilin-1 (NRP1) synergy (4),3a as agents with antibacterial, antiviral, anti-inflammatory properties,3b–d as well as phosphodiesterase V inhibitors (5),3e histamine H3 receptor antagonists,3f or as selective chemical probes for the bromodomains BAZ2A and BAZ2B.3g Indolizine derivatives have also been investigated for their suitability as dyes for dye sensitized solar cells (DSSC) (6)4a or organic light emitting devices (OLEDs),4b,c owing to their appreciable photophysical properties.
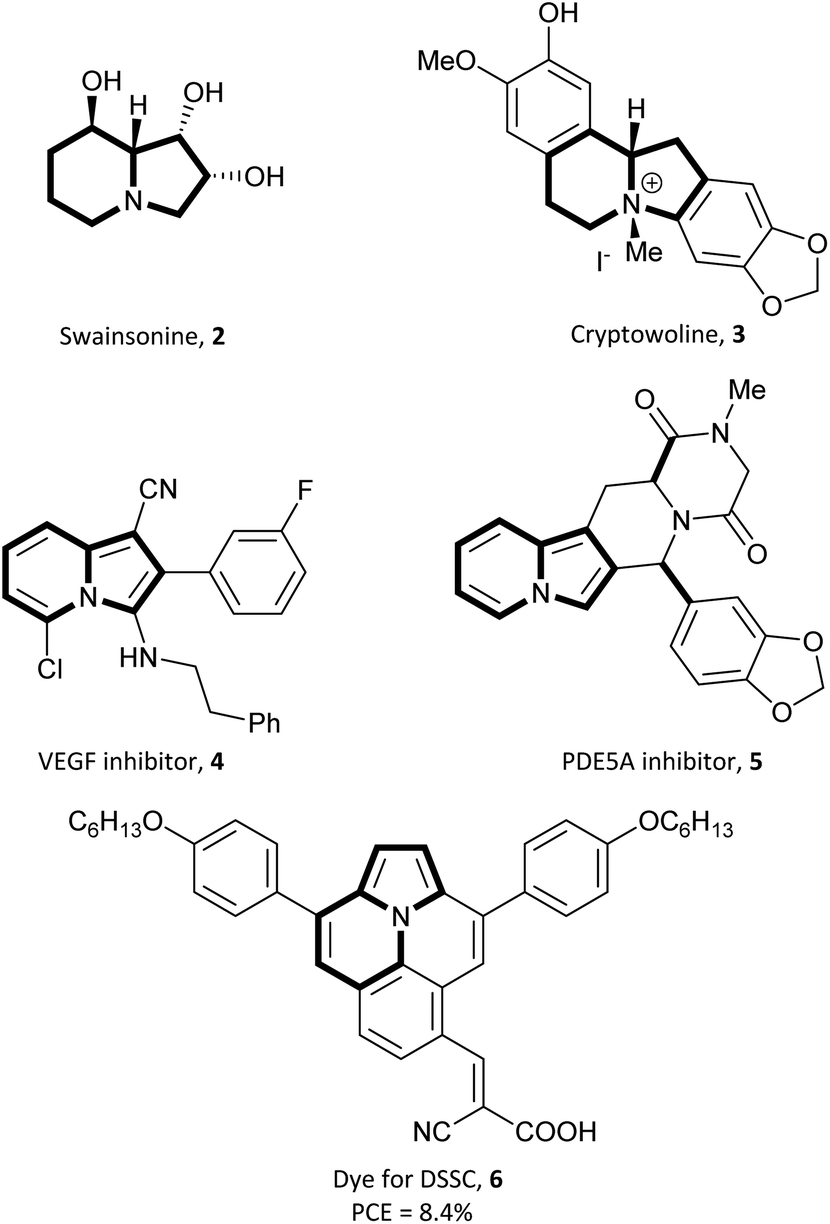 | ||
| Fig. 2 Examples of useful indolizine derivatives. | ||
The first report on indolizine comes from the 19th century.5 Since that time, numerous studies on their synthesis, properties, and applications have been published, which have been partially collected in earlier reviews.6 In this contribution, we have highlighted the most important methodologies leading to the formation of the indolizine core developed over the past ten years. The number of valuable contributions has compelled us to be selective. Our choices featured in this review were based on a combination of the novelty and utility of these novel approaches. Synthetic strategies leading to indolizines possessing additional exocyclic conjugated C–C double or triple bonds, as well as functionalization of the indolizine core7 have been omitted here for the sake of brevity.
2. Methods based on pyridine derivatives
Starting the indolizine synthesis from pyridine derivatives has the fundamental advantage that pyridine derivatives are easily available via numerous methods and >2000 of them are commercially available. Due to their electron-deficient nature, they are typically stable under ambient conditions. Although the pyridine ring is stable due to its aromaticity, it readily undergoes various transformations of its core as long as the final product is also aromatic.2.1 From pyridines with a free C2 position
Basavaiah et al. synthesized a series of indolizines and their benzofused analogues (7) starting from pyridine, quinoline, or isoquinoline and Baylis–Hillman bromides (i.e., allyl bromides which can be prepared directly from products of the Baylis–Hillman reaction) via a one pot sequence, which includes salt formation and subsequent 1,5-cyclization of the nitrogen ylides (Scheme 1).8 The described reaction is a convenient way to access 1-aryl-2-cyanoindolizines and their π-expanded derivatives under mild conditions despite moderate yields.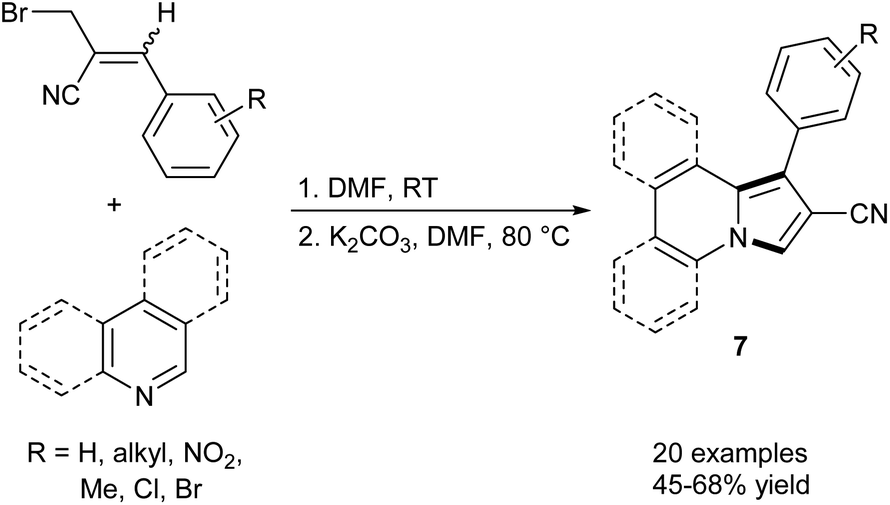 | ||
| Scheme 1 | ||
Attempting to use eco-friendly copper as a less expensive alternative to rhodium-based catalysts, Barluenga, Tomás and co-workers developed a direct annulation of pyridine derivatives with diazoacetates possessing carbon–carbon double bonds, forming indolizine derivatives 8 (Scheme 2).9 The broad scope of substrates and mild reaction conditions caused this method to be very useful in the synthesis of highly functionalized indolizines. Furthermore, fused pyridines such as quinoline, isoquinoline, and phenanthridine also reacted easily with ethyl isopropenyldiazoacetate under these conditions to give π-expanded analogues of indolizines in 65, 80, and 30% yield, respectively. Additionally, the selectivity of this method, depending on the type of substituent located at position 3 of pyridine, was investigated. It appeared, that in the case of 3-methylpyridine, the C3![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C4 bond is preferred for the coordination with the metal centre because of the electron donating influence of the methyl group, while for 3-nitropyridine the C4C5 bond is much more preferable for the coordination owing to the strong electron-withdrawing effect of the NO2 group, thus forming more stable complex intermediates I (Scheme 2, bottom).
C4 bond is preferred for the coordination with the metal centre because of the electron donating influence of the methyl group, while for 3-nitropyridine the C4C5 bond is much more preferable for the coordination owing to the strong electron-withdrawing effect of the NO2 group, thus forming more stable complex intermediates I (Scheme 2, bottom).
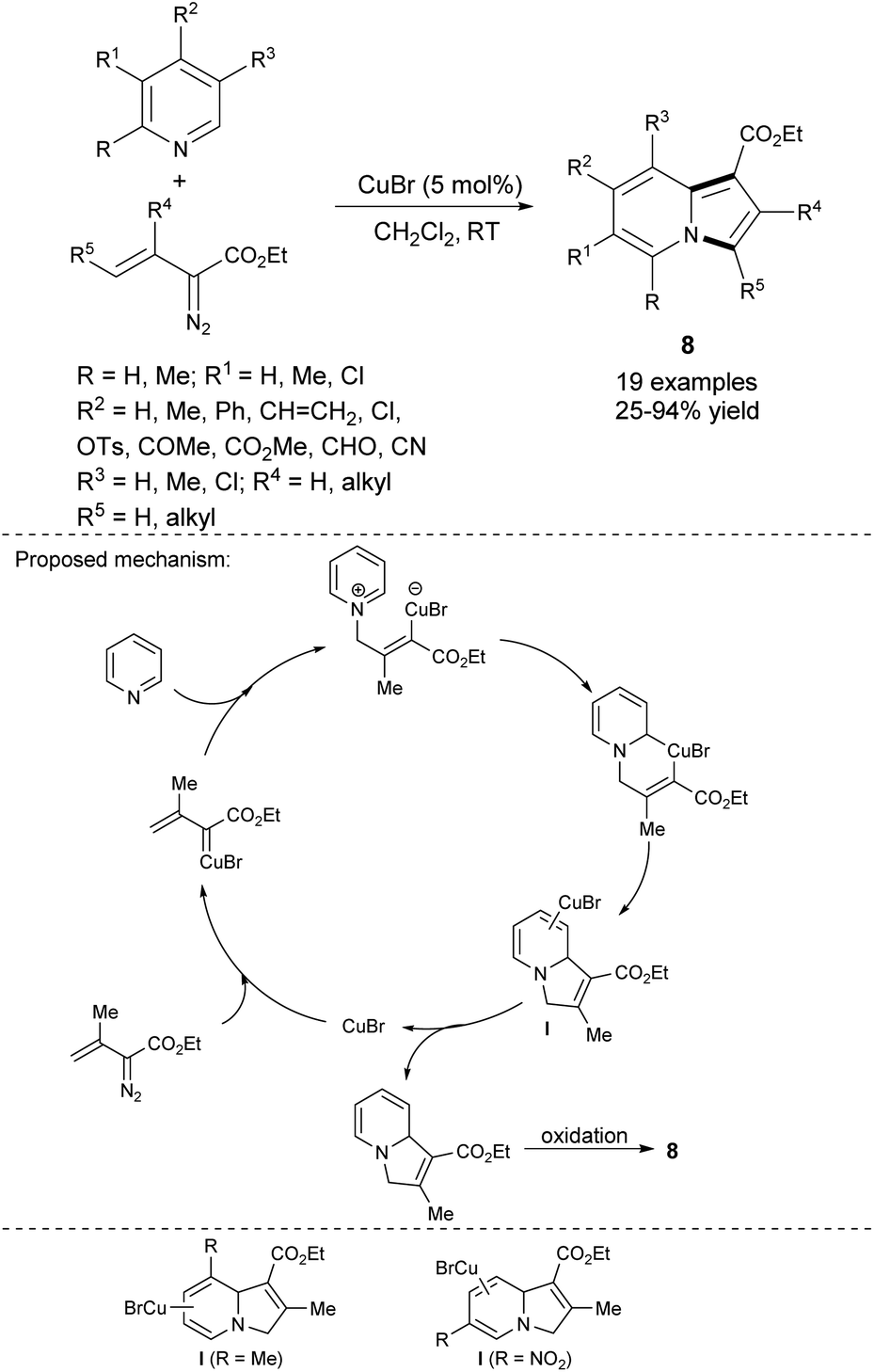 | ||
| Scheme 2 | ||
Recently, Wang's group proposed an elegant route leading to highly-substituted 1-cyanoindolizines (9) and 3-cyanoindolizines (10) starting from esters of 1-cyanocyclopropane-1-carboxylic acid (Scheme 3).10 This iodine-catalyzed reaction also proceeded well with quinoline and isoquinoline to give π-expanded indolizines 11 and 12, respectively, in good yields. The application of pyridine as a solvent led to heterocycle 10, suggesting that different reaction mechanisms apply in the case of either toluene or pyridine. During the course of the reaction, 1-cyanocyclopropane-1-carboxylic acid is converted under the influence of a base and iodine into a carboanion, which can exist in two mesomeric forms – A and A′. The stability of these two species strongly depends on both solvent polarity and steric hindrances presented therein. The α-carboanion A with two strongly electron-withdrawing groups (cyano and ester) is stable in a weak polar solvent such as toluene and due to large steric hindrances is solvated with difficulty by pyridine. Consequently, the 1,3-dipolar cycloaddition reaction of A and the pyridine derivative in toluene gave 9. In contrast, the benzyl anion A′, which is a stable resonance in pyridine,10b reacted easily in pyridine to give 10. The position of CN, aryl and aroyl substituents at C1, C2 and C3 in compounds 9 and 10 was confirmed by X-ray analyses. Surprisingly, esters of 1-cyanocyclopropane-1-carboxylic acid showed no reactivity toward Lewis acids such as aluminum(III) chloride, zinc(II) chloride, iron(III) chloride or boron trifluoride.
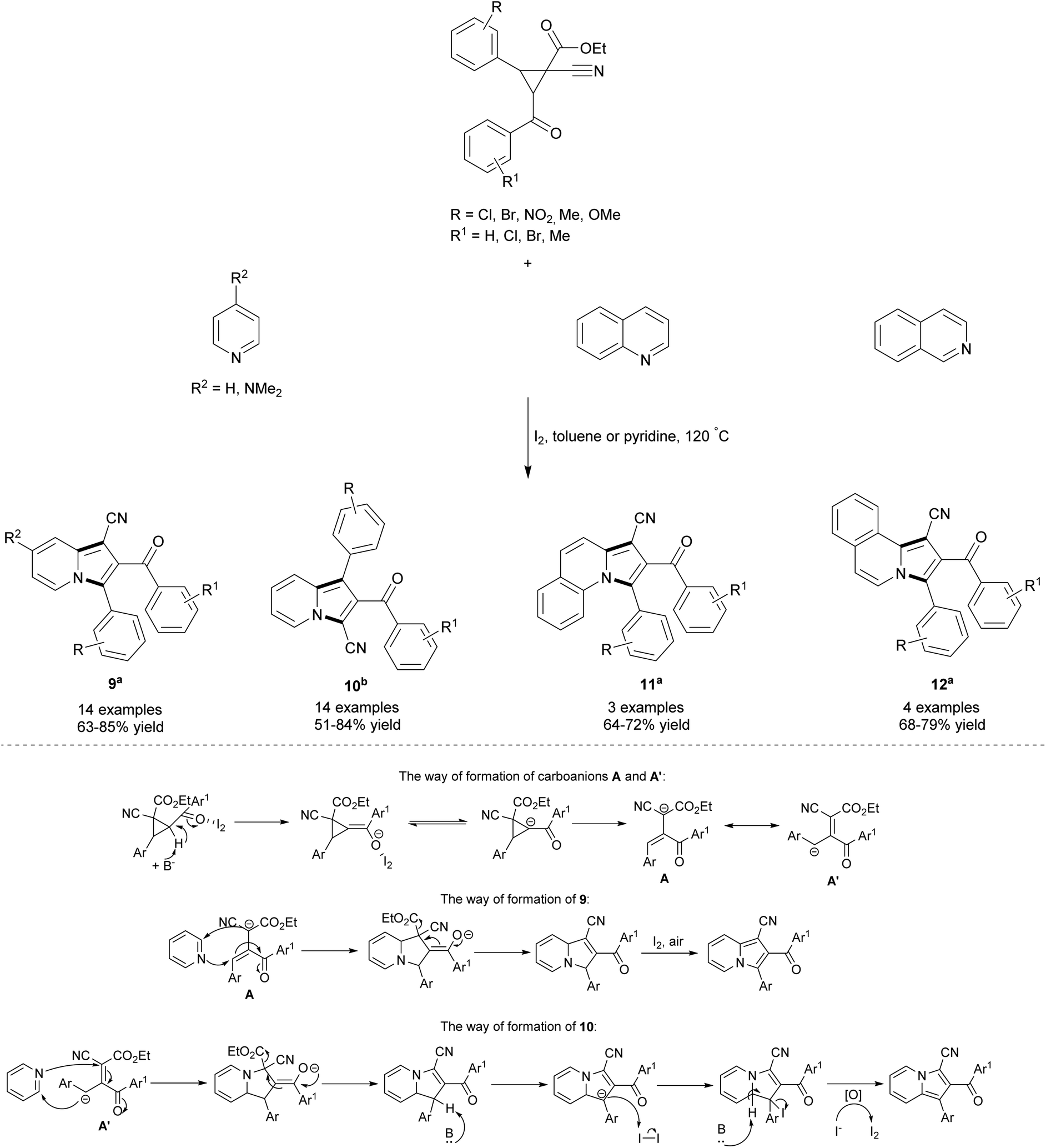 | ||
| Scheme 3 aToluene was used as a solvent. bPyridine was used as a solvent. | ||
2.2 From pyridines bearing formyl or keto functionalities at position 2
Although the sequence of the reaction leading to the desired product starting from unsubstituted pyridine has the advantage of simplicity, only very few such processes are available as far as indolizine synthesis is concerned. The overwhelming majority of strategies require mono-substituted pyridines as starting materials.Rao and Basavaiach reported the first example of an electrophile-induced Baylis–Hillman reaction between pyridine-2-carboxaldehyde and activated alkenes such as alkyl vinyl ketones or cyclic enones under the influence of trimethylsilyl trifluoromethanesulfonate (TMSOTf) (Scheme 4).11 The authors assumed that this reaction would proceed according to the most generally accepted Baylis–Hillman reaction mechanism (Scheme 4, bottom).12
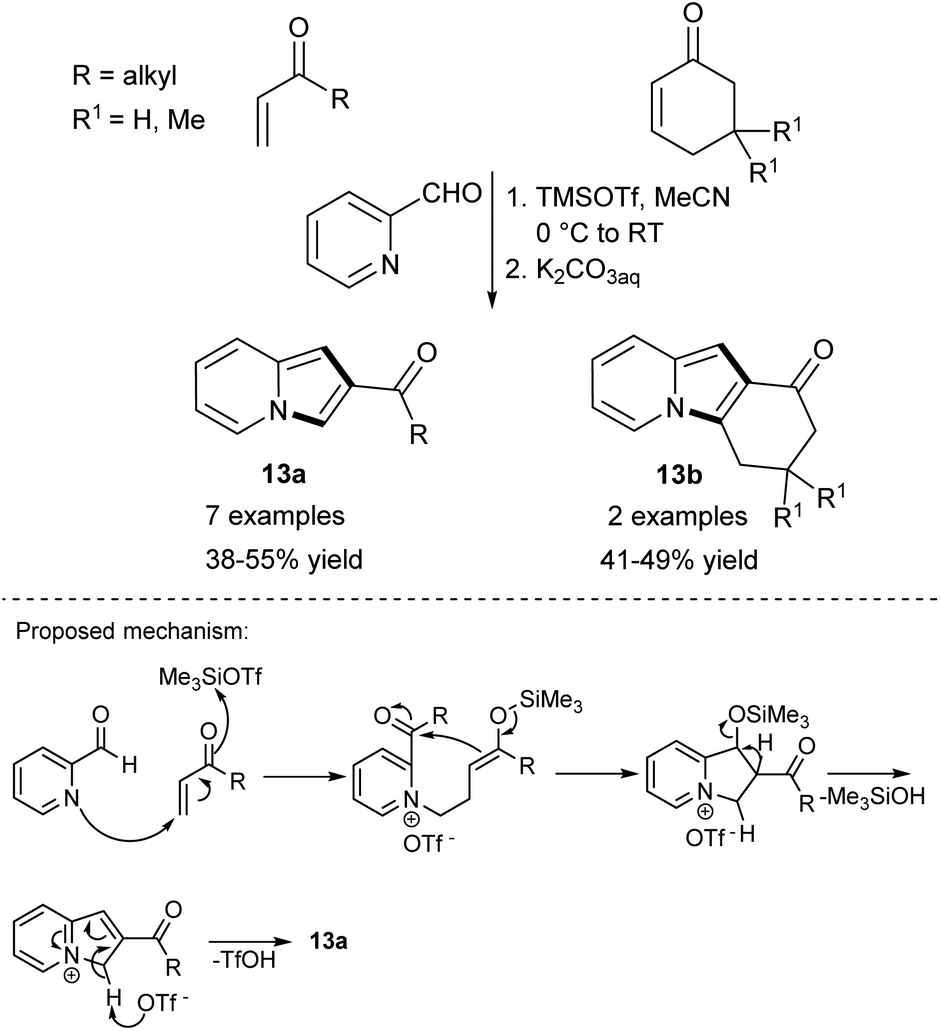 | ||
| Scheme 4 | ||
Later, the same group extended application of this method employing 2-alkanoyl- or 2-aroylpyridines and vinyl ketones in refluxing acetonitrile to produce 14a–b in moderate yields (Scheme 5).13 Unfortunately, the subsequent Michael addition of 14a and the corresponding vinyl ketone resulted in a decrease in the overall yield. The reaction of isoquinolin-1-yl phenyl ketone and cyclohex-2-enone gave the corresponding π-expanded indolizine in 45% yield.
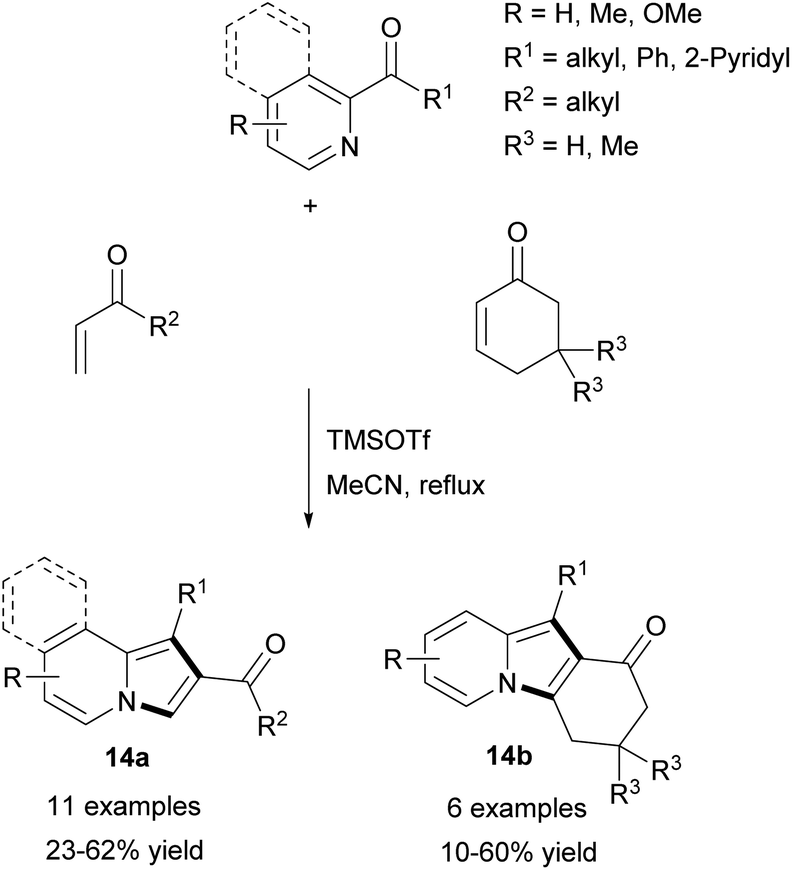 | ||
| Scheme 5 | ||
Liu and Yan were the first to report the Au-catalyzed reaction between aldehydes, secondary amines, and terminal alkynes leading to 1-aminoindolizines in a simple and high-atom economical route (Scheme 6).14 This reaction proceeds either under solvent-free conditions or in water. Importantly, use of aromatic amines in this transformation resulted in relatively low yields of the corresponding products. A series of N-benzyl amino acid derivatives was used as the amine partner according to this protocol, giving 15 in good yields without loss of enantiomeric purity. It is noteworthy that while unsubstituted indolizine is not stable under ambient conditions, its derivatives 15 bearing an amino group attached to a five-membered ring are very unstable both in solution and in the solid state and they must be handled with care. This multicomponent reaction was a breakthrough that prompted other research groups to seek less expensive catalysts. Eventually it was found that the same type of reaction can be realized under the influence of AgBF4,15 Fe(acac)3/TBAOH,16 or copper nanoparticles supported on activated carbon (CNPs/C)17 systems. Recently, fully recyclable, green, and highly efficient conditions (CuI/CSP in ethylene glycol) have been proposed for this process with high atom economy (94%), and a small environmental (E) factor (0.06).18
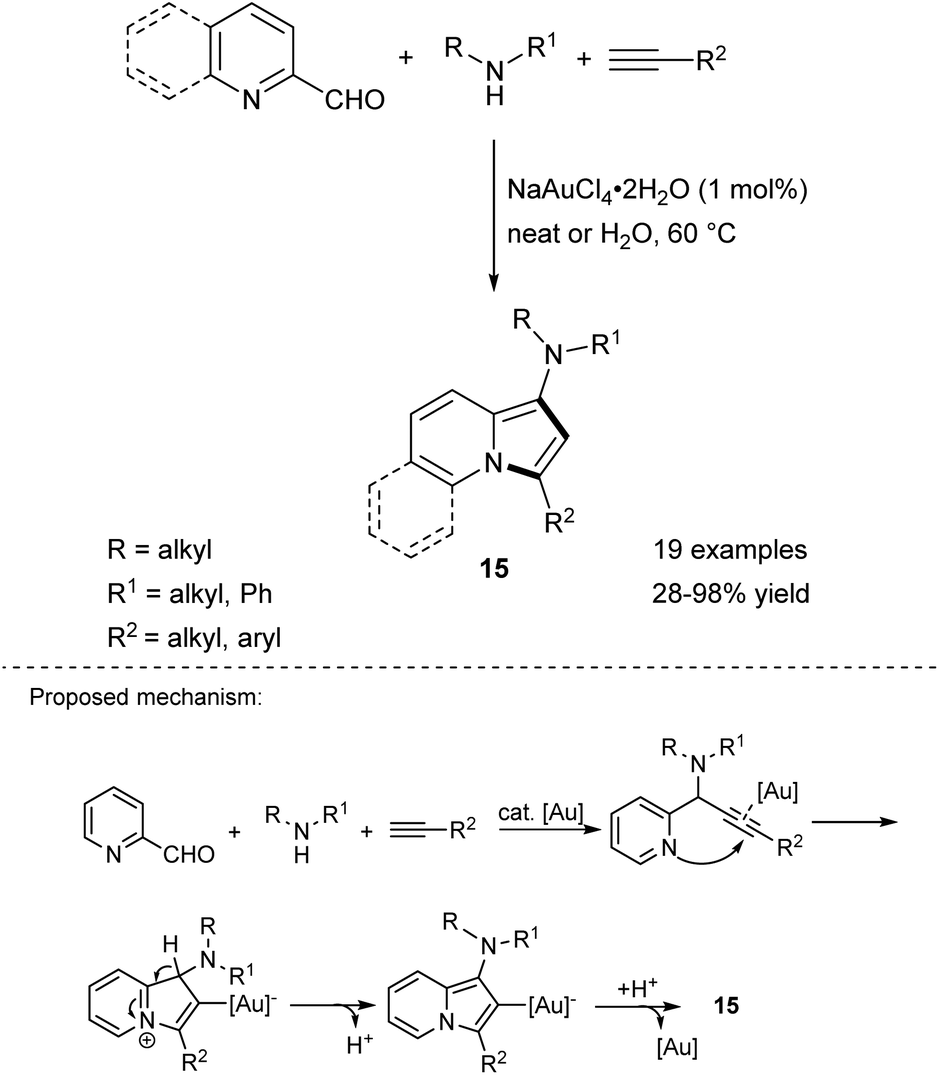 | ||
| Scheme 6 | ||
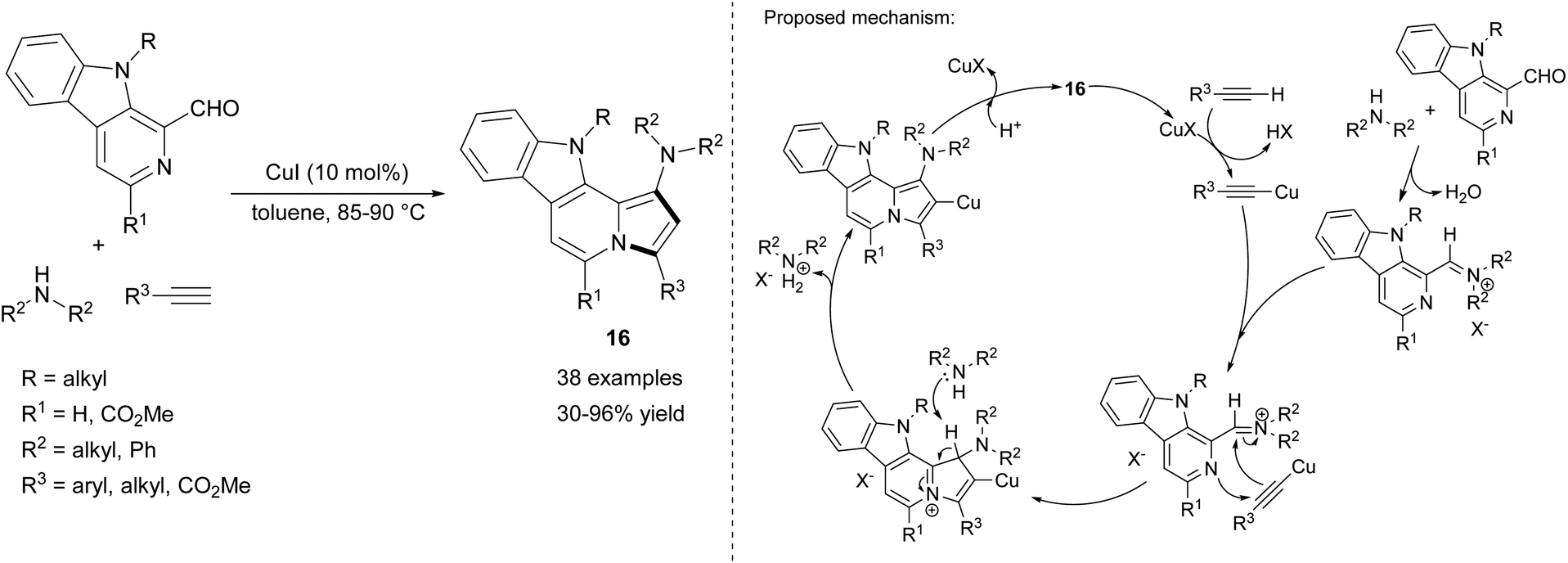
The series of compounds 16 containing the indolizino[8,7-b]indole's skeleton, occurring in many alkaloids, was obtained similarly by Batra's group via a coupling/cycloisomerization sequence of N-substituted 1-formyl-9H-β-carbolines, secondary amines, and terminal alkynes (Scheme 7).19 The method was equally efficient for both aromatic and aliphatic alkynes, however the authors obtained no product in the case of trimethylsilylacetylene. Moreover, while aliphatic amines gave the corresponding products in good yields, diphenylamine showed no reactivity in this reaction.
 | ||
| Scheme 7 | ||
2.3 From 2-halo- or 2-triflylpyridines and alkynes
The ability to introduce an amino group at any aromatic hydrocarbon or heterocycle opens a plethora of possibilities for further functionalization via electrophilic aromatic substitution. An efficient one-pot route leading to 3-aminoindolizines and its expanded analogues 17 has been developed via a Pd/Cu-catalyzed sequential Sonogashira cross-coupling/cycloisomerization process (Scheme 8).20 Interestingly, a few other derivatives (18 and 19a–c) containing two indolizine moieties within a molecule were also synthesized according to the above protocol in satisfactory yields, demonstrating its high synthetic potential. The use of flow technology in this process allowed linear scale-up from milligram- to gram-scale in the synthesis of a broad range of 3-aminoindolizines.21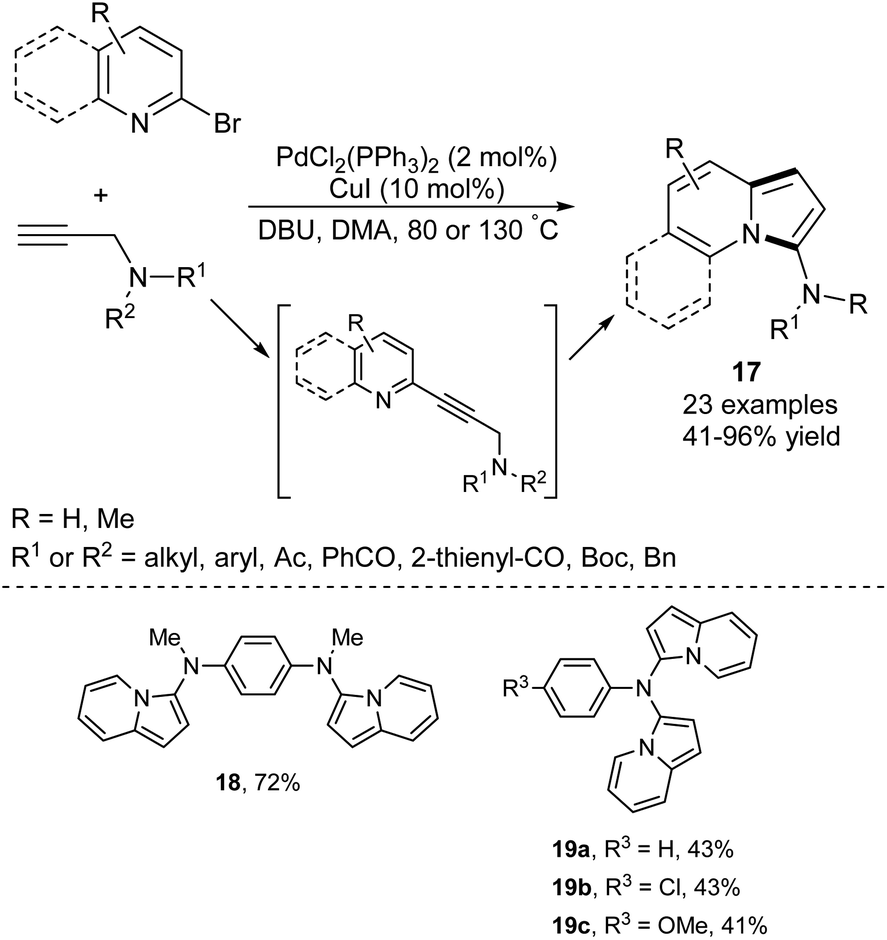 | ||
| Scheme 8 | ||
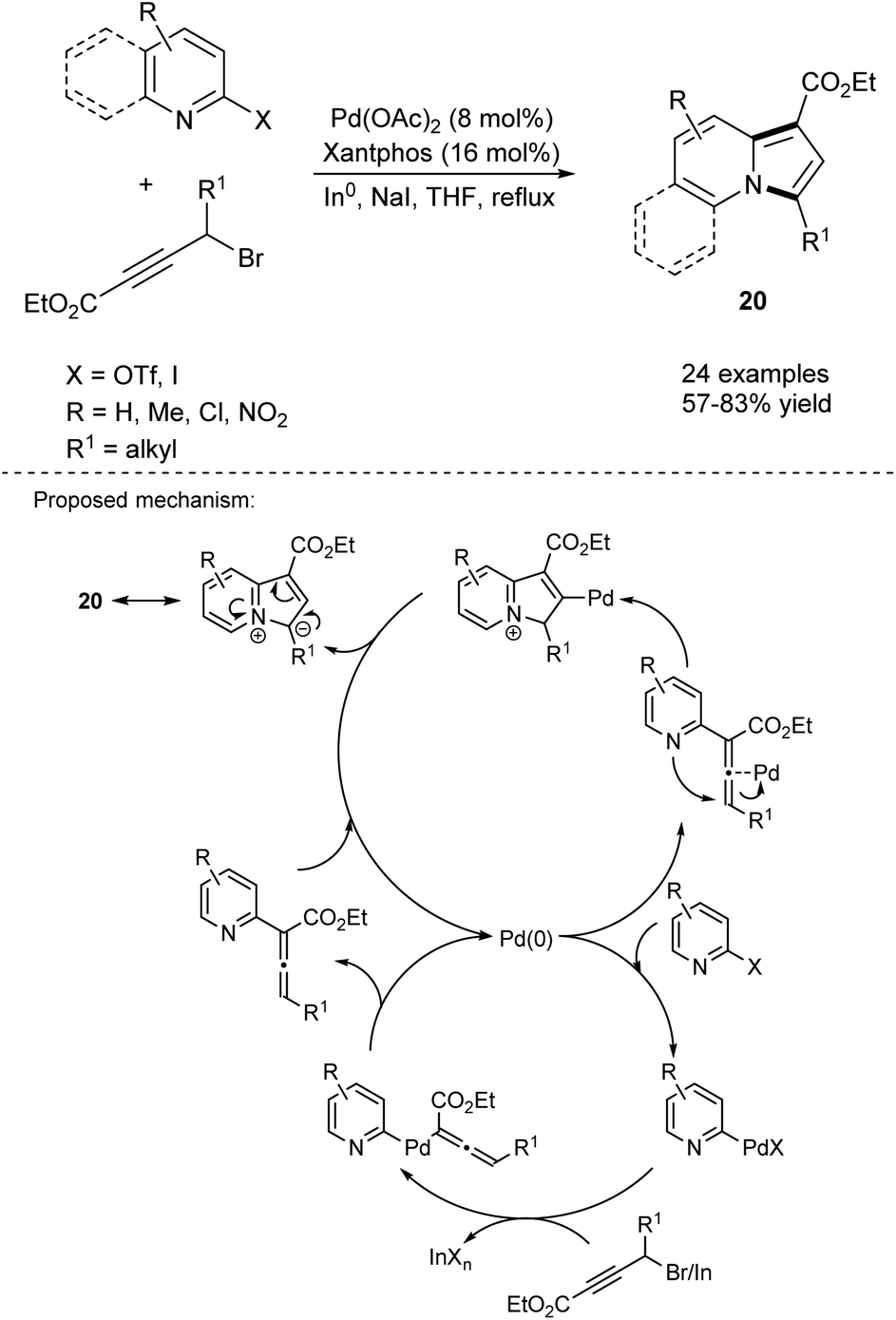
Kim et al. reported the direct formation of indolizines 20 through a tandem one-pot Pd-catalyzed cross-coupling reaction using organoindium reagents formed in situ, followed by subsequent cycloisomerization of the allenylpyridine intermediate (Scheme 9).22 The role of NaI has not yet been established, but the authors suspect it facilitates the formation of the organoindium reagent through the substitution of bromine. The presence of a strong electron-withdrawing group in the substrate had a negative influence on the reaction outcome.
 | ||
| Scheme 9 | ||
2.4 From 2-alkylpyridines
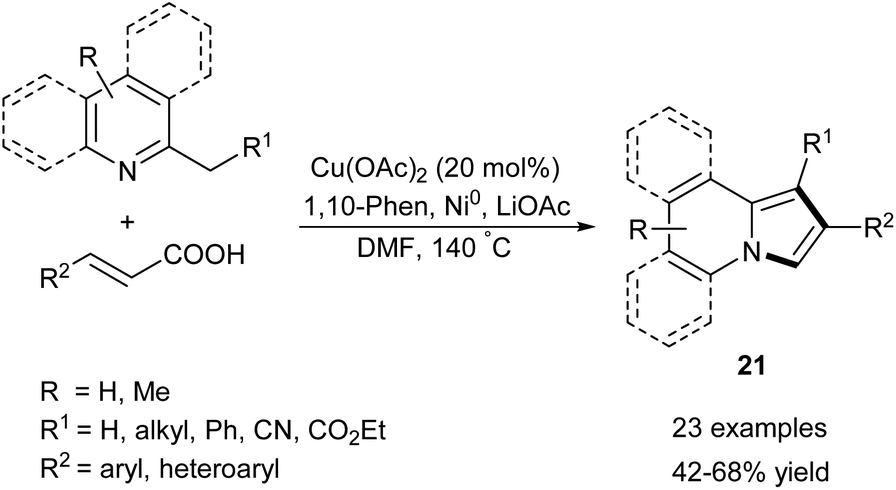
2-Alkylpyridines (especially α-picolines) have long been exploited as valuable substrates to access the indolizine skeleton (e.g., Scholtz or Chichibabin methods for indolizine synthesis). In stark contrast to these older methodologies, the new approaches toward indolizines starting from 2-alkylpyridines rely on oxidation of the initially formed intermediate. A survey of the more recent advances within this area is presented in the following section.Zhang and co-workers developed a protocol for the annulation of substituted 2-alkylpyridines and cinnamic acids in the presence of copper(II) acetate, powdered nickel, 1,10-phenanthroline and lithium acetate (Scheme 10).23 Although the method gave good results for the broad scope of substrates, 2-alkylindolizines cannot be obtained according to this method. It is worth noting that this approach leads to the same pattern of substituents as the Chichibabin reaction.
 | ||
| Scheme 10 | ||
2-Arylated indolizines (22) can also be synthesized by the reaction between 2-alkylpyridines and Morita–Baylis–Hillman acetates (MBHAs) only in the presence of triethylamine (Scheme 11).24 2-Pyridylacetonitrile reacted well also as did ethyl 2-pyridylacetate, although the yields were found to be slightly lower. Notably, this strategy bears a resemblance to the pyrrole synthesis from imines and nitroolefins, which also proceeds with extrusion of HNO2.25
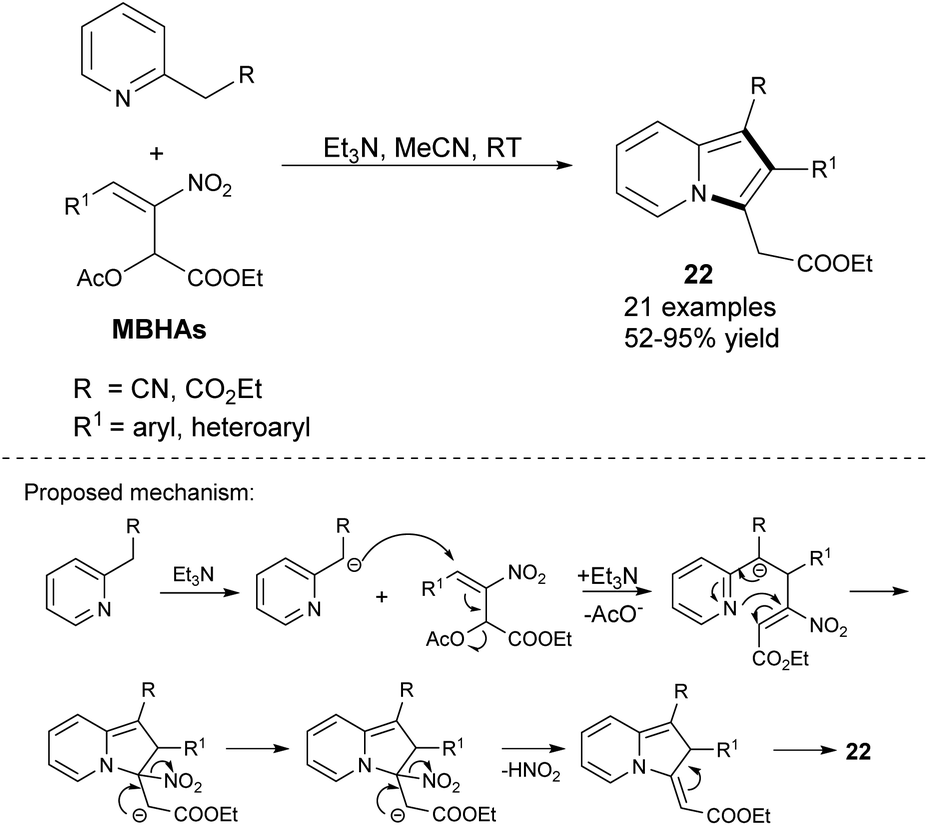 | ||
| Scheme 11 | ||
The synthesis of π-expanded indolizines starting from 2-(quinolin-2-yl)acetates and nitroolefins in the presence of cerium(III) salt has been reported (Scheme 12).26 The method was almost equally efficient when either CoCl2·6H2O or ZrCl4 was used as a catalyst. The authors proposed that the reaction was initiated by the Michael addition to give an adduct, which after cyclization and aromatization with loss of a water molecule and a nitroxyl group (HNO) afforded the final product.
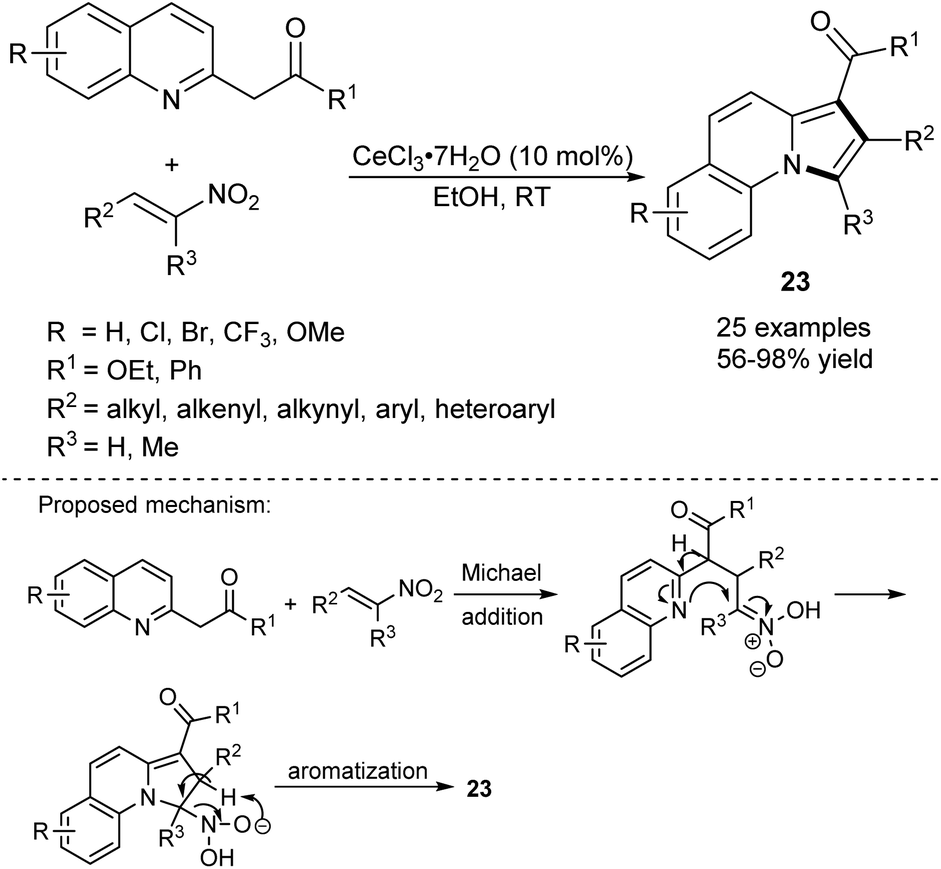 | ||
| Scheme 12 | ||
Lei's group proved that the reaction between 2-(pyridin-2-yl)acetates and nitroolefins can also proceed smoothly without any catalyst.27 Heating the substrates only in DMSO under a nitrogen atmosphere at 100 °C gave various indolizine derivatives in moderate to good yield. However, when 10 mol% copper(II) acetate was used, the rate of the reaction was 2.5-fold faster at the same temperature.
The iodine-mediated, direct oxidative cyclization between 2-alkylpyridines and enolizable aldehydes was reported by Yan's group (Scheme 13).28 Except for n-butanal, all tested aldehydes exhibited good reactivity, giving 1,3-disubstituted indolizines 24 in moderate to good yields. The complementary methodology exploiting either alkyl-alkenes or styrenes instead of aldehydes was developed by the same group one year later (Scheme 13).29 Generally, styrenes react with activated 2-alkylpyridines much easier than alkenes, providing better yields of 24. In two cases, the authors obtained only traces of the expected products (unsubstituted styrene and cyclohexene). An analogous protocol utilizing 2-alkylpyridine derivatives and styrenes was proposed by Tang et al.30 This iodine-catalyzed radical oxidative annulation afforded 3-arylindolizines in yields of 25–59% (9 examples) with good regioselectivity. Aliphatic alkenes were not evaluated in this report.
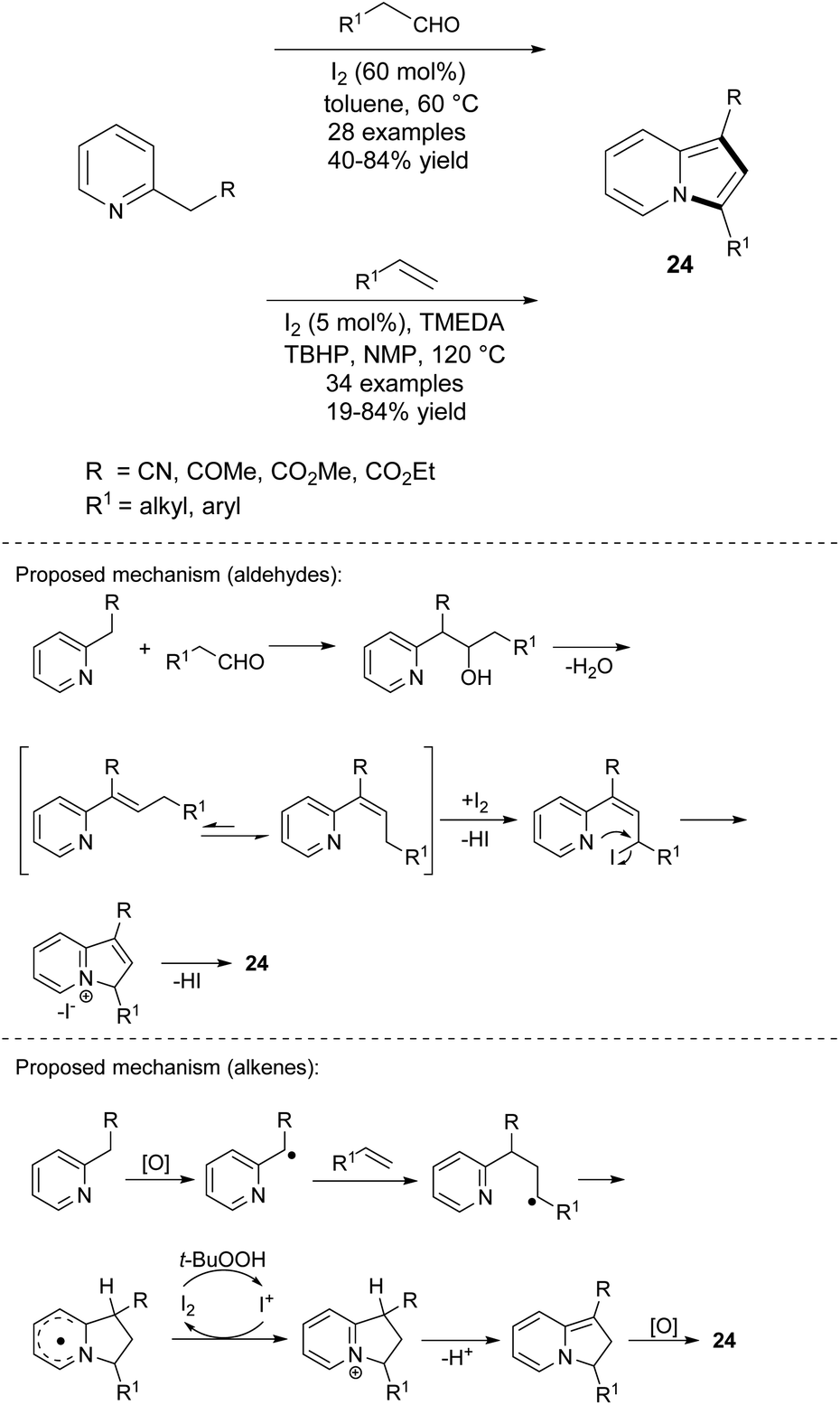 | ||
| Scheme 13 | ||
Very recently, Sahoo et al. reported the visible-light-mediated preparation of polycyclic indolizines without use of organometallic photosensitizers (Scheme 14a).31 The reaction between 2-bromomethyl-pyridine derivatives and enol carbamates gave tetra- and pentacyclic products 25 in moderate to good overall yields. The lowest yield was obtained for a pyridine substrate bearing the CN group in the side chain. The attempts to obtain 2,3-dialkylindolizine were not successful either under the standard conditions or in the presence of the photosensitizer – [Ir(ppy)2(dtbbpy)](PF6). Moreover, 25 could be easily oxidized or partially reduced in excellent yields, showing the great synthetic potential of this method (Scheme 14b).
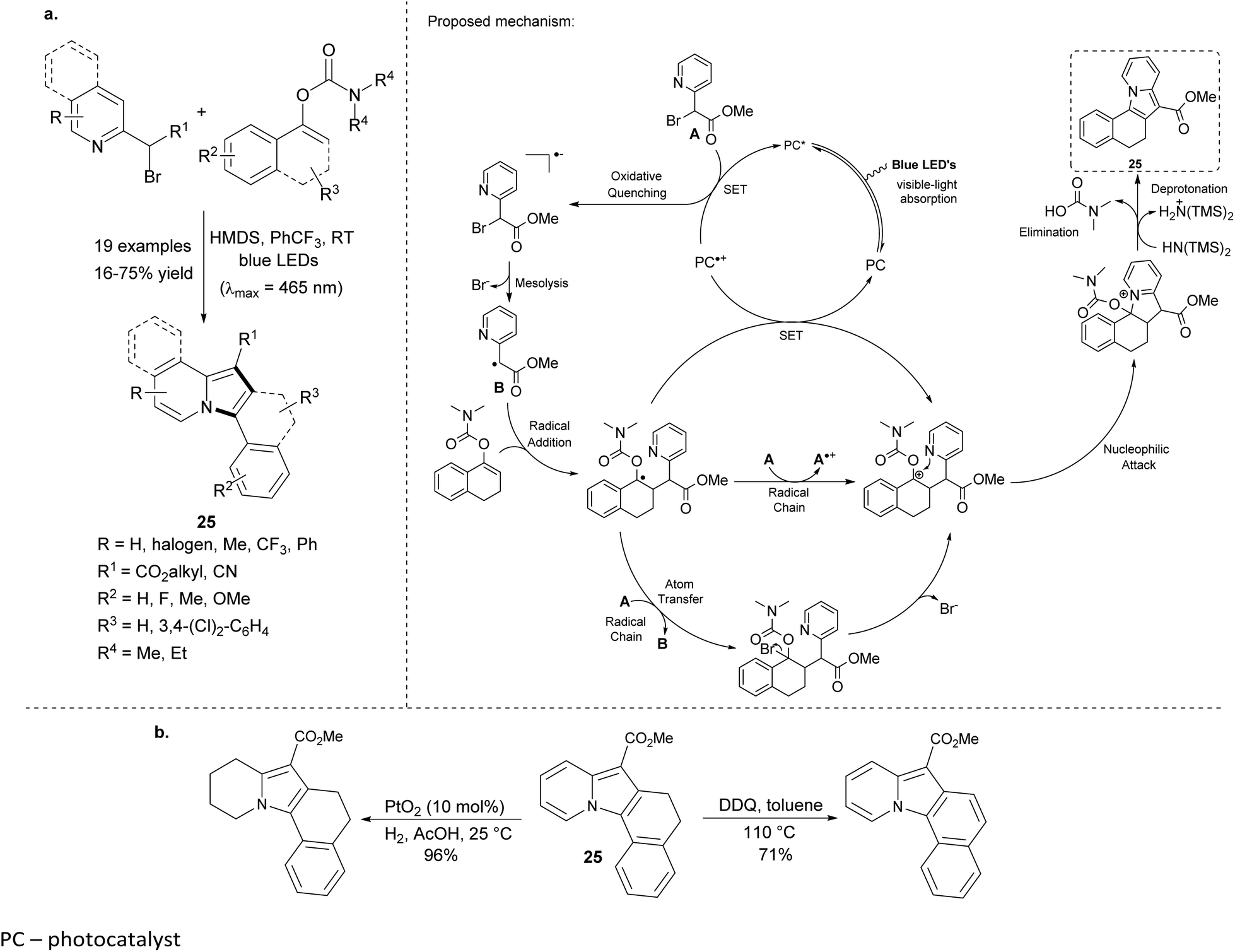 | ||
| Scheme 14 | ||
Wang et al. proved that propargyl alcohols are also good substrates in the preparation of 2-alkylindolizines (Scheme 15).32 This samarium(III) triflate-catalyzed reaction proceeded under solvent free conditions. Depending on the substituent R2, one of the regioisomers 26 or 27 could be obtained over the reaction course.
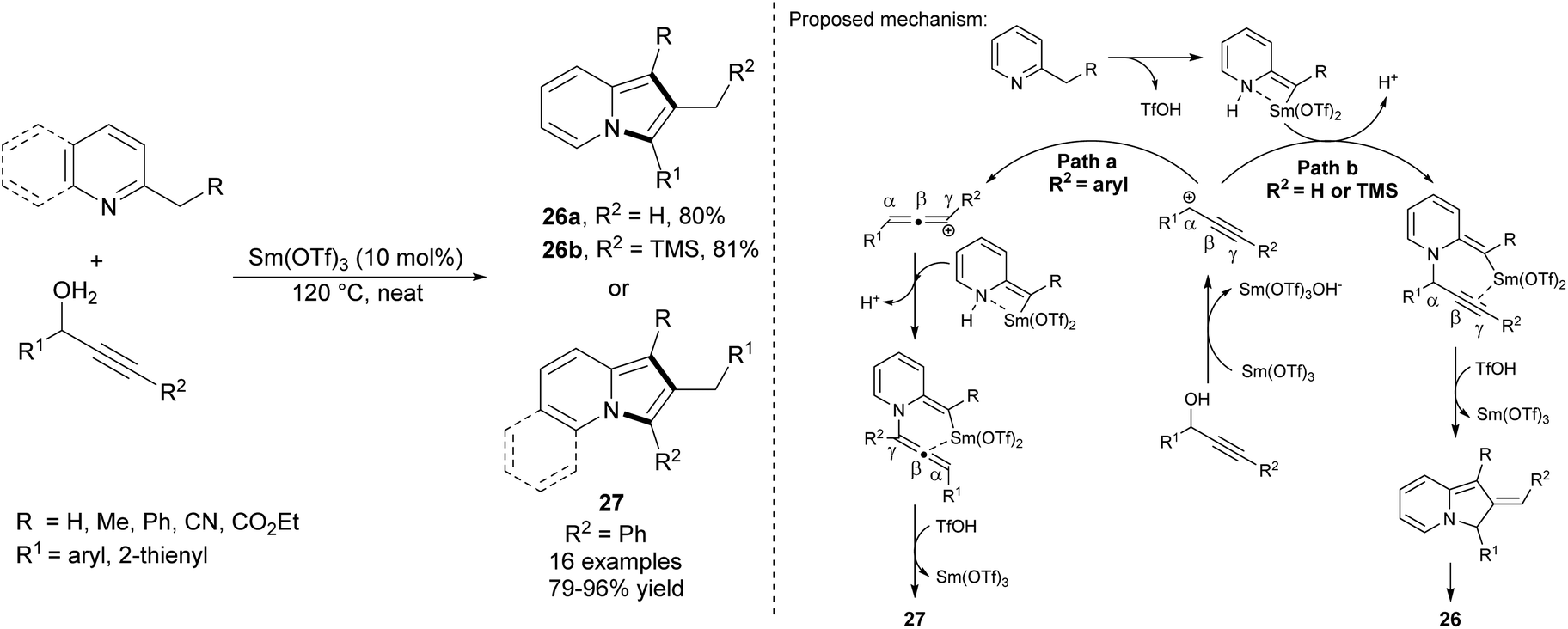 | ||
| Scheme 15 | ||
The silver-mediated cyclization between 2-alkylpyridines and terminal alkynes was investigated by Agrawal's group (Scheme 16).33 The presence of an ester group in the side chain of pyridine provided generally higher yields of 28. The authors have stated that the electronic character of substituents of the alkynes had no influence on the reaction outcome. Interestingly, due to the required substantial excess and cost of Ag2CO3, the possibility of the salt's regeneration was investigated. It appeared that the recovered salt possessed analogous activity to the commercially available salt.
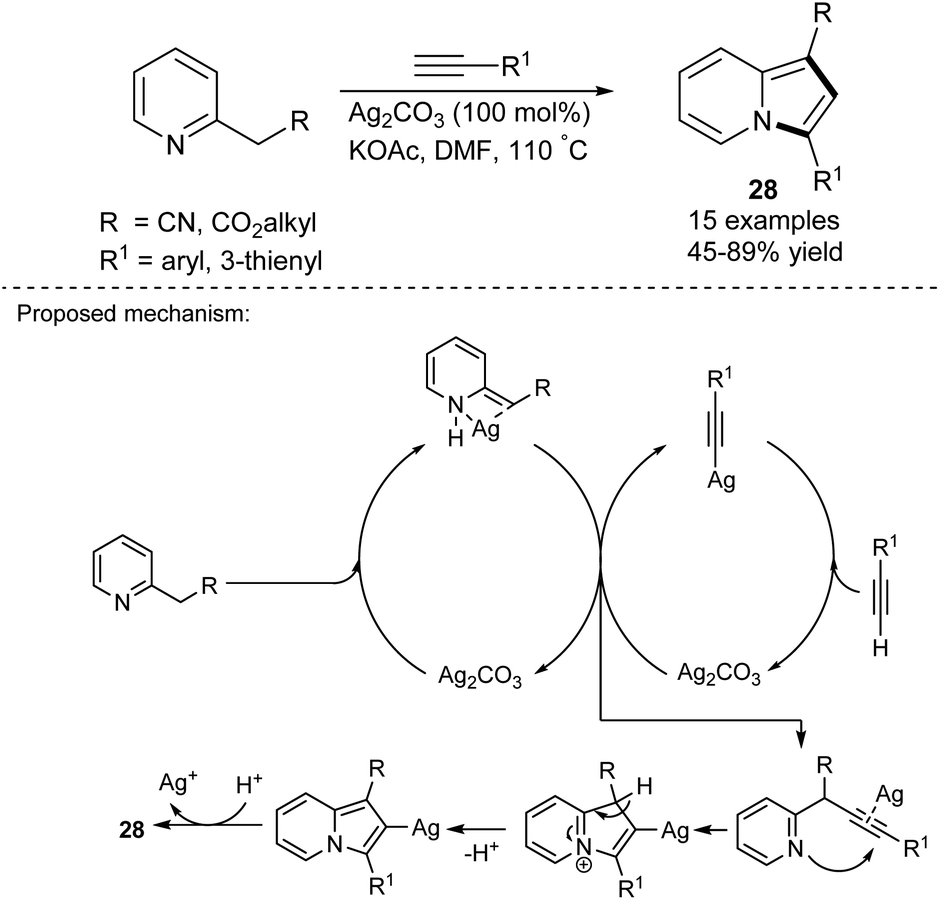 | ||
| Scheme 16 | ||
Tan et al. proved that disubstituted alkynes also reacted smoothly with 2-alkylpyridines in the presence of silver(I) carbonate to give 1,2,3-trisubstituted indolizines 29 with excellent regioselectivity in moderate to good yields (Scheme 17).34 In contrast to the previous work,33 higher yields were obtained when electron-rich alkynes were used as a starting material. In the case of both 1-octyne and 4-octyne, the method gave only traces of the corresponding products. The excellent regioselectivity of this method is probably due to the presence of either the aryl or heteroaryl moiety as R2, which enhanced the stability of radical A as well as carbocations B and B′ (Scheme 17, bottom).
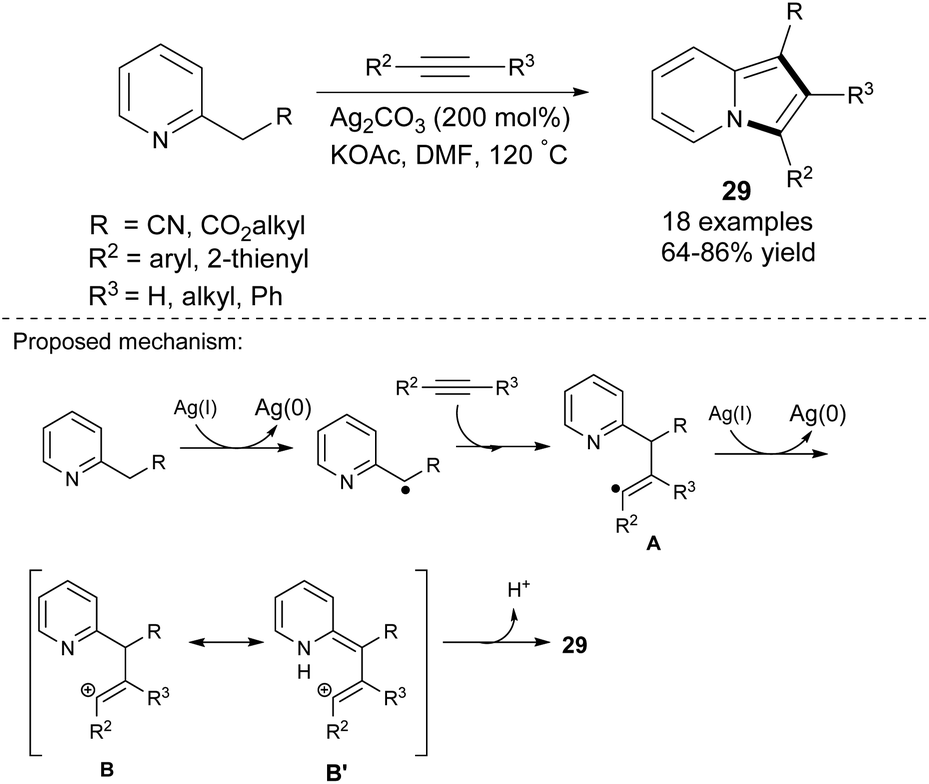 | ||
| Scheme 17 | ||
One major drawback of the above reactions is the significant amount of Ag2CO3 that was required. Tang et al. solved this problem partially by investigating TM-free catalysts in the reaction of 2-alkylpyridines and terminal alkynes. These substrates in the presence of molecular iodine, cumyl hydroperoxide (CHP), and sodium acetate gave indolizines 30 in low to excellent yields (Scheme 18).35 Electron-donating and electron-withdrawing substituents on the aromatic ring in the alkyne were tolerated, while aliphatic alkynes showed poor reactivity under these conditions.
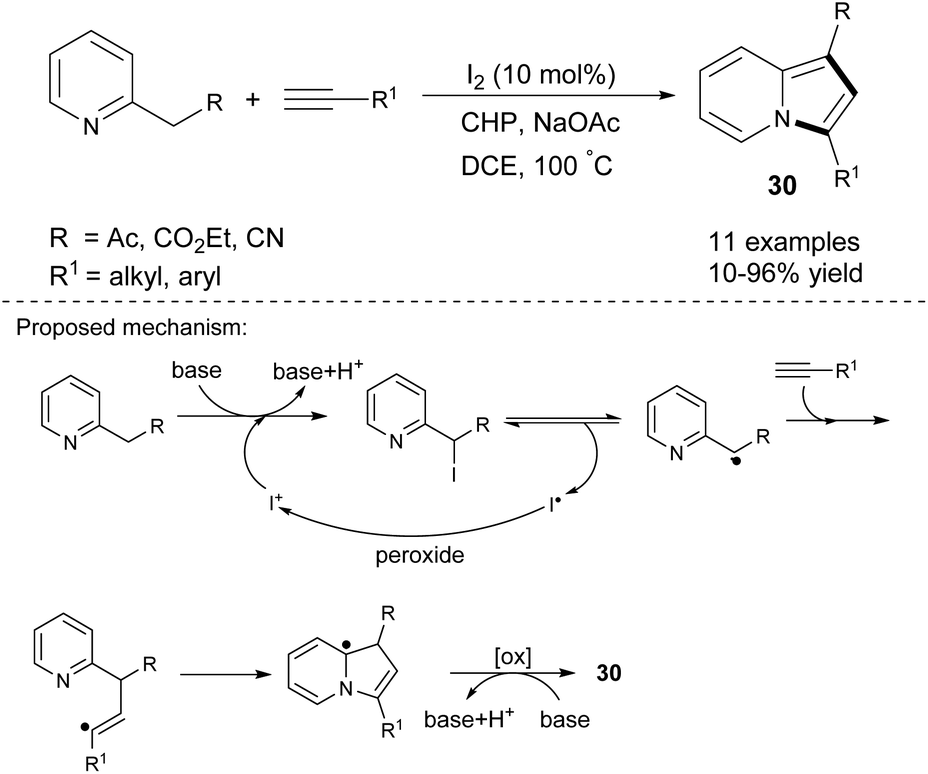 | ||
| Scheme 18 | ||
A breakthrough approach toward the synthesis of 2-substituted indolizines was made by Yudin's group (Scheme 19).36 According to their protocol, MIDA-protected bromomethyl acylboronates 31 reacted smoothly with 2-alkylpyridines giving 2-borylated indolizines 32 in good to excellent yields. The latter compounds may serve as useful substrates in Suzuki coupling, which makes compounds 32 very promising intermediates in the synthesis of a broad range of 2-functionalized indolizines in a simple manner.
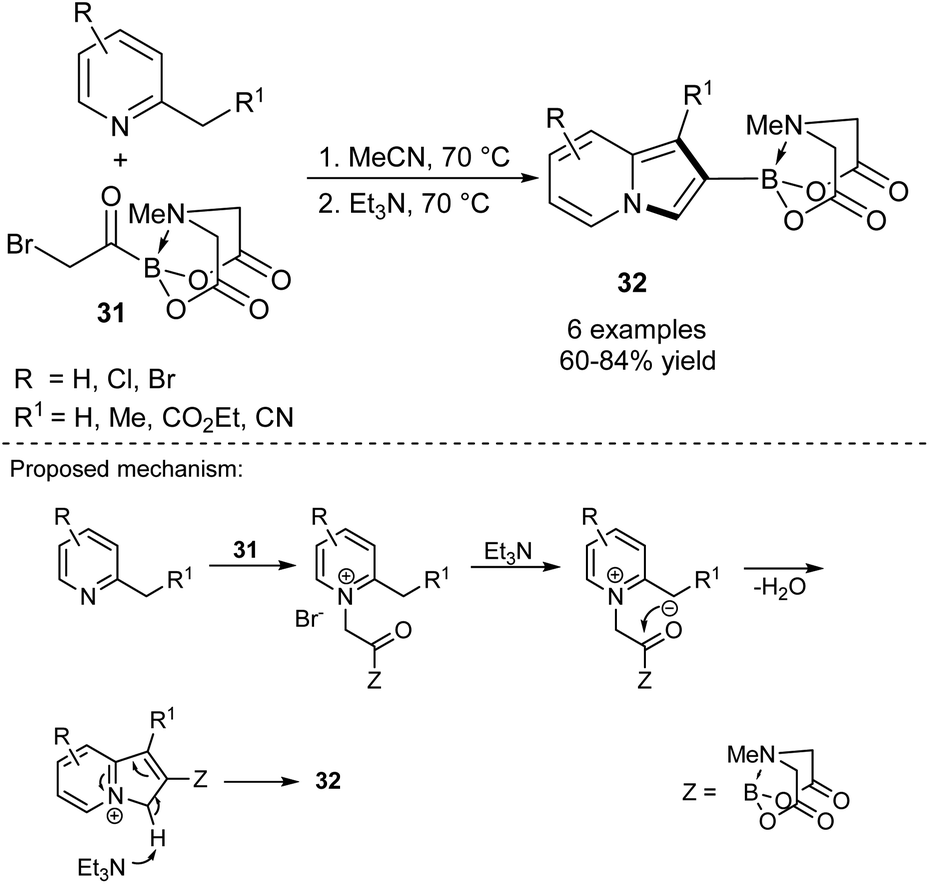 | ||
| Scheme 19 | ||
In 2006, Bienaymé and co-workers published the highly efficient, three-component synthesis of diversely substituted indolizines (33) by the condensation of 2-(pyridin-2-yl)acetonitrile with aldehydes and isonitriles (Scheme 20).37 An advantage of this methodology is that it gives rise to indolizines possessing a readily functionalizable primary amino group available after removal of a benzyl group. The significant disadvantage of this strategy laid in the use of isonitriles – compounds possessing limited commercial availability and an unpleasant smell. Two years later, Okamoto and Niyomura successfully synthesized indolizine 34 starting from 2-(pyridin-2-yl)acetonitrile and selenium dioxide (Scheme 21).38 Interestingly, the authors found that 34 easily formed the NiL2 complex by simply heating with Ni(NO3)2·6H2O in ethanol.
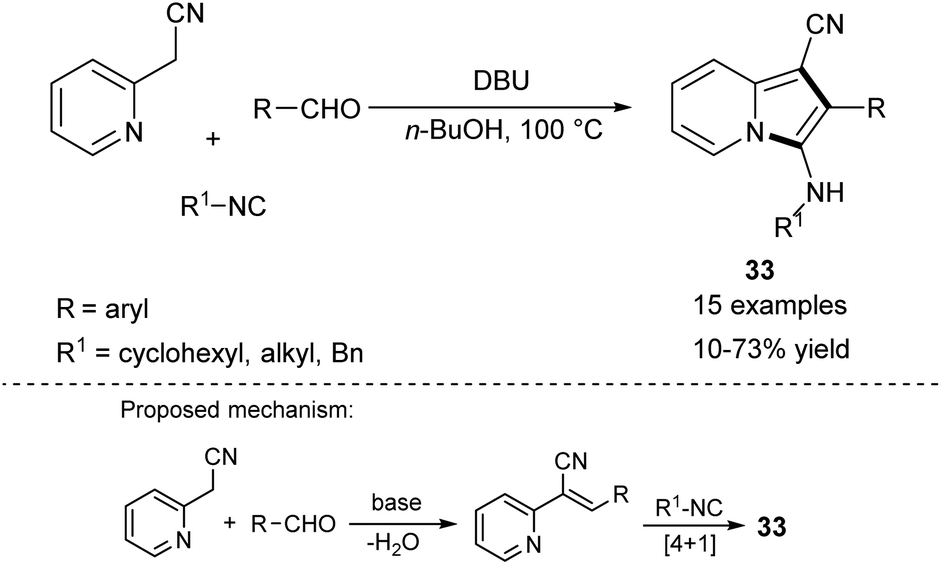 | ||
| Scheme 20 | ||
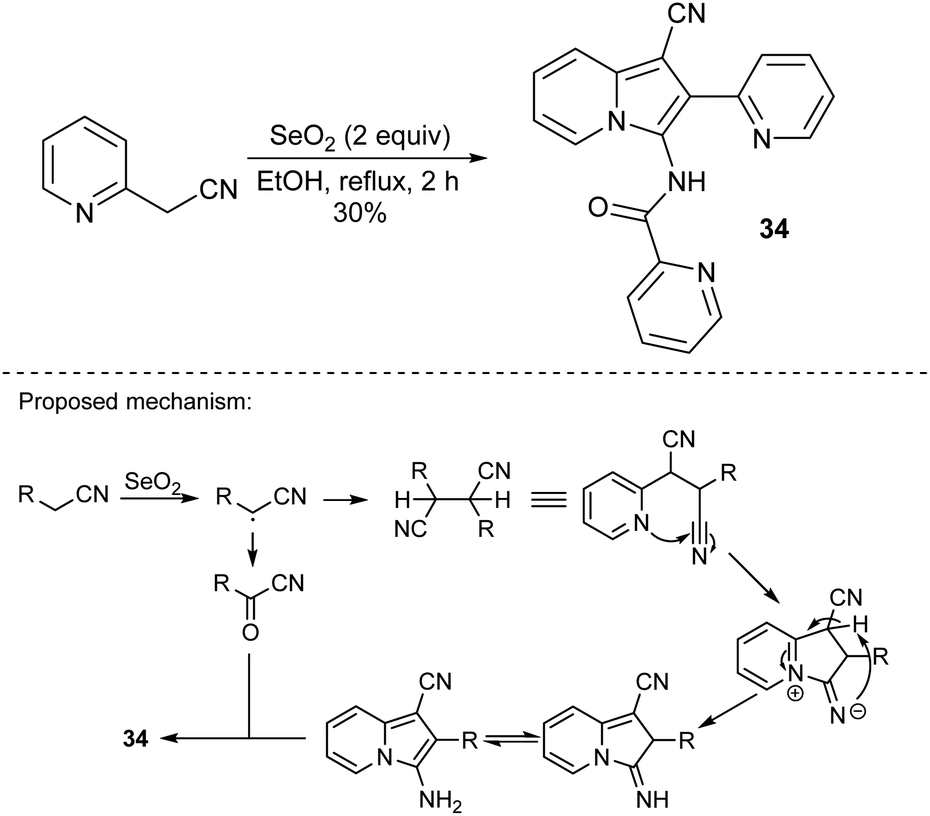 | ||
| Scheme 21 | ||
2.5 From pyridinium salts and dipolarophiles (via 1,3-dipolar cycloaddition)
The 1,3-dipolar cycloaddition reaction between pyridinium salts and dipolarophiles is a widely used method for the synthesis of indolizines with acceptor moieties at positions C1 and/or C3 (Scheme 22). Both activated alkenes and alkynes have been used in this approach, with the fundamental difference being that in the first case the addition of a very selective oxidant is required. The first example of successful realization of this type of strategy with alkenes was presented by Hu and co-workers, who used Cr(VI)-salts as oxidants.39 Taking into consideration the overall number of papers devoted to this topic, in this section we will focus only on the most important reports, which have appeared during the last decade.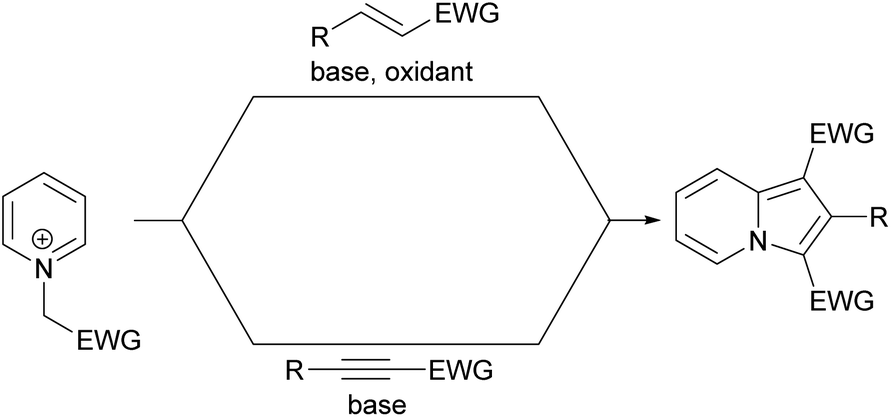 | ||
| Scheme 22 | ||
In 2010, Coffinier et al. succeeded in the synthesis of 2-aminoindolizines (36) using phosphorylated hydroxyketenimines (35) (Scheme 23).40 These reagents were prepared by the Nef-Perkov cascade, exploiting isocyanides as a starting material and they showed good stability in air and on silica. Interestingly, the authors did not detect any side product resulting from a cycloaddition to the carbon–nitrogen moiety of the ketenimine.
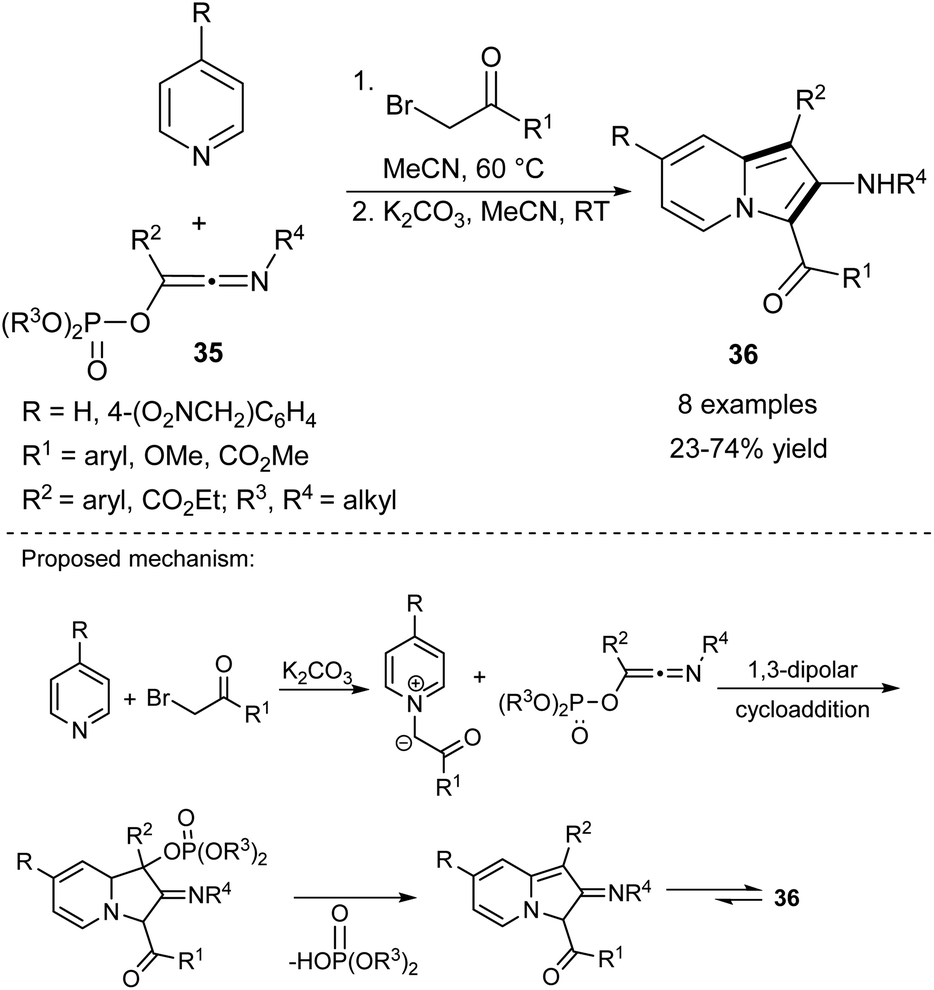 | ||
| Scheme 23 | ||
2-Aminoindolizines 38 can also be successfully obtained in the reaction of either pyridinium or isoquinolinium salts and electron-deficient ynamides 37 (Scheme 24).41 The synthetic potential of this method was confirmed in the synthesis of 1,3-dialkyl-2-aminoindolizines or a molecule possessing the indoloquinolizidine scaffold.
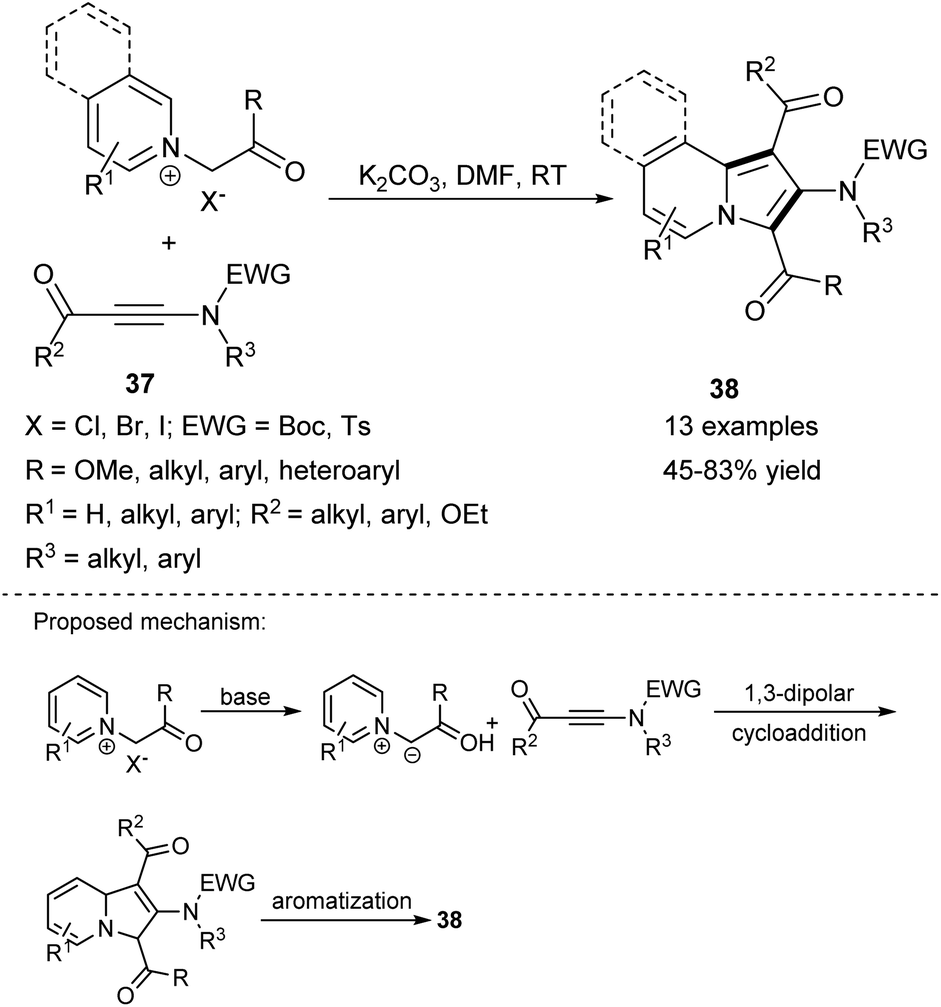 | ||
| Scheme 24 | ||
Another interesting finding by Chen et al. was that 4-acetoxyallenoates (39) are capable of undergoing 1,3-dipolar cycloaddition reactions with various pyridinium salts (Scheme 25).42 According to this protocol, a set of 2-styrylindolizines (40) as well as their π-expanded analogues was synthesized in good yields with an excellent tolerance of substituents. It is worth pointing out that the oxidation step occurs in the presence of the most environmentally preferred oxidant – air.
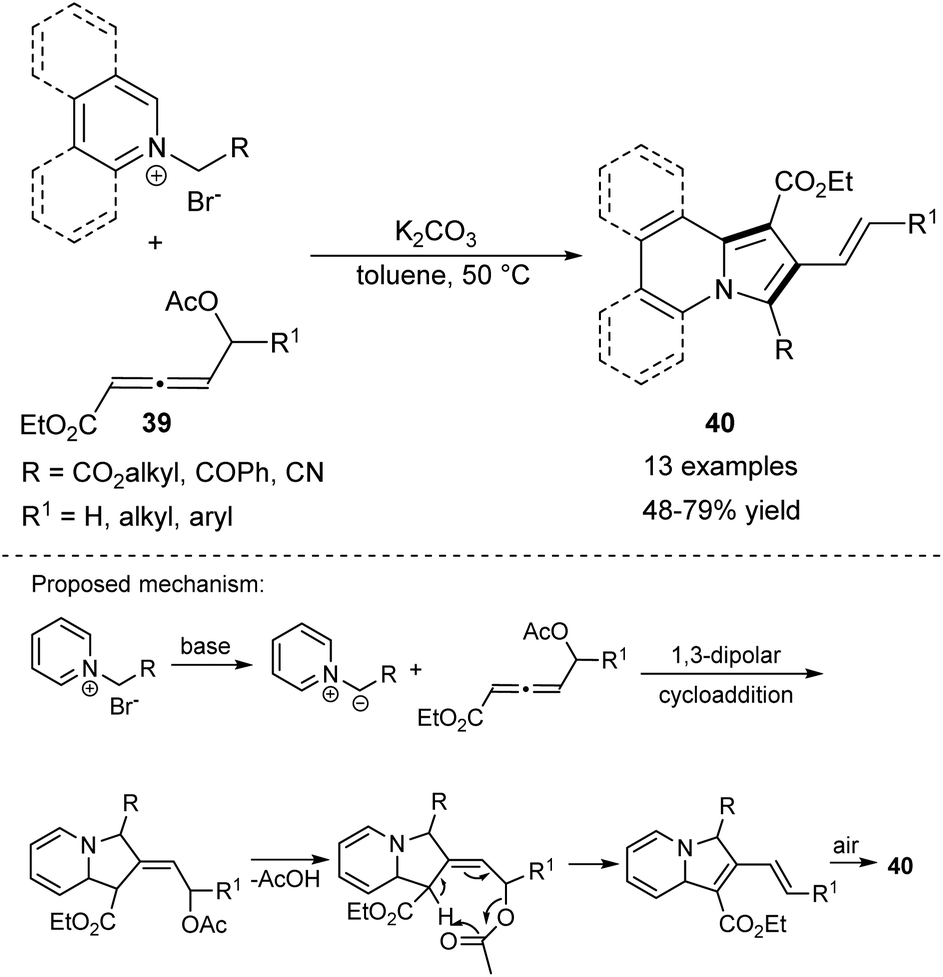 | ||
| Scheme 25 | ||
Park and co-workers designed an intramolecular version of that strategy with the aim of obtaining indolizine derivatives possessing additional saturated rings (Scheme 26).43 Pyridinium salts 42, which were prepared from the pyridine derivatives and the corresponding bromides 41, underwent an in situ cycloaddition/oxidation sequence to give 43.
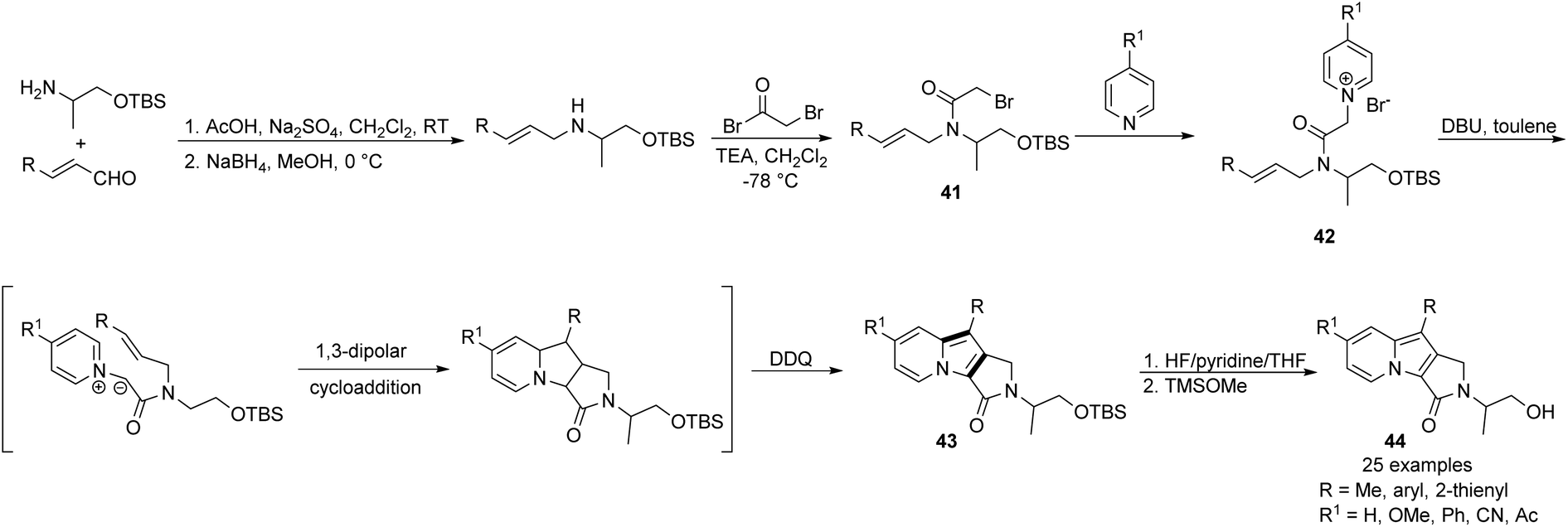 | ||
| Scheme 26 | ||
Subsequent deprotection led to rather complex and highly fluorescent 1,2-dihydropyrrolo[3,4-β]indolizin-3-ones (44) in a relatively simple manner. This method was limited only by the synthesis of building blocks 41, which could be completed in two steps from readily available chemicals. Advantages of compounds 44 such as pH-dependent fluorescence properties and drug-like lipophilicity for membrane permeability resulted in successful application as bioprobes in HeLa cells. Particular studies on the structure, photophysical property relationship, and potential applications of this intriguing skeleton, termed Seoul-Fluors, have appeared in other articles.44
2.6 From 2-pyridine derivatives via cycloisomerization process
In 2006, Gevorgyan and Seregin started a new chapter in the synthesis of indolizines. They developed an Au-catalyzed method for introduction of 2-silyl-, 2-stannyl-, and 2-germyl groups into the indolizine skeleton (Scheme 27).45 The reaction proceeded by an alkyne-vinylidene isomerization step followed by the 1,2-migration of the corresponding group G to give indolizines 45 in good to excellent yields. Silyl- and stannyl-based compounds were useful reagents in either Hiyama or Stille coupling, thus this method possessed enormous potential in the synthesis of C-2-functionalized indolizines. The detailed mechanism of this process has also been investigated.46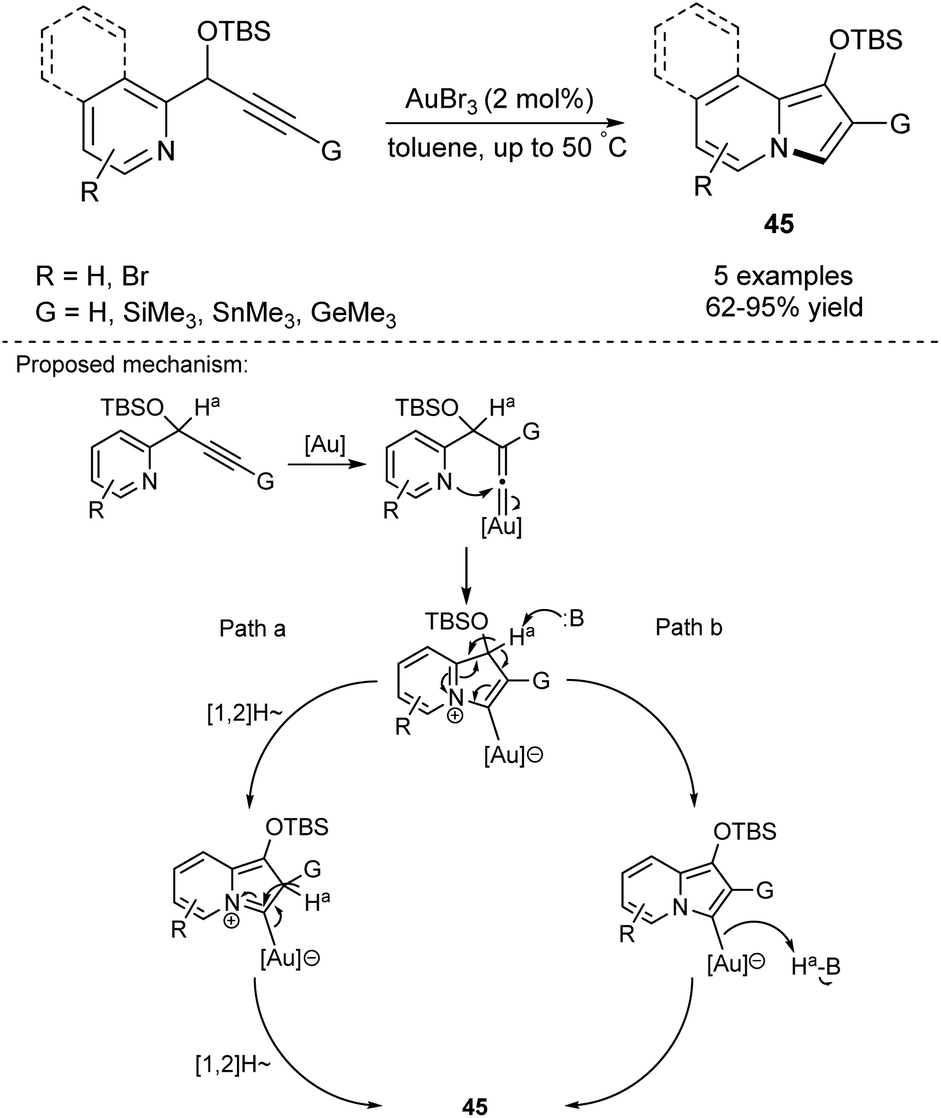 | ||
| Scheme 27 | ||
This initial report encouraged both Gevorgyan and co-workers as well as other researchers to further exploit this chemistry. Later it was shown that this type of reaction can be also catalyzed by Pt(II),47 Cu(I),48 or Ag(I)49 species and the scope of the reaction was significantly extended. Notably, Sarpong and Narayan proved that cycloisomerization of propargyl esters may be accomplished by simply heating in pure water.50 Other solvents such as methanol, ethanol, or 1,1,1-trifluoroethanol also promoted the formation of product in high yields, probably due to the beneficial effect of hydrogen bonding.
Later, Bi and co-workers studied the Ag-catalyzed cyclization of 2-pyridyl alkynyl carbinols in the presence of isocyanides leading to 1-alkyl- or 1-aryl-3-carboxyamidoindolizines (46) (Scheme 28).51 This reaction occurred smoothly in most cases, although propargyl alcohol bearing the i-Pr group cyclized less efficiently, giving the corresponding indolizine in 30% yield.
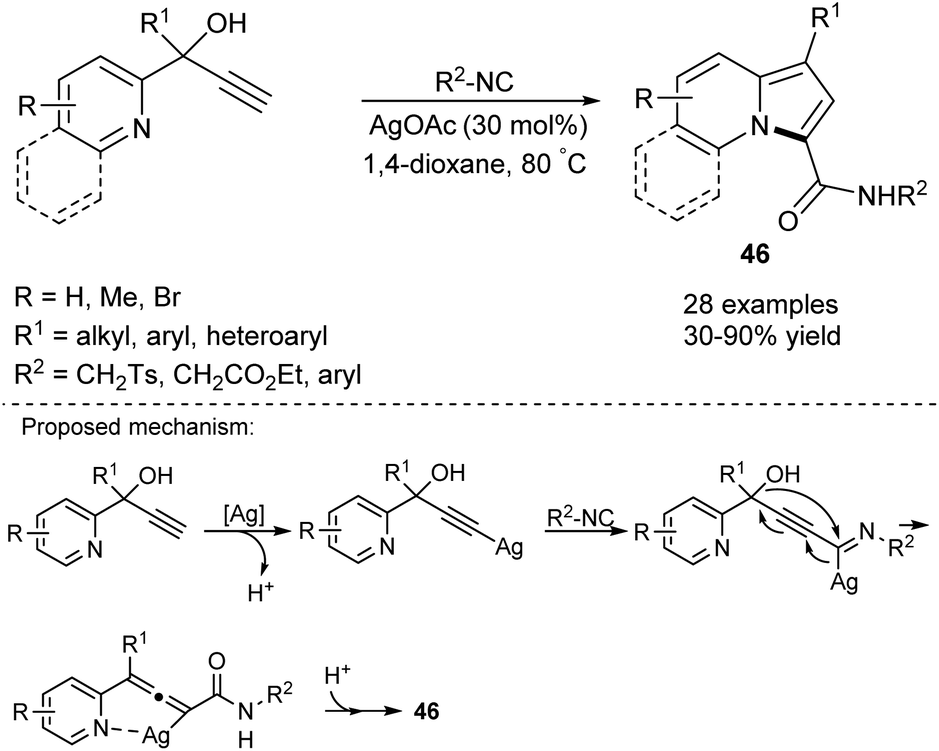 | ||
| Scheme 28 | ||
Based on their earlier report,45 Gevorgyan and co-workers proposed an efficient, one-pot route to 1,2,3-substituted indolizines 47, where an aryl moiety is introduced into position C2 (Scheme 29).52 This cycloisomerization/arylation cascade process exhibited tolerance against a wide range of functional groups present in the coupling partner (ArX) such as halo or nitro, trifluoromethyl, or ester groups. Notably, propargyl phenyl ethers were used as a substrate for the first time to give indolizines in high yields.
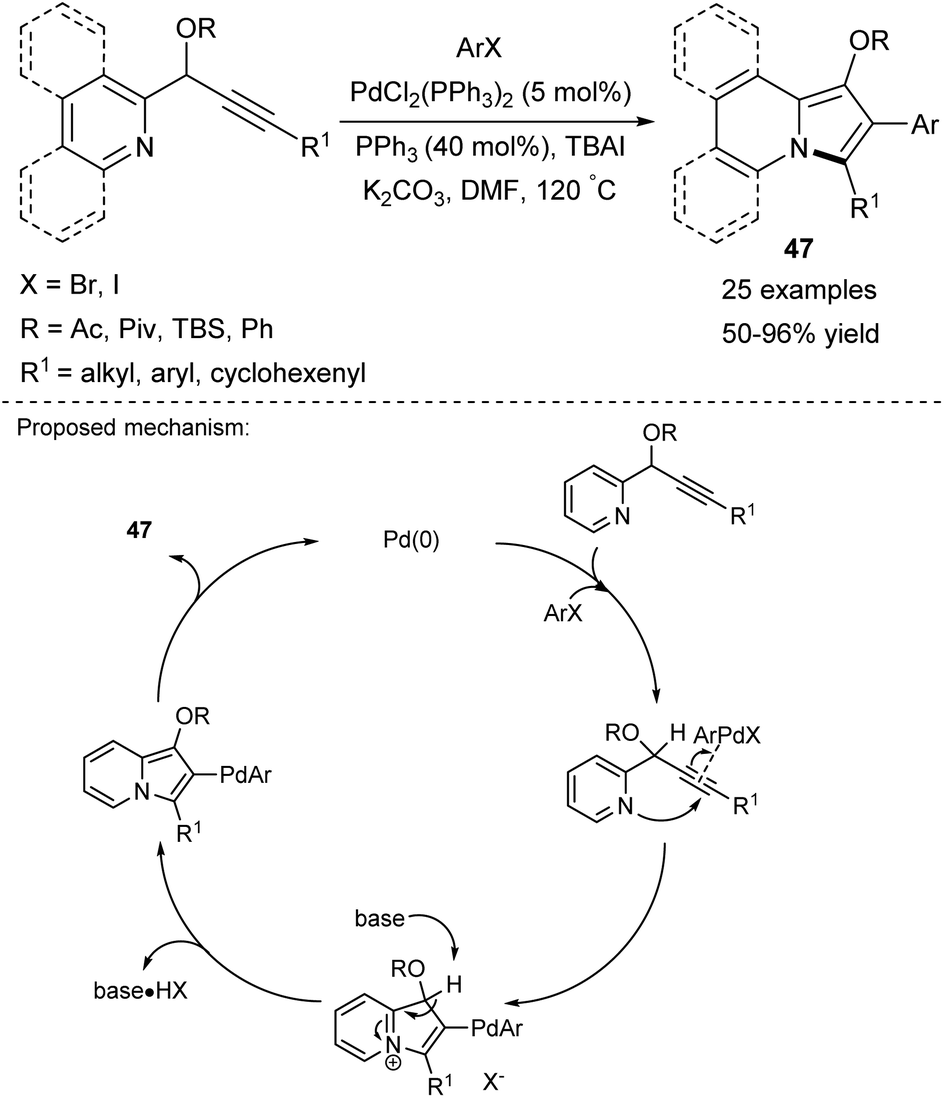 | ||
| Scheme 29 | ||
The series 2-aroylindolizines (48) can be obtained in a similar manner via the palladium-catalyzed carbonylative cyclization/arylation cascade utilizing carbon monoxide as a source of the carbonyl group (Scheme 30).53 This method fully tolerated electron-rich, neutral, or electron-deficient aryl iodides and various pyridine derivatives, although using a 3-methylpyridine derivative led to low yield (35%).
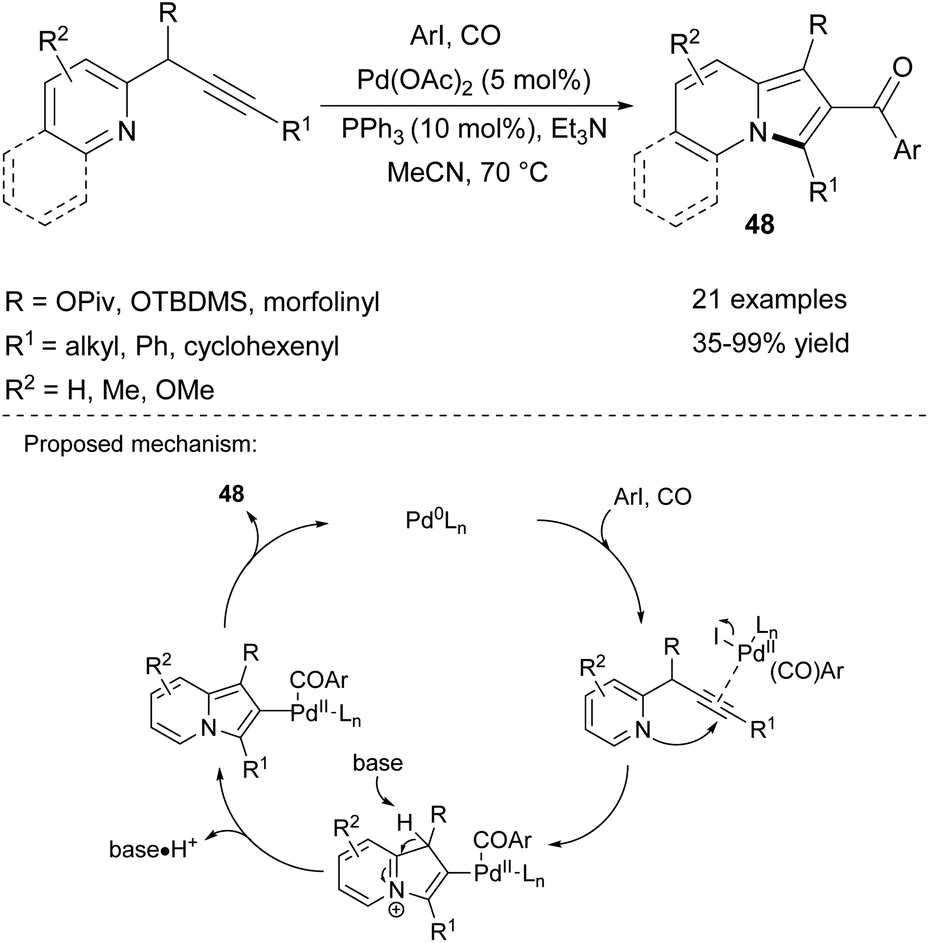 | ||
| Scheme 30 | ||
Very recently, Alper and Xu employed a similar carbonylative methodology leading to 2-alkoxycarbonylindolizines (49) instead of 2-aroylindolizines (Scheme 31).54 This reaction, catalyzed by either Pd2(dba)3 or Pd/C, proceeds easily at room temperature in the presence of carbon monoxide and benzoquinone as an oxidant. Pd2(dba)3 usually gave slightly higher yields in comparison with Pd/C, although palladium on carbon could be easily separated and recovered after the reaction. Relatively low yields were obtained for indolizines bearing the n-Bu group at position C3 or when i-PrOH was used as an alcohol substrate.
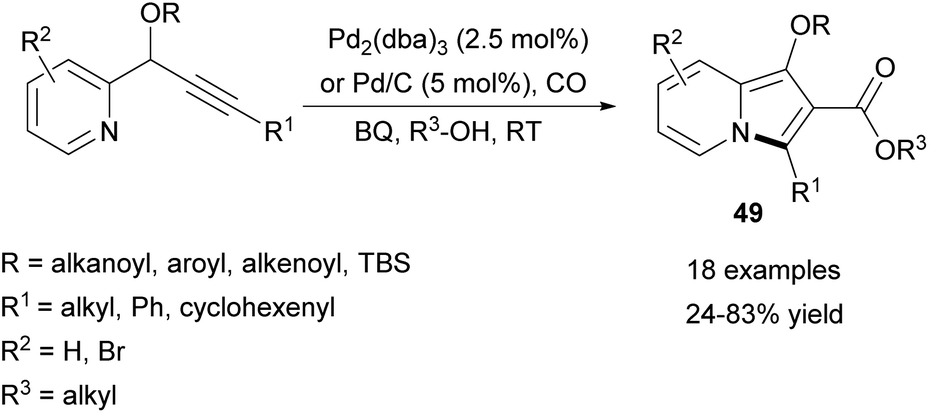 | ||
| Scheme 31 | ||
Chernyak and Gevorgyan proposed a palladium-catalyzed carbopalladation/cyclization cascade leading to polycyclic fused heterocycles 50 based on the indolizine skeleton as an intramolecular version of a previously described52 method (Scheme 32).55 The generality of the process was established by employing substituted propargyl esters, ethers, or silyl ethers to obtain molecules containing the indolizine skeleton fused with coumarin, benzofuran, or the novel silacyclic scaffold, respectively.
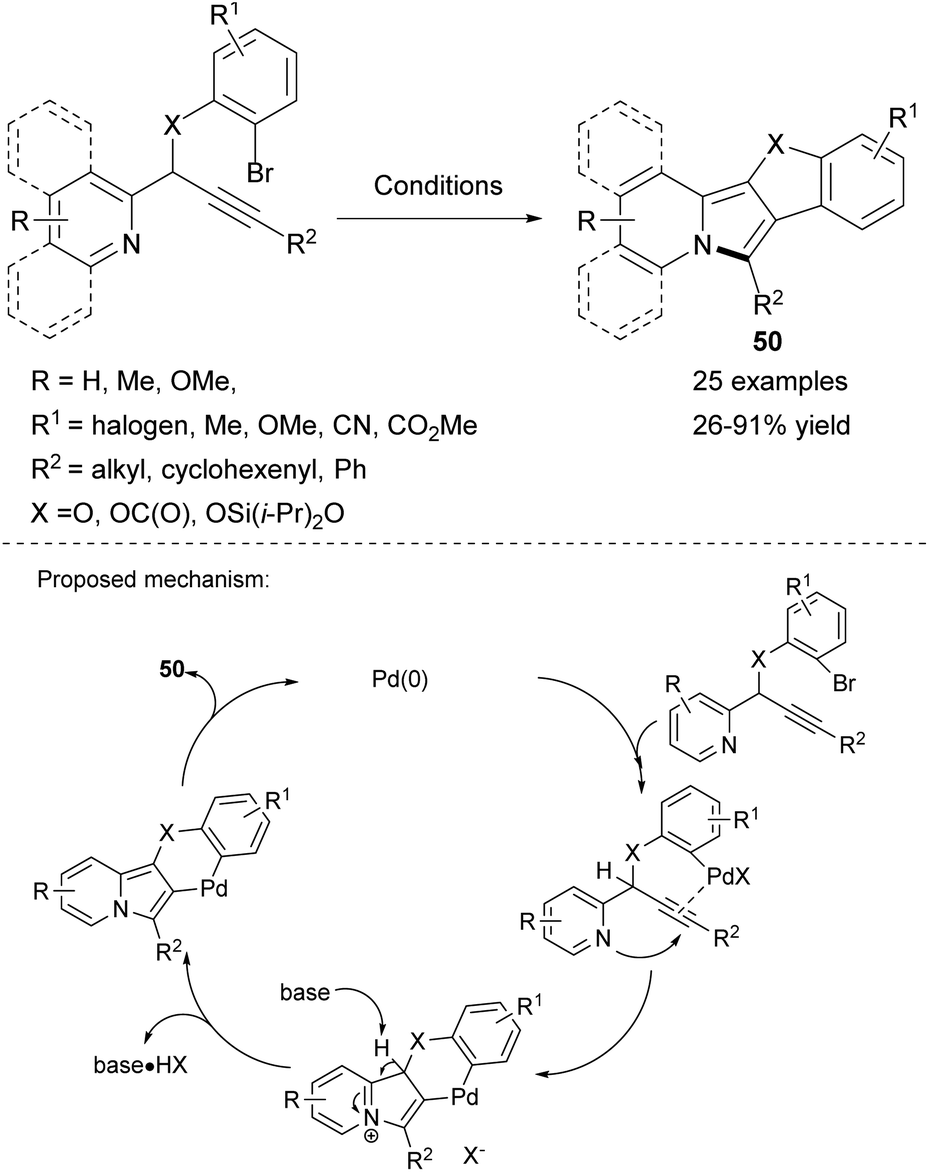 | ||
| Scheme 32 Reagents and conditions: X = OC(O): Pd(OAc)2 (5 mol%), K2CO3, DMA, 120–130 °C; X = O: Pd(OAc)2 (5 mol%), TBACl, K2CO3, DMF, 120 °C; X = OSi(i-Pr)2O: Pd(OAc)2 (5 mol%), LiCl, K2CO3, p-xylene, 135 °C. | ||
Alternatively, indolizines can be synthesized by cycloisomerization of 2-alkynylpyridines. Sarpong and Hardin utilized substrates of this type in the synthesis of 2,3- or 1,3-disubstituted indolizines 51 and 52, respectively (Scheme 33).56 The ratio of 51/52 (determined by comparison of 1H NMR spectra) was highly dependent on the electronic effect of both R and R1. The initial 5-exo-dig cyclization leading to A and then to 51 was more favored in the case of the electron-donating character of both R and R1, otherwise B is formed efficiently via 6-endo-dig cyclization with a substantial change in 51/52 from >20:1 to 2:1. The lowest yields were obtained for indolizines bearing the tert-butyl or 2-furyl moiety at position C3.
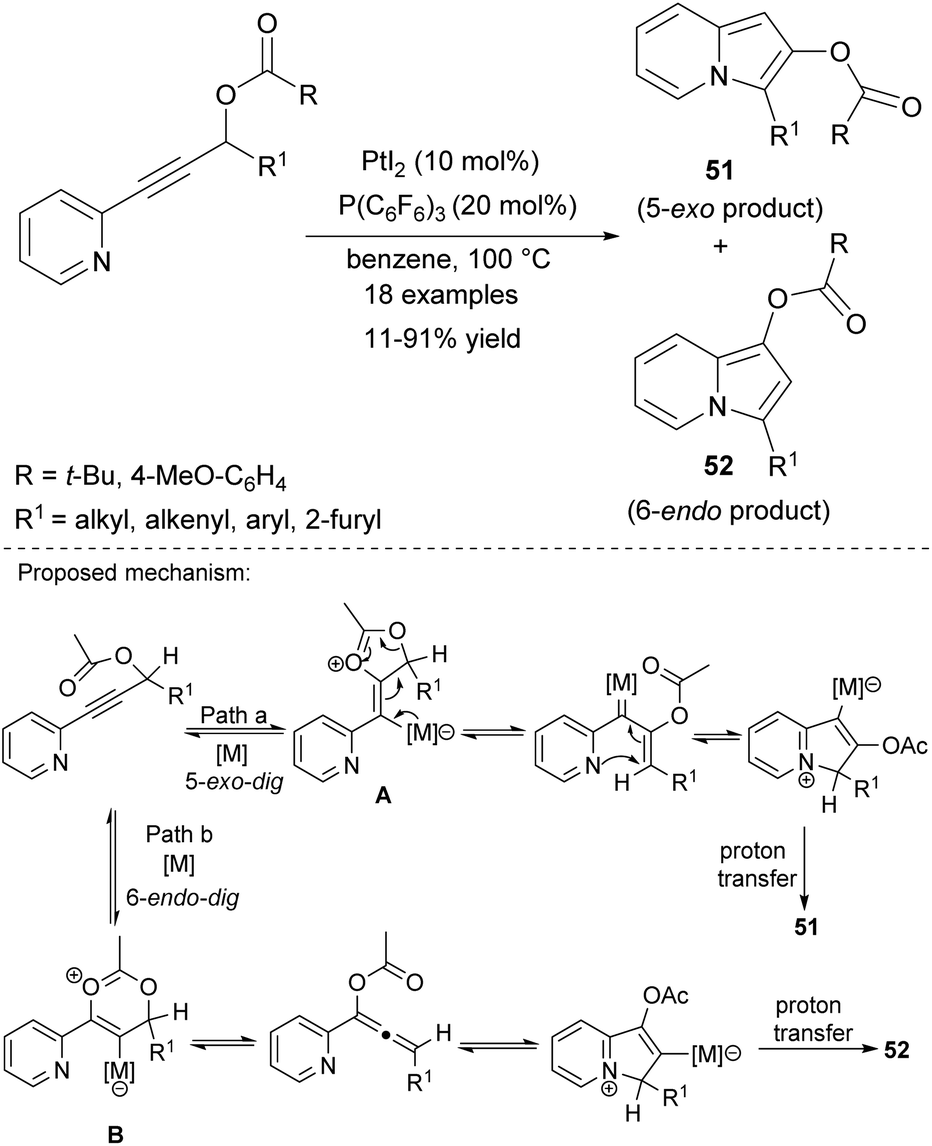 | ||
| Scheme 33 | ||
The organocopper-mediated cross coupling/cycloisomerization sequence at low temperatures, leading to 53 was proposed afterward (Scheme 34).57 During this process, the organocopper reagent acted as both a catalyst and a nucleophile. It is noteworthy that this methodology allowed the preparation of 1-alkyl or 1-arylindolizines, not accessible by previously described cycloisomerization-based methods. Moreover, the quinoline or isoquinoline derivative could be also used as a substrate leading to π-expanded indolizines.
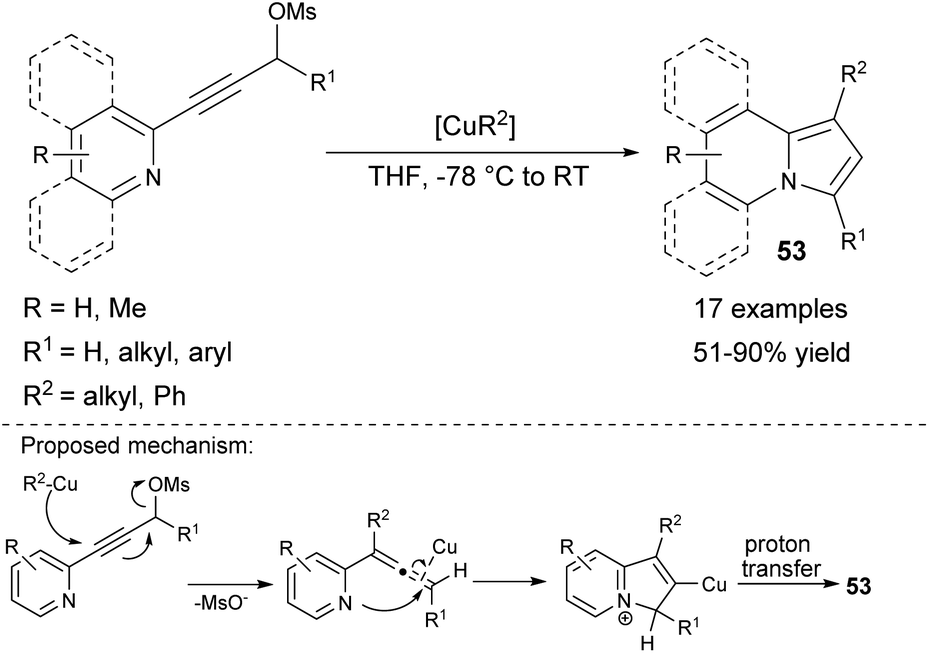 | ||
| Scheme 34 | ||
Very recently, the aryl group was introduced at position C1 via a palladium-catalyzed cascade (Scheme 35).58 At the initial step, Suzuki coupling of 3-(2-pyridyl)-propargyl carbonates with the corresponding boronic acids gave an allenyl pyridine intermediate, which subsequently underwent spontaneous cycloisomerization to 54. The above methodology was efficient for a wide range of boronic acids, although 2-furyl boronic acid proved to be less reactive. Taking into account the structure of the starting material, this method was less efficient for pyridines bearing 4-CF3-C6H4, 4-MeO-C6H4, or 2,4,6-(MeO)3-C6H4 groups.
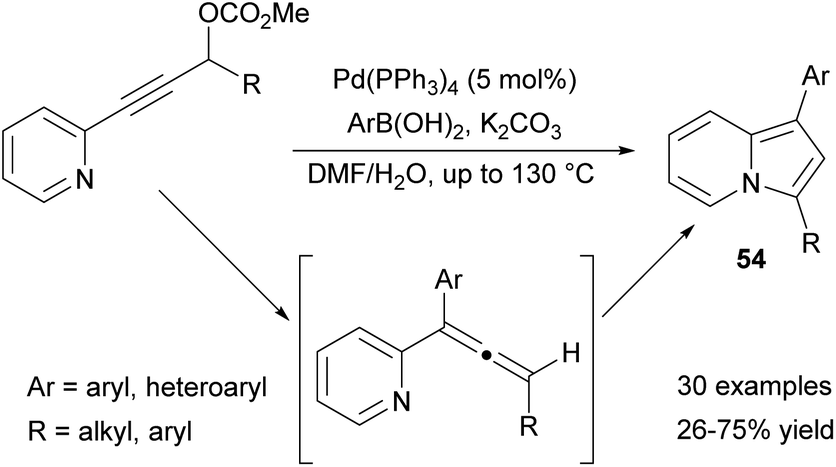 | ||
| Scheme 35 | ||
Further research has proven that not only 2-alkynylpyridines and 2-propargylpyridines but also α,β-unsaturated ketones derived from 2-acetylpyridines can serve as substrates for the synthesis of indolizines. In particular, Shi and co-workers demonstrated that 1-aminoindolizines 55 could be easily obtained via the titanium(IV)-mediated reaction of 2-enoylpyridines with 2-thienylsulfonamide (Scheme 36).59 Both electron-rich and electron-deficient substituents were suitable for this process with the exception of the cinnamyl group which gave no desired indolizine.
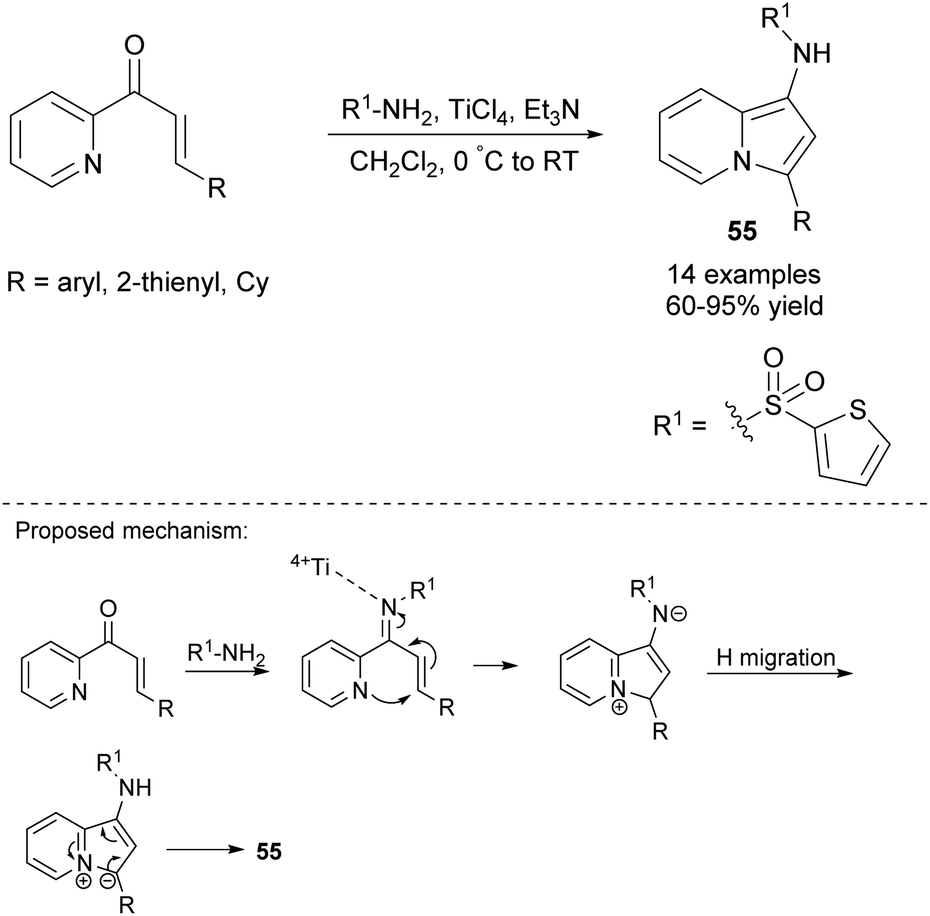 | ||
| Scheme 36 | ||
Another strategy to utilize 2-enoylpyridines was revealed by Liu and colleagues. These authors discovered that such α,β-unsaturated ketones can be effectively transformed into indolizines 56 under the influence of symmetrical acid anhydrides (Scheme 37).60 This method proceeded via TM-free C–H bond activation and afforded indolizines in moderate to excellent yields.
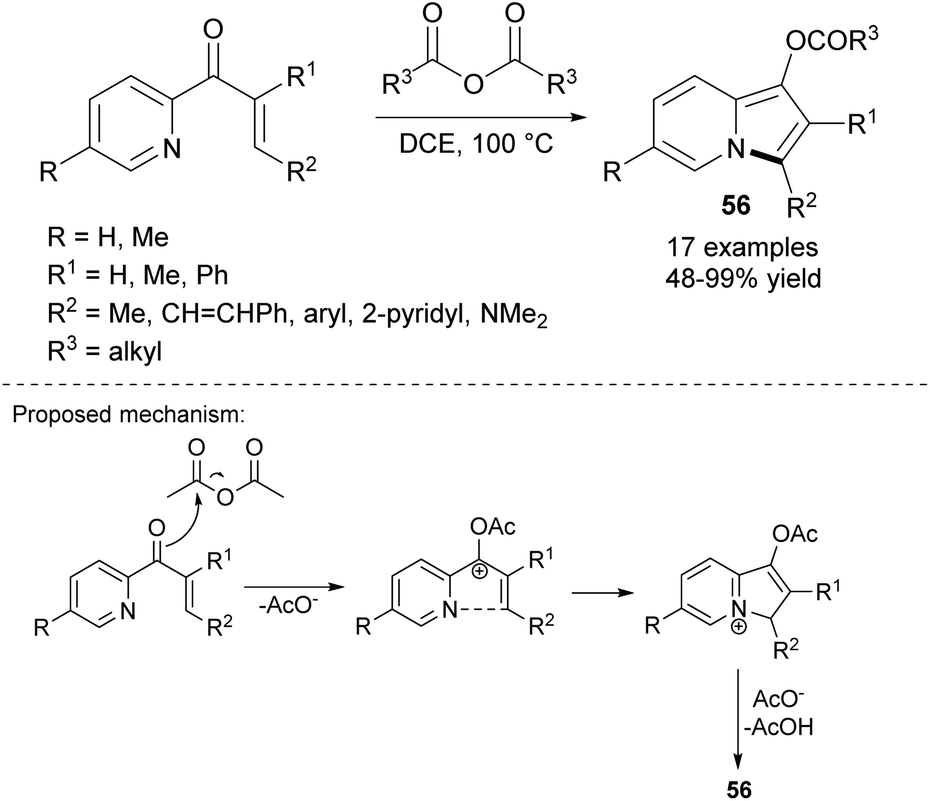 | ||
| Scheme 37 | ||
2-(2-Enynyl)pyridines are also effective substrates toward various indolizine molecules. Jia's group recently proved that this type of cyclization is catalyzed by copper(I) iodide and proceeded readily in the presence of a nucleophile (NuH) such as 1,3-dicarbonyl compounds, indoles, amides, alcohols, and even water and can achieve excellent yields (Scheme 38).61 In their next paper, they applied PdCl2(MeCN)2 as a catalyst and allyl chlorides as an electrophile and obtained 2-allylindolizines (58) in good yields (Scheme 39).62 Here, the same set of nucleophiles was used as in the initial study.
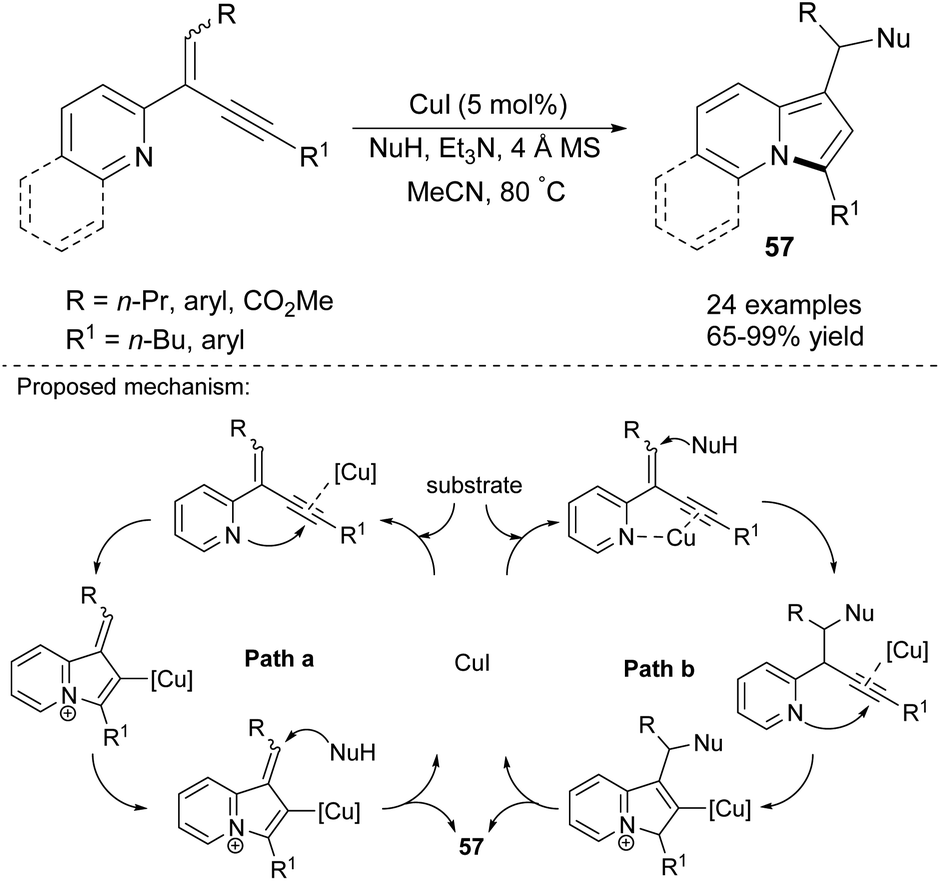 | ||
| Scheme 38 | ||
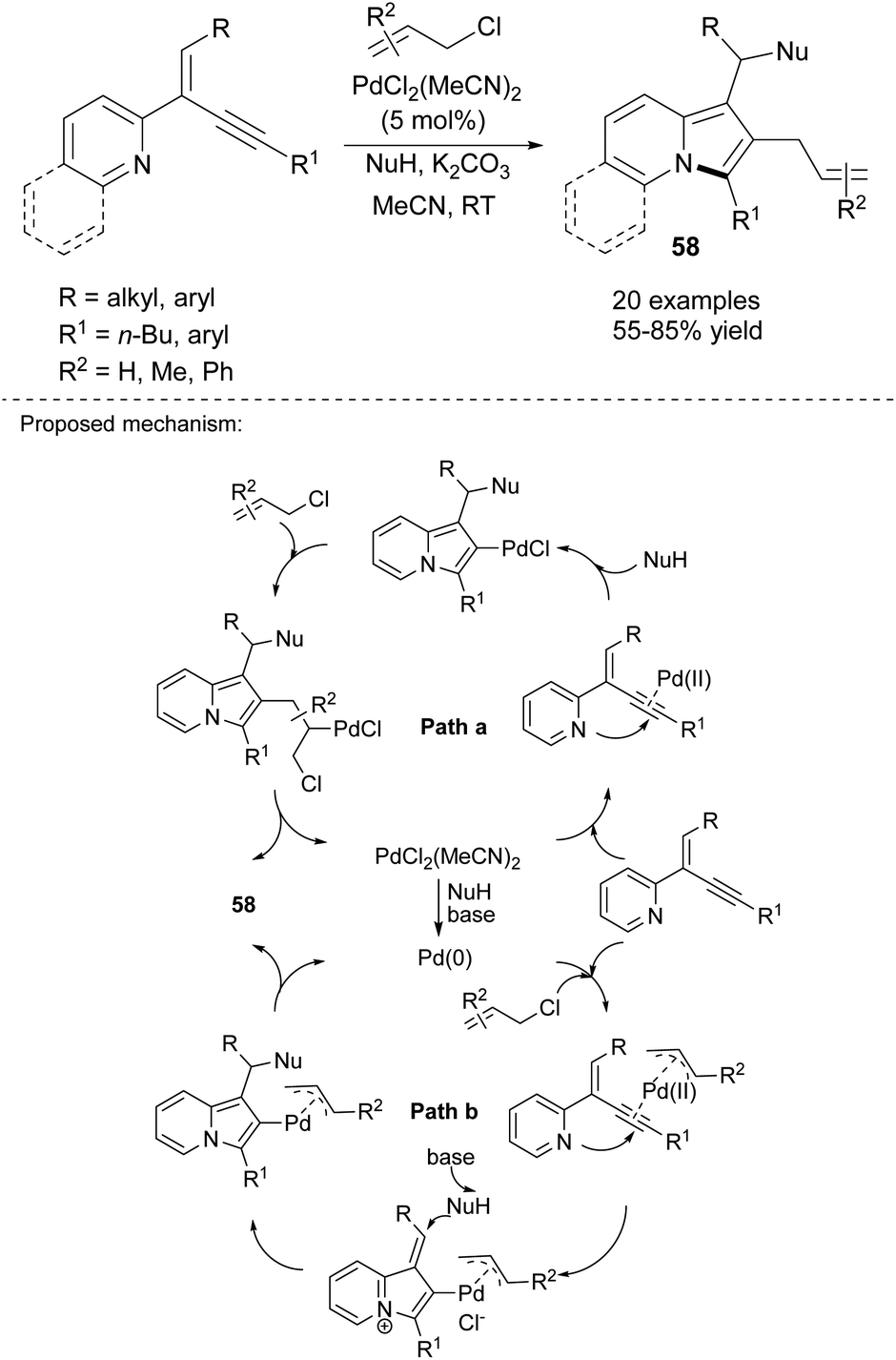 | ||
| Scheme 39 | ||
This same research group also developed Au-catalyzed cycloisomerization of 2-(1-alkynyl-cyclopropyl)pyridines in the presence of nucleophiles (Scheme 40).63 In this case, the IPrAuCl/AgNTf catalytic system proved to be very efficient and tolerated a variety of functional groups as well as different N-, C- and O-based nucleophiles such as amines, indoles, 1,3-dicarbonyl compounds, alcohols, carboxylic acids, etc.
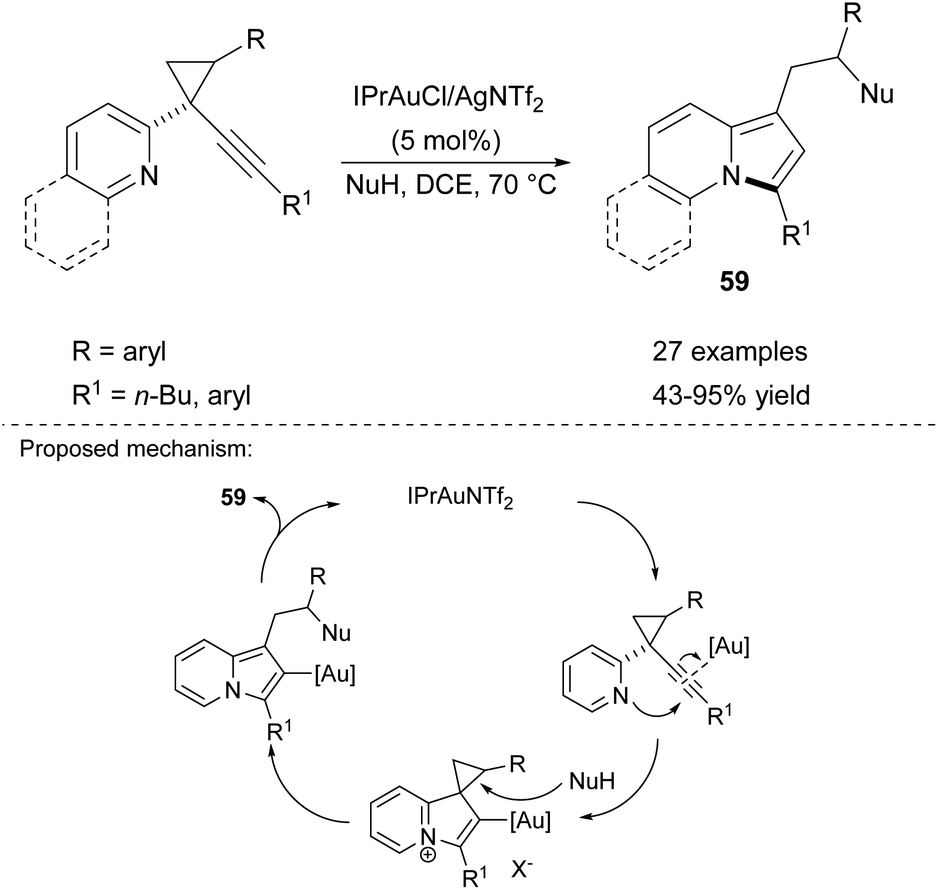 | ||
| Scheme 40 | ||
The cyclization of 2-pyridine derivatives was also achieved in the absence of transition metals. Kim and co-workers developed the iodocyclization of propargylic acetates toward indolizines 60 (Scheme 41a).64 The presence of an iodo substituent at position C2 opened new opportunities for the functionalization of 60. The authors successfully employed 60 as a halogen partner in Suzuki, Heck or Sonogashira couplings in excellent yields. Moreover, the radical reaction of 2-iodoindolizines, e.g., dehalogenation in the presence of the AIBN/n-Bu3SnH system was also possible and provided 1,3-disubstituted indolizines in excellent yields. Additionally, iodocyclization of allylic acetates was also achieved by the same group (Scheme 41b).65 In this particular case, the reaction system should be enriched with Et3N to achieve 61 in satisfactory yields. Later, the series of 3-acylated indolizines66 was synthesized by these investigators in a similar manner, providing 1,2,3-trisubstituted indolizines in satisfactory yields. All the aforementioned methodologies tolerated a wide range of functional groups and required mild reaction conditions.
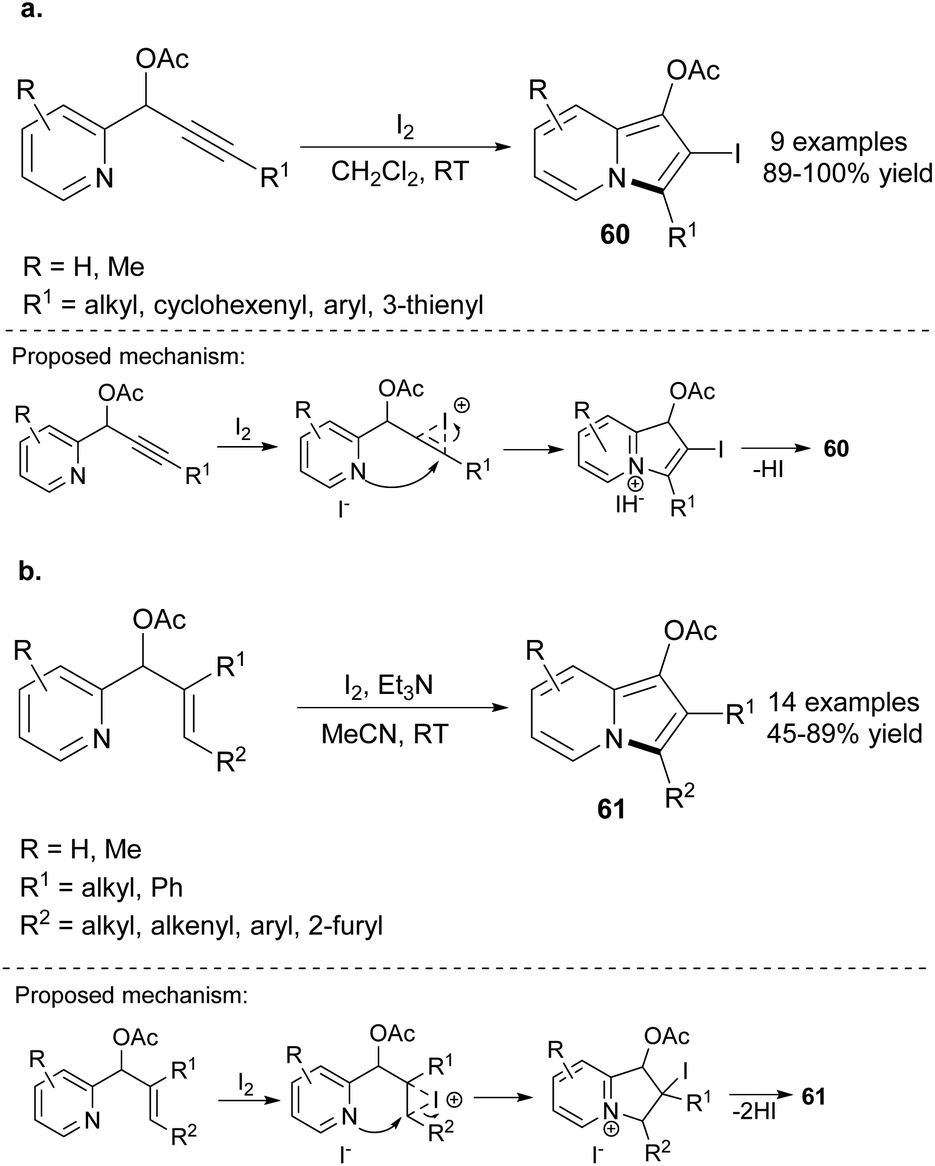 | ||
| Scheme 41 | ||
2.7 From pyridotriazoles and alkynes
Gevorgyan and co-workers employed Rh-catalyzed transannulation of activated pyridotriazoles with terminal alkynes for the synthesis of 3-arylindolizines 63 (Scheme 42).67 Pyridotriazoles themselves can be obtained either via reaction of pyridine-2-yl-acetates with 4-acetamidobenzenesulfonyl azide or from hydrazones via oxidative N–N coupling. This reaction proceeded smoothly to produce a mixture of cyclopropene 62 and indolizine 63, which could be easily separated by column chromatography. Unfortunately, the utilization of aliphatic alkynes resulted in a sluggish or messy reaction. Importantly, it was found that only 7-halopyridotriazoles could indeed serve as convenient substrates in this type of transannulation.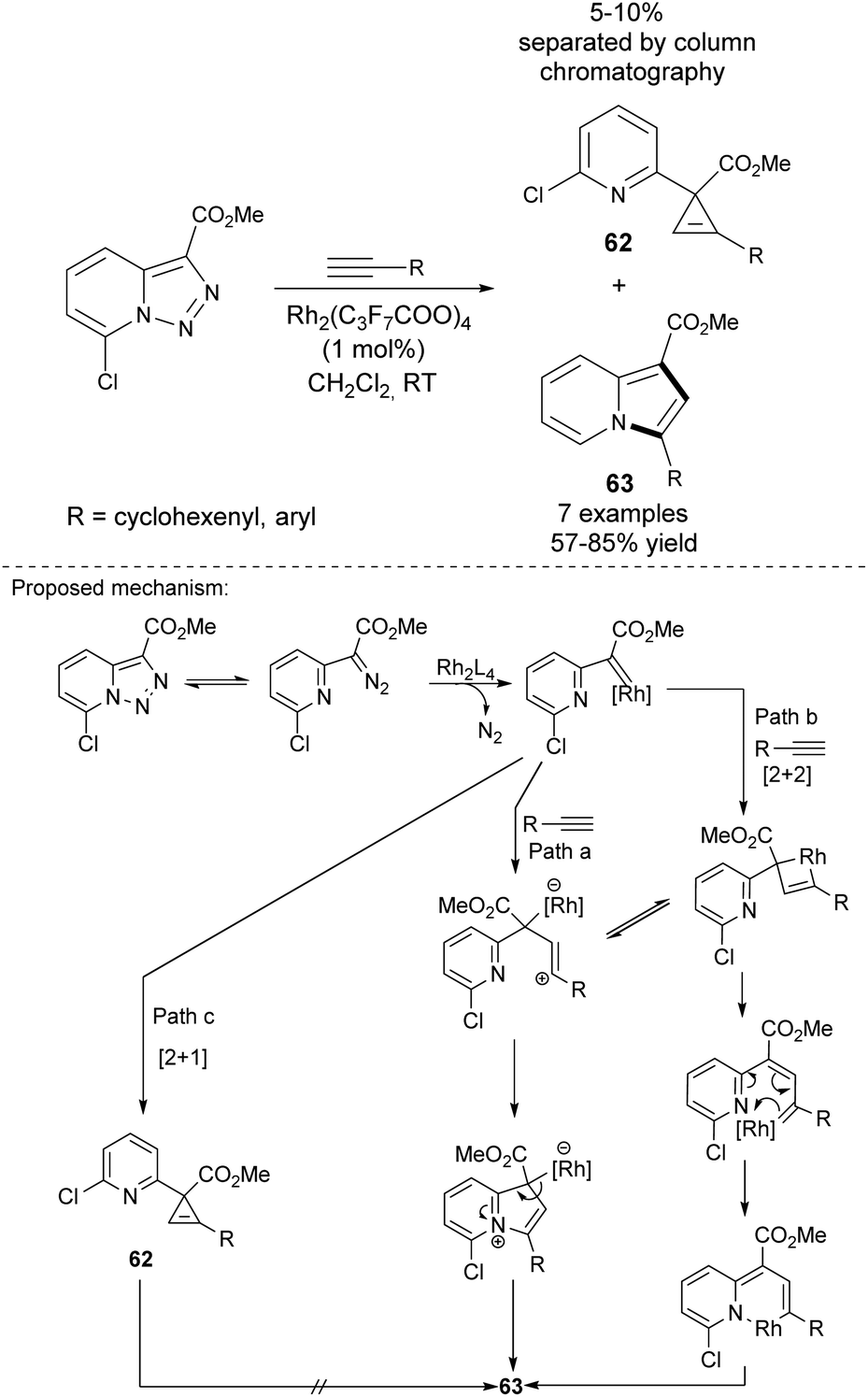 | ||
| Scheme 42 | ||
Later, the same group proved that cyclopropenes of type 64 could also be converted into indolizine molecules (Scheme 43a).68 They found that depending on the nature of the catalyst, 64 isomerized selectively to 1,3- or 1,2-disubstituted indolizines 65 or 66, respectively. Both methods tolerated electron-rich, electron-deficient, and sterically hindered aryl alkynes. Additionally, aliphatic alkynes were successfully applied in the Cu-catalyzed isomerization of 66 leading to 1,2-disubstituted indolizines 66. Excellent yields and the presence of halogen in the product provided many opportunities for further transformations, e.g., subjecting these compounds to the coupling reactions.
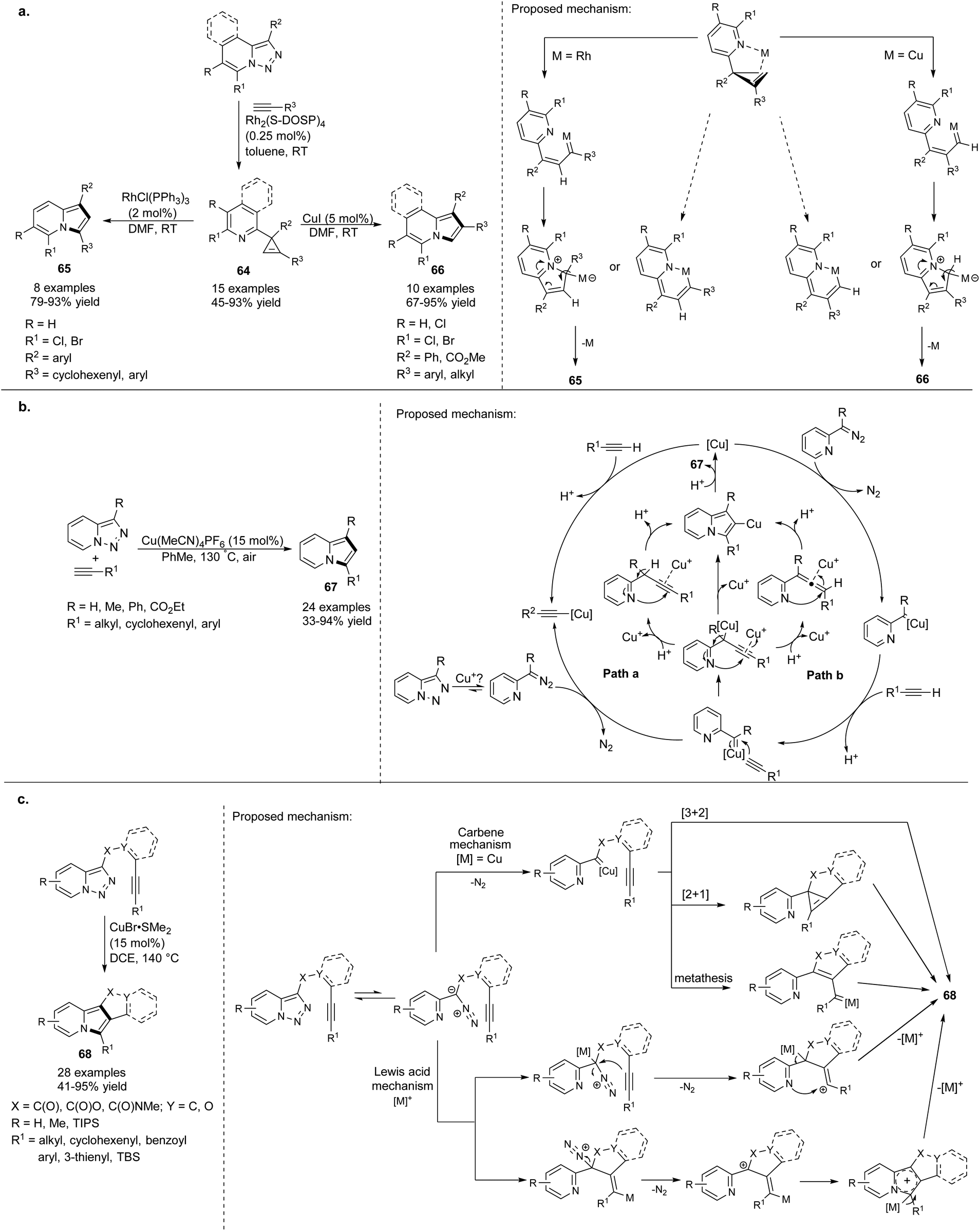 | ||
| Scheme 43 | ||
In the previously described methods67,68 7-halopyridotriazoles were only used as substrates for the Rh-catalyzed transannulation processes. Very recently, Helan et al. demonstrated that pyridotriazoles without a halo substituent at position C7 could also be converted into the indolizine skeleton under aerobic conditions (Scheme 43b).69 Among copper-based catalysts tested in this transformation, Cu(MeCN)4PF6 showed the best catalytic activity. The protocol fully tolerated both aryl and aliphatic alkynes giving 67 in good to excellent yields. A much broader scope of substrates and the lower cost of the catalyst in comparison with previously reported methodologies resulted in a great synthetic potential for this reaction.
Inspired by the aforementioned methodologies in which terminal alkynes were exclusively exploited, Gevorgyan and Shi proposed a simple approach to fused polycyclic indolizines 68via intramolecular transannulation reaction of the pyridotriazole core with an alkyne triple bond catalyzed by CuBr2·SMe2 (Scheme 43c).70 This reaction was complementary to a previously published method,55 and can be catalyzed also by other Lewis acids such as Sc(OTf)3 or TIPSOTf. Using the appropriately substituted pyridotriazole component, the synthesis of 6-substituted indolizines is possible.
3. Methods based on pyrrole derivatives
In theory, the alternative approach toward assembling the indolizine core starting from pyrrole derivatives and building six-membered rings is simply the mirror image of the previous concept. However, such a synthesis has been a challenge to realize. Numerous difficulties such as significantly fewer commercially available pyrrole derivatives, their lower oxidation potential, and hence their stability are reflected in an overall smaller number of such synthetic strategies.3.1 From N–H free pyrroles
An efficient and highly selective method for the synthesis of 69 starting from 2-formylpyrroles and ethyl γ-bromocrotonates was described by Ge et al. (Scheme 44).71 According to this protocol, it would be possible to obtain 2- or 3-bromoindolizines – useful substrates in a variety of cross-coupling reactions. Subsequently, this method was extended by Yang and co-workers to 2-acetylpyrrole derivatives.72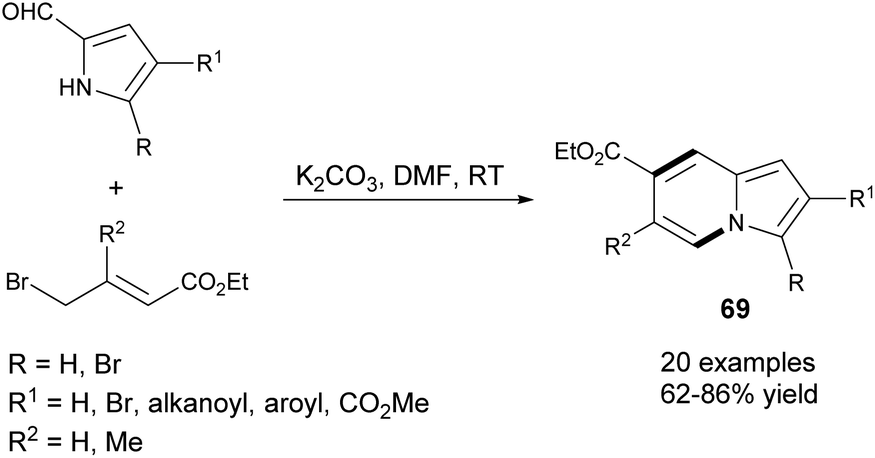 | ||
| Scheme 44 | ||
An analogous type of transformation was used to obtain indolizine-7-phosphonates 70 starting from 2-formylpyrrole derivatives and diethyl 3-bromoprop-1-enylphosphonate (Scheme 45a).73 Mechanistically, this process was similar to the above-mentioned methods71,72 with the exception of the last stage, in which elimination of a water molecule was more favored due to the conjugation between the indolizine skeleton and the diethyl phosphonate group (Scheme 45b). Moreover, this method was useful in the synthesis of other nitrogen bridgehead heterocycles such as pyrido[1,2-a]benzimidazole phosphonates and pyrazolo[1,5-a]pyridine phosphonate in moderate to good yields.
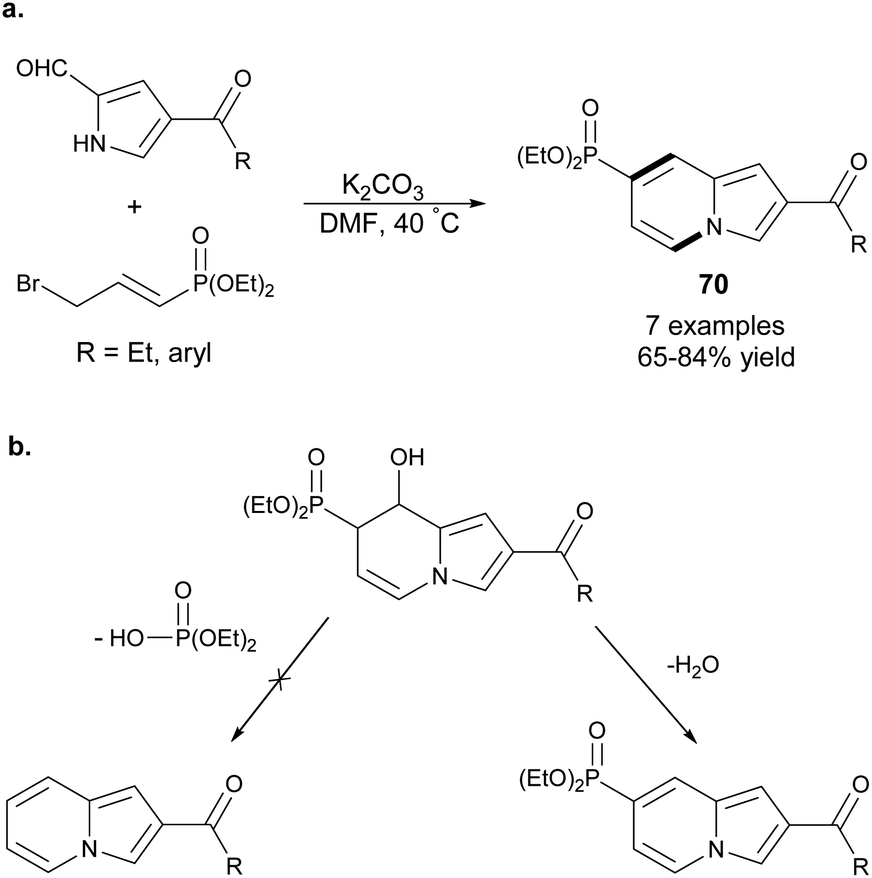 | ||
| Scheme 45 | ||
Later, Wang's group utilized β-aryl-γ-bromocrotonates as a synthon to build six-membered rings to give 6-arylindolizines (71) in moderate yields (Scheme 46).74 In contrast to the protocol used before,71 higher reaction temperatures (50 °C vs. room temperature) and extended reaction times were required due to the electronic effect of the aryl substituent in bromocrotonate.
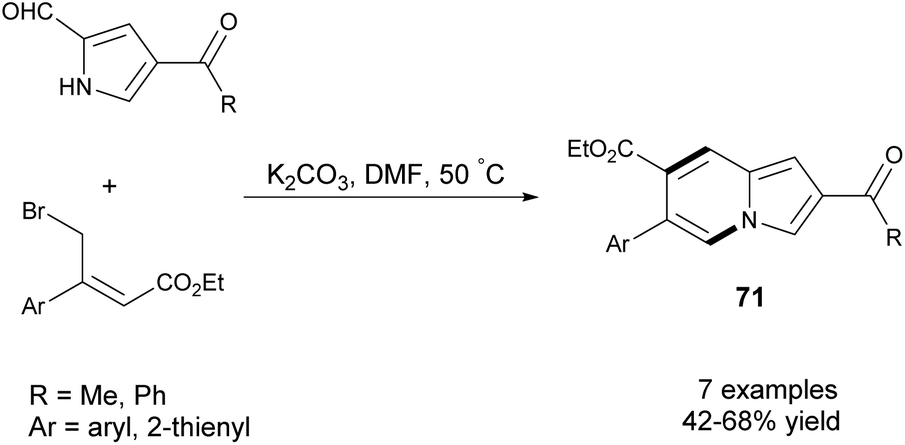 | ||
| Scheme 46 | ||
On the other hand, 7-arylindolizines 74 could be constructed employing the corresponding allyl bromides 72 as a source of the aryl substituent (Scheme 47).75 2-Formylindole or 2-formylpyrrole derivatives smoothly undergo N-alkylation with 7276 (pure isomer or E/Z mixture) to give 73, which after isolation were converted into various indolizine derivatives 74 in low to moderate yields. Many other heterocyclic skeletons, such as imidazo[1,2-a]pyridines and pyrido[1,2-a]indoles can be successfully prepared according to this protocol.
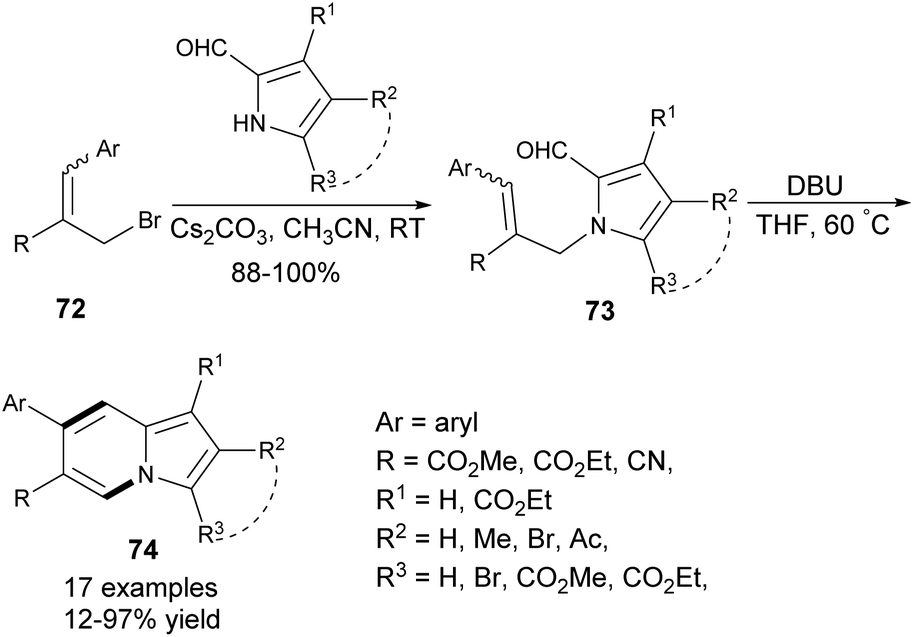 | ||
| Scheme 47 | ||
Recently, Townsend and co-workers proposed an elegant approach leading to 1,2,3-substituted 5-amino-7-cyanoindolizines 76–77 in good yields via a one-pot procedure exploiting 2-formyl pyrrole derivatives and fumaronitrile as substrates (Scheme 48).77 In the first step, Wittig olefination took place, producing (E)-alkene 75 predominately. Next, in the presence of a catalytic amount of KOH, cycloaromatization occurred, giving the corresponding indolizines. Interestingly, there is the possibility of introducing an additional substituent at position C6 by selective alkylation of the allylic position in 75 at low temperature, followed by cyclization induced by a second equivalent of LDA at room temperature (Table 1). This efficient protocol was also applicable to the synthesis of imidazo[1,2-a]pyridines and imidazo[1,5-a]pyridines in good yields.
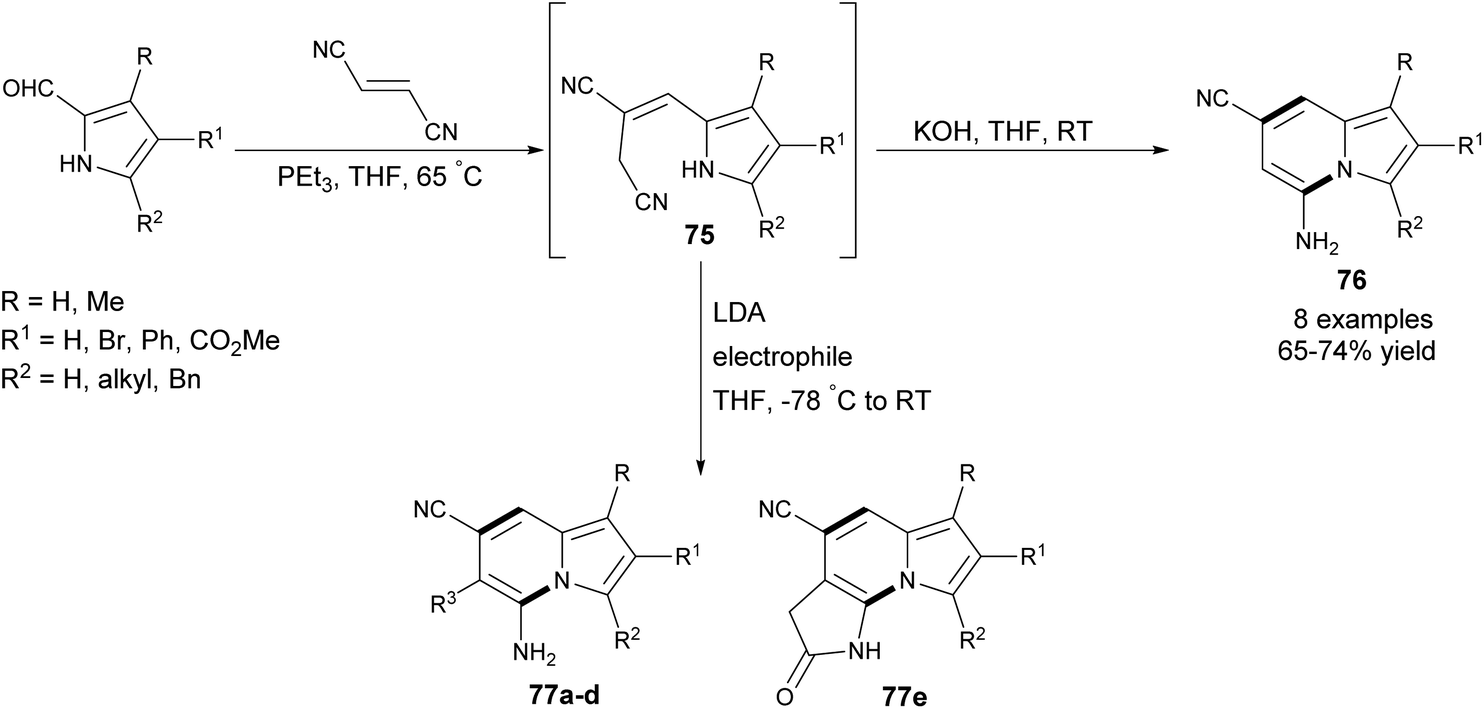 | ||
| Scheme 48 | ||
| Entry | Electrophile | Product | R3 | Yield/% |
|---|---|---|---|---|
| 1 | MeI | 77a | Me | 87 |
| 2 | H2CCHCH2Br |
77b | –CH2CHCH2 |
98 |
| 3 | HC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CCH2Br CCH2Br |
77c | –CH2CCH |
94 |
| 4 | PhCH2Br | 77d | –CH2Ph | 98 |
| 5 | BrCH2CO2Et | 77e | 90 |
Hao et al. demonstrated a facile Pd-catalyzed synthesis of indolizines 78 starting from pyrrole derivatives and 1,4-dibromo-1,3-butadienes (Scheme 49a).78 Importantly, in the case of ethyl 3-carboxypyrrole, the mixture of two possible products was obtained in a 1:1 ratio. Additionally, the authors reacted dibromides 79 with pyrrole to obtain π-expanded molecules 80a and 80b in 82 and 76% yield, respectively (Scheme 49b). The presence of the trimethylsilyl group (TMS) gave additional possibilities of extending the chromophore, e.g., by Hiyama-type coupling reactions.
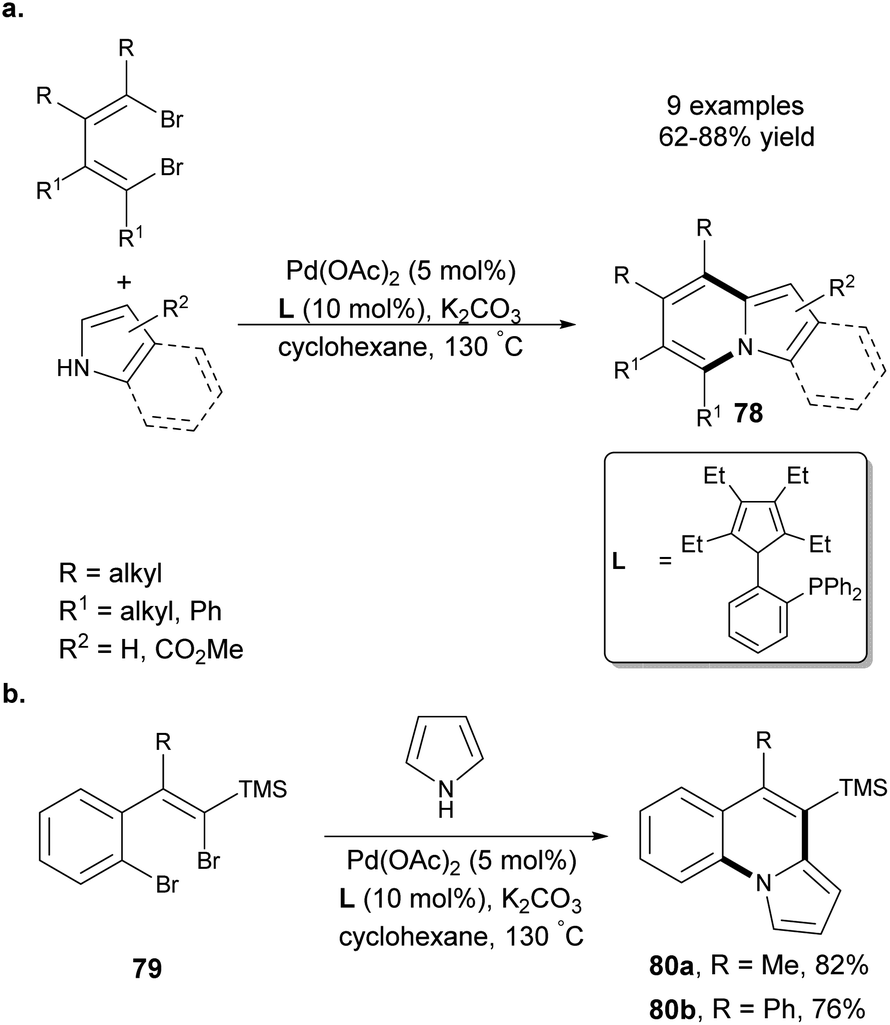 | ||
| Scheme 49 | ||
An interesting discovery was made recently by Katukojvala and co-workers, who examined the reactivity of 3-substituted indoles toward keto diazoenals in the presence of rhodium-based salts (Scheme 50).79 A set of 9,10-disubstituted pyrido[1,2-a]indoles 81 was obtained under mild conditions with a wide range of substituents. For example, 81 was selectively hydrogenated (H2/PtO2/MeOH) leading to 82 possessing the tetrahydropyrido[1,2-a]indole motif, a structure present in many biologically important polycyclic indole alkaloids.
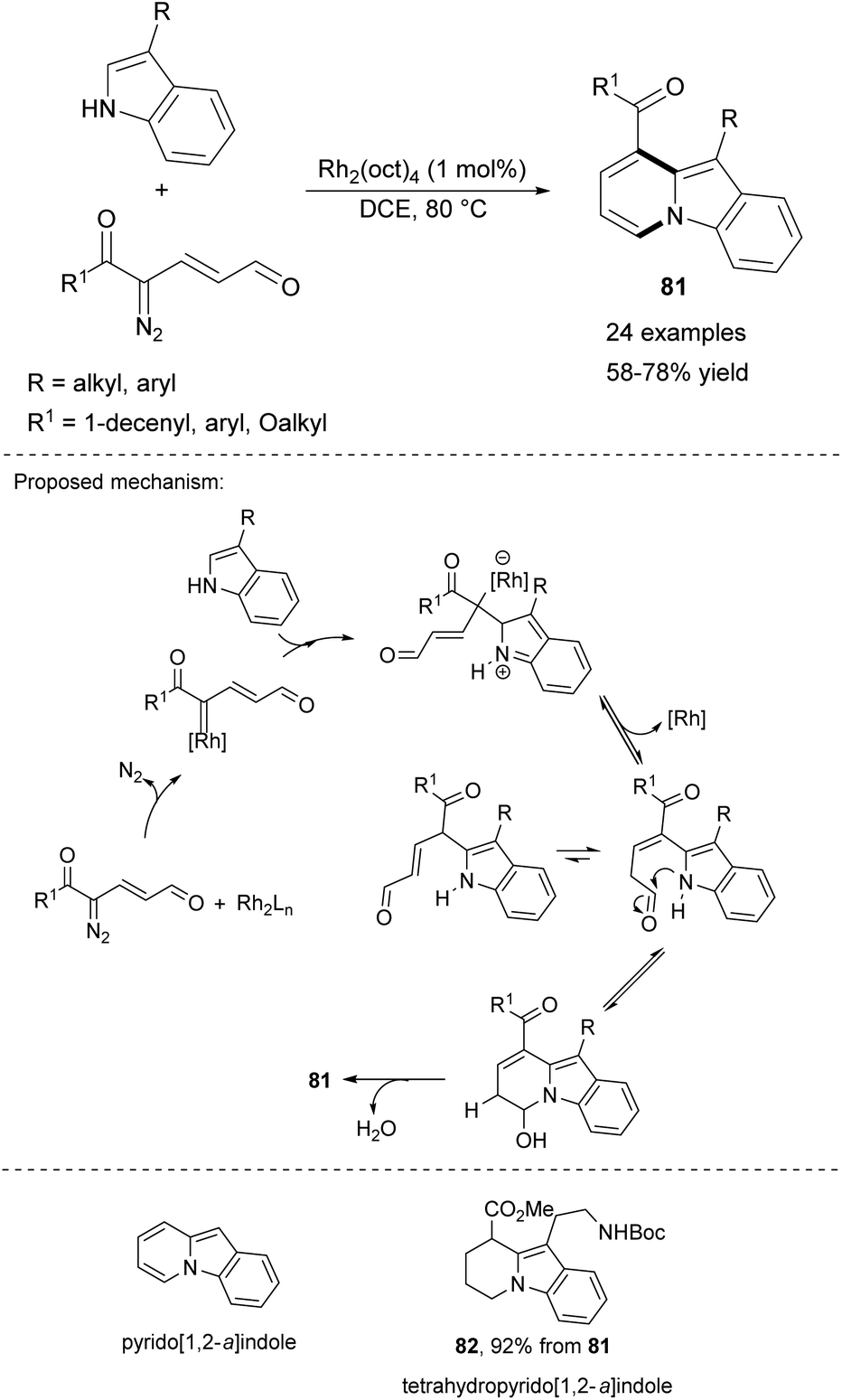 | ||
| Scheme 50 | ||
3.2 From N-substituted pyrroles
In 2013, Kim et al. developed a new synthetic methodology for the preparation of polysubstituted indolizines 83 (Scheme 51).80 This annulation process involves Knoevenagel condensation and intramolecular aldol cyclization and tolerates a broad scope of substrates. A significant advantage of this methodology is the possibility of introducing a variety of substituents into both pyridine-type and pyrrole-type rings. Furthermore, Opatz and Kucukdisli demonstrated a one-pot approach for the synthesis of indolizines 84 fully substituted in the pyridine-type ring (Scheme 52).81 This method required no expensive reagents or catalysts and afforded products in low to high yields.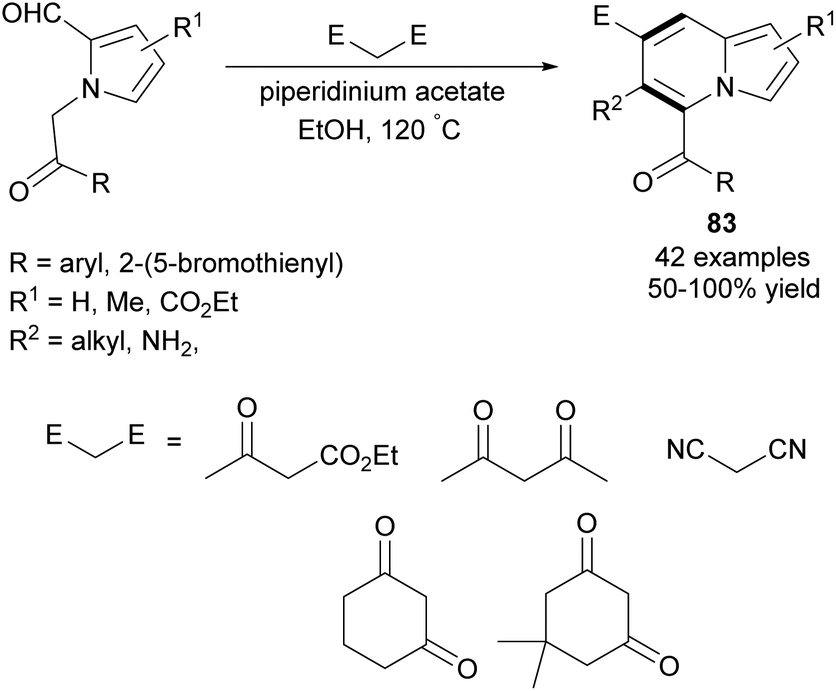 | ||
| Scheme 51 | ||
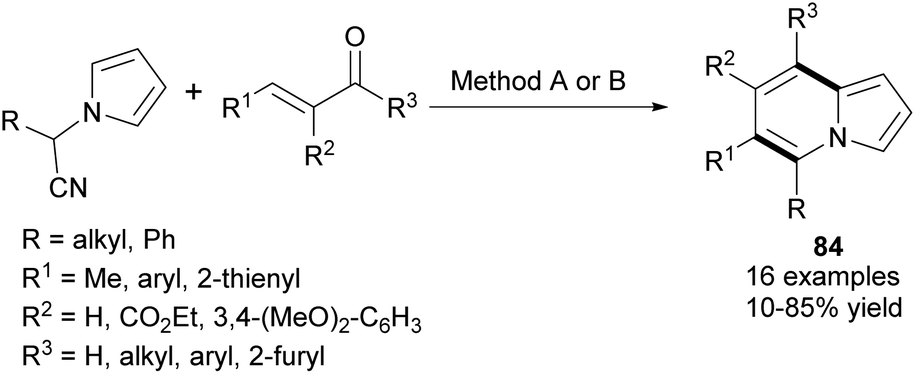 | ||
| Scheme 52 Reagents and conditions: (A) (i) t-BuOK, 0 °C, THF; (ii) AcOH, EtOH, BF3·OEt2, RT; (iii) DBU, reflux; (B) (i) t-BuOK, DMF, 0 °C; (ii) TfOH, RT; (iii) DBU, 90 °C. | ||
At the same time, an easy and efficient route to 6,8-disubstituted indolizines 85via intramolecular cycloaromatization was proposed (Scheme 53).82 Once obtained in this way, 8-triflyl indolizines could be further employed in a Pd-catalyzed Suzuki–Miyaura cross-coupling reaction leading to indolizines with various aryl groups at position C8 in 42 to 88% yield (12 examples). In order to maximize diversity, the products of cross-coupling reactions were successfully applied to the Heck coupling, Friedel–Crafts acylation, and Vilsmeier–Haack formylation.
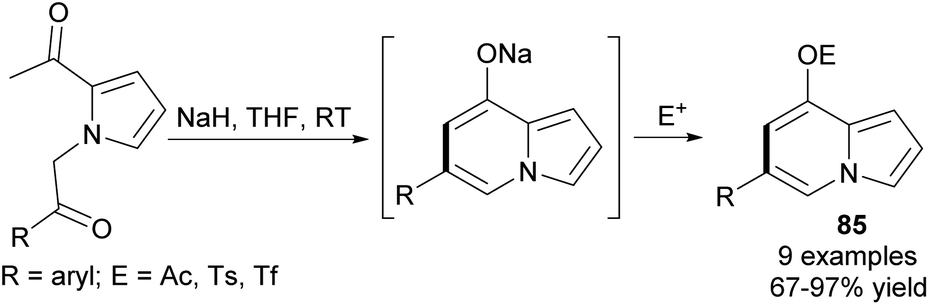 | ||
| Scheme 53 | ||
Microwave-assisted benzannulation of 1-propargyl-2-formylpyrrole in the presence of pyrrolidine gave 6-(pyrrolidin-1-yl)indolizine (86) in 88% yield (Scheme 54).83 The reaction proceeded under mild conditions without a metal catalyst and additives. Moreover, the liquid amine acted as a solvent. Undoubtedly, the scope of this process should be examined in detail.
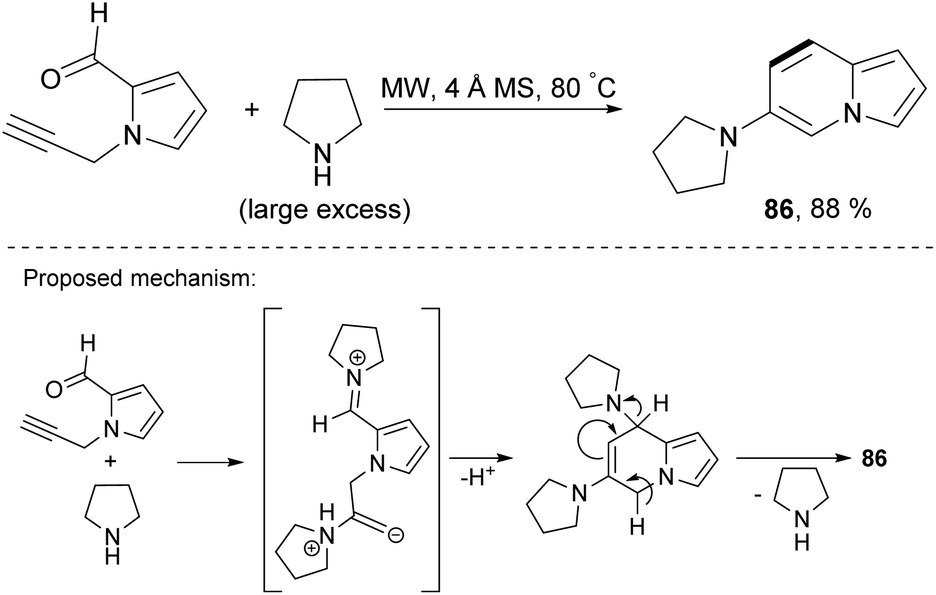 | ||
| Scheme 54 | ||
More recently, an intramolecular alkyne–carbonyl metathesis approach toward benzo-fused indolizines 88 was proposed by Kim's group (Scheme 55).84 The parent pyrrole derivative 87 was prepared via Sonogashira coupling and subsequently transformed into the desired indolizine by heating in TFA. Although a number of TM-based catalysts were tested, none of them proved to be efficient. Substrates bearing alkenyl groups led to the corresponding products in low yields, whereas those possessing alkyl groups were unreactive.
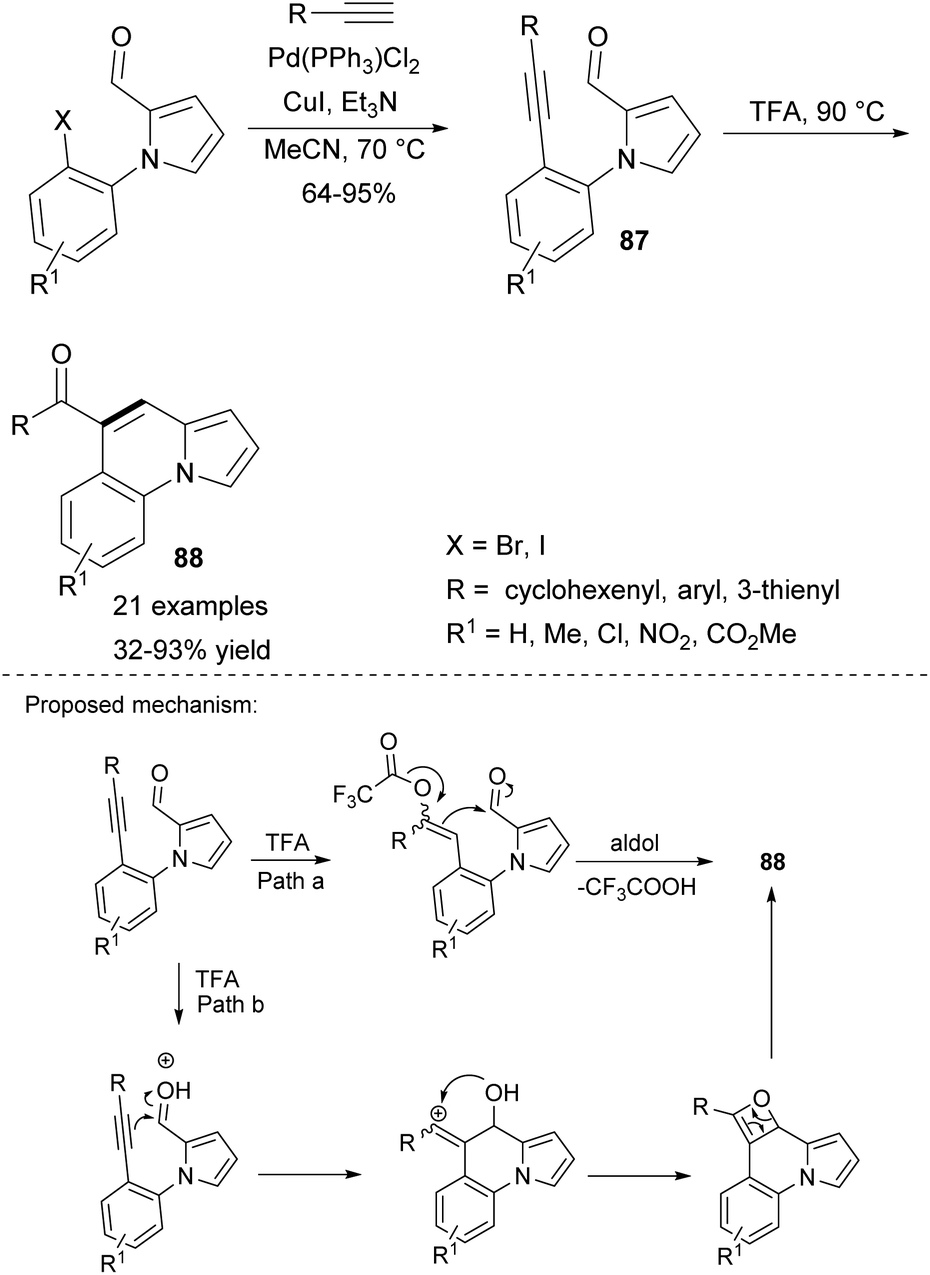 | ||
| Scheme 55 | ||
4. Selected examples of methodologies leading to π-expanded indolizines
The intramolecular cycloaddition reactions catalyzed by transition metal species are a popular means to construct complex aromatic molecules. Bis(propargylphenyl)carbodiimides can be converted smoothly into L-shaped π-extended compounds with pyrrolo[1,2-a][1,8]naphthyridine units 89–92via a [2 + 2 + 2] cycloaddition reaction followed by subsequent oxidation (Scheme 56).85 Interestingly, these molecules exhibited sky-blue to golden-orange fluorescence with high quantum yields, even in polar solvents.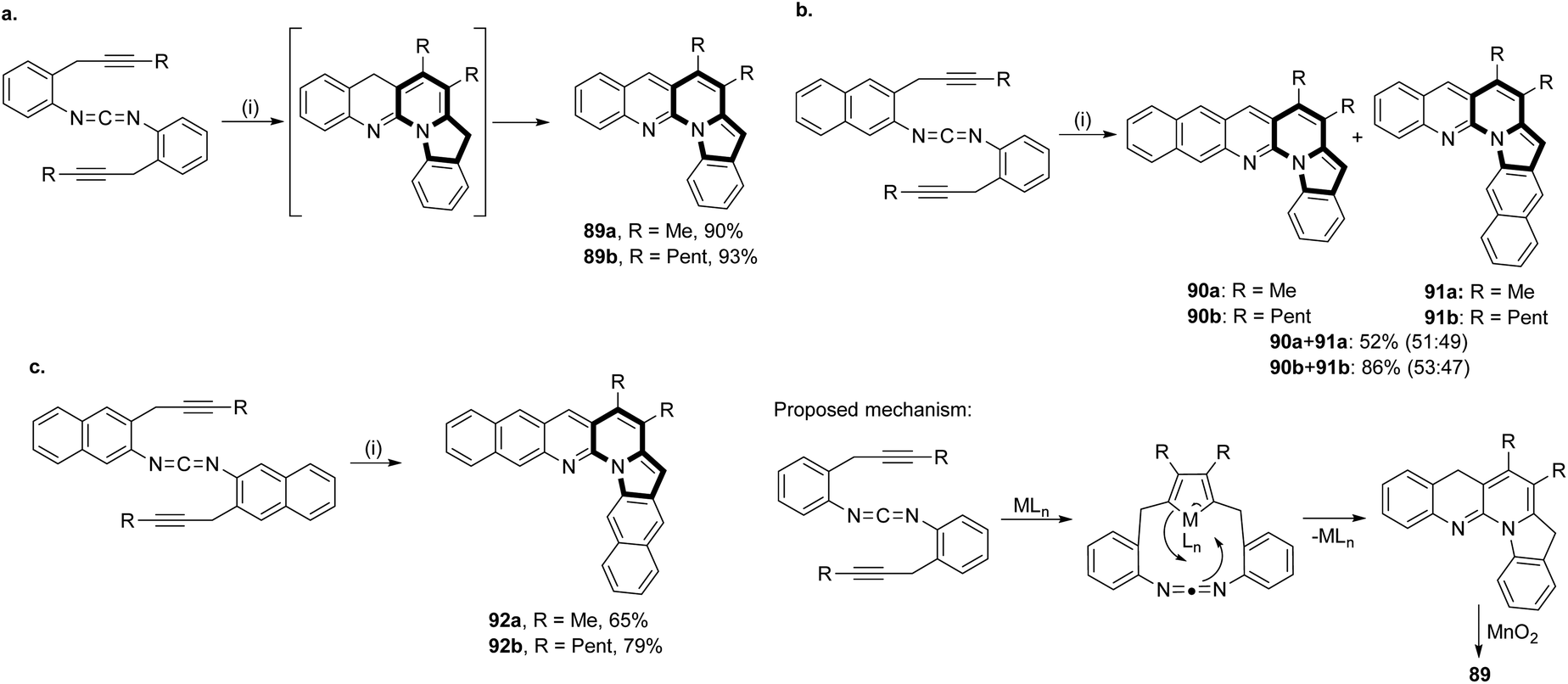 | ||
| Scheme 56 Conditions: (i) (1) RhCl(PPh3)3 (10 mol%), toluene, reflux; (2) MnO2, RT. | ||
In 2015, Kim's group proved that the same type of cyclization used by them previously84 could be successfully applied in the synthesis of much more complex derivatives – benzo[e]pyrido[1,2-a]indoles 93–95 (Scheme 57).86 The starting material was obtained in a three step synthesis involving indolizine formation, Vilsmeier–Haack formylation, and Sonogashira coupling. This intramolecular hydroarylation with concomitant extrusion of a formyl group was catalyzed by TFA and the mixture of two products 94–95 was formed. When the reaction was also applied to the substrates bearing the alkyl group at R2, the substrates that possessed no formyl group at position C3 gave only 93 as the sole product in the presence of AgOTf. Importantly, applying the substrates bearing an alkyl group such as R2 to acidic conditions resulted in a complex mixture, while TM-based conditions gave good yields of products.
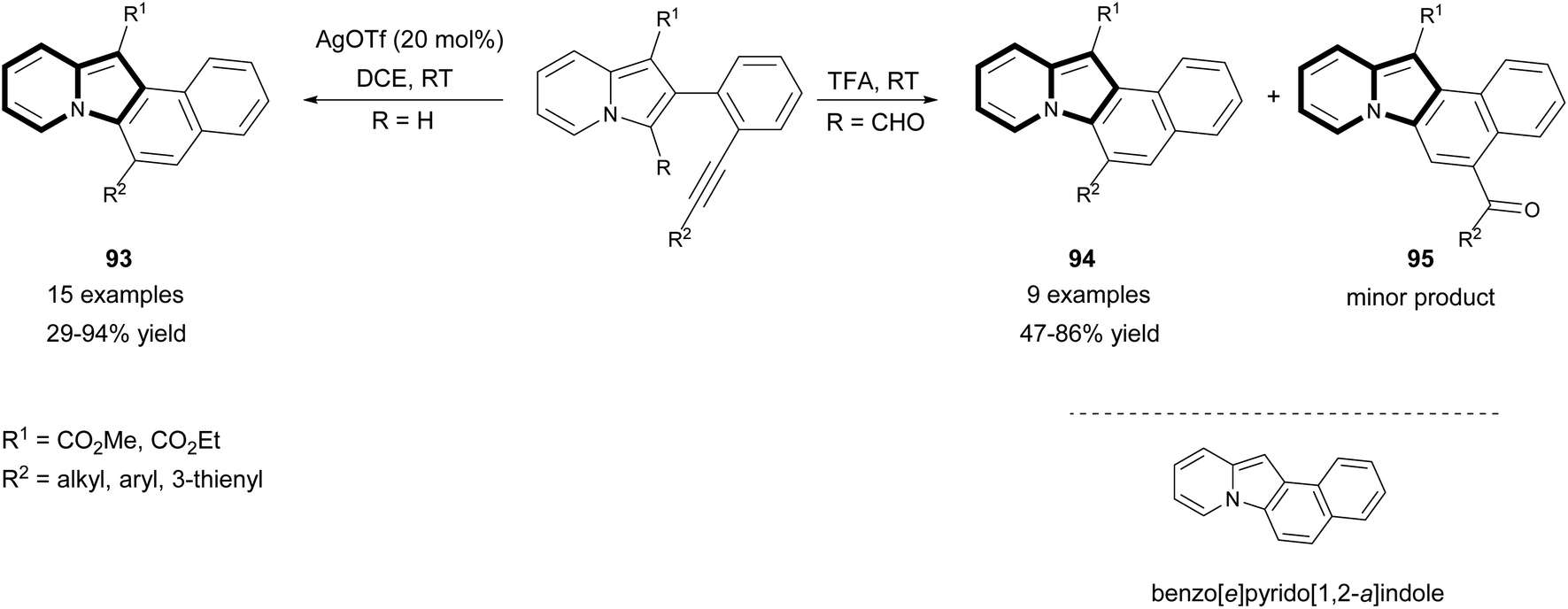 | ||
| Scheme 57 | ||
Very recently, the 6-endo-dig iodocyclization leading to benzo[e]pyrido[1,2-a]indoles (96) bearing an iodo substituent was developed (Scheme 58).87 Some derivatives were applied in Sonogashira or Suzuki coupling, and thus the diversity of products was maximized by introducing two different substituents at positions C5 and C6.
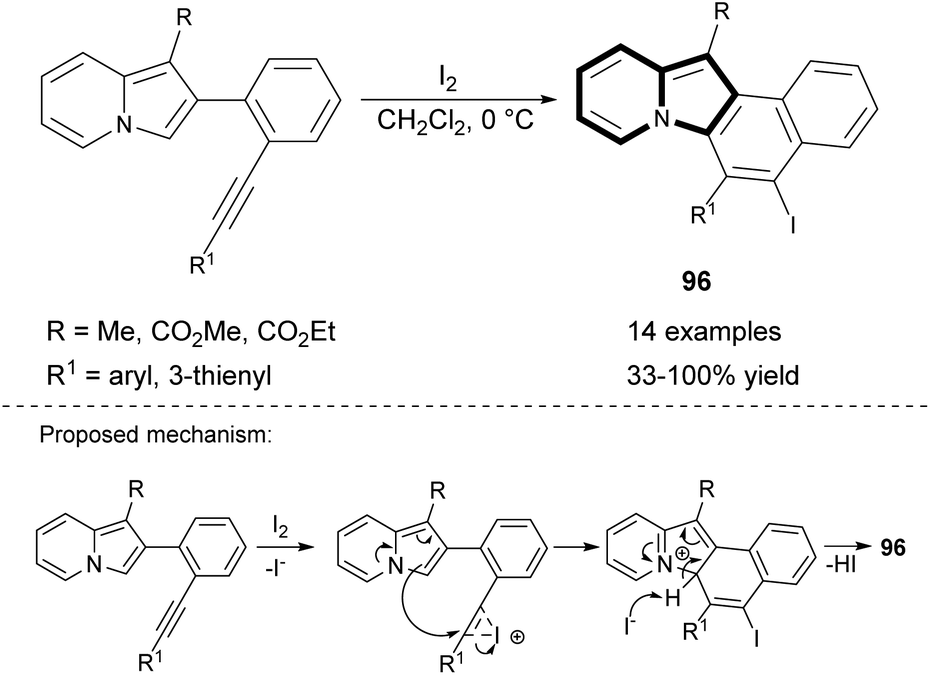 | ||
| Scheme 58 | ||
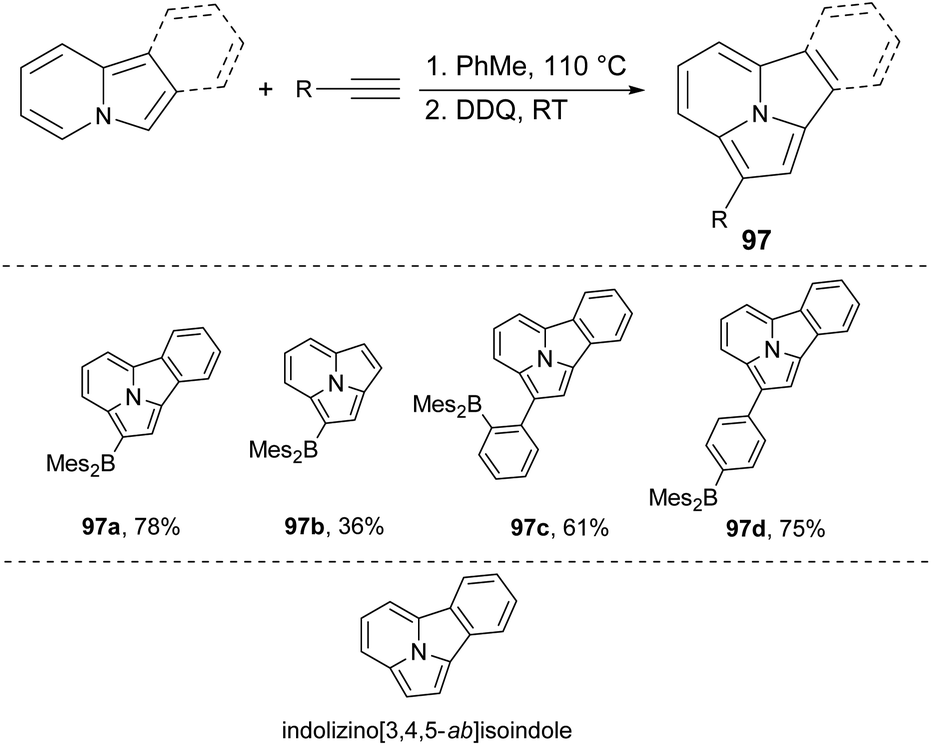
It was recently shown88 that the dehydrogenative Heck annelation reaction is a good tool for the synthesis of fused indolizines in a relatively simple manner. Four structurally unique brightly blue or blue-green fluorescent indolizino[3,4,5-ab]isoindoles 97a–d can be obtained via the above mentioned method (Scheme 59).89 The X-ray diffraction data of 97a revealed that there were discrete π-stacked dimers in the crystal lattice, in contrast to previously reported indolizino[3,4,5-ab]isoindole derivatives which showed extended π-stacking interactions.90
 | ||
| Scheme 59 | ||
5. Conclusions
Although the history of indolizine synthesis dates back to the nineteenth century and many methods have been explored to date, there is still a need to develop more versatile and/or eco-friendly methodologies. This review summarizes the greatest achievements in this field over the past ten years and we hope that the advancements presented here, combined with many opportunities in synthetic organic chemistry will be helpful in the development of completely new reactions toward indolizines as well as other heterocyclic motifs.During the last decade many methods for the synthesis of indolizine molecules have been explored and several prevailing trends can be identified:
(1) while the majority of strategies still have been based on pyridine derivatives as a starting material (with stand-alone contributions from Gevorgyan and co-workers), there have also been a rising number of methodologies employing pyrrole derivatives. Many of these methods are catalyzed by transition metal species, which is undesirable from the viewpoint of medicinal chemistry, but allows, in most cases, the construction of much more diversified molecules.
(2) The unsurpassed simplicity of the Chichibabin synthesis has only one drawback – the substituent in the five-membered ring must always be at position 2. As a consequence, many recent efforts have been concentrated on methods which allow the insertion of substituents at positions 1 and 3. This alteration bears multiple implications, not only for purely academic reasons, but also because these highly reactive positions are responsible for lower stability under ambient conditions. Blocking these sites with substituents will afford more stable indolizines.
(3) One of the recent trends in indolizine synthesis mirrors some more general trends in organic chemistry, namely the attempt to replace a prefunctionalization-based approach with oxidation-based strategies.
(4) In the past, indolizines attracted attention as unsaturated analogs of naturally present indolizidine alkaloids. Interest has shifted in recent years toward fluorescence-related applications. This is perhaps the most profoundly visible in the work of Park's group on the so-called Seoul-Fluor dyes. This, in turn, has influenced the direction of development. The synthesis of indolizines bearing simple alkyl and aryl groups became less important while it also became necessary to introduce substituents possessing a strong electron-donating or electron-withdrawing character. This change has been reflected in the most decisive manner in the emergence of methodologies leading to the formation of indolizines bearing the amino group (typically tertiary) in positions 1, 2, 3, 5 and 6. Such compounds were hardly available before 2005.
Needless to say, much additional effort is needed to explore the possibilities of some of these molecules. We hope that in addition to organizing knowledge on this topic, this Review will serve as a catalyst to spark further studies. The future target compounds can be extended to more densely substituted indolizines and push–pull indolizines.
Acknowledgements
The work was financially supported by the Polish National Science Centre, grant MAESTRO-2012/06/A/ST5/00216 and by the Global Research Laboratory Program (2014K1A1A2064569) through the National Research Foundation (NRF) funded by the Ministry of Science, ICT & Future Planning (Korea).References
- (a) Comprehensive Heterocyclic Chemistry, ed. A. R. Katritzky and C. W. Rees, Pergamon Press, Oxford, 1984 Search PubMed; (b) Comprehensive Heterocyclic Chemistry II, ed. A. R. Katritzky, C. W. Rees and E. F. V. Scriven, Pergamon Press, Oxford, 1996 Search PubMed.
- (a) J. P. Michael, Nat. Prod. Rep., 2008, 25, 139 RSC; (b) M. Lebceuf, A. Cave, A. Ranaivo and H. Moskowitz, Can. J. Chem., 1989, 67, 947 CrossRef CAS.
- (a) K. Bedjeguelal, H. Bienayme, A. Dumoulin, S. Poigny, P. Schmitt and E. Tam, Bioorg. Med. Chem. Lett., 2006, 16, 3998 CrossRef CAS PubMed; (b) L. L. Gundersen, C. Charnock, A. H. Negussie, F. Rise and S. Teklu, Eur. J. Pharm. Sci., 2007, 30, 26 CrossRef CAS PubMed; (c) W. L. Huang, T. Zuo, H. W. Jin, Z. M. Liu, Z. J. Yang, X. H. Yu, L. R. Zhang and L. H. Zhang, Mol. Diversity, 2013, 17, 221 CrossRef CAS PubMed; (d) K. M. Dawood, H. Abdel-Gawad, M. Ellithey, H. A. Mohamed and B. Hegazi, Arch. Pharm. Chem. Life Sci., 2006, 339, 133 CrossRef CAS PubMed; (e) M. W. Orme, J. S. Sawyer and L. M. Schultze, PCT Int. Appl, WO000657, 2002 Search PubMed; (f) W. Chai, J. G. Breitenbucher, A. Kwok, X. Li, V. Wong, N. I. Carruthers, T. W. Lovenberg, C. Mazur, S. J. Wilson, F. U. Axe and T. K. Jones, Bioorg. Med. Chem. Lett., 2003, 13, 1767 CrossRef CAS PubMed; (g) P. Chen, A. Chaikuad, P. Bamborough, M. Bantscheff, C. Bountra, C.-W. Chung, O. Fedorov, P. Grandi, D. Jung, R. Lesniak, M. Lindon, S. Müller, M. Philpott, R. Prinjha, C. Rogers, C. Selenski, C. Tallant, T. Werner, T. M. Willson, S. Knapp and D. H. Drewry, J. Med. Chem., 2016, 59, 1410 CrossRef CAS PubMed.
- (a) J. H. Delcamp, A. Yella, T. W. Holcombe, M. K. Nazeeruddin and M. Grätzel, Angew. Chem., Int. Ed., 2013, 52, 376 CrossRef CAS PubMed; (b) J. Wan, C.-J. Zheng, M.-K. Fung, X.-K. Liu, C.-S. Lee and X.-H. Zhang, J. Mater. Chem., 2012, 22, 4502 RSC; (c) Y. R. Song, C. W. Lim and T. W. Kim, Luminescence, 2015, 31, 364 CrossRef PubMed.
- (a) A. Angeli, Gazz. Chim. Ital., 1890, 26, 761 Search PubMed; (b) A. Angeli, Ber. Dtsch. Chem. Ges., 1890, 23, 1793 CrossRef; (c) A. Angeli, Ber. Dtsch. Chem. Ges., 1890, 23, 2154 CrossRef.
- (a) E. T. Borrows and D. O. Holland, Chem. Rev., 1948, 42, 611 CrossRef CAS PubMed; (b) Heterocyclic Compounds, ed. H. R. Ing and R. C. Elderfield, Wiley, New York, 1952, vol. 3 Search PubMed; (c) W. L. Mosby, Heterocyclic Systems with Bridgehead Nitrogen Atoms, Part I, Interscience, New York, 1961 Search PubMed; (d) N. S. Prostakov and O. B. Baktibaev, Usp. Khim., 1975, 44, 1649 CrossRef CAS; (e) T. Uchida and K. Matsumoto, Synthesis, 1976, 209 CrossRef CAS; (f) H. L. Blewitt, Chemistry of Heterocyclic Compounds: Special Topics in Heterocyclic Chemistry, John Wiley & Sons, Inc., Hoboken, NJ, USA, 1977, vol. 30 Search PubMed; (g) F. J. Swinbourne, J. H. Hunt and G. Klinkert, Advances in Indolizine Chemistry, in Advances in Heterocyclic Chemistry, 1978, vol. 23, p. 103 Search PubMed; (h) P. Shipman, Sci. Synth., 2001, 745 Search PubMed; (i) G. S. Singh and E. E. Mmatli, Eur. J. Med. Chem., 2011, 46, 523 Search PubMed.
- S. El Kazzouli, J. Koubachi, N. El Brahmia and G. Guillaumet, RSC Adv., 2015, 5, 15292 RSC.
- D. Basavaiah, B. Devendar, D. V. Lenin and T. Satyanarayana, Synlett, 2009, 411 CrossRef CAS.
- J. Barluenga, G. Lonzi, L. Riesgo, L. A. López and M. Tomás, J. Am. Chem. Soc., 2010, 132, 13200 CrossRef CAS PubMed.
- (a) J. Liu, L. Zhou, W. Ye and C. Wang, Chem. Commun., 2014, 50, 9068 RSC; (b) C. Reichardt, Solvents and Solvent Effects in Organic Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 3rd edn, 2003 Search PubMed.
- D. Basavaiach and A. J. Rao, Chem. Commun., 2003, 604 RSC.
- E. Ciganek, Organic Reactions, ed. L. A. Paquette, Wiley, New York, 1997, vol. 51, p. 201 Search PubMed.
- D. Basavaiah, G. Veeraraghavaiah and S. S. Badsara, Org. Biomol. Chem., 2014, 12, 1551 CAS.
- B. Yan and Y. Liu, Org. Lett., 2007, 9, 4323 CrossRef CAS PubMed.
- Y. Bai, J. Zeng, J. Ma, B. K. Gorityala and X.-W. Liu, J. Comb. Chem., 2010, 12, 696 CrossRef CAS PubMed.
- S. S. Patil, S. V. Patil and V. D. Bobade, Synlett, 2011, 2379 CAS.
- (a) M. J. Albaladejo, F. Alonso and M. Yus, Chem. – Eur. J., 2013, 19, 5242 CrossRef CAS PubMed; (b) M. J. Albaladejo, F. Alonso and M. J. González-Soria, ACS Catal., 2015, 5, 3446 CrossRef CAS.
- U. C. Rajesh, G. Purohit and D. S. Rawat, ACS Sustainable Chem. Eng., 2015, 3, 2397 CrossRef CAS.
- S. U. Dighe, S. Hutait and S. Batra, ACS Comb. Sci., 2012, 14, 665 CrossRef CAS PubMed.
- Y. Liu, Z. Song and B. Yan, Org. Lett., 2007, 9, 409 CrossRef CAS PubMed.
- P. P. Lange, A. R. Bogdan and K. James, Adv. Synth. Catal., 2012, 354, 2373 CrossRef CAS.
- H. Kim, K. Lee, S. Kim and P. H. Lee, Chem. Commun., 2010, 46, 6341 RSC.
- Y. Yang, C. Xie, Y. Xie and Y. Zhang, Org. Lett., 2012, 14, 957 CrossRef CAS PubMed.
- H. Zhu, N. Shao, T. Chen and H. Zou, Chem. Comunun., 2013, 49, 7738 RSC.
- H. Shiraishi, T. Nishitani, S. Sakaguchi and Y. Ishii, J. Org. Chem., 1998, 63, 6234 CrossRef CAS PubMed.
- C. Feng, Y. Yan, Z. Zhang, K. Xu and Z. Wang, Org. Biomol. Chem., 2014, 12, 4837 CAS.
- M. Gao, J. Tian and A. Lei, Chem. – Asian J., 2014, 9, 2068 CrossRef CAS PubMed.
- L. Xiang, Y. Yang, X. Zhou, X. Liu, X. Li, X. Kang, R. Yan and G. Huang, J. Org. Chem., 2014, 79, 10641 CrossRef CAS PubMed.
- L. Xiang, F. Zhang, B. Chen, X. Pang, X. Yang, G. Huang and R. Yan, RSC Adv., 2015, 5, 29424 RSC.
- S. Tang, K. Liu, Y. Long, X. Gao, M. Gao and A. Lei, Org. Lett., 2015, 17, 2404 CrossRef CAS PubMed.
- B. Sahoo, M. N. Hopkinson and F. Glorius, Angew. Chem., Int. Ed., 2015, 54, 15545 CrossRef CAS PubMed.
- X. Wang, S.-Y. Li, Y.-M. Pan, H.-S. Wang, H. Liang, Z.-F. Chen and X.-H. Qin, Org. Lett., 2014, 16, 580 CrossRef CAS PubMed.
- A. N. Pandya, J. T. Fletcher, E. M. Villa and D. K. Agrawal, Tetrahedron Lett., 2014, 55, 6922 CrossRef CAS PubMed.
- X.-C. Tan, Y. Liang, F.-P. Bao, H.-S. Wang and Y.-M. Pan, Tetrahedron, 2014, 70, 6717 CrossRef CAS.
- S. Tang, K. Liu, Y. Long, X. Qi, Y. Lan and A. Lei, Chem. Commun., 2015, 51, 8769 RSC.
- S. Adachi, S. K. Liew, C. F. Lee, A. Lough, Z. He, J. D. St. Denis, G. Poda and A. K. Yudin, Org. Lett., 2015, 17, 5594 CrossRef CAS PubMed.
- K. Bedjeguelal, H. Bienaymé, S. Poigny, P. Schmitt and E. Tan, QSAR Comb. Sci., 2006, 25, 504 CAS.
- O. Niyomura, Y. Yamaguchi, R. Sakao, M. Minoura and Y. Okamoto, Heterocycles, 2008, 75, 297 CrossRef CAS.
- J. Zhou, Y. Hu and H. Hu, Synthesis, 1999, 166 CrossRef CAS.
- D. Coffinier, L. El Kaim and L. Grimaud, Synlett, 2010, 2474 CrossRef CAS.
- J. Brioche, C. Meyer and J. Cossy, Org. Lett., 2015, 17, 2800 CrossRef CAS PubMed.
- F. Li, J. Chen, Y. Hou, Y. Li, X.-Y. Wu and X. Tong, Org. Lett., 2015, 17, 5376 CrossRef CAS PubMed.
- E. Kim, M. Koh, J. Ryu and S. B. Park, J. Am. Chem. Soc., 2008, 130, 12206 CrossRef CAS PubMed.
- (a) E. Kim, M. Koh, B. J. Lim and S. B. Park, J. Am. Chem. Soc., 2011, 133, 6642 CrossRef CAS PubMed; (b) E. Kim, Y. Lee, S. Lee and S. B. Park, Acc. Chem. Res., 2015, 48, 538 CrossRef CAS PubMed , and references therein.
- I. V. Seregin and V. Gevorgyan, J. Am. Chem. Soc., 2006, 128, 12050 CrossRef CAS PubMed.
- Y. Xia, A. S. Dudnik, Y. Li and V. Gevorgyan, Org. Lett., 2010, 12, 5538 CrossRef CAS PubMed.
- (a) C. R. Smith, E. M. Bunnelle, A. J. Rhodes and R. Sarpong, Org. Lett., 2007, 9, 1169 CrossRef CAS PubMed; (b) E. M. Bunnelle, C. R. Smith, S. K. Lee, S. W. Singaram, A. J. Rhodes and R. Sarpong, Tetrahedron, 2008, 64, 7008 CrossRef CAS PubMed.
- B. Yan, Y. Zhou, H. Zhang, J. Chen and Y. Liu, J. Org. Chem., 2007, 72, 7783 CrossRef CAS PubMed.
- (a) I. V. Seregin, A. W. Schammel and V. Gevorgyan, Org. Lett., 2007, 9, 3433 CrossRef CAS PubMed; (b) I. V. Seregin, A. W. Schammel and V. Gevorgyan, Tetrahedron, 2008, 64, 6876 CrossRef CAS PubMed.
- A. R. H. Narayan and R. Sarpong, Green Chem., 2010, 12, 1556 RSC.
- X. Meng, P. Liao, J. Liu and X. Bi, Chem. Commun., 2014, 50, 11837 RSC.
- D. Chernyak, C. Skontos and V. Gevorgyan, Org. Lett., 2010, 12, 3242 CrossRef CAS PubMed.
- Z. Li, D. Chernyak and V. Gevorgyan, Org. Lett., 2012, 14, 6056 CrossRef CAS PubMed.
- T. Xu and H. Alper, Org. Lett., 2015, 17, 4526 CrossRef CAS PubMed.
- D. Chernyak and V. Gevorgyan, Org. Lett., 2010, 12, 5558 CrossRef CAS PubMed.
- A. R. Hardin and R. Sarpong, Org. Lett., 2007, 9, 4547 CrossRef CAS PubMed.
- D. Chernyak, S. B. Gadamsetty and V. Gevorgyan, Org. Lett., 2008, 10, 2307 CrossRef CAS PubMed.
- L. Zhang, X. Li, Y. Liu and D. Zhang, Chem. Commun., 2015, 51, 6633 RSC.
- D. Wang, Y. Wei and M. Shi, Asian J. Org. Chem., 2013, 2, 480 CrossRef CAS.
- J. Wu, W. L. Leng, H. Liao, K. L. M. Hoang and X.-W. Liu, Chem. – Asian J., 2015, 10, 853 CrossRef CAS PubMed.
- R.-R. Liu, Z.-Y. Cai, C.-J. Lu, S.-C. Ye, B. Xiang, J. Gao and Y.-X. Jia, Org. Chem. Front., 2015, 2, 226 RSC.
- R.-R. Liu, C.-J. Lu, M.-D. Zhang, J.-R. Gao and Y.-X. Jia, Chem. – Eur. J., 2015, 21, 7057 CrossRef CAS PubMed.
- R.-R. Liu, S.-C. Ye, C.-J. Lu, B. Xiang, J. Gao and Y.-X. Jia, Org. Biomol. Chem., 2015, 13, 4855 CAS.
- I. Kim, J. Choi, H. K. Won and G. H. Lee, Tetrahedron Lett., 2007, 48, 6863 CrossRef CAS.
- I. Kim, H. K. Won, J. Choi and G. H. Lee, Tetrahedron, 2007, 63, 12954 CrossRef CAS.
- (a) I. Kim, S. G. Kim, Ji Y. Kim and G. H. Lee, Tetrahedron Lett., 2007, 48, 8976 CrossRef CAS; (b) Y. Jung and I. Kim, Tetrahedron, 2012, 68, 8198 CrossRef CAS.
- S. Chuprakov, F. W. Hwang and V. Gevorgyan, Angew. Chem., Int. Ed., 2007, 46, 4757 CrossRef CAS PubMed.
- S. Chuprakov and V. Gevorgyan, Org. Lett., 2007, 9, 4463 CrossRef CAS PubMed.
- V. Helan, A. V. Gulevich and V. Gevorgyan, Chem. Sci., 2015, 6, 1928 RSC.
- Y. Shi and V. Gevorgyan, Chem. Commun., 2015, 51, 17166 RSC.
- Y.-Q. Ge, J. Jia, H. Yang, G.-L. Zhao, F.-X. Zhan and J. Wang, Heterocycles, 2009, 78, 725 CrossRef CAS.
- B. Yang, Z. Huang, H. Guan, X. Niu, Y. Li, S. Fang and C. Ma, Tetrahedron Lett., 2013, 54, 5994 CrossRef CAS.
- Y.-F. Xie, Y.-Q. Ge, L. Feng, H.-Q. Xu, S. Meng, G.-L. Zhao, W.-R. Xu, J. Jia and J.-W. Wang, Heterocycles, 2013, 87, 815 CrossRef CAS.
- Y. Q. Ge, X. Y. Gong, G. J. Song, X. Q. Cao and J. W. Wang, Spectrochim. Acta, Part A, 2015, 135, 7 CrossRef CAS PubMed.
- S. Park and I. Kim, Tetrahedron, 2015, 71, 1982 CrossRef CAS.
- (a) C.-R. Yu, L.-H. Xu, S. Tu, Z.-N. Li and B. Li, J. Fluorine Chem., 2006, 127, 1540 CrossRef CAS; (b) R. Saxena, V. Singh and S. Batra, Tetrahedron, 2004, 60, 10311 CrossRef CAS.
- V. K. Outlaw, F. B. d'Andrea and C. A. Townsend, Org. Lett., 2015, 17, 1822 CrossRef CAS PubMed.
- W. Hao, H. Wang, Q. Ye, W.-X. Zhang and Z. Xi, Org. Lett., 2015, 17, 5674 CrossRef CAS PubMed.
- S. G. Dawande, B. S. Lad, S. Prajapati and S. Katukojvala, Org. Biomol. Chem., 2016, 14, 5569–5573 CAS.
- M. Kim, Y. Jung and I. Kim, J. Org. Chem., 2013, 78, 10395 CrossRef CAS PubMed.
- M. Kucukdisli and T. Opatz, J. Org. Chem., 2013, 78, 6670 CrossRef CAS PubMed.
- J. H. Lee and I. Kim, J. Org. Chem., 2013, 78, 1283 CrossRef CAS PubMed.
- M. Nagaraj, M. Boominathan, S. Muthusubramanian and N. Bhuvanesh, Synlett, 2012, 1353 CAS.
- M. Nayak and I. Kim, Org. Biomol. Chem., 2015, 13, 9697 CAS.
- T. Otani, T. Saito, R. Sakamoto, H. Osada, A. Hirahara, N. Furukawa, N. Kutsumura, T. Matsuo and K. Tamao, Chem. Commun., 2013, 49, 6206 RSC.
- Y. Jung and I. Kim, Org. Lett., 2015, 17, 4600 CrossRef CAS PubMed.
- Y. Jung and I. Kim, Asian J. Org. Chem., 2016, 5, 147 CrossRef CAS.
- (a) H. Hu, G. Li, W. Hu, Y. Liu, X. Wang, Y. Kan and M. Ji, Org. Lett., 2015, 17, 1114 CrossRef CAS PubMed; (b) N. Chernyak, D. Tilly, Z. Li and V. Gevorgyan, ARKIVOC, 2011, 76 CAS; (c) S. K. Ghosh, B.-C. Kuo, H.-Y. Chen, J.-Y. Li, S.-D. Liu and H. M. Lee, Eur. J. Org. Chem., 2015, 4131 CrossRef CAS.
- D.-T. Yang, J. Radtke, S. K. Mellerup, K. Yuan, X. Wang, M. Wagner and S. Wang, Org. Lett., 2015, 17, 2486 CrossRef CAS PubMed.
- T. Mitsumori, M. Bendikov, O. Dautel, F. Wudl, T. Shioya, H. Sato and Y. Sato, J. Am. Chem. Soc., 2004, 126, 16793 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |