
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of trifluoromethylated isoxazoles and their elaboration through inter- and intra-molecular C–H arylation†
Jian-Siang
Poh
a,
Cristina
García-Ruiz
a,
Andrea
Zúñiga
a,
Francesca
Meroni
a,
David C.
Blakemore
b,
Duncan L.
Browne
*ac and
Steven V.
Ley
a
aDepartment of Chemistry, University of Cambridge, Lensfield Road, Cambridge, CB2 1EW, UK. E-mail: dlbrowne@cardiff.ac.uk
bPfizer Worldwide Medicinal Chemistry, The Portway Building, Granta Park, Cambridge, CB21 6GS, UK
cSchool of Chemistry, Cardiff University, Main Building, Park Place, Cardiff, CF10 3AT, UK
First published on 20th May 2016
Abstract
We report conditions for the preparation of a range of trifluoromethylated isoxazole building blocks through the cycloaddition reaction of trifluoromethyl nitrile oxide. It was found that controlling the rate (and therefore concentration) of the formation of the trifluoromethyl nitrile oxide was Critical for the preferential formation of the desired isoxazole products versus the furoxan dimer. Different conditions were optimised for both aryl- and alkyl-substituted alkynes. In addition, the reactivity at the isoxazole 4-position has been briefly explored for these building blocks. Conditions for intermolecular C–H arylation, lithiation and electrophile quench, and alkoxylation were all identified with brief substrate scoping that signifies useful tolerance to a range of functionalities. Finally, complementary processes for structural diversification through either intramolecular cyclisation or intermolecular cross-coupling were developed.
Introduction
The addition of fluorine to organic molecules does a great deal to alter chemical properties and behavior.1 For example, in biologically active materials fluorine substituents can affect the charge distribution, electrostatic surface and solubility of chemical entities, often leading to positive outcomes.2 Perhaps the most attractive property however is the ability to reduce the rate of metabolism compared to non-fluorous congeners, which ultimately leads to reduced dosing rates for both patients and crops.3 Furthermore, in the area of materials chemistry fluorous substituents can impart advantages such as reduced band gaps and higher quantum efficiencies in OLED devices.4There are typically two strategic methods for introducing fluorous substituents. The late stage introduction, which has received much attention recently,5 is an important method, particularly for taking advantage of the radioactive properties of 18F labelled materials.6 Furthermore, with regards to trifluoromethylation, this doctrine serves as a convenient method to easily functionalise existing compound libraries, usually through a C–H, C–O, C–B, C–Sn or C–X bond transformation and more recently that of a C–N bond.7 Alternatively, a strategy involving the preparation of novel fluorinated building blocks which are then incorporated in a convergent manner (a fluorous synthon approach) is also of importance, particularly when it comes to scaling up the preparation of the compound of interest.8 Common to both strategic approaches is the fact that fluorine is not present in many naturally occurring organic compounds and so any reagents or building blocks must be derived from fluorine gas, or other mineral derived fluorines (such as HF, SF4, SF6 or HOF) thus requiring both highly skilled workers and specialised equipment and safety protocols.
Recently, we and others have been exploring cycloaddition methods to rapidly access a series of fluorinated intermediates, from simple fluorous synthon starting materials, that can further undergo a range of diverse modifications.9 Here we report a robust protocol for the preparation and cycloaddition of trifluoroacetonitrile oxide with alkynes, followed by diversification via palladium-catalysed inter- and intra-molecular C–H activation reactions. We have also assessed the further derivatisation of these systems via anion and cycloaddition chemistry.
Results and discussion
Our approach to the preparation of 3-trifluoromethyl isoxazoles hinged on a nitrile-oxide [3 + 2] cycloaddition reaction with alkyne substrates. Few examples of this approach are known. In order to synthesize the desired 5-substituted 3-trifluoromethylated isoxazoles via [3 + 2] cycloaddition reactions, a stable nitrile oxide precursor for trifluoroacetonitrile oxide was required. We decided to use hydroximoyl bromide 3, prepared using a two-step procedure (Scheme 1) from the commercially available trifluoromethylated hemiacetal 1.10 Owing to the volatility of both intermediate aldoxime 2 and hydroximoyl bromide 3, these materials were extracted into an ethereal solution and then stored and used from this stock in subsequent steps.11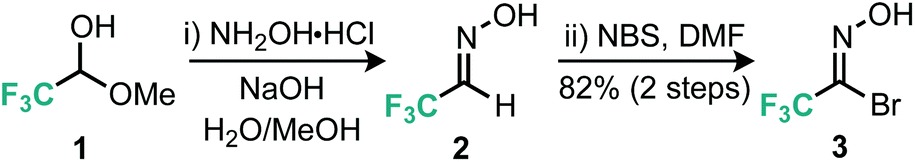 | ||
| Scheme 1 Preparation of CF3 nitrile oxide precursor. | ||
With the desired trifluoromethyl nitrile oxide precursor in hand, we then focused on the [3 + 2] cycloaddition reaction with terminal alkynes. Early attempts with the cycloaddition reaction under similar conditions reported by Tanaka et al. with a small variety of alkynes revealed that the yield of isoxazole was rather dependent on the electronic properties of the alkyne (Table 1).12 In general, more electron-deficient alkynes (in particular, alkyl alkynes) appeared to undergo slower cycloaddition with trifluoroacetonitrile oxide. Analysis of the 19F NMR spectra of the crude reaction mixture for some alkyl alkynes (Scheme 2) indicated the presence of furoxan resulting from the unwanted dimerization reaction of trifluoroacetonitrile oxide.13
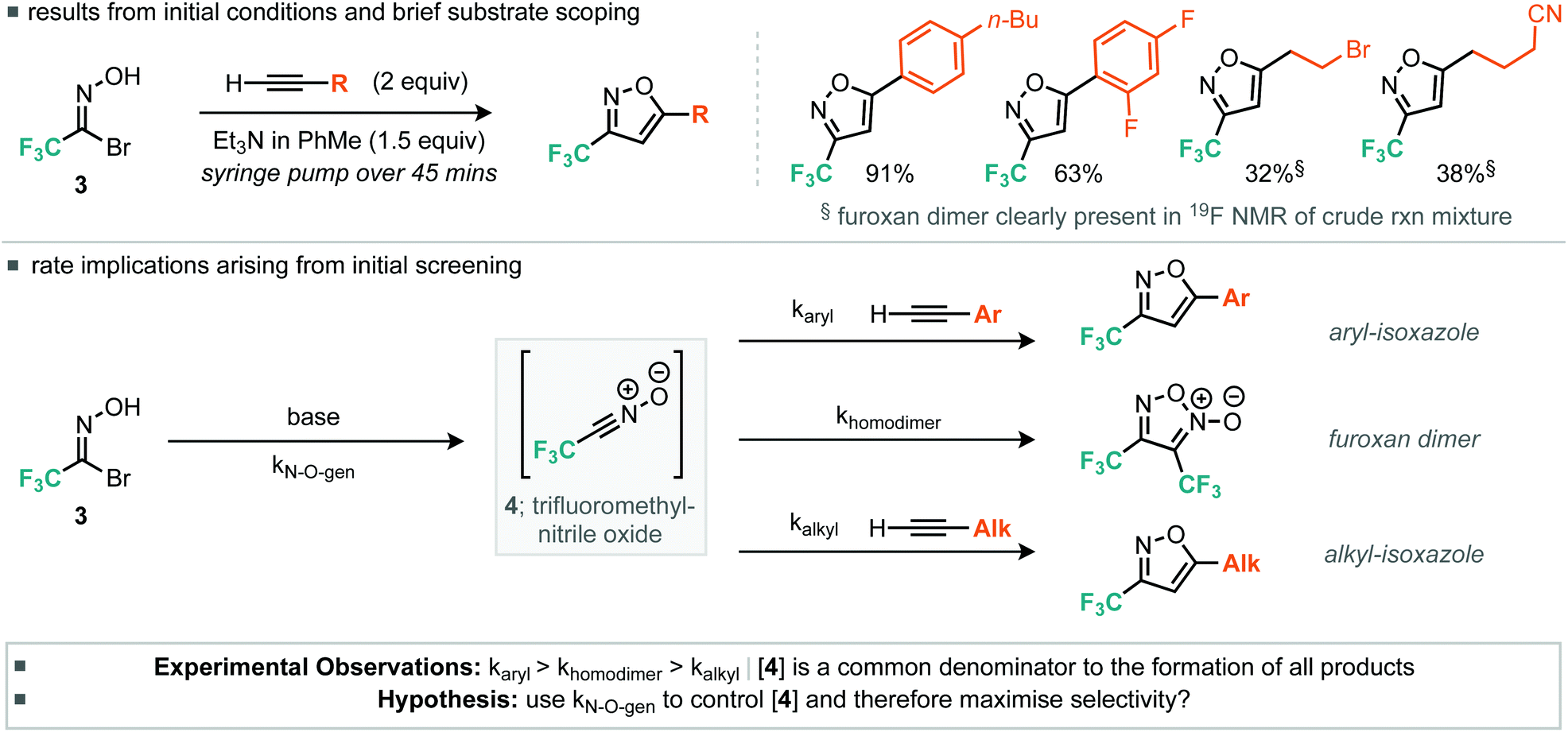 | ||
| Scheme 2 Initial cycloaddition experiments and considerations. | ||
|
|
||||
|---|---|---|---|---|
| Entry | Alkyne equiv. (X) | Addition rate (Y) | Base/solvent | Yielda |
| a Yield of isolated product. | ||||
| 1 | 1.0 | 45 | Et3N/PhMe | 70% |
| 2 | 2.0 | 45 | Et3N/PhMe | 90% |
| 3 | 3.8 | 45 | Et3N/PhMe | 93% |
| 4 | 2.0 | 45 | iPr2NEt/PhMe | 69% |
| 5 | 2.0 | 45 | Pyridine/PhMe | 9% |
| 6 | 2.0 | 45 | 2,6-Lutidine/PhMe | 5% |
| 7 | 2.0 | 45 | TBAF/THF | 2% |
| 8 | 2.0 | 45 | Na2CO3/H2O | 82% |
| 9 | 2.0 | 5 | Et3N/PhMe | 81% |
| 10 | 2.0 | 120 | Et 3 N/PhMe | 96% |
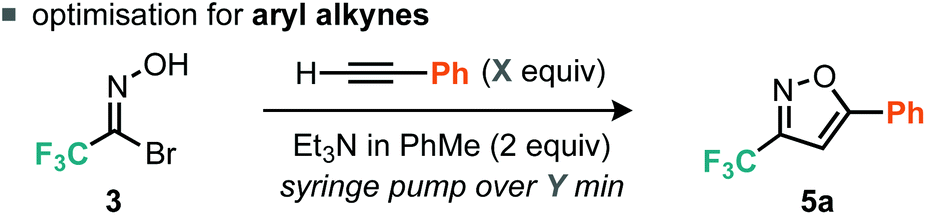
We considered the rates of the reactions explored in the context of these experimental observations. Empirically, the preliminary evidence suggests that the rate of nitrile oxide cycloaddition with aryl alkynes is greater or comparable to that of the dimerization of the CF3 nitrile oxide, whereas the rate of cycloaddition between the nitrile oxide and alkyl alkynes appears to be slower than the rate of furoxan formation. This suggested that control could be obtained by reducing the concentration of nitrile oxide relative to alkyne. Such control could potentially be achieved by invoking pseudo first-order conditions in nitrile oxide. This would be achieved by using a syringe pump to deliver either the base or the nitrile oxide precursor slowly to a flask containing all of the alkyne. With this in mind we therefore optimized procedures for aryl and alkyl alkynes separately, with particular attention to methods that limit formation of the furoxan. For the optimization studies of aryl alkyne cycloadditions, we used phenylacetylene as the substrate. Firstly, the number of equivalents of alkyne was assessed.
With 1.1 equiv. of alkyne and dropwise addition via syringe pump of 2 equiv. Et3N/PhMe over 45 min at r.t. (Table 1, entry 1), the corresponding isoxazole 5a was isolated in 70% yield. Analysis of the crude reaction mixture by 19F NMR spectroscopy indicated the presence of furoxan. Increasing the alkyne equivalents to 2.0 and 3.8 gave yields of 90% and 93% respectively (Table 1, entries 2 and 3), indicating that an excess of alkyne relative to the nitrile oxide would be advantageous to suppressing furoxan formation. We decided to use 2.0 equiv. of alkyne for subsequent reactions as it appeared that greater excesses of alkyne provided diminishing returns in terms of yield of desired isoxazole product. When a variety of bases were assessed only Et3N, iPr2NEt and Na2CO3 provided good yields of isoxazole 5a (Table 1, entries 3, 4 and 8), whereas weaker bases such as pyridine, 2,6-lutidine and TBAF produced negligible yields of 5a (Table 1, entries 5–7). Finally, the rate of base addition was assessed; when the time taken for addition of Et3N was decreased to 5 min, isoxazole 5a was isolated in 81% yield (Table 1, entry 9). However, when the time taken was increased to 2 h, isoxazole 5a was isolated in an improved yield of 96% (Table 1, entry 10).
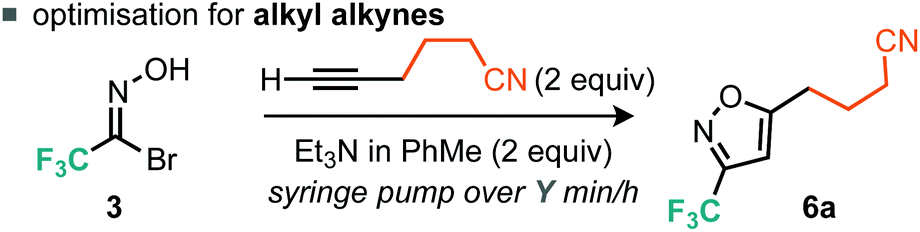
Evidently, using a slower rate of base addition to controllably unveil small amounts of nitrile oxide could further suppress the unwanted dimerisation reaction, since the relative concentration of the nitrile oxide compared to the alkyne was low. We then focused our attention on alkyl alkynes using 5-hexynenitrile as the substrate. Since it appeared that suppression of furoxan formation was required to obtain good yields of isoxazole, it was postulated that increasing the time taken for base addition would be a simple modification to the optimized procedure for aryl alkynes. Indeed, when the time taken for base addition was progressively increased to 16 h (Table 2, entries 1–3), higher yields of isoxazole 6a were obtained; further increasing the time taken to 24 h (Table 2, entry 4) had little effect on yield.14 With optimized conditions for the cycloaddition reaction with aryl and alkyl alkynes established, we decided to explore the scope of the reaction.
For the aryl alkyne examples explored, both electron-rich and electron-poor systems underwent cycloaddition, with the latter proceeding in lower yield, perhaps due to a mismatch in the electronics of the dipolarophile and 1,3-dipole. With regard to the alkyl alkynes a variety of functional groups were tolerated such as alkyl bromides, free alcohols, cyclopropanes, nitriles and ketones (Scheme 3). In addition, we explored some more exotic alkyl alkynes derived from nucleophilic aromatic substitution of heteroaromatic halides with propargyl alcohol. Of the three explored, all proceeded to the desired product in yields greater than 50% (Scheme 4, 6i, 6j and 6k). For all of the explored substrates the cycloaddition reaction showed exclusive regioselectivity for the formation of 5-substituted isoxazoles, an observation congruent with the literature.15 Several examples were also conducted on larger scale and afforded greater than 2.5 grams of product in comparable or greater yields to the smaller scale reactions (5a, 6d, 6e and 6f).
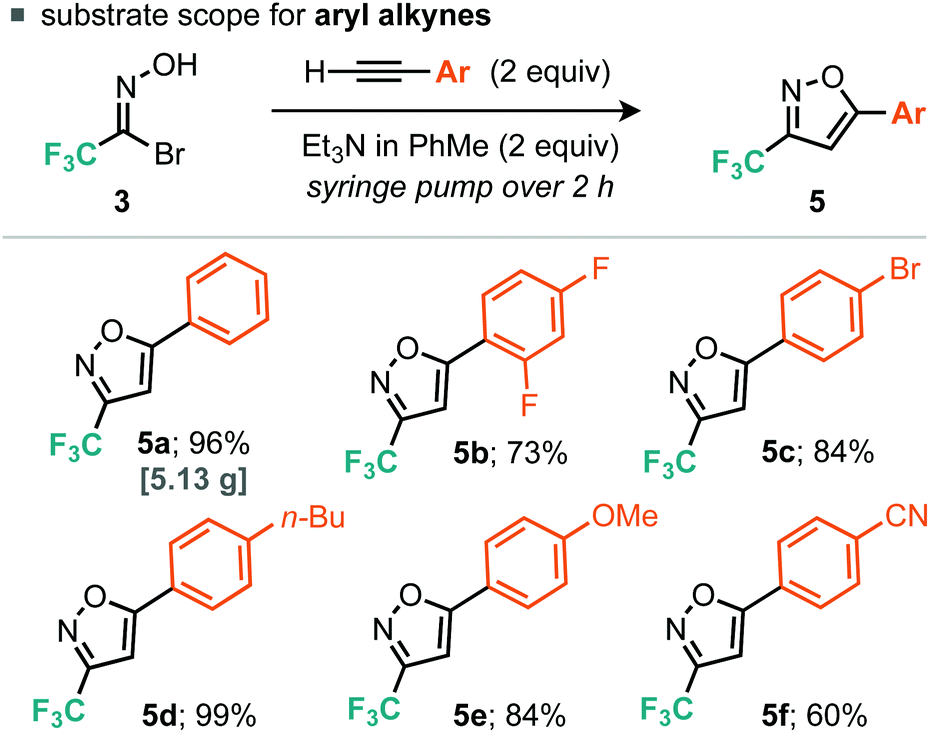 | ||
| Scheme 3 Scope of the aryl alkyne cycloaddition. | ||
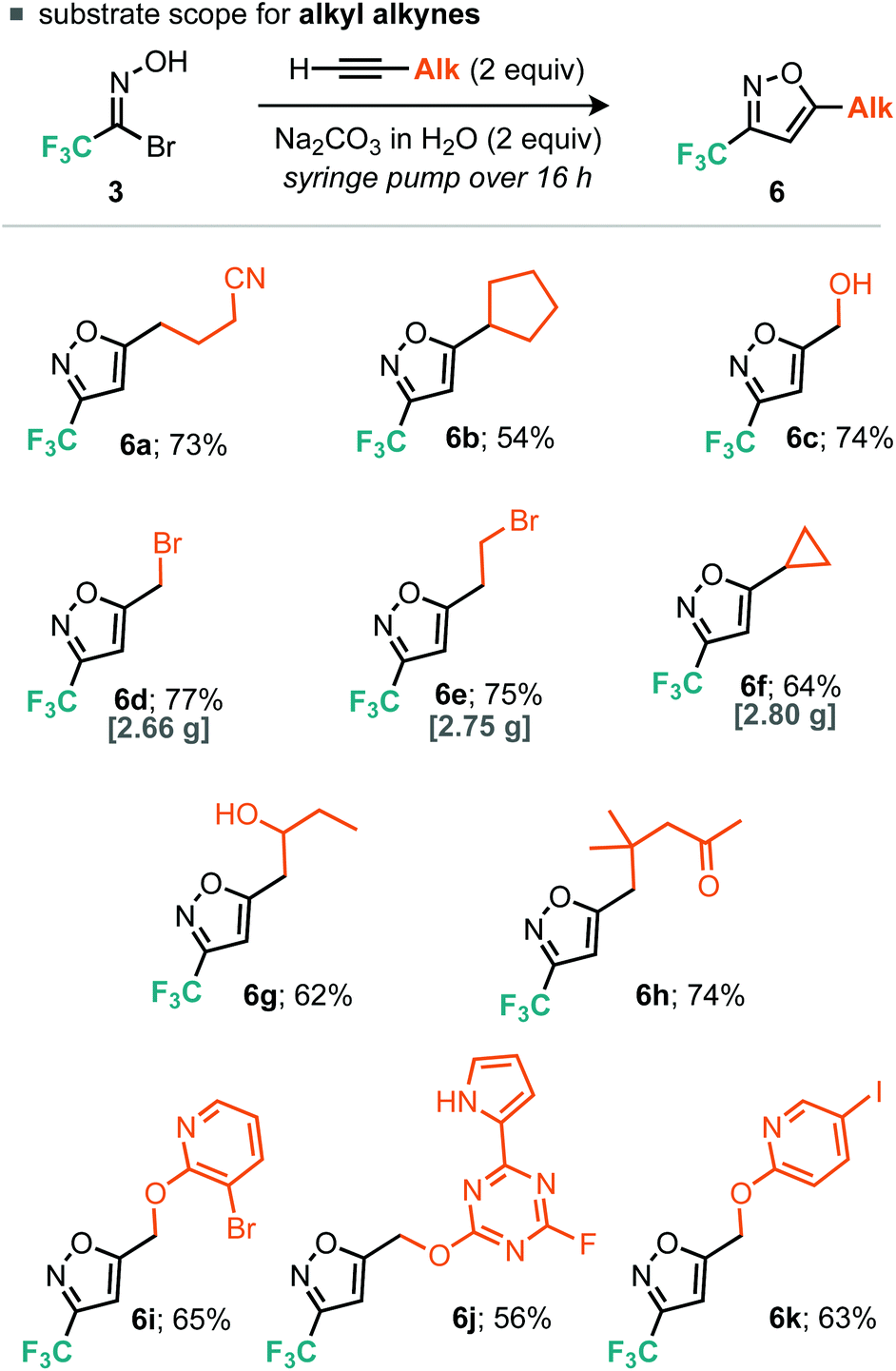 | ||
| Scheme 4 Scope of the alkyl alkyne cycloaddition. | ||
Having demonstrated a broad substrate scope we next wished to explore the possibility of further manipulation of these 5-substituted 3-trifluoromethylisoxazoles by the introduction of functionality at the 4-position. Palladium-catalyzed C–H activation of isoxazoles has been demonstrated by Fall et al. on 3,5-dimethylisoxazole and 3-methyl-5-phenylisoxazole, but they note that electron-deficient isoxazoles were coupled much less efficiently.16 Indeed, the intermolecular cross-coupling of 4-bromotoluene and 5-phenyl-3-trifluoromethylisoxazole (5a) resulted in only 38% conversion to the desired 4-arylated isoxazole (7a), despite use of 5 mol% PdCl2 and heating for 72 h. However, switching to 2 equiv. aryl bromide allowed the desired product 7a to be isolated in 85% yield. Alternative aryl bromide coupling partners for 5-phenyl-3-trifluoromethylisoxazole (5a), such as electron-rich, electron-deficient and heterocyclic bromides, were assessed (Scheme 5) and gave moderate to excellent yields. Further, 5-cyclopropyl-3-trifluoromethylisoxazole (5g) could also be coupled to 3-bromopyridine in 47% yield. Rather than undergoing cross-coupling of the C–H bond at the 4-position, lithiation at this position was also demonstrated using nBuLi.
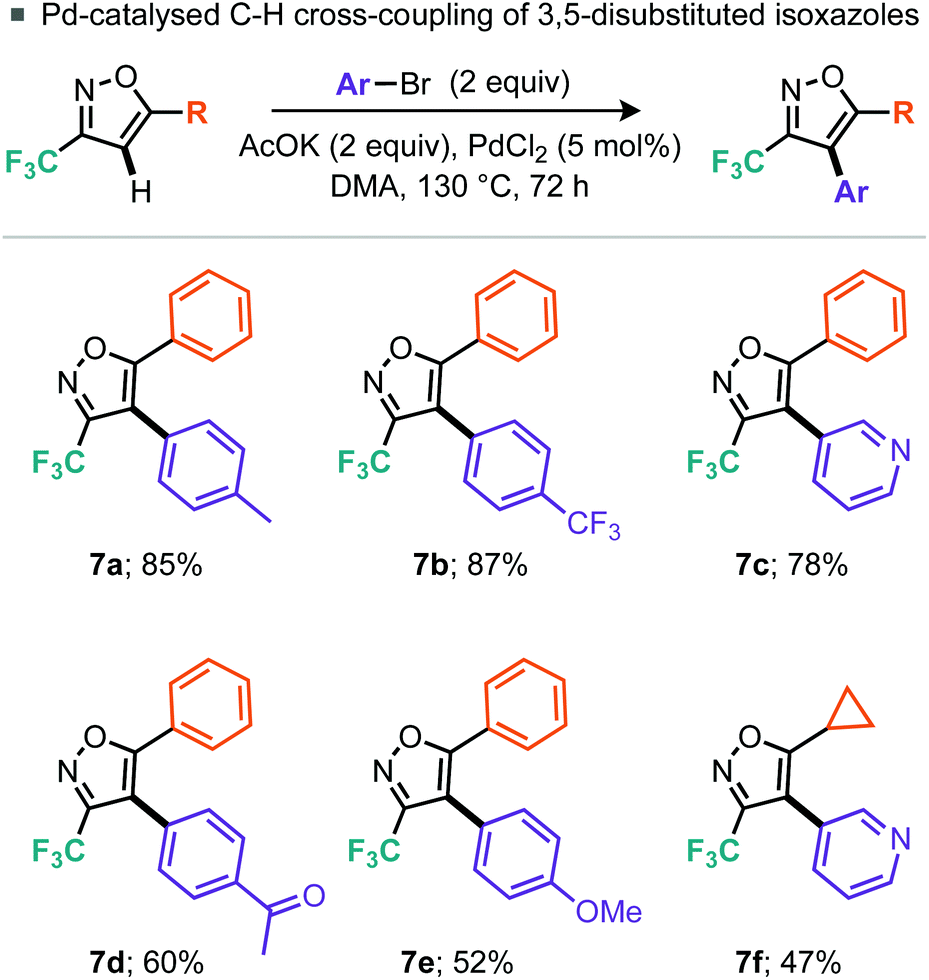 | ||
| Scheme 5 Intermolecular C–H arylation. | ||
Again for the 5-phenyl-3-trifluoromethylisoxazole (5a) substrate, lithiation was straightforward (Scheme 6). Quenching of the thus formed organometallic species with DMF led to the corresponding aldehyde whereas a boron electrophile (i-PrOBPin) could be used to afford the boronic ester product 8b in good yields. Notably, whilst the 5-cyclopropyl-3-trifluoromethylisoxazole (5g) also participated in this chemistry; the aldehyde product 8c was isolated in reduced yield compared to the phenyl analogue.
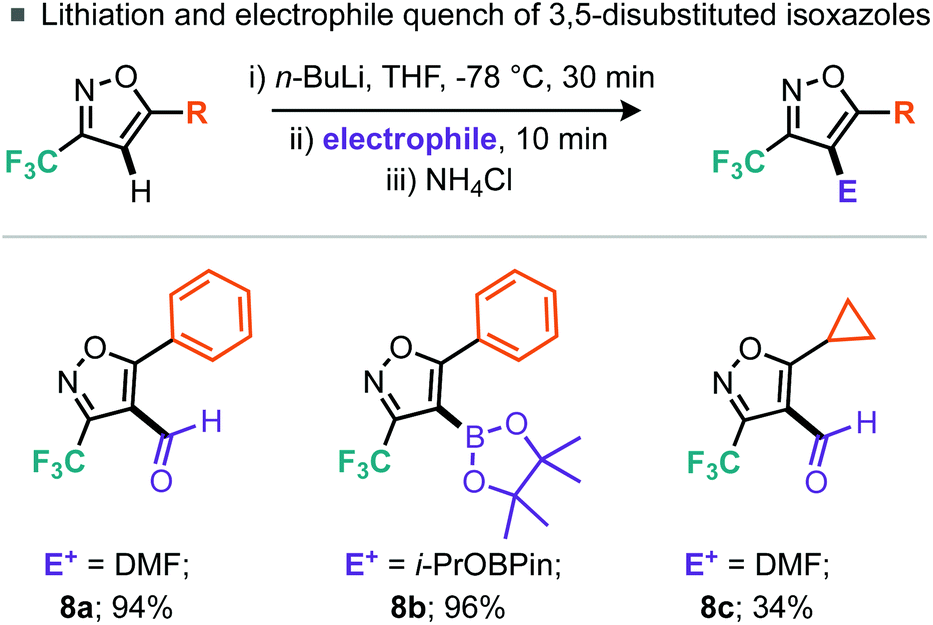 | ||
| Scheme 6 Lithiation and electrophilic quench. | ||
Other strategies to rapidly elaborate this ring system hinged around the versatile fluorinated, bromoalkyl building blocks 6d and 6e. Indeed, we found that homobenzylic bromide (6e) was an excellent surrogate for the 5-alkenyl-3-trifluoromethyl-isoxazole (9) through an elimination process. This alkene was most optimally accessed by stirring 6e in the presence of the polymer-supported base, Ambersep® OH resin, followed by filtration of the resin and storage of the resultant volatile alkene (9) in an ethereal solution. The reactivity of this versatile alkene building block (9) was then explored in the context of nitrile oxide cycloaddition chemistry (Scheme 7). In this instance it is well documented that the rate of alkene cycloaddition is greater than that of the nitrile oxide homo-dimerisation process. Indeed, the nitrile oxide derived from cinnamaldehyde underwent smooth cycloaddition to give the isoxazole–isoxazoline bicyclic moiety (10a). Electron-rich and electron-poor benzaldehyde derivatives also proceeded without incident affording the 4-methoxy and 4-trifluoromethyl variants 10b and 10c in excellent yield. The cycloaddition reaction of electron-poor alkene building block 9 with the electron-poor trifluoromethyl nitrile oxide (4) did also afford the desired isoxazole–isoxazoline bicyclic product, but in reduced yield (47%) relative to the other examples explored, again likely due to a mis-match in electronics.
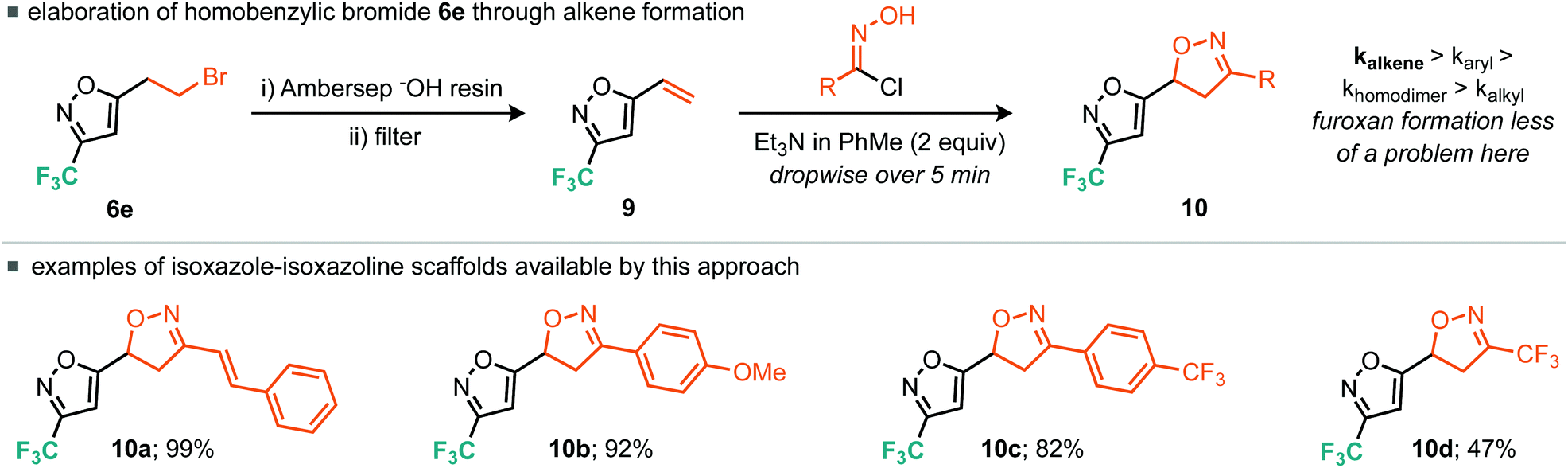 | ||
| Scheme 7 Elimination and nitrile oxide cyclisation. | ||
A strategy for derivatisation of the trifluoromethylated bromoalkyl building block 6d, is described in Scheme 8. The approach commences by using this material as an alkylating agent for an aromatic ring bearing a nucleophilic heteroatom and an adjacent halide function.
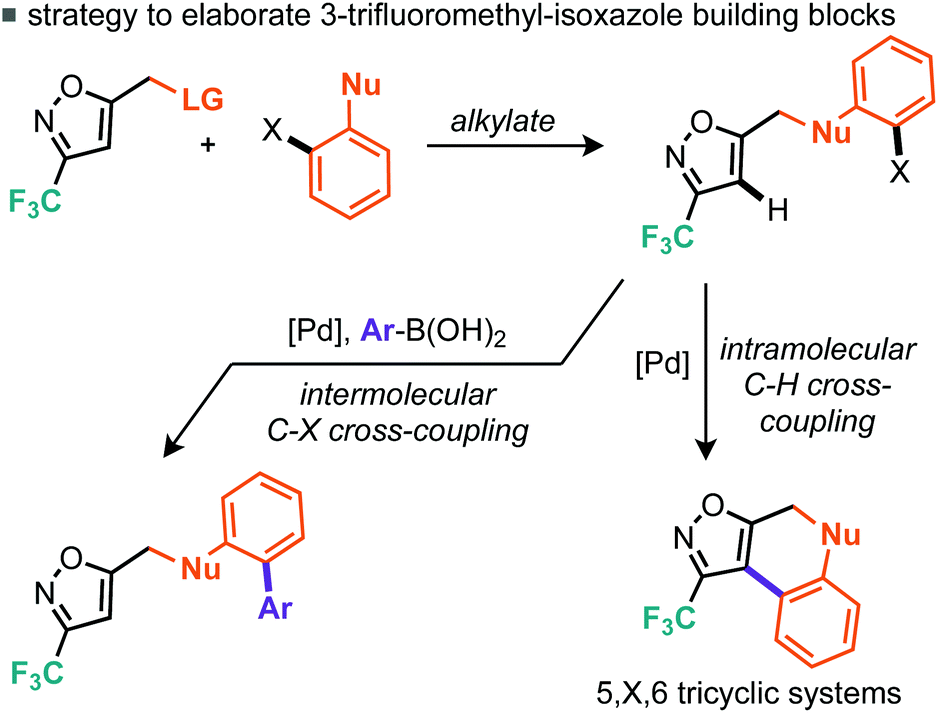 | ||
| Scheme 8 Proposed strategy for rapid structural diversification through complementary cross-coupling. | ||
The halide on these resulting products could then potentially undergo two different cross-coupling reactions leading to rapid structural diversification.17
One cross-coupling would be intermolecular and could likely be controlled by appropriate choice of base and inclusion of a coupling partner (boronic acid), whereas the other coupling would be an intramolecular C–H functionalizing ring formation leading to a novel series of trifluoromethylated tricyclic scaffolds. Indeed this approach also offers significant scope for varying linker size in the initial alkylation step and therefore the subsequent central ring size of the tricyclic system. Commencing with the alkylation of a variety of ortho-iodophenols with bromide 6d, we found that optimal conditions consisted of gentle heating to 60 °C in DMF using potassium carbonate as base (Scheme 9). Conditions for the alkylation of ortho-iodobenzyl alcohols were better conducted with sodium hydride in THF, which furnished several homologated products (Scheme 9). Next we turned to the intramolecular C–H arylation reaction and looked to further develop the cross-coupling conditions. Initially this was investigated on para-methoxyphenyl substrate 13. Repeating our previously successful conditions for the intermolecular case on this substrate afforded 12% isolated yield of the intramolecular product (14) but 98% conversion of the starting material. The main byproduct was that derived from deiodination (Table 3, entry 1). Exploring DMF and 1,4-dioxane as solvent options, it was found that in DMF the reaction afforded an improved 31% yield of the cyclised product (Table 3, entry 2). Replacement of the KOAc base with K2CO3 and Cs2CO3 did not lead to dramatic changes in yield. Taking inspiration from the literature on intramolecular C–H arylation reactions of benzene systems via proton abstraction, we made a more significant change in our approach.18
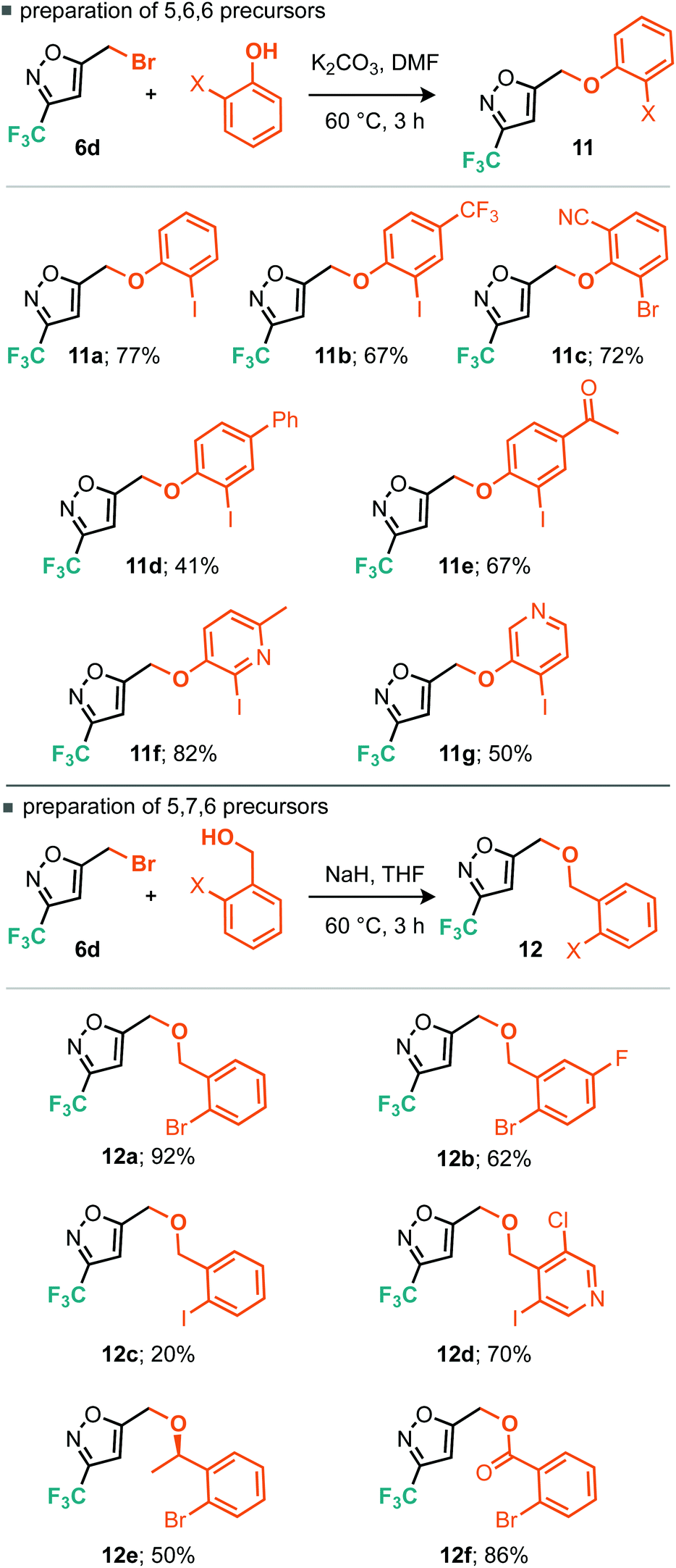 | ||
| Scheme 9 Conditions and scope for alkylation reaction. | ||
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Base | Solvent | Ligand | Additive | Time (h) | Conversion | Isol. yield | |
| a Starting material recovered. | ||||||||
| PdCl2 (5 mol%); base (2.0 equiv.); solvent, 130 °C | 1 | KOAc | DMA | — | — | 72 | 98 | 12 |
| 2 | DMF | — | — | 48 | 98 | 31 | ||
| 3 | Dioxane | — | — | 48 | —a | — | ||
| 4 | K2CO3 | DMA | — | — | 72 | —a | — | |
| 5 | DMF | — | — | 72 | — | 21 | ||
| 6 | Dioxane | — | — | 72 | —a | — | ||
| 7 | Cs2CO3 | DMA | — | — | 72 | —a | — | |
| 8 | DMF | — | — | 72 | — | 40 | ||
| 9 | Dioxane | — | — | 72 | —a | — | ||
| Pd(OAc)2 (5 mol%); base (4.0 equiv.); ligand, additive (2.0 equiv.); solvent, 100 °C | 10 | K2CO3 | DMF | PCy3 | TBAB | 24 | 98 | 57 |
| 11 | DMF | dppf | TBAB | 24 | 98 | 0 | ||
| 12 | DMA | PtBu3·HBF4 | TBAB | 24 | 98 | 42 | ||
| 13 | DMA | PCy3·HBF4 | TBAB | 24 | 98 | 36 | ||
| 14 | DMA | Xantphos | — | 24 | 98 | 52 | ||
| Pd(OAc)2 (5 mol%); base (4.0 equiv.); ligand, additive (0.3 equiv.); solvent, 65 °C | 15 | K 2 CO 3 | DMA | P(Ph)3 | TMBA | 16 | 98 | 47 |
| 16 | DMA | P(Ph)3 | PhCO2H | 16 | 98 | 38 | ||
| 17 | DMA | P(Ph)3 | PivOH | 16 | 98 | 51 | ||
| 18 | DMA | P(4-F-Ph) 3 | PivOH | 16 | 98 | 60 | ||
| 19 | DMA | P(4-Cl-Ph)3 | PivOH | 16 | 98 | 44 | ||
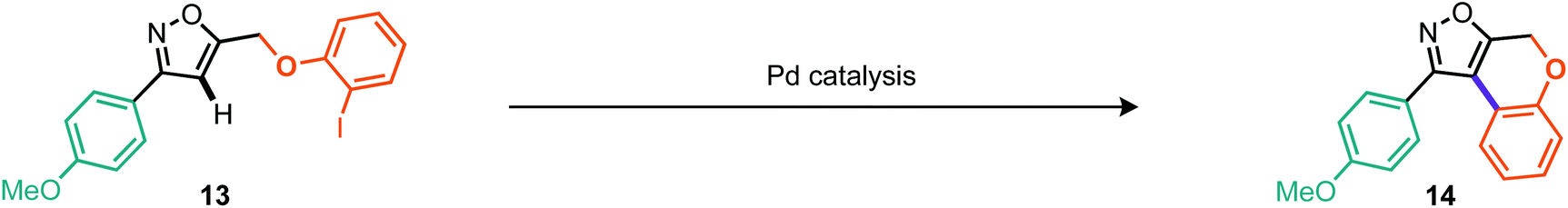
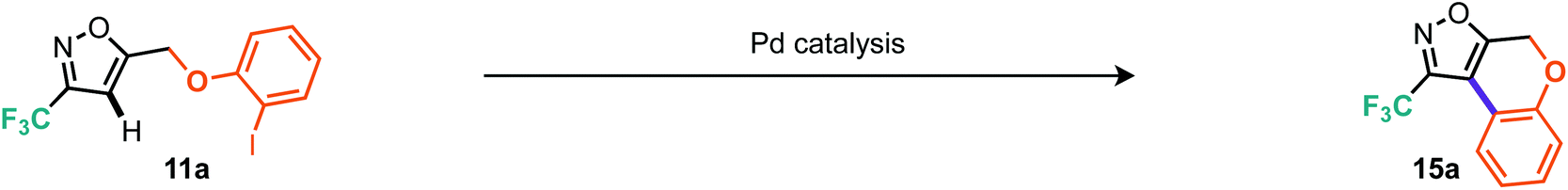
Table 3, entries 10–14 describe the results from the changed catalyst system, now including a ligand, additive and two further equivalents of base, whilst dropping the temperature by 30 °C and reaction time down to 1 day. Notably, in almost all cases the outcome was improved, with tricyclohexylphosphine ligand and tetrabutylammonium bromide as additive providing the superior result at 57% isolated yield. Indeed, we surveyed several other literature reports19 and found that the conditions reported by Fagnou et al. offered further improvements.19a Under these conditions, which included reduced loading of additive and use of triarylphosphines, we made gains on both reaction time and temperature where the optimal conditions consisted of heating to 65 °C over 16 hours (Table 3, entry 18). Notably, P(4-F-Ph)3 as ligand outperformed PPh3, providing a 10% improvement in isolated yield (Table 3, cf. entries 17 and 18). Applying these optimal conditions, as developed for the less reactive model substrate, to the 3-trifluoroisoxazole 11a we were pleased to find that the tricyclic product (15a) could be isolated in 90% yield (Table 4, entry 2). Control reactions showed that P(4-F-Ph)3 still provides notable benefit as does the presence of pivalic acid. With these optimal conditions, we then applied them to a range of the alkylated materials to successfully furnish a range of 5,6,6 tricyclic compounds including the pyridine derivatives 15c and 15f in moderate to excellent yields (Scheme 10). One example of a 5,7,6 tricycle (15h) was also prepared in 60% yield using these C–H arylation conditions.
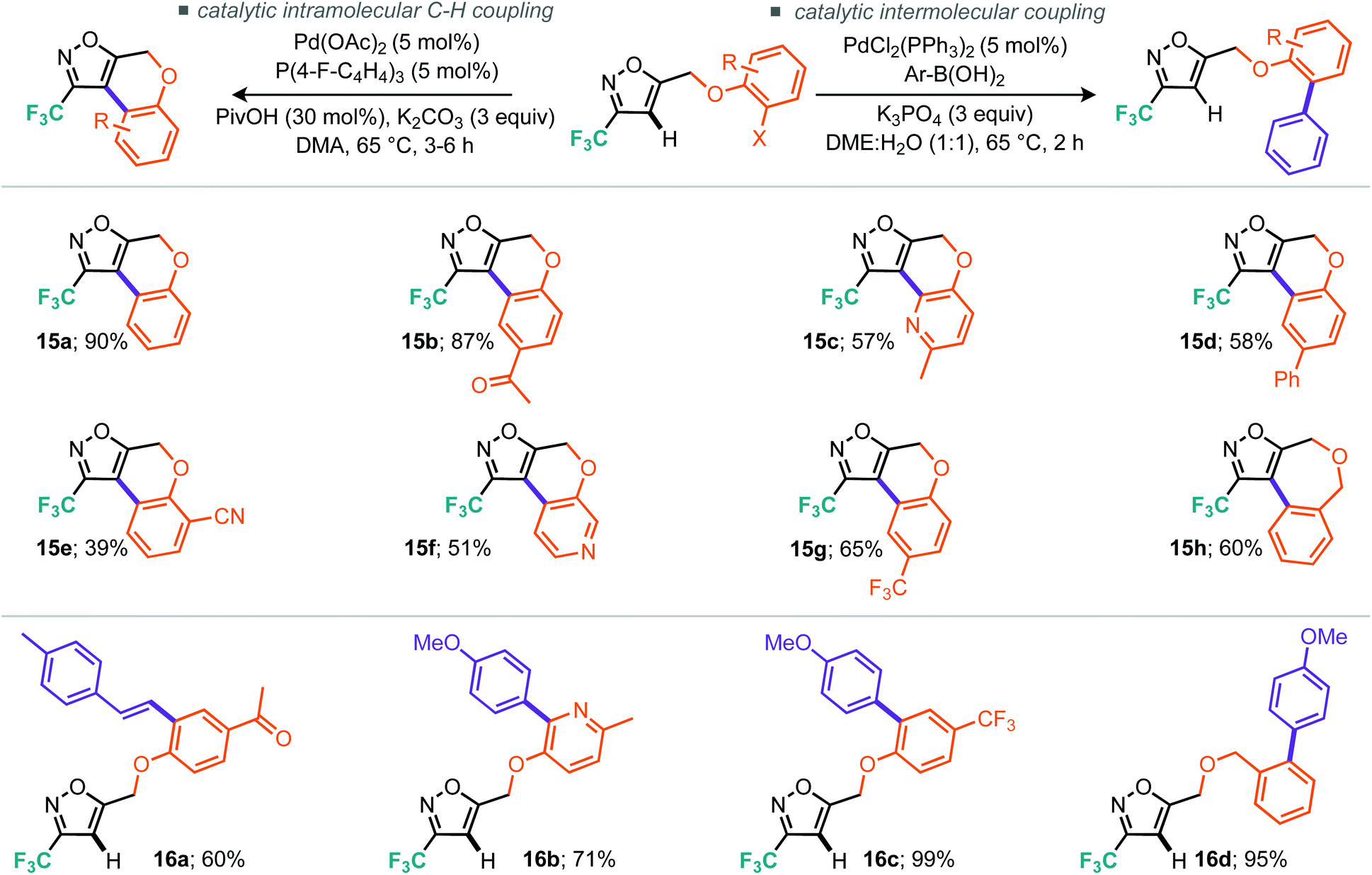 | ||
| Scheme 10 Conditions and scope for alkylation reaction. | ||
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Base | Solvent | Ligand | Additive | Time (h) | Conversion | Isol. yield | |
| Pd(OAc)2 (5 mol%); base (4.0 equiv.); ligand, additive (0.3 equiv.); solvent, 65 °C | 1 | K 2 CO 3 | DMA | P(Ph)3 | PivOH | 16 | 98 | 85 |
| 2 | DMA | P(4-F-Ph) 3 | PivOH | 16 | 98 | 90 | ||
| 3 | DMA | P(4-F-Ph)3 | — | 16 | 98 | 73 | ||
In order to demonstrate the diversity of this scaffold through cross-coupling we also found optimal conditions for the intermolecular Suzuki–Miyaura reaction. Inclusion of the requisite boronic acid and switching both the base and solvent system permitted smooth access to the linear products 16a–d in good to excellent yield. Notably in the latter cross-coupling cases, no intramolecular cyclised product was detected signalling that the reactivity and structural diversity can truly be switched by a simple change in reaction conditions.
Conclusions
In summary, we report conditions for the preparation of a range of trifluoromethylated isoxazole building blocks though trifluoromethyl nitrile oxide cycloaddition. It was found that controlling the rate (and therefore concentration) of the formation of the trifluoromethyl nitrile oxide was critical for the preferential formation of the desired isoxazole products versus the furoxan dimer. Different conditions were optimised for both aryl- and alkyl-substituted alkynes. In addition, the reactivity at the isoxazole 4-position was then briefly explored for these building blocks. Conditions for intermolecular C–H arylation, lithiation and electrophile quench, and alkoxylation were all identified with brief substrate scoping that signifies useful tolerance to a range of functionalities. Finally we developed complementary processes for structural diversification through either intramolecular cyclisation or intermolecular cross-coupling, with a series of 5,6,6 and 5,6,7 tricyclic systems being formed.Acknowledgements
We thank the Cambridge Home and European Scholarship Scheme (JSP) and the EPSRC (SVL grant numbers EP/K009494/1 and EP/M004120/1) for financial support.Notes and references
- For some overviews in the area of organofluorine chemistry see: (a) C. N. Neumann and T. Ritter, Angew. Chem., Int. Ed., 2015, 54, 3216 CrossRef CAS PubMed; (b) T. Liang, C. N. Neumann and T. Ritter, Angew. Chem., Int. Ed., 2013, 52, 8214 CrossRef CAS PubMed; (c) T. Furuya, C. A. Kuttruff and T. Ritter, Curr. Opin. Drug Discovery Dev., 2008, 11, 803 CAS; (d) J. Wang, M. Sánchez-Roselló, J. L. Aceña, C. del Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Chem. Rev., 2014, 114, 2432 CrossRef CAS PubMed; (e) J.-P. Bégué and D. Bonnet-Delpon, J. Fluorine Chem., 2006, 127, 992 CrossRef; (f) K. L. Kirk, Org. Process Res. Dev., 2008, 12, 305 CrossRef CAS; (g) S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320 RSC; (h) W. K. Hagmann, J. Med. Chem., 2008, 51, 4359 CrossRef CAS PubMed; (i) N. A. Meanwell, J. Med. Chem., 2011, 54, 2529 CrossRef CAS PubMed; (j) D. T. Wong, K. W. Perry and F. P. Bymaster, Nat. Rev. Drug Discovery, 2005, 4, 764 CAS; (k) X.-H. Xu, K. Matsuzaki and N. Shibata, Chem. Rev., 2015, 115, 731 CrossRef CAS PubMed; (l) P. Jeschke, ChemBioChem, 2004, 5, 570 CrossRef CAS PubMed; (m) K. Müller, C. Faeh and F. Diederich, Science, 2007, 317, 1881 CrossRef PubMed; (n) L. M. Yagupol'skii, A. Y. Il'chenko and N. V. Kondratenko, Russ. Chem. Rev., 1974, 43, 32 CrossRef; (o) D. L. Browne, Synlett, 2015, 33 CAS; (p) I. Hyohdoh, N. Furuichi, T. Aoki, Y. Itezono, H. Shirai, S. Ozawa, F. Watanabe, M. Matsushita, M. Sakaitani, P. S. Ho, K. Takanashi, N. Harada, Y. Tomii, K. Yoshinari, K. Ori, M. Tabo, Y. Aoki, N. Shimma and H. Iikura, Med. Chem. Lett., 2013, 4, 1059 CrossRef CAS PubMed; (q) J.-A. Ma and D. Cahard, Chem. Rev., 2004, 104, 6119 CrossRef CAS PubMed.
- (a) D. O’ Hagan, J. Fluorine Chem., 2010, 131, 1071 CrossRef; (b) M. Morgenthaler, E. Schweizer, A. Hoffmann-Röder, F. Benini, R. E. Martin, G. Jaeschke, B. Wagner, H. Fischer, S. Bendels, D. Zimmerli, J. Schneider, F. Diederich, M. Kansy and K. Müller, ChemMedChem, 2007, 2, 1100 CrossRef CAS PubMed; (c) R. Filler and R. Saha, Future Med. Chem., 2009, 1, 777 CrossRef CAS PubMed; (d) D. B. Berkowitz and M. Bose, J. Fluorine Chem., 2001, 112, 13 CrossRef CAS; (e) B. E. Smart, J. Fluorine Chem., 2001, 109, 3 CrossRef CAS.
- P. Jeschke, ChemBioChem, 2004, 5, 570 CrossRef CAS PubMed.
- (a) F. Guittard, E. T. de Givenchy, S. Geribaldi and A. Cambon, J. Fluorine Chem., 1999, 100, 85 CrossRef CAS; (b) M. G. Dhara and S. Banerjee, Prog. Polym. Sci., 2010, 35, 1022 CrossRef CAS; (c) S. Schlögl, R. Kramer, D. Lenko, H. Schröttner, R. Schaller, A. Holzner and W. Kern, Eur. Polym. J., 2011, 47, 2321 CrossRef; (d) Y. Li, Acc. Chem. Res., 2012, 45, 723 CrossRef CAS PubMed; (e) M. Cametti, B. Crousse, P. Metrangolo, R. Milani and G. Resnati, Chem. Soc. Rev., 2012, 41, 31 RSC.
- (a) X. Yang, T. Wu, R. J. Phipps and F. D. Toste, Chem. Rev., 2015, 115, 826 CrossRef CAS PubMed; (b) M. G. Campbell and T. Ritter, Org. Process Res. Dev., 2014, 18, 474 CrossRef CAS PubMed.
- (a) S. Preshlock, M. Tredwell and V. Gouverneur, Chem. Rev., 2016, 116, 719 CrossRef CAS PubMed; (b) F. Buckingham and V. Gouverneur, Chem. Sci., 2016, 7, 1645 RSC.
- (a) X. Wang, Y. Xu, F. Mo, G. Ji, D. Qiu, J. Feng, Y. Ye, S. Zhang, Y. Zhang and J. Wang, J. Am. Chem. Soc., 2013, 135, 10330 CrossRef CAS PubMed; (b) G. Danoun, B. Bayarmagnai, M. F. Grünberg and L. J. Gooßen, Angew. Chem., Int. Ed., 2013, 52, 7972 CrossRef CAS PubMed; (c) J.-J. Dai, C. Fang, B. Xiao, J. Yi, J. Xu, Z.-J. Liu, X. Lu, L. Liu and Y. Fu, J. Am. Chem. Soc., 2013, 135, 8436 CrossRef CAS PubMed; (d) D. L. Browne, Angew. Chem., Int. Ed., 2014, 53, 1482 CrossRef CAS PubMed.
- J. Wang, M. Sánchez-Roselló, J. L. Aceña, C. del Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Chem. Rev., 2014, 114, 2432 CrossRef CAS PubMed.
- (a) G. Tran, R. Meier, L. Harris, D. L. Browne and S. V. Ley, J. Org. Chem., 2012, 77, 11071 CrossRef CAS PubMed; (b) D. M. Allwood, D. L. Browne and S. V. Ley, Org Synth., 2014, 91, 162 CrossRef CAS; (c) G. Tran, D. G. Pardo, T. Tsuchiya, S. Hillebrand, J.-P. Vors and J. Cossy, Org. Lett., 2015, 17, 3414 CrossRef CAS PubMed; (d) G. A. Molander and L. N. Cavalcanti, Org. Lett., 2013, 15, 3166 CrossRef CAS PubMed.
- S. K. Bagal, K. R. Gibson, M. I. Kemp, C. Poinsard, B. L. Stammen, S. M. Denton and M. S. Glossop, Pfizer Limited, Patent WO2008/135826 A2, 2008 Search PubMed.
- See the ESI† for a modified procedure for the preparation of the hydroxyimoyl bromide 3.
- K. Tanaka, H. Masuda and K. Mitsuhashi, Bull. Chem. Soc. Jpn., 1984, 57, 2184 CrossRef CAS.
- W. J. Middleton, J. Org. Chem., 1984, 49, 919 CrossRef CAS.
- A change in base was made between entries 2 and 3. We found that over prolonged syringe pump addition times that some disposable syringes would degrade and become very stiff in the presence of Et3N and toluene, thus causing the syringe pump to stall against the pressure, this does not occur in the case of Na2CO3 in water.
- For an excellent review on the recent advances in the field of isoxazole synthesis see: F. Hu and M. Szostak, Adv. Synth. Catal., 2015, 357, 2583 CrossRef CAS.
- (a) Y. Fall, C. Reynaud, H. Doucet and M. Santelli, Eur. J. Org. Chem., 2009, 4041 CrossRef CAS; (b) D. Roy, S. Mom, S. Royer, D. Lucas, J.-C. Hierso and H. Doucet, ACS Catal., 2012, 2, 1033 CrossRef CAS.
- (a) M. D. Burke and S. L. Schreiber, Angew. Chem., Int. Ed., 2004, 43, 46 CrossRef PubMed; (b) S. L. Schreiber, Science, 2000, 287, 1964 CrossRef CAS PubMed; (c) D. R. Spring, Org. Biomol. Chem., 2003, 1, 3867 RSC; (d) R. J. Spandl, A. Bender and D. R. Spring, Org. Biomol. Chem., 2008, 6, 1149 RSC.
- (a) D. García-Cuardrado, A. A. C. Braga, F. Maseras and A. M. Echavarren, J. Am. Chem. Soc., 2006, 128, 1066 CrossRef PubMed; (b) D. García-Cuardrado, P. de Mendoza, A. A. C. Braga, F. Maseras and A. M. Echavarren, J. Am. Chem. Soc., 2007, 129, 6880 CrossRef PubMed; (c) S. Pascual, P. de Mendoza, A. A. C. Braga, F. Maseras and A. M. Echavarren, Tetrahedron, 2008, 128, 1066 Search PubMed.
- (a) M. Lafrance, D. Lapointe and K. Fagnou, Tetrahedron, 2008, 64, 6015 CrossRef CAS; (b) B. Gûmez-Lor, E. González-Cantalapiedra, M. Ruiz, Ó. de Frutos, D. J. Cárdenas, A. Santos and A. M. Echavarren, Chem. – Eur. J., 2004, 10, 2601 CrossRef PubMed; (c) D. Kim, J. L. Petersen and K. K. Wang, Org. Lett., 2006, 8, 2313 CrossRef CAS PubMed; (d) C. Huang and V. Gevorgyan, Org. Lett., 2010, 12, 2442 CrossRef CAS PubMed; (e) S. Hernández, R. SanMartin, I. Tellitu and E. Domínguez, Org. Lett., 2003, 5, 1095 CrossRef PubMed; (f) J. K. Laha, P. Petrou and G. D. Cuny, J. Org. Chem., 2009, 74, 3152 CrossRef CAS PubMed; (g) Z. Ma, Z. Xiang, T. Luo, K. Lu, Z. Xu, J. Chen and Z. Yang, J. Comb. Chem., 2006, 8, 696 CrossRef CAS PubMed; (h) S. Hostyn, B. U. W. Maes, G. Van Baelen, A. Gulevskaya, C. Meyers and K. Smits, Tetrahedron, 2006, 62, 4676 CrossRef CAS; (i) B. Delest, J.-Y. Tisserand, J.-M. Robert, M.-R. Nourrisson, P. Pinson, M. Duflos, G. Le Baut, P. Renard and B. Pfeiffer, Tetrahedron, 2004, 60, 6079 CrossRef CAS; (j) Y.-M. Zhang, T. Razler and P. F. Jackson, Tetrahedron Lett., 2002, 43, 8235 CrossRef CAS; (k) N. Ramkumar and R. Nagarajan, J. Org. Chem., 2013, 78, 2802 CrossRef CAS PubMed; (l) L. Basolo, E. M. Beccalli, E. Borsini, G. Broggini and S. Pellegrino, Tetrahedron, 2008, 64, 8182 CrossRef CAS; (m) A. L. Gotumukkala and H. Doucet, Adv. Synth. Catal., 2008, 350, 2183 CrossRef; (n) O. René, D. Lapointe and K. Fagnou, Org. Lett., 2009, 11, 4560 CrossRef PubMed; (o) M. Yagoubi, A. C. F. Cruz, P. L. Nichols, R. L. Elliott and M. C. Willis, Angew. Chem., Int. Ed., 2010, 49, 7958 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ob00970k |
| This journal is © The Royal Society of Chemistry 2016 |