
Directed electrostatic activation in enantioselective organocatalytic cyclopropanation reactions: a computational study†
Abstract
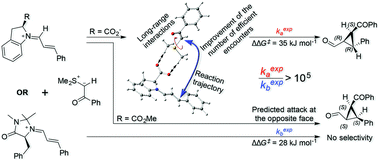
Cyclopropane rings are versatile building blocks in organic chemistry. Their synthesis, by the reaction of sulfur ylides with α,β-unsaturated carbonyl compounds, has recently aroused renewed interest after the discovery of efficient catalysis by using (S)-indoline-2-carboxylic acid. In order to rationalize the behavior of this catalyst, MacMillan proposed a directed electrostatic activation (DEA) mechanism, in which the negative carboxylate group interacts with the positive thionium moiety, thus reducing the activation energy and increasing the reaction rate. More recently, Mayr refuted some of MacMillan conclusions, but accepted the DEA mechanism as a justification for the experimental high reaction rates. In contrast, our results indicate that the selectivity obtained in the process seems to result from several strong hydrogen bond interactions between the two reacting species, while no strong evidence for a DEA mechanism was found. We also concluded that the hydrogen bonds don't improve the reaction rate by lowering the activation energy of the rate-determining step, but can do it by promoting efficient reaction trajectories due to long-range complexation of the reagents. Finally, our results confirm that the cyclopropanation reaction occurs by a two-step mechanism, and that the overall enantioselectivity depends on the relative energies of the two steps, averaged by the relative populations of the iminium intermediates that are initially formed in the reaction.
 Please wait while we load your content...
Please wait while we load your content...

