
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRegioselective addition of Grignard reagents to tosylazafulleroid and derivatization to 1,2-disubstituted [60]fullerene†
Naohiko
Ikuma
*,
Koji
Nakagawa
,
Ken
Kokubo
and
Takumi
Oshima
Division of Applied Chemistry, Graduate School of Engineering, Osaka University, Suita, Osaka 565-0871, Japan. E-mail: ikuma@chem.eng.osaka-u.ac.jp
First published on 29th June 2016
Abstract
Grignard reagents (RMgBr: R = Et, p-tolyl) selectively attacked the β-position of the bridgehead double bond of tosylazafulleroid through interaction of Mg with the S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group. The following [5,6] ring closure and C–N bond scission led to aryl/alkyl aminylfullerenes with 1,2-configuration. Tolyl-substituted aminylfullerene was further converted into 1,4-di(p-tolyl)fullerene on treatment in acidic toluene.
O group. The following [5,6] ring closure and C–N bond scission led to aryl/alkyl aminylfullerenes with 1,2-configuration. Tolyl-substituted aminylfullerene was further converted into 1,4-di(p-tolyl)fullerene on treatment in acidic toluene.
Introduction
Multiple functionalization of fullerene C60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 1–8 is an important process for the materials application of fullerenes, because the introduced substituents modify their electronic properties and enhance their solubility to make them suitable for wet-processing. For example, introduction of multiple substituents improves the open-circuit voltage (Voc) of a fullerenyl photovoltaic cell due to the elevation of its LUMO level.2,3 However, regiocontrolled sequential multi-functionalization is still a challenging topic because fullerene C60 has the equivalent of 30 double bonds; the second addition to the monoadduct often leads to various regioisomeric diadducts with a statistic ratio.1 To overcome this difficulty, facile regioselective multiaddition has eagerly been explored using copper reagents,2,5 halogenations,6 some radical reactions,7 and tether-directed procedures.1c,8
1–8 is an important process for the materials application of fullerenes, because the introduced substituents modify their electronic properties and enhance their solubility to make them suitable for wet-processing. For example, introduction of multiple substituents improves the open-circuit voltage (Voc) of a fullerenyl photovoltaic cell due to the elevation of its LUMO level.2,3 However, regiocontrolled sequential multi-functionalization is still a challenging topic because fullerene C60 has the equivalent of 30 double bonds; the second addition to the monoadduct often leads to various regioisomeric diadducts with a statistic ratio.1 To overcome this difficulty, facile regioselective multiaddition has eagerly been explored using copper reagents,2,5 halogenations,6 some radical reactions,7 and tether-directed procedures.1c,8
Nevertheless, it would also be desired to improve the regioselectivity of the second addition to the monoadducts since the selective combination of two reactants allows a wide variety of introduced substituents with different roles. In fact, difunctionalized fullerenes with aryl and alkyl groups brought about a change in both electronic properties and solubility.4 Moreover, an unprecedented reaction can be found for the monoadduct as its reactivity is rather different from the pristine C60 caused by the first introduced substituents. In order to attain highly regioselective multiaddition, [5,6] open azafulleroid,9 has been employed as such a synthetic intermediate with ambident reactivity at the bridged nitrogen and the adjacent strained bridgehead double bonds. So far, regioselective reactions of azafulleroids have been reported for the [2 + 2] addition of singlet oxygen,10 acidic arylation11 and oxidation with peracids.12 In addition to these electrophilic additions, the nucleophilic Grignard reaction, a useful reaction for the introduction of alkyl and aromatic groups with high yields,13 can also be regioselective in the reaction of azafulleroid on account of the chelation of magnesium with the SO2N unit of the tosylamino group.
Here, we report the regioselective additions of Grignard reagents to the β-position of the bridgehead double bond of tosylazafulleroid 1, followed by [5,6] ring closure and C–N bond scission leading to 1,2-adducts.14 As compared to the usual 1,4-adducts arising from Grignard13d/lithium15 diaddition and from acid-catalyzed arylation,11,16,17 the substituents of C60 1,2-diadducts are limited to hydrogen and less bulky groups such as fluoride/chloride,18 hydroxyl/ester,17a,19 ethynyl,20 methylene-connected alkyl,21 cyano19c,22 and azide group.23 In order to explore the mechanistic preference for 1,2-addition, we have carried out the DFT calculations. For preparative extension, we also attempted the acid-catalyzed reaction of the initially obtained 1,2-substituted aminylfullerene.
Results and discussion
Grignard reaction to azafulleroid 1
As seen in Table 1, Grignard reaction of RMgBr (R = Et, p-tolyl) with azafulleroid 1 gave Cs-symmetric monoadducts 2a,bvia introduction of the R group at the β-carbon and the ring closure of the [5,6] open bridge associated with C–N bond scission. At room temperature, the alkyl Grignard reagent gave multiadducts or regioisomeric monoadducts as byproducts, while at low temperature 2a was isolated in moderate yield in spite of lower conversion (Fig. S1†). On the other hand, the aryl Grignard reagent has high regioselectivity at room temperature. Nevertheless, excess amounts of Grignard reagents gave inseparable multiadducts because the fullerene sphere can undergo further Grignard additions. The regioselectivity could be ascribed to the steric hindrance and Mg-coordination with the tosyl group (Ts) as well as its electron-withdrawing effects. In the expected intermediary, the negative charge appearing on Cα would accelerate the following ring-closure and C–N scission. The appearance of the TsNH-group was confirmed by deuteration with D2O (Fig. S2, in ESI†).
Theoretical study on regioselective addition
To reveal the regioselectivity of azafulleroid 1, we have relied on the DFT energy calculations for the possible geometric transition states. To simplify calculations, only Cα/Cβ additions of 1 with PhMgBr (in place of p-tolylMgBr) and EtMgBr were considered and clusterization of the Grignard reagent in the initial state was ignored.24 Due to the S–N bond rotation and the Mg coordination of the TsN group,25 four possible TS structures were examined for the Cα and Cβ additions, respectively. As seen in Fig. 1(a) and (b), regioselectivity for Cβ-addition can be explained by the DFT calculations (Tables S1–S4†). The smallest ΔE‡ value was attained in TS(2-Cβ) both for PhMgBr (16.0 kJ mol−1, Fig. 1(c)) and EtMgBr (25.6 kJ mol−1, Table S4†). The noticeable difference of activation energy (18 kJ mol−1, vs. 3-Cα) between Cα/Cβ would explain the high regioselectivity for aryl Grignard reagents, while the small difference (8 kJ mol−1, vs. 1-Cα) for the less hindered ethyl reagent may result in the reduced Cβ-selectivity. As compared to the reaction of C60 (ΔE‡ = +75.8 kJ mol−1 for PhMgBr, Fig. 1(d)), the higher reactivity of 1 can be rationalized by the lower LUMO (Fig. 2) and release of strain energy of the anti-Bredt double bond.10,11,26 The Cβ-selectivity is mainly attributed to the steric effect of both the tosyl group and Grignard substituents as well as the O⋯Mg coordination effect, rather than the electronic effect because neither charge values (both Mulliken and NBO, Fig. S3†) nor LUMO distribution are remarkable on Cβ in comparison with Cα. | ||
| Fig. 1 TS energies (kJ mol−1) with several configurations (top view) for reaction of 1 and (a) PhMgBr and (b) EtMgBr by B3LYP/6-31G(d)/IEFPCM(o-DCB). Circled-S means the stacking of S and N atoms. Ball and stick model of (c) TS(2-Cβ) with the lowest energy. (d) TS structure of C60 and PhMgBr. | ||
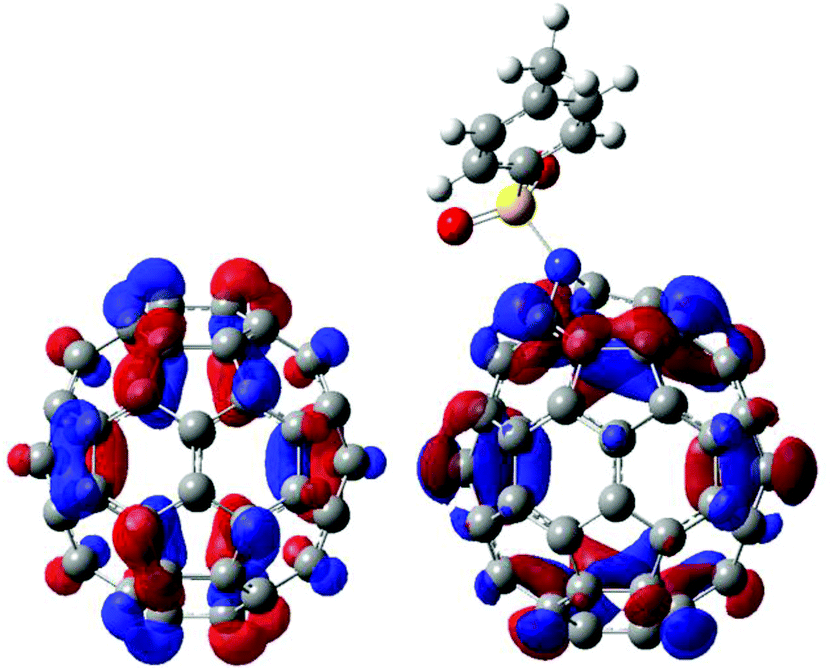 | ||
| Fig. 2 LUMO orbital distribution of C60 (left, −3.14 eV) and 1 (right, −3.22 eV) calculated with B3LYP/6-31G(d)/IEFPCM (o-DCB). | ||
Theoretical study on ring-opening
From the product analysis, the generated Cβ-addition intermediate would undergo [5,6] ring-closure and cleavage of one C–N bond (see scheme in Table 1). Since the bond reorganization is essential for obtaining relatively-limited 1,2-disubstituted fullerenes, TS calculations for this process for the PhMgBr adduct were carried out with DFT (Fig. 3 and 4). The addition of PhMgBr through the TS(2-Cβ) configuration resulted in the formation of INT1 with interaction of Mg and the anionic Cα-center. Ring-closure and C–N bond scission seem to be almost concerted processes due to the negligible energy difference between INT2 and TS(α2 scission), although INT2 is a stationary point without an imaginary frequency. The slightly larger energy barrier of ring-closure is partly ascribed to the destabilization of Mg–Cα1 dissociation. If the Ts group rotates, Cβ2⋯Mg coordination becomes possible with high energetic stabilization during 5,6-closure and α2 scission processes as shown in Fig. S4.† The C–N cleaved INT3 obtains very large energy of stabilization due to the coordination of the anionic nitrogen on Mg+Br as well as the cleavage of the strained aziridine ring (Fig. 4). Finally, INT3 leads to 1,2-diadduct 2 on acidic quenching. | ||
| Fig. 3 (a) Plausible reaction pathway from Cβ-substituted intermediate INT1 to 1,2-adduct 2 (R = Ph) via 5,6-bond closure and N–C bond scission (energy: kJ mol−1). (b) Cα2-cleaved and (c) Cα1-cleaved TS structures (phenyl group except for ipso carbon is omitted for clarity). | ||
 | ||
| Fig. 4 Full energy diagram via Grignard reaction, 5,6-ring closure and CN bond scission. The detail of rotated pathway via INT2′ and TS′ is shown in ESI.† | ||
Further substitution via 1,2-aminylfullerene
We attempted acid catalyzed substitution of 1-aryl-2-aminylfullerene to induce further functionalization by replacing the tosylamino group. Similar to the reaction of 1-aryl-4-aminylfullerene,11 treatment of 2b in o-DCB with trifluoromethanesulfonic acid (TfOH) and toluene (5 equiv.) provided 1,4-ditolylfullerene 3via elimination of tosylamide (Scheme 1). This compound has 1,4-configuration with C1 symmetry,27 indicating that steric hindrance of the tolyl group inhibits the formation of the 1,2-diadduct. | ||
| Scheme 1 | ||
Conclusions
Regioselective addition of Grignard reagents was performed on the β-carbon of the bridgehead double bond of tosyl substituted azafulleroid 1. The high regioselectivity was attained by the steric and coordination effect of the tosyl group. The Mg-coordinated intermediate was degraded into 1-alkyl/2-aminyl fullerene via [5,6] ring-closure and C–N bond scission. The 1,2-adducts are expected to undergo further derivatization owing to the presence of the tosylamino group. The reaction mechanism was supported by the DFT calculations. These results will provide significant information regarding regioselective heterodifunctionalization of fullerenes with the relatively rare 1,2-configuration.Experimental
Synthesis of azafulleroid 1
Tosyl azafulleroid 1 was prepared by the previously reported method.9eGrignard reaction of tosyl azafulleroid 1
Grignard reagent (3 M ethyl magnesium bromide in Et2O or 1 M tolylmagnesium bromide in THF) was added to a solution of azafulleroid 1 (20 mg) in dichloromethane (10 mL) at −70 °C (for 2a) or in anhydrous o-DCB at room temperature (2b), respectively, and stirred for 5–15 min. The reaction solution was washed with saturated aqueous NH4Cl solution and fractionated. The organic layer was dried over MgSO4 and evaporated in vacuo. The residue was purified by silica gel column chromatography (CS2/CHCl3 eluent) to give product 2a,b as a dark brown solid.
2a: 1H-NMR (270 MHz, CDCl3:CS2 = 1:1): δ 1.92 (t, 3H, J = 7.3 Hz), 2.33 (s, 3H), 3.92 (q, 2H, J = 7.3 Hz), 6.90(s, 1H), 7.13(d, 2H, J = 7.9 Hz), 7.80 (d, 2H, J = 8.2 Hz) ppm; 13C-NMR (68 MHz, CDCl3:CS2 = 1:1): δ 14.49, 21.46, 34.85, 66.00, 127.46, 129.22, 134.33, 137.32, 138.66, 139.35, 140.47, 141.16, 141.37, 142.01, 142.19, 142.45, 142.56, 142.91, 143.17, 144.00, 144.21, 144.75, 145.10, 145.16, 145.47, 145.63, 145.80, 146.05, 146.12, 146.21, 146.58, 147.45, 148.18, 149.96, 155.94 ppm. One sp3 carbon connected to the tosyl group is ambiguous probably due to the rotation of the tosyl group. HRMS (FAB-MS) m/z calcd for C69H13NO2S+ [M+]: 919.0662, found: 919.0646.
2b: 1H-NMR (270 MHz, CDCl3): δ 2.34 (s, 3H), 2.61 (s, 3H), 6.29 (s, 1H), 7.17 (d, 2H, J = 8.2 Hz), 7.67 (dd, 4H, J = 7.9, 8.2 Hz), 8.37 (d, 2H, J = 7.9 Hz) ppm; 13C-NMR (68 MHz, CDCl3): δ 21.44, 21.54, 74.96, 127.77, 129.36, 130.36, 131.17, 135.00, 136.97, 137.26, 138.56, 139.13, 140.08, 141.20, 141.48, 142.20, 142.29, 142.35, 142.65, 142.79, 143.18, 143.59, 144.54, 144.84, 144.90, 145.04, 145.27, 145.47, 145.64, 145.80, 146.08, 146.34, 146.39, 146.55, 146.80, 148.46, 149.38, 155.15 ppm. One sp3 carbon connected to the tosyl group may be ambiguous probably due to the rotation of tosyl group. HRMS (FAB-MS) m/z calcd for C74H15NO2S+ [M+]: 981.0818, found: 981.0827.
Acid-catalyzed substitution of 2b
The compound 2b (10 mg) was dissolved in o-DCB (10 mL) containing 5 equiv. of toluene. A catalytic amount of trifluoromethanesulfonic acid (TfOH) was added at room temperature. After 30 minutes stirring, the reaction was quenched with water. The organic layer was separated, dried over MgSO4 and evaporated. The residue was purified by silica gel column chromatography (CS2 eluent) to give product 3 as a dark brown solid.Compound 3: 1H-NMR (270 MHz, CDCl3:CS2 = 1:1): δ 2.47 (s, 6H), 7.33(d, 4H, J = 8.2 Hz), 8.00(d, 4H, J = 8.2 Hz) ppm; 13C-NMR (68 MHz, CDCl3:CS2 = 1:1): δ 21.06, 62.38, 128.11, 128.89, 137.29, 137.53, 137.56, 137.83 (2 × 2C), 138.78, 142.18, 142.51, 142.56, 142.67, 143.09, 143.14, 143.82, 143.88, 143.94, 144.17, 144.31 (2 × 2C), 144.70, 144.82, 144.97, 145.18, 145.49, 146.77, 146.91, 146.95, 147.05, 148.47, 148.60, 151.06, 156.77, 156.82 ppm.
Calculation procedure
DFT calculations were carried out with Gaussian 09. The full citation is in the ESI.† The calculations were carried out with the B3LYP/6-31G(d) level, with o-DCB solvent parameter (IEFPCM). In the initial state, tosylazafulleroid 1 has four geometries,12 and the most stable isomer is 5-exo-1 (Fig. 2). Relative energies of the transition states were obtained as shown in Tables S1–S4† based on the total energies of initial 5-exo-1 and PhMgBr. In all TS calculations, only one imaginary frequiency was obtained by IR calculations as shown in ESI.†Acknowledgements
This work was supported by Grant-in-Aid for Young Scientist (B) (no. 24750039 and 15K21132) from Japan Society for the Promotion of Science (JSPS).Notes and references
- (a) A. Hirsch, I. Lamparth and H. Karfunkel, Angew. Chem., Int. Ed. Engl., 1994, 33, 437–438 CrossRef; (b) F. Djojo, A. Herzog, I. Lamparth, F. Hampel and A. Hirsch, Chem. – Eur. J., 1996, 2, 1537–1547 CrossRef CAS; (c) Y. Nakamura, K. O-Kawa and J. Nishimura, Bull. Chem. Soc. Jpn., 2003, 76, 865–882 CrossRef CAS.
- (a) R. D. Kennedy, A. L. Ayzner, D. D. Wanger, C. T. Day, M. Halim, S. I. Khan, S. H. Tolbert, B. J. Schwartz and Y. Rubin, J. Am. Chem. Soc., 2008, 130, 17290–17292 CrossRef CAS PubMed; (b) T. Niinomi, Y. Matsuo, M. Hashiguchi, Y. Sato and E. Nakamura, J. Mater. Chem., 2009, 19, 5804–5811 RSC.
- (a) J. H. Choi, K. I. Son, T. Kim, K. Kim, K. Ohkubo and S. Fukuzumi, J. Mater. Chem., 2009, 20, 475–482 RSC; (b) M. Lenes, S. W. Shelton, A. B. Sieval, D. F. Kronholm, J. C. K. Hummelen and P. W. M. Blom, Adv. Funct. Mater., 2009, 19, 3002–3007 CrossRef CAS; (c) Y. He, H. Y. Chen, J. Hou and Y. Li, J. Am. Chem. Soc., 2010, 132, 1377–1382 CrossRef CAS PubMed; (d) C. Z. Li, S. C. Chien, H. L. Yip, C. C. Chueh, F. C. Chen, Y. Matsuo, E. Nakamura and A. K. Y. Jen, Chem. Commun., 2011, 47, 10082–10084 RSC; (e) Y. Zhen, N. Obata, Y. Matsuo and E. Nakamura, Chem. – Asian J., 2012, 7, 2644–2649 CrossRef CAS PubMed; (f) T. Mikie, A. Saeki, N. Ikuma, K. Kokubo and S. Seki, ACS Appl. Mater. Interfaces, 2015, 7, 12894–12902 CrossRef CAS PubMed.
- Y. Matsuo, H. Oyama, I. Soga, T. Okamoto, H. Tanaka, A. Saeki, S. Seki and E. Nakamura, Chem. – Asian J., 2012, 8, 121–128 CrossRef PubMed.
- For a review, see: Y. Matsuo and E. Nakamura, Chem. Rev., 2008, 108, 3016–3028 CrossRef CAS PubMed.
- For reviews, see: (a) S. I. Troyanov and E. Kemnitz, Eur. J. Org. Chem., 2005, 4951 CrossRef CAS; (b) R. Taylor, J. Fluorine Chem., 2004, 125, 359–368 CrossRef CAS.
- For a review, see: M. D. Tzirakis and M. Orfanopoulos, Chem. Rev., 2013, 113, 5262–5321 CrossRef CAS PubMed.
- (a) J. F. Nierengarten, V. Gramlich, F. Cardullo and F. Diederich, Angew. Chem., Int. Ed. Engl., 1996, 35, 2101–2103 CrossRef CAS; (b) F. Diederich and R. Kessinger, Acc. Chem. Res., 1999, 32, 537–545 CrossRef CAS; (c) R. K. M. Bouwer and J. C. Hummelen, Chem. – Eur J., 2010, 16, 11250–11253 CrossRef CAS PubMed; (d) W. Yan, S. M. Seifermann, P. Pierrat and S. Bräse, Org. Biomol. Chem., 2014, 13, 25–54 RSC.
- (a) M. Prato, Q. C. Li, F. Wudl and V. Lucchini, J. Am. Chem. Soc., 1993, 115, 1148–1150 CrossRef CAS; (b) C. J. Hawker, P. M. Saville and J. W. White, J. Org. Chem., 1994, 59, 3503–3505 CrossRef CAS; (c) J. Averdung and J. Mattay, Tetrahedron, 1996, 52, 5407–5420 CrossRef CAS; (d) A. Hirsch and B. Nuber, Acc. Chem. Res., 1999, 32, 795–804 CrossRef CAS; (e) T. Nagamachi, Y. Takeda, K. Nakayama and S. Minakata, Chem. – Eur. J., 2012, 18, 12035–12045 CrossRef CAS PubMed; (f) C. B. Miao, X. W. Lu, P. Wu, J. Li, W. L. Ren, M. L. Xing, X. Q. Sun and H. T. Yang, J. Org. Chem., 2013, 78, 12257–12262 CrossRef CAS PubMed.
- J. C. Hummelen, M. Prato and F. Wudl, J. Am. Chem. Soc., 1995, 117, 7003–7004 CrossRef CAS.
- N. Ikuma, Y. Doi, K. Fujioka, T. Mikie, K. Kokubo and T. Oshima, Chem. – Asian J., 2014, 9, 3084–3088 CrossRef CAS PubMed.
- N. Ikuma, K. Fujioka, Y. Misawa, K. Kokubo and T. Oshima, Org. Biomol. Chem., 2015, 13, 5038–5043 CAS.
- (a) A. Hirsch, A. Soi and H. R. Karfunhel, Angew. Chem., Int. Ed. Engl., 1992, 31, 766–768 CrossRef; (b) A. Hirsch, T. Grösser, A. Skiebe and A. Soi, Chem. Ber., 1993, 126, 1061–1067 CrossRef CAS; (c) H. Nagashima, H. Terasaki, E. Kimura, K. Nakajima and K. Itoh, J. Org. Chem., 1994, 59, 1246–1248 CrossRef CAS; (d) H. Nagashima, H. Terasaki and Y. Saito, J. Org. Chem., 1995, 60, 4966–4967 CrossRef CAS.
- Very recently, Gan reported C–N scission of aziridino-C70 with tetraamino substituents in associated with the introduction of amine. The introduced amine and remaining phenylsulfonimide are in the 1,2-configuration. See: Y. Li, D. Xu and L. Gan, Angew. Chem., Int. Ed., 2016, 55, 2483–2487 CrossRef CAS PubMed.
- Y. Murata, K. Komatsu and T. S. M. Wan, Tetrahedron Lett., 1996, 37, 7061–7064 CrossRef CAS . Addition of Cu reagents into Grignard/lithium reaction leads to pentaadducts (in ref. 5 where two of the adjacent addends usually have 1,4-configuration).
- A. Iwashita, Y. Matsuo and E. Nakamura, Angew. Chem., Int. Ed., 2007, 46, 3513–3516 CrossRef CAS PubMed.
- Acid catalytic 1,4-diaryl products have also been obtained from activated fullerenes such as epoxide, aziridinofullerene and hydroxyarylfullerene. (a) Y. Tajima, T. Hara, T. Honma, S. Matsumoto and K. Takeuchi, Org. Lett., 2006, 8, 3203–3205 CrossRef CAS PubMed; (b) R. Tsuruoka, T. Nagamachi, Y. Murakami, M. Komatsu and S. Minakata, J. Org. Chem., 2009, 74, 1691–1697 CrossRef CAS PubMed; (c) G. W. Wang, Y. M. Lu and Z. X. Chen, Org. Lett., 2009, 11, 1507–1510 CrossRef CAS PubMed; (d) M. Nambo, Y. Segawa and K. Itami, J. Am. Chem. Soc., 2011, 133, 2402–2405 CrossRef CAS PubMed.
- (a) O. V. Boltalina, A. Y. Lukonin, J. M. Street and R. Taylor, Chem. Commun., 2000, 1601–1602 RSC; (b) I. E. Kareev, G. S. Quiñones, I. V. Kuvychko, P. A. Khavrel, I. N. Ioffe, I. V. Goldt, S. F. Lebedkin, K. Seppelt, S. H. Strauss and O. V. Boltalina, J. Am. Chem. Soc., 2005, 127, 11497–11504 CrossRef CAS PubMed; (c) I. V. Kuvychko, A. V. Streletskii, N. B. Shustova, K. Seppelt, T. Drewello, A. A. Popov, S. H. Strauss and O. V. Boltalina, J. Am. Chem. Soc., 2010, 132, 6443–6462 CrossRef CAS PubMed; (d) Y. Li, N. Lou and L. Gan, Org. Lett., 2015, 17, 524–527 CrossRef CAS PubMed.
- (a) M. S. Meier and J. Kiegiel, Org. Lett., 2001, 3, 1717–1719 CrossRef CAS PubMed; (b) G. W. Wang, F. B. Li and Y. Xu, J. Org. Chem., 2007, 72, 4774–4778 CrossRef CAS PubMed; (c) H. Irngartinger and A. Weber, Tetrahedron Lett., 1997, 38, 2075–2076 CrossRef CAS; (d) M. Hashiguchi, N. Obata, M. Maruyama, K. S. Yeo, T. Ueno, T. Ikebe, I. Takahashi and Y. Matsuo, Org. Lett., 2012, 14, 3276–3279 CrossRef CAS PubMed.
- (a) Y. Murata, K. Motoyama, K. Komatsu and T. S. M. Wan, Tetrahedron, 1996, 52, 5077–5090 CrossRef CAS; (b) K. Fujiwara, Y. Murata, T. S. M. Wan and K. Komatsu, Tetrahedron, 1998, 54, 2049–2058 CrossRef CAS.
- (a) C. Caron, R. Subramanian, F. D'Souza, J. Kim, W. Kutner, M. T. Jones and K. M. Kadish, J. Am. Chem. Soc., 1993, 115, 8505–8506 CrossRef CAS; (b) E. Allard, L. Rivière, J. Delaunay, D. Dubois and J. Cousseau, Tetrahedron Lett., 1999, 40, 7223–7226 CrossRef CAS; (c) Y. Zhang, Y. Matsuo and E. Nakamura, Org. Lett., 2011, 13, 6058–6061 CrossRef CAS PubMed.
- M. Keshavarz-K, B. Knight, G. Srdanov and F. Wudl, J. Am. Chem. Soc., 1995, 117, 11371–11372 CrossRef CAS.
- U. M. Dzhemilev, A. R. Tuktarov, I. R. Yarullin and A. R. Akhmetov, Mendeleev Commun., 2013, 23, 326–328 CrossRef CAS.
- Although the ΔE‡ values based on monomeric Grignard reagents may include systematic error due to the cluster stabilization, the relative energies of transition states among regioisomers are comparable to each other. Clusterizations of Grignard reagents are well known as below. (a) A. W. Ehlers, G. P. M. van Klink, M. J. van Eis, F. Bickelhaupt, P. H. J. Nederkoorn and K. Lammertsma, J. Mol. Model., 2000, 6, 186–194 CrossRef CAS; (b) S. Yamazaki and S. Yamabe, J. Org. Chem., 2002, 67, 9346–9353 CrossRef CAS PubMed; (c) A. Tuulmets, J. Tammiku-Taul and P. Burk, J. Mol. Struct. (THEOCHEM), 2004, 674, 233–239 CrossRef CAS; (d) J. O. C. Jimenez-Halla, F. M. Bickelhaupt and M. Solà, J. Organomet. Chem., 2011, 696, 4104–4111 CrossRef CAS.
- As shown in Tables S1–S4,† Grignard reagents seem to coordinate oxygen rather than nitrogen of tosyl amide compounds due to the low N-basicity. Such results were found in the following papers. (a) B. Z. Lu, C. Senanayake, N. Li, Z. Han, R. P. Bakale and S. A. Wald, Org. Lett., 2005, 7, 2599–2602 CrossRef CAS PubMed; (b) S. D. Kuduk, C. N. D. Marco, S. M. Pitzenberger and N. Tsou, Tetrahedron Lett., 2006, 47, 2377–2381 CrossRef CAS; (c) G. Jiménez-Osés, A. J. Brockway, J. T. Shaw and K. N. Houk, J. Am. Chem. Soc., 2013, 135, 6633–6642 CrossRef PubMed.
- High reactivity at the anti-Bredt double bonds has also been exhibited in carbon analog fulleroids. See: (a) B. R. Weedon, R. C. Haddon, H. P. Spielmann and M. S. Meier, J. Am. Chem. Soc., 1999, 121, 335–340 CrossRef CAS; (b) N. Ikuma, Y. Susami and T. Oshima, Org. Biomol. Chem., 2010, 8, 1394–1398 RSC; (c) N. Ikuma, Y. Susami and T. Oshima, Eur. J. Org. Chem., 2011, 6452–6458 CrossRef CAS.
- Product 3 has 1,4-ditolyl structure (C2 symmetry) rather than 1,2-ditolyl product (C2v symmetry), because the 13C NMR of 4 has ca. 30 peaks of sp2 region similar to the previously obtained 1,4-ditolyl product by acidic arylation in ref. 11 (ESI†).
Footnote |
| † Electronic supplementary information (ESI) available: HPLC and NMR charts, calculation results and full citation of calculation software. See DOI: 10.1039/c6ob00869k |
| This journal is © The Royal Society of Chemistry 2016 |