
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRegioselective oxidation of unprotected 1,4 linked glucans†
Niek N. H. M.
Eisink
,
Jonas
Lohse
,
Martin D.
Witte
* and
Adriaan J.
Minnaard
*
Stratingh Institute for Chemistry, University of Groningen, Nijenborgh 7, 9747 AG Groningen, The Netherlands. E-mail: A.J.Minnaard@rug.nl; m.d.witte@rug.nl
First published on 4th May 2016
Abstract
Palladium-catalyzed alcohol oxidation allows the chemo- and regioselective modification of unprotected 1,4 linked glucans. This is demonstrated in the two-step bisfunctionalization of 1,4 linked glucans up to the 7-mer. Introduction of an anomeric azide is followed by a highly regioselective mono-oxidation of the terminal C3–OH functionality. The resulting orthogonal bis-functionalized oligosaccharides are a viable alternative to PEG-spacers as demonstrated in the conjugation of a cysteine mutant of 4-oxalocrotonate tautomerase with biotin.
Introduction
Complex molecule functionalization including complex molecule diversification is one of the frontiers of contemporary chemistry. Control over chemical reactivity and predictable selectivity are the key goals in this field. Important strategies that currently see considerable development are C–H bond oxidation1 and C–H bond alkylation,2 given the large number of C–H bonds present in most organic molecules.The regioselective functionalization, and in particular oxidation, of oligosaccharides should be placed in the same ball park. In oligo- and polysaccharides, the number of hydroxyl groups roughly equals the number of C–H bonds, and whereas in C–H activation the actual bond cleavage is energetically costly, that is, control over chemical reactivity is challenging, in carbohydrate oxidation, (regio)selectivity is the crux. Indeed, hardly any studies have appeared on this topic,3,4 apart from those focusing on the anomeric hemiacetal, which obviously stands out reactivity-wise.5
Although based on the available literature the picture seems bleak, it is well known that in monosaccharides acetal formation is often highly regioselective. This feature has been exploited to selectively oxidize hydroxyl groups via tin-acetals.6 In the field of palladium-catalyzed alcohol oxidation, Waymouth and coworkers have shown that 1,2-vicinal diols are selectively oxidized to hydroxyl ketones, e.g. the secondary hydroxyl group is oxidized preferentially over the primary hydroxyl group, via the palladium diol-complex.7 We recently extended this approach by demonstrating that this catalyst also discriminates between multiple secondary hydroxyl groups and oxidizes mono- and diglucosides selectively at the C3 position.8
We here demonstrate that palladium-catalyzed alcohol oxidation is able to modify a series of azido-β-1,4-glucans (“azido oligomaltoses”) with extreme regioselectivity as shown in the oxidation of maltoheptaosyl azide, containing 15 secondary and 7 primary hydroxyl groups (Fig. 1). The compatibility of the oxidation with the presence of an azide makes it a powerful tool to prepare orthogonal bis-functionalized oligosaccharides. Like PEG chains, oligomaltoses, have shown to stabilize proteins,9 and may serve as spacers for the preparation of protein–drug conjugates. We illustrate this concept with a biotin conjugation to 4-oxalocrotonate tautomerase. In addition, oligomaltoses, may well be used as molecular rulers and building blocks for copolymers.
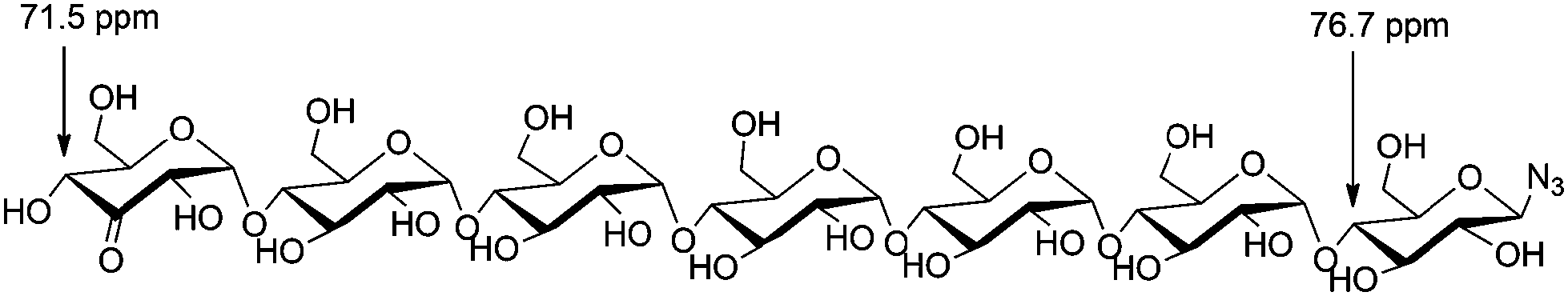 | ||
| Fig. 1 β-D-3-Ketomaltoheptaosyl azide + 13C-NMR shifts. | ||
Results and discussion
The synthesis of the required glycosyl azides as starting materials for the catalytic oxidation was carried out according to literature. Shoda recently pioneered the application of 2-chloro-1,3-dimethylimidazolinium chloride (DMC) for the synthesis of anomeric azides in unprotected oligosaccharides.10–14 For scalability reasons, we aimed to replace the described preparative HPLC purification. Silica gel chromatography with 10–15% water in acetonitrile as the eluent provided NMR-pure glucosyl azides, but all oxidation experiments failed to give conversion. Further study showed that 1,3-dimethyl-imidazolidin-2-one did not hamper the but small amounts of NaCl, NaBr, KI and NaN3 led to complete inhibition. Apparently, salts in the reaction mixture co-eluted during purification.15 To lower the amount of chloride present, DMC chloride was replaced by DMC PF6. Additional advantage is that this reagent is less hygroscopic and therefore easier to handle. The key measure, however, turned out to be the application of column chromatography on charcoal. Improving upon the procedure of Whistler et al.,16 a ratio of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (w/w charcoal to product) and elution with water followed by a gradient of ethanol/water turned out sufficient to purify the glucosyl azides on preparative scale.
:1 (w/w charcoal to product) and elution with water followed by a gradient of ethanol/water turned out sufficient to purify the glucosyl azides on preparative scale.
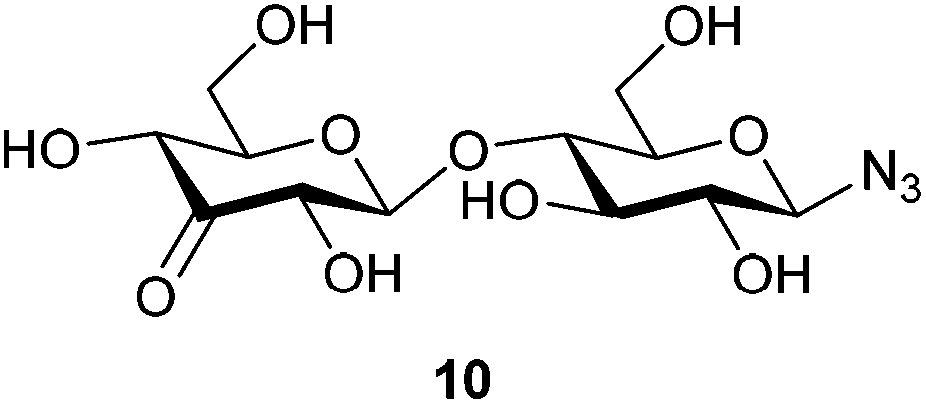
With this purification method in hand, we prepared the desired range of maltosyl to maltoheptaosyl azides. We subsequently subjected the obtained glucosyl azides to palladium catalyzed regioselective oxidation. Hitherto, its compatibility with azides had not been studied, and the substrate scope was limited to mono- and disaccharides. β-D-Cellobiosyl azide 2 was selected as starting point for the oxidation due to its straightforward comparison to the reported oxidation of methyl-α-D-cellobioside. When 2 was reacted with 0.5 mol% of catalyst, no conversion was observed. An increased catalyst loading of 7.5 mol%, however, led to full conversion (see Scheme 1). Ketone 10 was readily purified by charcoal column chromatography to give 61% isolated yield (Table 1, entry 1). According to NMR, the C3–OH of the non-azido glucose residue had been oxidized selectively.
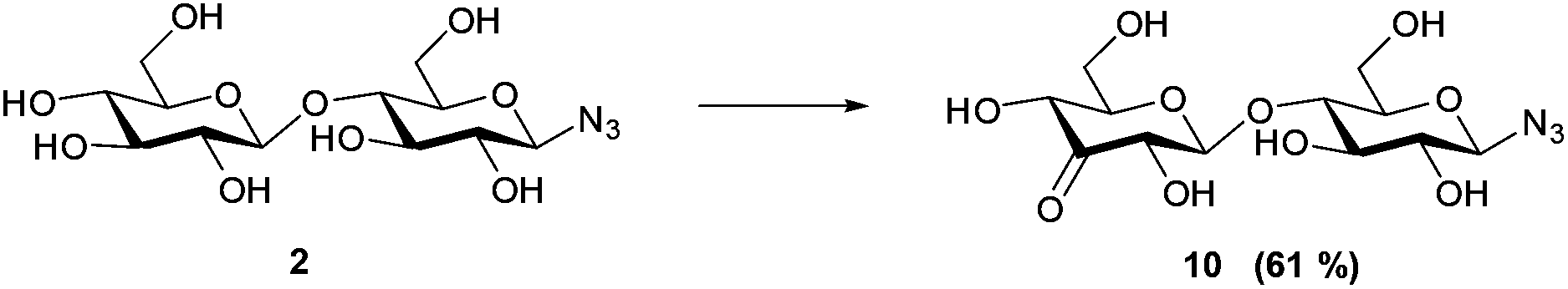 | ||
| Scheme 1 Oxidation of β-D-cellobiosyl azide 2 reaction conditions: 7.5 mol% of [(neocuproine)PdOAc]2OTf2, 3 eq. benzoquinone, 0.3 M in DMSO/dioxane 1:4, r.t. 7 h. | ||
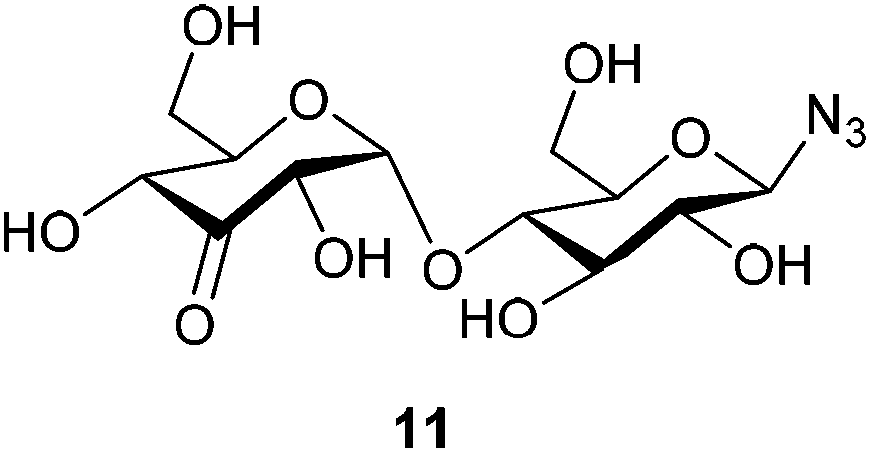
The H2 and H4 of this ring shifted significantly downfield, separating them from the other signals in the 1H-NMR and showed a long-range coupling (∼1.6 Hz), for an example of these shifts in β-D-3-ketomaltotriosyl azide (13) see the 1H-NMR in Fig. 2. Furthermore, the H2 of the oxidized ring coupled to the H1 of the O-glycoside and not to the H1 of the N-glycoside. Also β-D-3-ketomaltosyl azide 11 could be produced in this fashion in a similar yield (Table 1, entry 2). Apparently, the Pd-catalyzed oxidation is compatible with anomeric azides although an increased amount of catalyst is required. In the same way, β-D-maltotriosyl azide was oxidized, which provided 12 in 60% isolated yield. Although 1H-NMR readily identified oxidation at a C3 position, identifying which glucose residue had been oxidized proved to be more challenging.
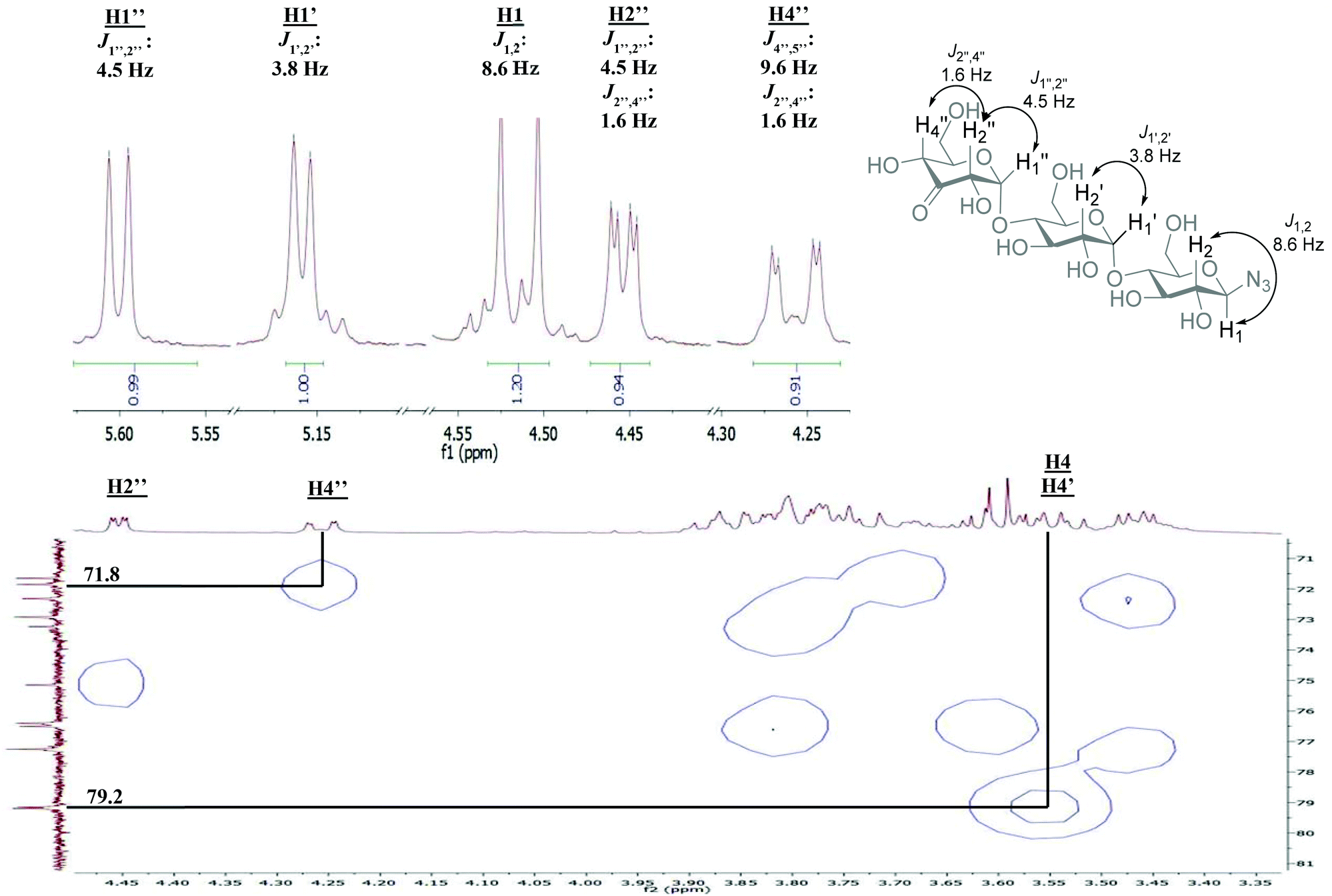 | ||
| Fig. 2 NMR analysis of β-D-3-ketomaltotriosyl azide (13) relevant sections of the spectrum are shown, for full spectrum see ESI.† HMQC: correlation of H4′′ with the corresponding carbon signal. H4 and H4′ were determined via COSY/TOCSY. | ||
An empirical study by Bock and coworkers17,18 had revealed that the C4 carbon involved in a glycosidic bond has a distinct downfield shift in 13C-NMR (∼77–80 ppm) compared to the other carbon signals (55–76 ppm, apart from the anomeric carbons). The straightforward assignment of the H4 next to the ketone in 1H-NMR enabled identification of the corresponding carbon signal with HMQC, see Fig. 2. The signal of this C4 appeared at 71.8 ppm, identifying the carbon as a CHOH moiety, which confirmed that oxidation had taken place at the terminal non-azido end (Fig. 1). To verify this analysis, the C4 of the glucosyl azide moiety was also determined. Using TOCSY and COSY NMR techniques H4 of this ring was readily identified. As described above, the corresponding C4 could be found using HMQC, giving a signal typically around 77–80 ppm. Furthermore, the chemical shifts of the synthesized β-D-3-ketomaltotriosyl azide were in agreement with the reported values of the oxidation at the terminal C3 position of maltotriose.19
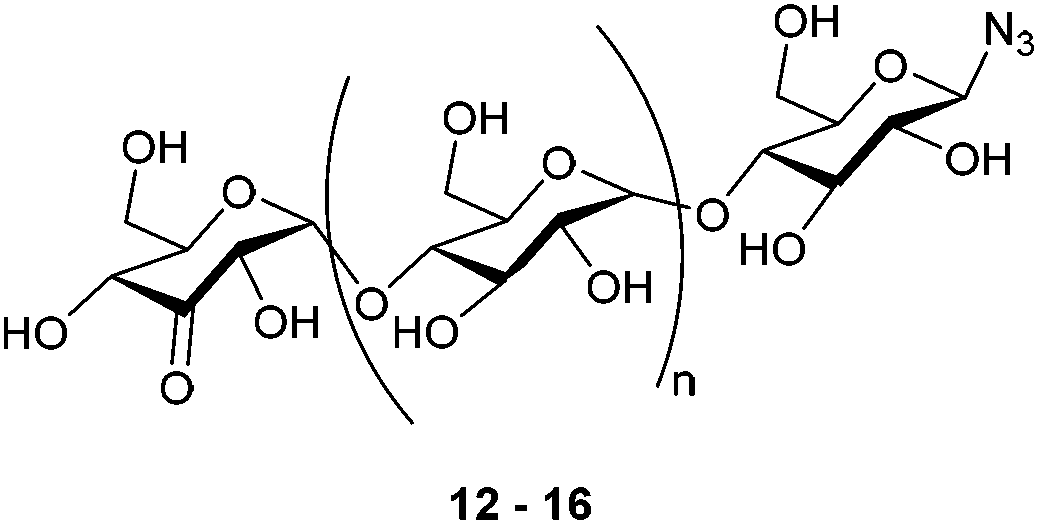
With selective oxidation on the terminal glucose moiety in this trisaccharide established, the scope was extended to even larger oligosaccharides. β-D-Maltotetraosyl azide was oxidized to 13 in 38% isolated yield upon increasing the catalyst loading to 15 mol%. With this protocol, we demonstrated that azido-β-1,4-glucans (“azido oligomaltoses”) up to maltoheptaose were readily and in high selectivity converted into their C3-keto derivatives (see Table 1). NMR analysis shows that in all cases oxidation takes place selectively at the C3-position of the terminal non-reducing glucose unit. The reaction proceeds with exceptional regioselectivity and only very small amounts of regio-isomers and products resulting from over-oxidation were observed in the crude reaction mixture (see the ESI† for the 1H-NMR spectrum of β-D-3-ketomaltoheptaosyl azide before purification). Although the reactions proceed with full conversion of the starting material, purification of these highly polar compounds is challenging. Charcoal column chromatography effectively removed the impurities, however resulting in a somewhat decreased isolated yield.
To further expand the scope of the oxidation reaction, readily available sucrose, although not a 1,4-linked glucan, was studied as it consists of glucose 1,1-linked to fructose. The reaction was monitored by quantitative 1H-NMR (Q-NMR). The reaction gave 50% of the expected 3-keto sucrose together with several side products in smaller amounts (Scheme 2).
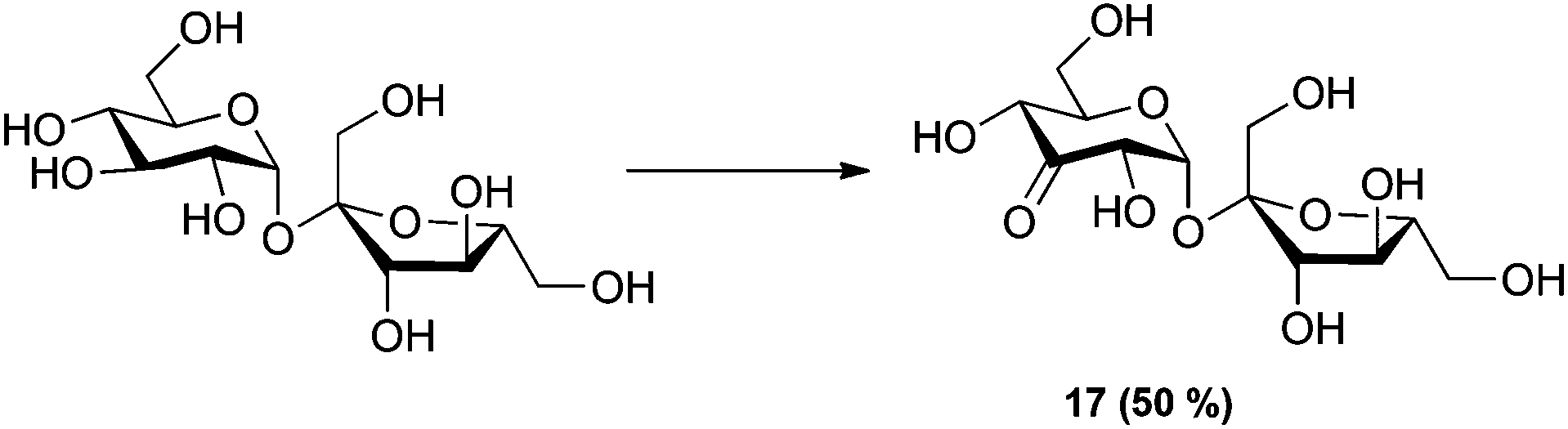 | ||
| Scheme 2 Oxidation of sucrose. Reaction conditions: 2.5 mol% of [(neocuproine)PdOAc]2OTf2, 3 eq. benzoquinone, 0.3 M in DMSO-d6, r.t. 1 h. Conversion determined by Q-NMR. | ||
Why the C3–OH oxidizes selectively over the other secondary positions in the ring is under further investigation, but we hypothesize that the selective oxidation of the terminal glucose residue is due to steric effects. The substituent at the C4 position of the other glucose units probably retards the oxidation. Upon prolonged reaction, over-oxidation on different positions is observed. As an indication of the extreme selectivity of the reaction, the 47% yield in the oxidation of β-D-maltoheptaosyl azide translates in a selectivity ratio of >10.
Bis-functionalized oligosaccharides, and in particular oligo-maltoses, are potentially highly effective molecular rulers and spacers.20–22 Oligomaltoses share a high water solubility with poly-ethylene glycol (PEG), but contrary to the latter they have a well-defined length and stiffness due to their internal structure.23 As the two introduced functional groups, a ketone and an azide, are orthogonal to each other, and bio-orthogonal, one of these handles can be used for the glycosylation of a protein, and the second one for subsequent modification of the glycoprotein conjugate with a molecule of interest (Fig. 3). To validate the feasibility of this application, such a protein–glycan conjugate was prepared. A cysteine mutant of 4-oxalocrotonate tautomerase, denoted 4-OT R61C-1, coupled to a terminal alkyne at the cysteine residue via a maleimide linker, was selected as a model protein.24 We decided to ligate biotin hydrazide to the ketone functionality of the saccharide residue, for straightforward visualization of the bisfunctionalized construct by western blotting. After hydrazone formation, the modified oligosaccharides β-D-maltotriosyl azide-biotin and β-D-maltoheptaosyl azide-biotin were incubated with the protein in the presence of CuSO4/tris (3-hydroxypropyltriazolylmethyl)amine and sodium ascorbate (Fig. 3). As a control, the same reaction was performed in the absence of the saccharides. Tricine SDS-PAGE analysis of the conjugation reaction visualized by Coomassie stain showed in the cases with saccharide present the appearance of new bands. The molecular weight of these bands is increased compared to the unmodified protein and corresponds with the respective functionalization of 4-OT with β-D-maltotriosyl azide-biotin and β-D-maltoheptaosyl azide-biotin (Fig. 3B). In the control reaction this particular shift was not observed, further confirming that these new bands originate from the biotin–carbohydrate–protein adduct. To verify the bis-functionalization of the oligosaccharide, we visualized the biotinylated protein adducts via western blotting. As depicted in Fig. 3C, a strong chemi-luminescence signal arising from the protein–oligosaccharide–biotin conjugate was observed at the expected molecular weight. Inversion of the ligation steps, e.g. first ligation of the protein to the azido carbohydrate, followed by biotinylation was also effective, albeit with a lower efficiency.
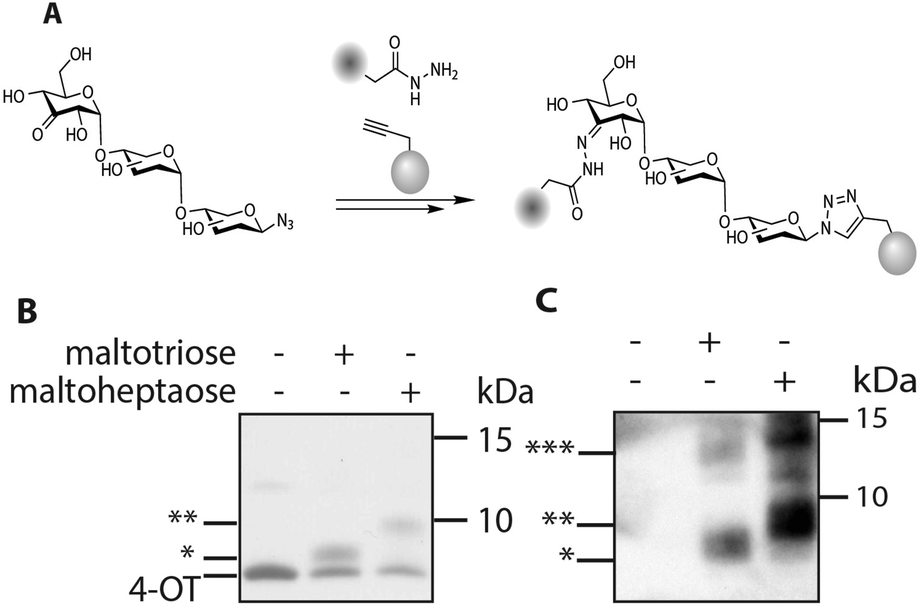 | ||
| Fig. 3 A: Introduction of a protein–alkyne and a biotin hydrazide onto β-D-ketomaltoheptaosyl azide. B: Tricine SDS Page, visualized by Coomassie Brilliant Blue stain. C: Western blot using strep-HRP and ECL (* 4-OT modified with maltotriose biotin, ** 4-OT modified with maltoheptaose biotin, *** 4-OT oligomers). | ||
Conclusions
In conclusion, palladium-catalyzed alcohol oxidation allows the regioselective modification of azido-β-1,4-glucans, in casu azido oligomaltoses. The chemo- and regioselectivity of the catalyst system [(neocuproine)PdOAc]2OTf2 is extremely high and unprecedented; in azido maltoheptaose one secondary hydroxyl group is oxidized in the presence of 7 primary and 15 nearly identical secondary hydroxyl groups to provide the product in 47% isolated yield. Oxidation takes place at the C3-position of the terminal residue at the non-azido end. As the oxidation is compatible with an azide functionality in the substrate this allows the synthesis of well-defined orthogonal bisfunctionalized 1,4-linked glucans. Column chromatography on charcoal allows purification on a synthetically useful scale. Among the many foreseeable applications as biocompatible spacers, molecular rulers, and building blocks for copolymers, it has been demonstrated that a protein–oligomaltose adduct can be prepared and subsequently visualized via biotinylation at the other terminus of the glycan.Experimental section
β-D-3-Ketocellobiosyl azide (10)
β-D-Cellobiosyl azide (40 mg, 0.109 mmol, 1 eq.) was dissolved in a dioxane/DMSO mixture (4:1, 370 μl, 0.3 M), before benzoquinone (35 mg, 0.327 mmol, 3 eq.) and [(2,9-dimethyl-1,10-phenanthroline)-Pd(μ-OAc)]2(OTf)2 (9 mg, 8.6 μmol, 7.5 mol%, added in 3 portions over 6 h) were added. The reaction was stirred at room temperature till complete consumption of starting material (indicated by TLC (eluent: CHCl3:MeOH:EtOAc:H2O 2:2:4:0.75)). Upon completion, the reaction mixture was diluted with H2O (7 ml) and the resulting aqueous solution was purified by charcoal column chromatography (12% EtOH/H2O eluted the desired product). The product was freeze-dried to yield 26 mg (0.071 mmol, 65%) of an off-white solid. 1H NMR (400 MHz, CD3OD) δ 4.56 (d, J = 8.7 Hz, 1H), 4.56 (d, J = 7.9 Hz, 1H), 4.24 (dd, J = 10.2, 1.7 Hz, 1H), 4.18 (dd, J = 8.0, 1.8 Hz, 1H), 3.97–3.86 (m, 3H), 3.78 (dd, J = 12.1, 5.0 Hz, 1H), 3.72–3.65 (m, 1H), 3.58 (t, J = 9.0 Hz, 1H), 3.52 (ddd, J = 9.7, 3.7, 2.3 Hz, 1H), 3.38 (ddd, J = 10.1, 5.0, 2.1 Hz, 1H), 3.21 (appt, J = 8.9 Hz, 1H). 13C NMR (101 MHz, CD3OD) δ 206.6, 105.7, 91.9, 79.6, 78.6, 78.2, 78.2, 76.4, 74.5, 73.4, 62.3, 61.3. HRMS (ESI) calculated for C12H19O10N3Na ([M + Na]+): 388.096, found: 388.096 IR νmax/cm−1: 3368 (OH), 2888 (C–H), 2118 (N3), 1734 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 1028 (C–O) [α]20D = −20 (c 0.6, H2O).
O), 1028 (C–O) [α]20D = −20 (c 0.6, H2O).
β-D-3-Ketomaltosyl azide (11)
β-D-Maltosyl azide (76 mg, 0.207 mmol, 1 eq.) was dissolved in a dioxane/DMSO mixture (4:1, 700 μl, 0.3 M), before benzoquinone (67 mg, 0.620 mmol, 3 eq.) and [(2,9-dimethyl-1,10-phenanthroline)-Pd(μ-OAc)]2(OTf)2 (16 mg, 15.5 μmol, 7.5 mol%, added in 3 portions over 6 h) were added. The reaction was stirred at room temperature till complete consumption of starting material (indicated by TLC (eluent: CHCl3:MeOH:EtOAc:H2O 2:2:4:0.75)). Upon completion, the reaction mixture was diluted with H2O (14 ml) and the resulting aqueous solution was purified by charcoal column chromatography (7% EtOH/H2O eluted the desired product). The product was freeze dried to yield 46 mg (0.122 mmol, 59%) as an off-white solid (contains ∼10% hydroquinone by NMR integration, isolated yield corrected for this value). 1H NMR (400 MHz, CD3OD) δ 5.64 (d, J = 4.5 Hz, 1H), 4.50 (d, J = 8.7 Hz, 1H), 4.46 (dd, J = 4.5, 1.5 Hz, 1H), 4.26 (dd, J = 9.5, 1.6 Hz, 1H), 3.91–3.76 (m, 5H), 3.64–3.58 (m, 2H), 3.43 (ddd, J = 9.2, 4.4, 1.9 Hz, 1H), 3.17 (appt, J = 8.7 Hz, 1H). 13C NMR (101 MHz, CD3OD) δ 207.0, 104.7, 91.9, 79.7, 78.4, 77.9, 77.6, 76.5, 74.4, 73.3, 62.5, 61.8. HRMS (ESI) calculated for C12H19O10N3Na ([M + Na]+): 388.096, found: 388.096 IR νmax/cm−1: 3343 (OH), 2928 (C–H), 2118 (N3), 1736 (CO), 1028 (C–O) [α]20D = +89.6 (c 1.00, H2O).
β-D-3-Ketomaltotrioside (12)
β-D-Maltotriosyl azide (190 mg, 0.360 mmol, 1 eq.) was dissolved in DMSO (2.4 ml, 0.3 M), before benzoquinone (117 mg, 1.080 mmol, 3 eq.) and [(2,9-dimethyl-1,10-phenanthroline)-Pd(μ-OAc)]2(OTf)2 (28 mg, 27 μmol, 7.5 mol%) were added. The reaction was stirred at room temperature till complete consumption of starting material (indicated by TLC (eluent: 15% H2O/CH3CN)). Upon completion the reaction mixture was diluted with H2O (10 ml) and the resulting aqueous solution was purified by charcoal column chromatography (20% EtOH/H2O eluted the desired product). The product was freeze dried to yield 121 mg (0.23 mmol, 60%) as an off-white solid. 1H NMR (400 MHz, CD3OD) δ 5.60 (d, J = 4.5 Hz, 1H), 5.16 (d, J = 3.8 Hz, 1H), 4.51 (d, J = 8.6 Hz, 1H), 4.45 (dd, J = 4.4, 1.6 Hz, 1H), 4.26 (dd, J = 9.6, 1.6 Hz, 1H), 3.93–3.71 (m, 8H), 3.65–3.43 (m, 6H), 3.18 (appt, J = 8.9 Hz, 1H). 13C NMR (101 MHz, CD3OD) δ 207.1, 104.8, 102.6, 91.9, 80.6, 80.5, 78.7, 77.9, 77.8, 76.6, 74.6, 74.3, 73.7, 73.3, 73.0, 62.5, 62.0, 61.9. HRMS (ESI) calculated for C18H29O15N3Na ([M + Na]+): 550.149, found: 550.148 IR νmax/cm−1: 3338 (OH), 2925 (C–H), 2118 (N3), 1737 (CO), 1025 (C–O), [α]20D = +46.6 (c 1.00, H2O).
β-D-3-Ketomaltotetraosyl azide (13)
β-D-Maltotetraosyl azide (55 mg, 0.08 mmol, 1 eq.) was dissolved in DMSO (530 μl, 0.15 M), before benzoquinone (26 mg, 0.240 mmol, 3 eq.) and [(2,9-dimethyl-1,10-phenanthroline)-Pd(μ-OAc)]2(OTf)2 (12 mg, 12 μmol, 15 mol%) were added. The reaction was stirred at room temperature till complete consumption of starting material (indicated by TLC (eluent: 20% H2O/CH3CN)). Upon completion the reaction mixture was diluted with H2O (15 ml) and the resulting aqueous solution was purified by charcoal column chromatography (2.5% tBuOH/H2O eluted the desired product). The product was freeze-dried to yield an off white solid containing traces of hydroquinone. Hydroquinone was removed by washing the product in water with diethyl ether to yield 21 mg (0.03 mmol, 38%) of an off-white solid. 1H NMR (400 MHz, D2O) δ 5.68 (d, J = 4.6 Hz, 1H), 5.27 (d, J = 4.0 Hz, 1H), 5.24 (d, J = 4.1 Hz, 1H), 4.62 (d, J = 9.1 Hz, 1H), 4.52 (dd, J = 4.6, 1.5 Hz, 1H), 4.32 (dd, J = 9.6, 1.5 Hz, 1H), 3.85–3.50 (m, 18H), 3.48 (dd, J = 9.8, 4.0 Hz, 2H), 3.17 (appt, J = 9.0 Hz, 1H). 13C NMR (101 MHz, D2O) δ 207.2, 102.1, 99.5, 99.3, 89.8, 76.7, 76.6, 76.4, 76.2, 76.1, 75.6, 74.6, 73.2, 73.0, 72.6, 71.5, 71.4, 71.4, 71.1, 70.8, 60.4, 60.4, 60.3, 60.1. HRMS (ESI) calculated for C24H39O20N3Na ([M + Na]+): 712.202, found: 712.201 IR νmax/cm−1: 3340 (OH), 2932 (C–H), 2121 (N3), 1738 (CO), 1027 (C–O), [α]20D = +102.6 (c 1.00, H2O).
β-D-3-Ketomaltopentaosyl azide (14)
β-D-Maltopentaosyl azide (58 mg, 0.068 mmol, 1 eq.) was dissolved in DMSO (450 μl, 0.15 M), before benzoquinone (22 mg, 0.20 mmol, 3 eq.) and [(2,9-dimethyl-1,10-phenanthroline)-Pd(μ-OAc)]2(OTf)2 (6.8 mg, 11 μmol, 15 mol%) were added. The reaction was stirred at room temperature till complete consumption of starting material (indicated by TLC (eluent: 25% H2O/CH3CN)). Upon completion the reaction mixture was diluted with H2O (15 ml) and the resulting aqueous solution was purified by charcoal column chromatography (3.0% tBuOH/H2O eluted the desired product). The product was freeze-dried to yield an off white solid containing traces of hydroquinone. Hydroquinone was removed by washing the product in water with diethyl ether to yield 17 mg (0.02 mmol, 30%) of a white solid. 1H NMR (400 MHz, D2O) δ 5.83 (d, J = 4.7 Hz, 1H), 5.44–5.35 (m, 3H), 4.76 (d, J = 8.4 Hz, 1H). 4.66 (d, J = 4.5 Hz, 1H), 4.46 (d, J = 9.6 Hz, 1H), 4.02–3.74 (m, 18H), 3.74–3.58 (m, 7H), 3.31 (appt, J = 9.0 Hz, 1H). 13C NMR (101 MHz, D2O) δ 207.1, 102.0, 99.5, 99.4, 99.3, 89.8, 76.7, 76.7, 76.6, 76.5, 76.3, 76.1, 76.0, 75.6, 74.5, 73.2, 73.1, 72.9, 72.5, 71.5, 71.4, 71.3, 71.0, 71.0, 70.8, 60.3, 60.3, 60.2, 60.2, 60.1. HRMS (ESI) calculated for C30H49O25N3Na ([M + Na]+): 874.255, found: 874.253 IR νmax/cm−1: 3339 (OH), 2927 (C–H), 2121 (N3), 1737 (CO), 1027 (C–O), [α]20D = +105.6 (c 1.00, H2O).
β-D-3-Ketomaltohexaosyl azide (15)
β-D-Maltohexaosyl azide (80 mg, 0.079 mmol, 1 eq.) was dissolved in DMSO (530 μl, 0.15 M), before benzoquinone (26 mg, 0.240 mmol, 3 eq.) and [(2,9-dimethyl-1,10-phenanthroline)-Pd(μ-OAc)]2(OTf)2 (13 mg, 12 μmol, 15 mol%) were added. The reaction was stirred at room temperature till complete consumption of starting material (indicated by TLC (eluent: 25% H2O/CH3CN)). Upon completion the reaction mixture was diluted with H2O (15 ml) and the resulting aqueous solution was purified by charcoal column chromatography (3.75% tBuOH/H2O eluted the desired product). The product was freeze dried to yield an off white solid containing traces of hydroquinone. Hydroquinone was removed by washing the product in water with diethyl ether to yield 24 mg (0.024 mmol, 30%) of a white solid. 1H NMR (400 MHz, D2O) δ 5.60 (d, J = 4.6 Hz, 1H), 5.23–5.14 (m, 4H), 4.54 (d, J = 9.1 Hz, 1H), 4.44 (dd, J = 4.7, 1.5 Hz, 1H), 4.24 (dd, J = 9.6, 1.6 Hz, 1H), 3.78–3.53 (m, 22H), 3.52–3.38 (m, 10H), 3.09 (appt, J = 9.0 Hz, 1H). 13C NMR (101 MHz, D2O) δ 207.1, 102.0, 99.5, 99.4, 99.4, 99.3, 89.8, 76.7, 76.7, 76.7, 76.6, 76.5, 76.3, 76.2, 76.1, 76.1, 76.0, 75.6, 74.5, 73.1, 73.1, 72.9, 72.5, 71.4, 71.4, 71.3, 71.0, 71.0, 70.7, 69.1, 60.3, 60.3, 60.2, 60.2, 60.2, 60.0. HRMS (ESI) calculated for C36H59O30N3Na ([M + Na]+): 1036.31, found: 1036.31 IR νmax/cm−1: 3340 (OH), 2929 (C–H), 2123 (N3), 1739 (CO), 1026 (C–O), [α]20D = +122.4 (c 1.00, H2O).
β-D-3-Ketomaltoheptaosyl azide (16)
β-D-Maltoheptaosyl azide (62 mg, 52.6 μmol, 1 eq.) was dissolved in DMSO (350 μl, 0.15 M), before benzoquinone (17 mg, 158 μmol, 3 eq.) and [(2,9-dimethyl-1,10-phenanthroline)-Pd(μ-OAc)]2(OTf)2 (8 mg, 8 μmol, 15 mol%) were added. The reaction was stirred at room temperature till complete consumption of starting material (indicated by TLC (eluent: 40% H2O/CH3CN)). Upon completion the reaction mixture was diluted with H2O (5 ml) and the resulting aqueous solution was purified by charcoal column chromatography (3.5% tBuOH/H2O eluted the desired product). The product was freeze-dried to yield an off white solid containing traces of hydroquinone. Hydroquinone was removed by washing the product in water with diethyl ether to yield 29 mg (24.5 μmol, 47%) of an off-white solid. 1H NMR (400 MHz, CD3OD) δ 5.69 (d, J = 4.6 Hz, 1H), 5.32–5.22 (m, 5H), 4.63 (d, J = 8.1 Hz, 1H), 4.53 (dd, J = 4.6, 1.6 Hz, 1H), 4.32 (d, J = 9.8 Hz, 1H), 3.89–3.57 (m, 36H), 3.52 (m, 16H), 3.18 (appt, J = 9.1 Hz, 1H). 13C NMR (101 MHz, CD3OD) δ 207.2, 102.1, 99.5, 99.5, 99.5, 99.3, 89.8, 76.7, 76.5, 76.4, 76.2, 76.1, 75.6, 74.8, 74.6, 73.2, 73.2, 73.0, 72.6, 71.5, 71.5, 71.4, 71.1, 71.1, 70.8, 60.4, 60.4, 60.3, 60.3, 60.1 (12 signals are missing due to severe overlap). HRMS (ESI) calculated for C42H69O35N3Na ([M + Na]+): 1198.36, found: 1198.36 IR νmax/cm−1: 3343 (OH), 2924 (C–H), 2119 (N3), 1738 (CO), 1025 (C–O), [α]20D = +120.2 (c 1.00, H2O).
3-Keto-sucrose (17)
Sucrose (62 mg, 0.18 mmol, 1 eq.) and benzoquinone (58 mg, 0.54 mmol, 3 eq.) were dissolved in DMSO-d6 (600 μl, 0.3 M) and transferred to a NMR tube. The T1 relaxation time was determined followed by a spectrum at t = 0 to determine the ratio of DMSO:starting material. [(Neocuproine)PdOAc]2OTf2 (4.7 mg, 4.5 μmol, 2.5 mol%) was added to the NMR tube, mixed and the reaction monitored till completion (1 h). 50% of the product mixture was 3-keto-sucrose 1H NMR (400 MHz, DMSO-d6) δ 5.60 (d, J = 4.5 Hz, 1H, H1), 4.27 (d, J = 4.4 Hz, 1H, H2), 4.14 (d, J = 9.7 Hz, 1H, H4), 3.94–3.86 (m, 2H, H3′ + H5), 3.75–3.61 (m, 3H, H4′ + H6), 3.61–3.55 (m, 3H, H5′ + H6′), 3.50–3.41 (m, 1H, H1a′), 3.40–3.32 (m, 1H, H1b′). 13C NMR (101 MHz, DMSO-d6) δ 206.6 (C3), 104.5 (C2′), 94.5 (C1), 82.8 (C5′), 76.2 (C3′), 75.6 (C5), 74.2 (C2), 74.1 (C4′), 71.6 (C4), 62.3 (C6′), 61.8 (C1′), 60.4 (C6).
Acknowledgements
Financial support from The Netherlands Organization for Scientific Research (NWO-CW) is acknowledged. Dr S. S. van Leeuwen is acknowledged for his support in the NMR analysis of the oligosaccharides. Prof. Dr F. J. Dekker and prof. Dr G. J. Poelarends are kindly acknowledged for providing the 4-OT R61C-1 protein mutant.Notes and references
- M. White, Synlett, 2012, 23, 2746 CAS.
- J. L. Jeffrey, J. A. Terrett and D. W. C. MacMillan, Science, 2015, 349, 1532 CrossRef CAS PubMed.
- M. Jäger and A. J. Minnaard, Chem. Commun., 2016, 52, 656 RSC.
- P. A. Jordan and S. J. Miller, Angew. Chem., Int. Ed., 2012, 51, 2907 CrossRef CAS PubMed.
- M. Bierenstiel and M. Schlaf, Eur. J. Org. Chem., 2004, 2004, 1474 CrossRef.
- W. Muramatsu, Org. Lett., 2014, 16, 4846 CrossRef CAS PubMed.
- R. M. Painter, D. M. Pearson and R. M. Waymouth, Angew. Chem., Int. Ed., 2010, 49, 9456 CrossRef CAS PubMed.
- M. Jäger, M. Hartmann, J. G. de Vries and A. J. Minnaard, Angew. Chem., Int. Ed., 2013, 52, 7809 CrossRef PubMed.
- T. J. Styslinger, N. Zhang, V. S. Bhatt, N. Pettit, A. F. Palmer and P. G. Wang, J. Am. Chem. Soc., 2012, 134, 7507 CrossRef CAS PubMed.
- T. Tanaka, T. Matsumoto, M. Noguchi, A. Kobayashi and S. Shoda, Chem. Lett., 2009, 38, 458 CrossRef CAS.
- T. Tanaka, W. C. Huang, M. Noguchi, A. Kobayashi and S. I. Shoda, Tetrahedron Lett., 2009, 50, 2154 CrossRef CAS.
- M. Noguchi, T. Tanaka, H. Gyakushi, A. Kobayashi and S. Shoda, J. Org. Chem., 2009, 74, 2210 CrossRef CAS PubMed.
- D. Lim, M. a. Brimble, R. Kowalczyk, A. J. a. Watson and A. J. Fairbanks, Angew. Chem., Int. Ed., 2014, 53, 11907 CrossRef CAS PubMed.
- T. Tanaka, H. Nagai, M. Noguchi, A. Kobayashi and S.-I. Shoda, Chem. Commun., 2009, 3378 RSC.
- The presence of salts was confirmed by positive test with AgNO3 solution.
- R. L. Whistler and D. F. Durso, J. Am. Chem. Soc., 1950, 72, 677 CrossRef CAS.
- K. Bock and C. Pedersen, Adv. Carbohydr. Chem. Biochem., 1983, 41, 27 CrossRef CAS.
- K. Bock, C. Pedersen and H. Pedersen, Adv. Carbohydr. Chem. Biochem., 1984, 42, 193 CrossRef CAS.
- P. Sedmera, P. Halada, E. Kubátová, D. Haltrich, V. Přikrylová and J. Volc, J. Mol. Catal. B: Enzym., 2006, 41, 32 CrossRef CAS.
- F. Pertici, N. J. de Mol, J. Kemmink and R. J. Pieters, Chem. – Eur. J., 2013, 19, 16923 CrossRef CAS PubMed.
- M. Petitou, J.-P. Herault, A. Bernat, P.-A. Driguez, P. Duchaussoy, J.-C. Lormeau and J.-M. Herbert, Nature, 1999, 398, 417 CrossRef CAS PubMed.
- T. Fyrner, K. Magnusson, K. P. R. Nilsson, P. Hammarström, D. Aili and P. Konradsson, Bioconjugate Chem., 2012, 23, 1333 CrossRef CAS PubMed.
- M. F. Schneider, G. Mathe, M. Tanaka and R. R. Schmidt, J. Phys. Chem. B, 2001, 105, 5178 CrossRef CAS.
- M. E. Ourailidou, P. Dockerty, M. Witte, G. J. Poelarends and F. J. Dekker, Org. Biomol. Chem., 2015, 13, 3648 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ob00608f |
| This journal is © The Royal Society of Chemistry 2016 |