
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
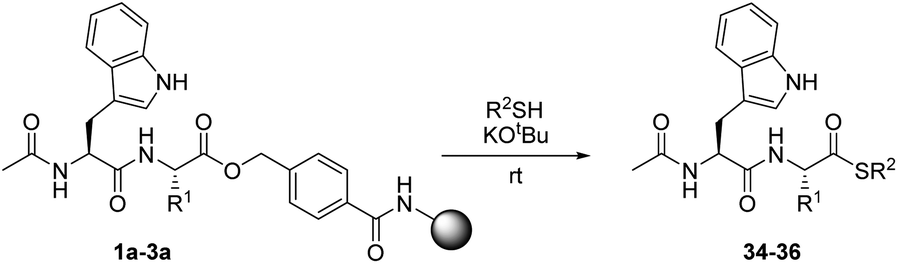
C-Terminally modified peptides via cleavage of the HMBA linker by O-, N- or S-nucleophiles†
J.
Hansen
,
F.
Diness
and
M.
Meldal
*
Center for Evolutionary Chemical Biology, Department of Chemistry University of Copenhagen, Universitetsparken 5, 2100 Copenhagen, Denmark. E-mail: meldal@nano.ku.dk
First published on 18th February 2016
Abstract
A large variety of C-terminally modified peptides was obtained by nucleophilic cleavage of the ester bond in solid phase linked peptide esters of 4-hydroxymethyl benzamide (HMBA). The developed methods provided peptides, C-terminally functionalized as esters, amides and thioesters, with high purity directly from the resin in a single reaction step. A comprehensive screening of the reaction conditions and scope for nucleophilic cleavage of peptides from the HMBA linker was performed.
Introduction
The ever expanding applications of solid phase peptide synthesis (SPPS)1 for the preparation of bioactive peptides and small proteins have been a driving force in the development of numerous linkers for C-terminal attachment of peptides to polymer resins.2,3 However, most linkers are only effectively cleaved under one set of cleavage conditions and they produce only a single functionality in the cleavage step. Hence, when several C-terminal derivatives of a particular peptide are required, a different linker is needed for each derivative. This is in contrast to modification of the N-terminus of peptides, which may be freely functionalized while these are still attached to the resin. C-Terminally modified peptides are nevertheless interesting for a variety of reasons. The nature of the C-terminal is important in medicinal chemistry, as it impacts peptide binding, affects stability and provides selectivity in binding to enzymes.4,5 Of particular interest is access to C-terminal peptide carboxylic acids, esters, thioesters and amides, as these are found in natural peptides, in their analogues or may even be needed as precursors for protein synthesis.6–9 The majority of synthesized peptides are attached to the solid support, either through amide or ester linkers. Amide linkers are almost exclusively cleaved by an acid whereas ester linkers can be cleaved by either a base or an acid, depending on the nature of the linker. Because acid labile ester linkers are cleaved via a SN1 reaction to release the linker as a carbonium ion, the cleavage occurs between the acidic oxygen and linker carbon. Commonly, base labile ester linkers cleave via a nucleophilic substitution reaction by attack on the carbonyl group of the ester, which leads to cleavage between the oxygen and the carbonyl group (Scheme 1). Hence, the applied nucleophile is attached to the peptide carbonyl group after the release and thereby provides the opportunity for modifying the C-terminal with various nucleophiles in the cleavage step.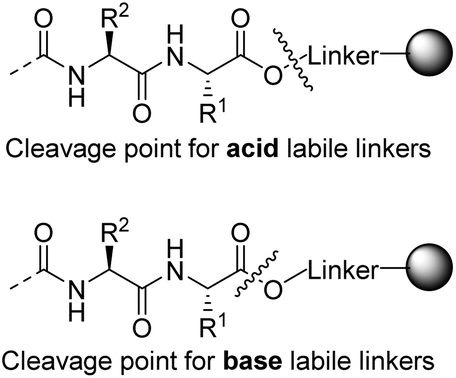 | ||
| Scheme 1 The different modes of reaction observed for acid labile vs. base labile linkers. | ||
A commonly used base labile linker is 4-hydroxymethyl benzamide (HMBA).10 One major benefit of the HMBA-linker is that peptides can be deprotected with TFA, while remaining attached to the resin. This then allows the deprotected peptides to be screened for activity on the resin and the hit compounds can later be cleaved for analysis, using a low concentration of an aqueous base. The purity of the cleaved peptides is usually very high and purification is frequently not required prior to characterization and use.11 Other base labile linkers have been developed over the years for specialized purposes.12–15 Concerning the functionalization of peptides, only a few examples of cleaving the HMBA linker with other nucleophiles than hydroxide have been reported, including the use of ammonia/THF vapor16,17 and a study using a few nucleophiles to produce esters and amides.18 However, a thorough investigation on the scope of the HMBA linker, in cleavage reactions using a variety of different nucleophiles, has not been performed.
Results and discussion
In order to evaluate the reaction of HMBA–esters with nucleophiles, three resin-bound dipeptide substrates were prepared, in which the ester linked amino acid was varied: Ac-Trp-Gly-HMBA-PEGA800 (1a), Ac-Trp-Ala-HMBA-PEGA800 (2a) and Ac-Trp-Ile-HMBA-PEGA800 (3a). These substrates allowed the steric influence at Cα on nucleophilic ester cleavage to be determined. The cleavage products were synthesized in sub-milligram amounts and identified by HPLC and MS. As HMBA linked esters are rapidly cleaved by hydroxide, it was hypothesized that alkoxides would only cleave by transesterification under strictly water free reaction conditions. Initial tests with 1,8-diazabicycloundec-7-ene (DBU) as the catalytic base in combination with methanol or ethanol led to partial release of the peptides as the corresponding esters. Further screening of non-nucleophilic bases proved that potassium tert-butoxide was superior, as it was sufficiently strong to facilitate the formation of the alkoxide and thereby, the reaction between all the substrates and the investigated alcohols. Using these conditions, four different alcohols were reacted with three different resin-bound peptides (Table 1). It was found that primary alkoxides readily effect transesterification reactions of the peptide–linker ester bond, as seen by short reaction times and complete conversion with methanol and ethanol respectively (Table 1, entries 1–6). However, under prolonged reaction times, the irreversible hydrolysis of the products was also frequently observed. As expected the reaction time needed for full conversion was longer for the more sterically hindered amino acid derivatives and the more substituted alcohol nucleophiles. When the combination of the nucleophile and substrate became sterically unfavorable for the reaction, conversion proceeded more slowly and prevention of the competing hydrolysis, presumably by trace amounts of water in the resin, proved difficult.|
|
|||||
|---|---|---|---|---|---|
| Entry | R1 | R2OH (product) | Time (min) | Conversion (%) | Purity (%) |
Resin (5 mg, 1 μmol) was treated with alcohol/alcoholate (200 μL, 89 μmol). Reactions were quenched (200 μL, 1% TFA/ACN). The solution and subsequent washings (H2O/CH3CN, 7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) were analyzed by HPLC. :1) were analyzed by HPLC. |
|||||
| 1 | H | MeOH (4) | 15 | 100 | >95 |
| 2 | Me | MeOH (5) | 30 | 100 | >95 |
| 3 | (S)sBu | MeOH (6) | 60 | 100 | >95 |
| 4 | H | EtOH (7) | 15 | 100 | >95 |
| 5 | Me | EtOH (8) | 30 | 100 | >95 |
| 6 | (S)sBu | EtOH (9) | 60 | 100 | >95 |
| 7 | H | iPrOH (10) | 15 | 30 | >90 |
| 8 | Me | iPrOH (11) | 30 | 20 | >90 |
| 9 | (S)sBu | iPrOH (12) | 120 | 100 | >90 |
| 10 | H | iBuOH (13) | 15 | 40 | >90 |
| 11 | Me | iBuOH (14) | 30 | 20 | >90 |
| 12 | (S)sBu | iBuOH (15) | 120 | 100 | >90 |
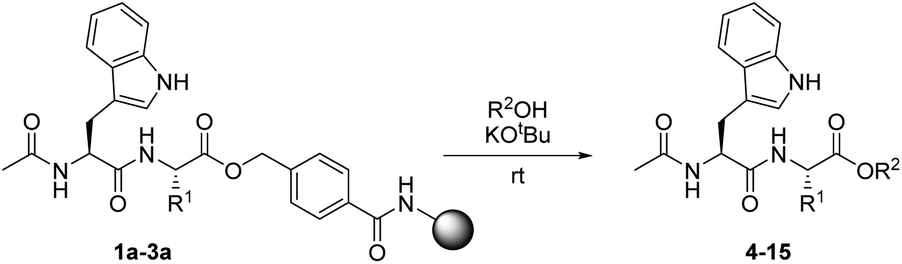
A balanced reaction time was determined where the product was obtained in high yield with negligible or little hydrolysis. For instance, reactions of 10 and 13 (Table 1, entries 7 and 10), with sterically hindered alcohols required 30 min for complete conversion. However, this period of time resulted in approximately 80% hydrolysis, while stopping the reactions at 15 min ensured purity of the product above 90% with about 30% conversion. A unique effect was observed for the isoleucine substrate, as it appeared to be more resistant to hydrolysis of the product ester.
Few other methods for obtaining peptides with C-terminal esters from resin bound amino acids have been reported. One method relies on a one pot two-step procedure from the Wang linker, forming esters under acidic condensation conditions.19 Another technique utilizes preformed C-terminal esters combined with acid labile linking to the resin through the side-chains, e.g. through the thiol of cysteine.20 However, the method presented here allows variation of the alcohol/alkoxide in the ester formation combined with cleavage in a one-step fashion.
In order to quantify the reactivity of the HMBA linked esters compared to simple alkyl esters, the transesterification reactions were studied in the solution phase (Scheme 2). To determine the reaction rates, the dipeptide Ac-Trp-Ala was prepared as either 4-carbamoylbenzyl ester (16) or isobutyl ester (17).
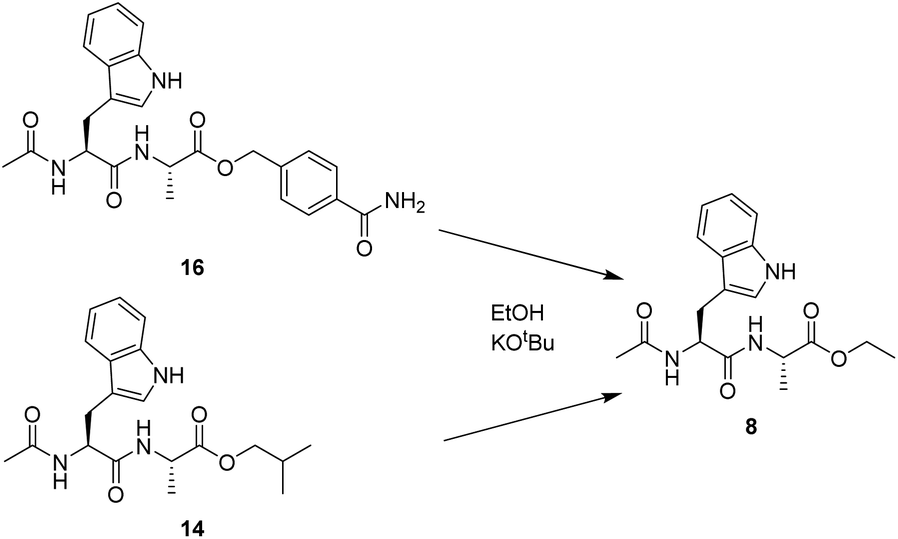 | ||
| Scheme 2 Comparison of transesterification kinetics for an aliphatic ester and the HMBA linker. | ||
The two esters were then used for transesterification with ethoxide, generated by addition of potassium tert-butoxide to ethanol. Both compounds 16 and 17 formed exclusively the ethyl ester product (8), but at significantly different rates. The halftime t1/2 for the transesterification reaction was 12 seconds for 16 and 2.25 minutes for 17, respectively. The more than 10-fold increase in the rate of transesterification of the HMBA ester demonstrates that the application of the HMBA is superior to alkyl esters as a leaving group in nucleophilic substitution reactions.
Aminolysis of simple alkyl esters is typically a slow process that often requires heating. However, in light of the described reactivity studies, aminolysis of the HMBA linked esters seemed feasible. A series of amines were applied in reactions with the resin bound peptide esters to produce the corresponding amides. Initially sodium amide in dioxane was tested as a nucleophile, which however resulted in complex mixtures of products. Therefore, all reactions with amines were performed with the neat primary amine, without application of an additional base (Table 2). The differences in reaction times and optimal conversion conditions were more pronounced with the amines, compared to those of the alcohols. Heating to 40 °C was attempted to increase the rate of the reactions and to reach complete conversion. A higher degree of conversion was observed, but heating also resulted in undesirable formation of byproducts. Most amines studied, proved to be sufficiently nucleophilic for amides to be produced. Particularly when using the glycine ester substrate, amides were readily formed (Table 2, entries 1 and 7).
|
|
|||||
|---|---|---|---|---|---|
| Entry | R1 | R2NH2 | Time (h) | Conversion (%) | Purity (%) |
| Resin (5 mg, 1 μmol) was treated with amine (200 μL, neat). The solution was drained and the resin was washed with (H2O/CH3CN/TFA, 7:1:0.1) for HPLC analysis. |
|||||
| 1 | H | PrNH2 (19) | 2 | 100 | >95 |
| 2 | Me | PrNH2 (20) | 4 | 99 | >95 |
| 3 | (S)sBu | PrNH2 (21) | 6 | 5 | — |
| 4 | H | iPrNH2 (22) | 2 | 40 | >95 |
| 5 | Me | iPrNH2 (23) | 4 | 5 | — |
| 6 | (S)sBu | iPrNH2 (24) | 6 | 0 | — |
| 7 | H | iBuNH2 (25) | 2 | 100 | >95 |
| 8 | Me | iBuNH2 (26) | 4 | 60 | >95 |
| 9 | (S)sBu | iBuNH2 (27) | 6 | 0 | — |
| 10 | H | (CH2NH2)2 (28) | 2 | 100 | >90 |
| 11 | Me | (CH2NH2)2 (29) | 4 | 100 | >90 |
| 12 | (S)sBu | (CH2NH2)2 (30) | 6 | 60 | >90 |
| 13 | H | HO(CH2)2NH2 (31) | 2 | 100 | >90 |
| 14 | Me | HO(CH2)2NH2 (32) | 4 | 100 | >90 |
| 15 | (S)sBu | HO(CH2)2NH2 (33) | 8 | 50 | >90 |
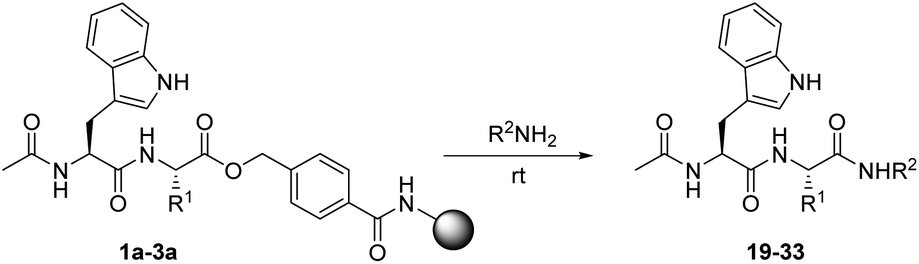
On the other hand, isoleucine proved to be too sterically hindered to facilitate complete conversion in less than 6 hours and only unbranched amines were sufficiently reactive towards the isoleucine substrate to yield peptide amides. It is noteworthy that ethylene diamine and ethanolamine react much faster with the isoleucine resin probably through anchimeric assistance by the distal amine or hydroxyl group, respectively (Table 2, entries 12 and 15). Furthermore, even though amines are basic, no issues with hydrolysis were observed indicating that no competing hydroxide was formed.
Reports on synthesis of N-alkyl amides of peptides are quite scarce. However, primary amides have previously been reported, synthesized from esters using ammonia vapor.16 A few alkyl amides have been synthesized in 13–16% yield using the HMBA linker.18 A more elaborated alternative, has been N-alkylation of the PAL linker with alkyl halides followed by acid cleavage.4 The synthesis requires three steps and several reagents, while in the present approach, the amine is the sole reagent in a one-step reaction.
Due to the general interest in peptide thioesters for chemical ligation, 1-propanethiol was investigated as a nucleophile in reactions with the three model substrates, 1a–3a. In this study, an approach similar to that used for alkoxides, was employed. Under optimized conditions, the corresponding thioester products were obtained in high purity from peptides 1a and 2a (Table 3, entries 1 and 2). Only a limited amount of hydrolysis was observed, presumably because deprotonated thiols do not generate significant amounts of hydroxide in contact with traces of water.
|
|
|||||
|---|---|---|---|---|---|
| Entry | R1 | R2SH | Time (h) | Conversion (%) | Purity (%) |
| Resin (5 mg, 1 μmol) was treated at 20 °C with thiol/thiolate (200 μL, 89 μmol). Reactions were quenched (200 μL, 1% TFA/ACN). The solution and subsequent washings (H2O/CH3CN, 7:1) were analyzed by HPLC. |
|||||
| 1 | H | PrSH (34) | 6 | 70 | 60 |
| 2 | Me | PrSH (35) | 6 | 70 | 60 |
| 3 | (S)sBu | PrSH (36) | 6 | 0 | — |
It was observed that in the presence of traces of oxygen, the thiols formed disulfides under base catalysis by potassium tert-butoxide. The problem was however significantly reduced by passing nitrogen through the solution, during these reactions.
Originally thioesters were reported to be synthesized using Boc/Bzl chemistry.21,22 The progress in the synthesis of this class of molecules has recently been reviewed, including different methods compatible with Fmoc-SPPS.23 A popular method is application of the sulfonamide “safety catch” linker, which after activation can be converted to thioesters by nucleophilic cleavage.24 The BAL linker may also be used to produce thioesters but only from peptides starting with a C-terminal glycine.25 The rearrangements O→S26 and N→S have also been explored in linker strategies,27 where a thiol functionalized linker and thiol exchange are used to facilitate the release of peptides as thioesters.
None of these methods combine the ability to deprotect the peptide on resin followed by it being released as a thioester into solution. The present nucleophilic cleavage of the HMBA linker with thiolates, allows for this convenient combination and simultaneously reduces the overall number of synthetic steps required for the synthesis of thioesters.
To demonstrate the preparative synthesis of esters, amides and thioesters by the present approach, 150 mg aliquots of Ac-Trp-Ala-HMBA (2a) resin were cleaved using ethanol, propylamine or 1-propanethiol, respectively. Excess nucleophile was evaporated and purification was performed by reverse phase flash column chromatography (for further details, see the ESI†). The products were analyzed by HPLC, NMR, and MS. The purified compounds were isolated in yields of 77%, 90% and 40%, respectively. Reaction with 1-propanethiol induced racemization and 24 hour treatment resulted in complete racemization and a 50:50 distribution of the two isomers. As a consequence, glycine was preferred as the C-terminal amino acid when thiols were used as nucleophiles.
Conclusions
In summary, we have provided a method for the synthesis of a variety of C-terminally modified peptides, prepared by nucleophilic cleavage of fully deprotected, resin bound peptides. In the absence of hydroxide ions, the HMBA–esters presented good reactivity in substitution reactions with a large variety of nucleophiles. Generally alcoholates react fast and efficiently, with high purity of the target peptide esters, and are less sensitive to steric bulk than the corresponding amines. Care needs to be taken to prevent hydrolysis, which in some cases is difficult. The amines are less reactive nucleophiles but at a high concentration they still react to form the corresponding amides when less hindered, sterically. The reactions of the amines do not require any additional base or strictly anhydrous conditions. Thioesters of peptides, valuable for native chemical ligation reactions were successfully formed by reaction of the HMBA linked peptides with thiolates. Because of racemization in these reactions the C-terminal amino acid used should preferably be glycine. Thus utilizing the different reactivities offered by the HMBA linker, the present methodology alleviates the urgent need for specialized linkers, dedicated to the introduction of a particular functionality.Experimental section
General
All purchased chemicals were used without further purification. All solvents were HPLC-grade. Solid phase synthesis was performed on a VA800 resin, bead size 200–400 (loading 0.200 mmol g−1) from Versa matrix. Flash chromatography (FC) was carried out on Merck silica gel 60 (0.040–0.063 mm) and analytical TLC was performed using Merck silica gel 60 F254 aluminum plates. Vacuum liquid chromatography (VLC) was carried out on Merck silica gel 60 (0.015–0.040 mm) or on IST Isolute Sorbent C18 gel 60 (0.040–0.070 mm). All HPLC analyses were performed on an analytical Agilent 1100 HPLC-MS using a 100 mm XBridge C18 column. A linear gradient of acetonitrile in water with 0.1% TFA was used, running from 0% to 90% acetonitrile, 1 mL min−1 over 10 min. The detection was performed by measurement of the absorbance of 254 nm UV-light. NMR spectra were recorded on a Bruker ADVANCE III 500 MHz CRYO probe instrument. Chemical shifts are reported in ppm relative to residual solvent signals (CDCl3, 7.26 ppm; DMSO-d6, 2.50 ppm) for 1H NMR spectra and relative to the central solvent resonance (CDCl3, 77.0 ppm; DMSO-d6, 39.5 ppm) for 13C NMR spectra. The following abbreviations are used to indicate the multiplicity in 1H and 13C NMR spectra: s, singlet; d, doublet; t, triplet; q, quartet; dd, double doublet; ddd, double double doublet; dddd, double double double doublet; dt, double triplet; m, multiplet. 13C spectra were acquired in broadband decoupled mode. High resolution mass spectrometry (HRMS) was performed on a Bruker micro-TOF using positive electrospray ionization.Synthesis of test substrates
Esterification of the resin with Fmoc amino acids in dichloromethane (DCM) was carried out using the conditions reported by Blankemeyer-Menge et al.28 All peptide couplings followed the standard Fmoc based synthesis conditions with preactivation (3 min) of Fmoc-amino acid (3 equiv.), with 2-(1-H-benzotriazolyl-1-yl)-1,1,3,3-tetramethyluronium tetrafluoroborate (TBTU) (2.9 equiv.) and 4-ethylmorpholine (NEM) (4 equiv.) in N,N-dimethylformamide (DMF). These conditions were also used for N-terminal acetylation using acetic acid. Fmoc-cleavage was achieved with 20% piperidine in DMF and between reactions the resin was washed with 10 resin volumes of DMF.General procedure for cleavage using sodium hydroxide
Resin (5 mg, 1 μmol) was transferred to a filtration column (2 mL). Aqueous sodium hydroxide (200 μL, 0.1 M) was added to the resin and the mixture was allowed to react for 15 min. The solution was neutralized using aqueous hydrochloric acid (200 μL, 0.1 M) and the eluate was collected. Any remaining product was extracted from the resin by washing with 7:3 H2O/CH3CN containing TFA (1%, 2 × 200 μL). The combined solution was analysed by quantitative HPLC.
Method 1. General procedure for cleavage using alcohols or thiols
Vacuum dried resin (5 mg, 1 μmol) was transferred to a 2 mL SPE filtration column. The cleavage solution (200 μL, 0.089 mmol, 89 equiv.) of alcoholate or thiolate in the corresponding solvent prepared by addition of potassium t-butoxide (10 mg, 0.089 mmol per mL of the solvent) was added to the resin. The full conversion varied according to the structure of the peptide and nucleophile, from 15 min to hours. The reaction mixture was then acidified using TFA (1% in acetonitrile, 200 μL) and the eluate was collected. The remaining product was extracted from the resin by washing with H2O/CH3CN (7:3) containing TFA (1%, 2 × 200 μL). The combined eluate was analysed by quantitative HPLC.
Method 2. General procedure for cleavage using amines
Vacuum dried resin (5 mg, 1 μmol) was transferred to a 2 mL SPE filtration column. Amine (200 μL, 2.00–3.30 mmol, 2000–3300 equiv.) was added to the resin and the mixture was allowed to react for 2–6 hours depending on the substrate resin. The solution was quenched using TFA (1% in acetonitrile 200 μL) and the eluate was collected. Any remaining product was extracted from the resin by washing with H2O/CH3CN (7:3) containing TFA (1%, 2 × 200 μL). The combined solution was analysed by quantitative HPLC.
NMR Data of crude compounds
:5:5:2) and reduced on a freeze dryer. The compound was purified by reverse phase flash column chromatography and isolated in 50% yield, compared to loading after first amino acid coupling and was used without further purification. 1H NMR (500 MHz, DMSO-d6) δ 10.78 (s, 1H), 8.56 (d, J = 6.9 Hz, 1H), 8.04 (d, J = 8.3 Hz, 1H), 7.97 (s, 1H), 7.90–7.84 (m, 2H), 7.59 (dd, J = 7.9, 1.0 Hz, 1H), 7.44 (d, J = 8.3 Hz, 2H), 7.37 (s, 1H), 7.32 (dt, J = 8.2, 0.9 Hz, 1H), 7.13 (d, J = 2.4 Hz, 1H), 7.06 (ddd, J = 8.1, 6.9, 1.2 Hz, 1H), 6.97 (ddd, J = 8.0, 6.9, 1.1 Hz, 1H), 5.18 (s, 2H), 4.59 (ddd, J = 9.8, 8.3, 4.4 Hz, 1H), 4.38 (p, J = 7.2 Hz, 1H), 3.12–3.03 (m, 1H), 2.85 (dd, J = 14.8, 9.8 Hz, 1H), 1.76 (s, 3H), 1.34 (d, J = 7.4 Hz, 3H). 13C-NMR DMSO-d6δ: 172.3, 171.9, 169.0, 167.4, 139.2, 136.0, 133.8, 127.5, 127.2, 127.2, 123.5, 120.8, 118.4, 118.1, 111.2, 110.1, 65.3, 52.8, 47.7, 27.8, 22.5, 16.7. m/z [M + H+], calc.: 451.1976, found: 451.1980.
Relative reactivity between HMBA and ethylester
Dry compound 16 (1 mg, 2.2 μmol) or 17 (1 mg, 2.7 μmol) was dissolved in dry ethanol (100 μL). Dry ethanol (100 μL) containing potassium tert-butoxide (1 mg) was added. Samples (50 μL) were collected and quenched using 2% TFA in CH3CN (200 μL,) after 15, 30 and 60 s for compound 16 and 2, 5 and 10 min for compound 17. The reaction temperatures were at all times fixed at 20 °C and conversion was assessed by quantitative HPLC. The 4-carbamoyl peptide 16 was reduced by 75% within 30 seconds and verified using LC-MS, whereas the iBu-ester 17 was still present in 15% after 10 min. The halftimes t1/2 of the reaction with ethanolate were 12 seconds for 16 and 2.25 min for 17 respectively.:3) containing TFA (1%, 2 × 1000 μL). The combined eluate was analyzed by quantitative HPLC, the volume was reduced and the compound purified by reverse phase, flash chromatography. 1H NMR (500 MHz, DMSO-d6) δ 10.80 (s, 1H), 8.47 (d, J = 7.0 Hz, 1H), 8.02 (d, J = 8.3 Hz, 1H), 7.64 (dd, J = 7.8, 1.0 Hz, 1H), 7.33 (dt, J = 8.1, 0.9 Hz, 1H), 7.15 (d, J = 2.4 Hz, 1H), 7.06 (ddd, J = 8.1, 6.9, 1.2 Hz, 1H), 6.98 (ddd, J = 8.0, 7.0, 1.1 Hz, 1H), 4.58 (ddd, J = 9.5, 8.3, 4.5 Hz, 1H), 4.27 (p, J = 7.2 Hz, 1H), 4.09 (qd, J = 7.1, 3.1 Hz, 2H), 3.11 (ddd, J = 14.7, 4.5, 0.9 Hz, 1H), 2.96–2.80 (m, 1H), 1.76 (s, 3H), 1.30 (d, J = 7.3 Hz, 3H), 1.18 (t, J = 7.1 Hz, 3H). 13C NMR (126 MHz, DMSO) δ 172.4, 171.8, 169.0, 136.0, 127.3, 123.5, 120.8, 118.4, 118.1, 111.2, 110.1, 60.4, 52.9, 47.7, 27.8, 22.5, 16.8, 14.0. m/z [M + H+], calc.: 346.1761, found: 346.1783.
:3) containing TFA (1%, 2 × 1000 μL). The combined eluate was analyzed by quantitative HPLC, the volume was reduced and the compound purified by reverse phase, flash chromatography. 1H NMR (500 MHz, DMSO-d6) δ 10.82 (s, 1H), 8.05 (t, J = 7.2 Hz, 2H), 7.60 (dd, J = 7.7, 1.0 Hz, 1H), 7.53 (t, J = 5.8 Hz, 1H), 7.32 (dt, J = 8.1, 0.9 Hz, 1H), 7.16 (d, J = 2.4 Hz, 1H), 7.06 (ddd, J = 8.1, 6.9, 1.2 Hz, 1H), 6.98 (ddd, J = 8.0, 7.0, 1.1 Hz, 1H), 4.54 (ddd, J = 8.8, 7.8, 5.1 Hz, 1H), 4.21 (p, J = 7.1 Hz, 1H), 3.17–3.08 (m, 1H), 2.96 (qd, J = 7.0, 2.2 Hz, 2H), 2.93–2.88 (m, 1H), 1.79 (s, 3H), 1.36 (h, J = 7.3 Hz, 2H), 1.19 (d, J = 7.1 Hz, 3H), 0.82 (t, J = 7.4 Hz, 3H). 13C NMR (126 MHz, DMSO) δ 171.7, 171.3, 169.3, 158.4, 158.1, 136.0, 127.3, 123.5, 120.8, 118.4, 118.1, 111.2, 110.1, 53.4, 48.3, 27.6, 22.5, 22.2, 18.2, 11.2. m/z [M + H+], calc.: 359.2078, found: 359.2078.
:3) containing TFA (1%, 2 × 1000 μL). The combined eluate was analyzed by quantitative HPLC, the volume was reduced and the compound purified by reverse phase, flash chromatography. 1H NMR (500 MHz, DMSO-d6) δ 10.72 (s, 1H), 8.71 (d, J = 7.2 Hz, 0.5H), 8.52 (d, J = 7.4 Hz, 0.5H), 8.00 (dd, J = 8.3, 2.8 Hz, 1H), 7.61 (d, J = 7.8 Hz, 0.5H), 7.51 (d, J = 7.8 Hz, 0.5H), 7.30–7.22 (m, 1H), 7.08 (dd, J = 14.5, 2.4 Hz, 1H), 6.99 (m, J = 8.1, 6.6, 5.3, 1.2 Hz, 1H), 6.95–6.86 (m, 1H), 4.62–4.51 (m, 1H), 4.34 (p, J = 7.2 Hz, 0.5H), 4.23 (p, J = 7.2 Hz, 0.5H), 3.16 (dd, J = 14.7, 4.1 Hz, 0.5H), 3.01 (dd, J = 14.5, 5.6 Hz, 0.5H), 2.83 (m, J = 14.7, 9.4 Hz, 1H), 2.69 (dd, J = 7.8, 6.4 Hz, 2H), 1.72 (s, 1.5H), 1.67 (s, 1.5H), 1.43 (m, J = 7.3, 2.2 Hz, 2H), 1.22 (d, J = 7.3 Hz, 1.5H), 1.08 (d, J = 7.3 Hz, 1.5H), 0.82 (td, J = 7.3, 3.1 Hz, 3H). 13C NMR (126 MHz, DMSO) δ 202.0, 171.8, 168.8, 136.0, 127.3, 123.7, 120.8, 118.4, 111.2, 110.1, 99.5, 53.0, 29.6, 28.0, 27.4, 22.5, 22.4, 17.3, 13.1. m/z [M + H+], calc.: 376.1689, found: 376.1660 m/z [M + H+], calc.: 376.1689, found: 376.1660.
Acknowledgements
The University of Copenhagen supported this work with the Center for Evolutionary Chemical Biology. We thank Theis Borch-Nannestad for support with LC-MS equipment, and Dr Sanne Schoffelen and Ming Li for obtaining high resolution MS data.References
- G. B. Fields and R. L. Noble, Int. J. Pept. Protein Res., 1990, 35, 161 CrossRef CAS PubMed.
- F. Guillier, D. Orain and M. Bradley, Chem. Rev., 2000, 100, 2091 CrossRef CAS PubMed.
- Peptide Synthesis and Applications, ed. K. J. Jensen, P. T. Shelton and S. L. Pedersen, Humana Press Inc, 999 Riverview Dr, Ste 208, Totowa, Nj 07512-1165 USA, 2nd edn, 2013, vol. 1047 Search PubMed.
- W. J. Fang, T. Yakovleva and J. V. Aldrich, Biopolymers, 2011, 96, 715 CrossRef CAS PubMed.
- M. M. Meijler, R. Arad-Yellin, Z. I. Cabantchik and A. Shanzer, J. Am. Chem. Soc., 2002, 124, 12666 CrossRef CAS PubMed.
- L. Andersson, L. Blomberg, M. Flegel, L. Lepsa, B. Nilsson and M. Verlander, Biopolymers, 2000, 55, 227 CrossRef CAS PubMed.
- A. K. Ghose, V. N. Viswanadhan and J. J. Wendoloski, J. Comb. Chem., 1999, 1, 55 CrossRef CAS PubMed.
- A. M. Bray, A. G. Jhingran, R. M. Valerio and N. J. Maeji, J. Org. Chem., 1994, 59, 2197 CrossRef CAS.
- C. P. R. Hackenberger and D. Schwarzer, Angew. Chem., Int. Ed., 2008, 47, 10030 CrossRef CAS PubMed.
- E. Atherton, C. J. Logan and R. C. Sheppard, J. Chem. Soc., Perkin Trans. 1, 1981, 538 RSC.
- M. Meldal and I. Svendsen, J. Chem. Soc., Perkin Trans. 1, 1995, 1591 RSC.
- S. B. Katti, P. K. Misra, W. Haq and K. B. Mathur, J. Chem. Soc., Chem. Commun., 1992, 843 RSC.
- F. Rabanal, E. Giralt and F. Albericio, Tetrahedron, 1995, 51, 1449 CrossRef CAS.
- C. Garcia-Echeverria, Tetrahedron Lett., 1997, 38, 8933 CrossRef CAS.
- J. J. N. Veerman, F. P. J. T. Rutjes, J. H. van Maarseveen and H. Hiemstra, Tetrahedron Lett., 1999, 40, 6079 CrossRef CAS.
- A. M. Bray, R. M. Valerio and N. J. Maeji, Tetrahedron Lett., 1993, 34, 4411 CrossRef CAS.
- J. M. Brown, W. D. Hoffmann, C. M. Alvey, A. R. Wood, G. F. Verbeck and R. A. Petros, Anal. Biochem., 2010, 398, 7 CrossRef CAS PubMed.
- W. R. Abd-Elgaliel, F. Gallazzi and S. Z. Lever, J. Pept. Sci., 2007, 13, 487 CrossRef CAS PubMed.
- R. A. Turner, R. J. Weber and R. S. Lokey, Org. Lett., 2010, 12, 1852 CrossRef CAS PubMed.
- V. Diaz-Rodriguez, D. G. Mullen, E. Ganusova, J. M. Becker and M. D. Distefano, Org. Lett., 2012, 14, 5648 CrossRef CAS PubMed.
- H. Hojo and S. Aimoto, Bull. Chem. Soc. Jpn., 1991, 64, 111 CrossRef CAS.
- A. Toften, K. Sørensen, K. Conde-Frieboes, T. Hoeg-Jensen and K. Jensen, Angew. Chem., Int. Ed., 2009, 48, 7411 CrossRef PubMed.
- F. Mende and O. Seitz, Angew. Chem., Int. Ed., 2011, 50, 1232 CrossRef CAS PubMed.
- R. Ingenito, E. Bianchi, D. Fattori and A. Pessi, J. Am. Chem. Soc., 1999, 121, 11369 CrossRef CAS.
- J. Brask, F. Albericio and K. Jensen, Org. Lett., 2003, 5, 2951 CrossRef CAS PubMed.
- J. D. Warren, J. S. Miller, S. J. Keding and S. J. Danishefsky, J. Am. Chem. Soc., 2004, 126, 6576 CrossRef CAS PubMed.
- T. Kawakami, M. Sumida, K. Nakamura, T. Vorherr and S. Aimoto, Tetrahedron Lett., 2005, 46, 8805 CrossRef CAS.
- B. Blankemeyer-Menge, M. Nimtz and R. Frank, Tetrahedron Lett., 1990, 31, 1701 CrossRef CAS.
- T. J. Trivedi, K. S. Rao, T. Singh, S. K. Mandal, N. Sutradhar, A. B. Panda and A. Kumar, ChemSusChem, 2011, 4, 604 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ob00213g |
| This journal is © The Royal Society of Chemistry 2016 |