
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTrihaloethenes as versatile building blocks for organic synthesis
Adriana S.
Grossmann
and
Thomas
Magauer
*
Department of Chemistry, Ludwig-Maximilians-Universität München, Butenandtstrasse 5-13, 81377 München, Germany. E-mail: thomas.magauer@lmu.de
First published on 26th January 2016
Abstract
This review highlights the chemistry of trihaloethene building blocks with a special focus on commercially available 1,1,2-trichloroethene. The topics surveyed herein include the use of trihaloethenes as C2-building blocks for transition metal-catalyzed coupling reactions, addition, elimination and cycloaddition reactions as well as natural product syntheses.
 Adriana S. Grossmann | Adriana S. Grossmann was born in Tegernsee, Germany in 1989. She studied chemistry at the LMU München and joined the group of Thomas Magauer for her Master's thesis in 2013. Since 2014, she is working as a PhD student on the development of novel ring-expansion reactions, sigmatropic rearrangements based on trihaloethenes and their application in the synthesis of novel antibiotics. |
 Thomas Magauer | Thomas Magauer was born in Linz, Austria in 1983. He grew up in Steyr and moved to Vienna in 2002 to study chemistry at the University of Vienna. In 2007, he joined the laboratories of Prof. Johann Mulzer and under his guidance he developed enantioselective syntheses of the complex polyketide kendomycin and the sesquiterpenoid echinopines A and B. After graduating in 2009, he moved to Harvard University, USA, to begin postdoctoral studies with Prof. Andrew G. Myers. At Harvard University he worked on carbohydrates, chiral silicon protecting groups and developed a synthesis of natural and diverse unnatural antiproliferative trioxacarcins. In 2012, he started his independent research as a Liebig-junior research group leader at the LMU Munich. Since 2013, his group has been supported by an Emmy Noether fellowship of the DFG. |
1. Introduction
Within the family of commercially available trihaloethene derivatives, 1,1,2-trichloroethene (1, TCE, b.p. 87 °C) is by far the most commonly used reagent. Dr E. Fischer (not Hermann Emil Fischer) achieved the first synthesis of 1 in 1864 by reductive dehalogenation of hexachloroethane with hydrogen.1 Since then, TCE (1) has gained major importance in various industrial applications.2 The ease of preparation of large quantities of this non-flammable, volatile organic compound led to a staggering global consumption of 428.6 × 103 t in 2011. Of this, 84% of the total production volume in the USA were used as an intermediate for manufacturing the refrigerant 1,1,1,2-tetrafluoroethane (HFC-134a) and 15% as a solvent for metal degreasing.2,3 The preparation of HFC-134a from 1 occurs by addition of hydrogen fluoride to the alkene, followed by halogen-exchange reactions to afford 2-chloro-1,1,1-trifluoroethane (HCFC-133a) (eqn (1)). By further treatment with hydrogen fluoride, HCFC-133a is then converted into HFC-134a (eqn (2)). These transformations can be mediated by several catalysts, which are typically derived from chromium salts.4 TCE (1) is classified as human carcinogen, and its toxicity has been investigated and discussed in detail.3,5 Amongst other factors, the toxicity associated with 1 has led to continuously decreasing production rates in the industry.2,6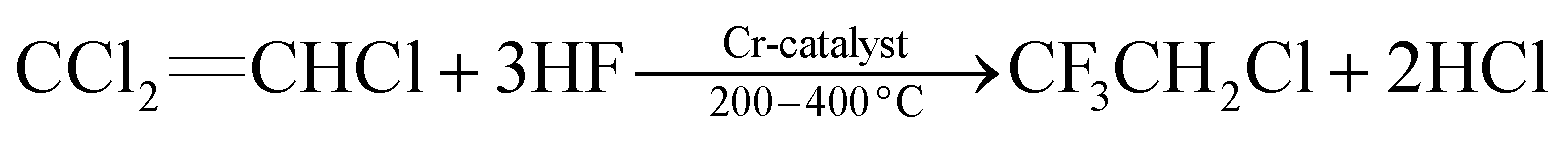 | (1) |
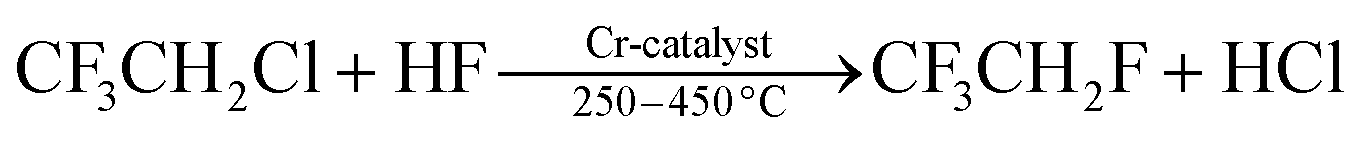 | (2) |
In the field of synthetic organic chemistry, TCE (1) serves as a chlorinated C2-building block7,8 that found very interesting applications beyond cross coupling reactions.9
The corresponding bromo- and fluoro-derivatives, 1,1,2-tribromoethene (2, TBE, b.p. 163 °C) and 1,1,2-trifluoroethene (3, TrFE, b.p. −51 °C), respectively, have received much less attention from synthetic chemists. Although tribromoethene (2) has been known for over 150 years, its use in synthesis has been relatively limited.10 Trifluoroethene (3), on the other hand, has found application in the synthesis of copolymers in combination with polyvinylidene fluoride (PVDF). This copolymer, named P(VDF-TrFE), is a very important organic ferroelectric material.4a,11 The addition of TrFE (3) to the polymer, usually in a composition of P(VDF-TrFE) 70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :30, enhances the all-trans conformation associated with the crystalline β-phase. This conformation and the dipole moment generated thereby, have a strong influence on the ferroelectric and also piezoelectric properties.11
:30, enhances the all-trans conformation associated with the crystalline β-phase. This conformation and the dipole moment generated thereby, have a strong influence on the ferroelectric and also piezoelectric properties.11
Since the preparation of 1 and 3 has been extensively studied over the years, we will provide the reader only with a selection of approaches. There are several feedstock chemicals that can be used to synthesize TCE (1). One such feedstock is acetylene, which is converted to 1via a chlorination–dehydrochlorination pathway.2 A different approach is the oxychlorination of ethene to afford 1. In this reaction, the chlorine source is hydrogen chloride, which is oxidized according to the Deacon process.2,12,13 Tetrachloroethene can also be converted into TCE (1) via hydrogenolysis with transition metal catalysts.14 TrFE (3) has also been prepared using various approaches: 1,1,2-trichloro-1,2,2-trifluoroethane (CFC-113) can be used as starting material in a vapor-phase reduction,4a hydrodechlorination15 or direct controlled-potential (bulk) electrolysis.16 Alternatively, chlorotrifluoroethene (CTFE) has been used in hydrodechlorinations,17 and 1,1,1,2-tetrafluoroethane in dehydrofluorination reactions.18 In contrast, the preparation of TBE (2) has received less attention and one of the very few examples is based on the elimination of hydrogen bromide from 1,1,2,2-tetrabromoethane via phase-transfer conditions (PTC).19
A selection of further commercially available trihaloethenes is depicted in Fig. 1, although their use is less prevalent compared to TCE (1) or TrFE (3). Some trihaloethenes are used as monomers for the synthesis of halogenated polymers (2-bromo-1,1-difluoroethene (4, BDFE)),20 or for radical addition reactions (2-chloro-1,1-difluoroethene (6)).21
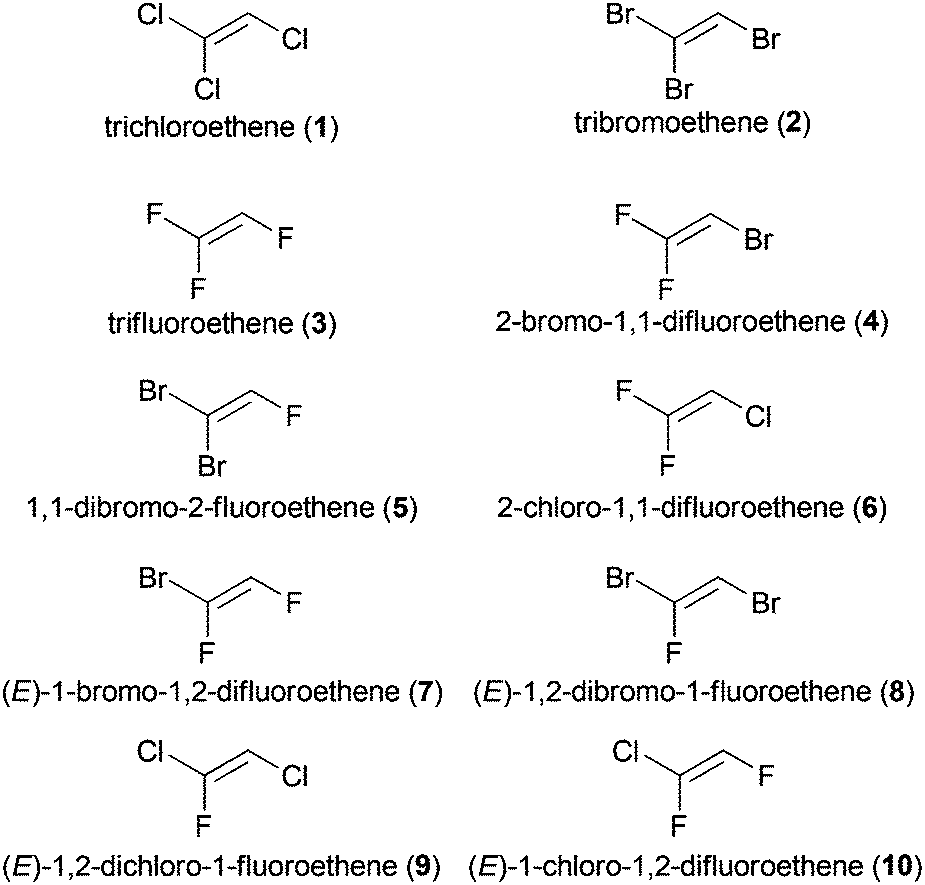 | ||
| Fig. 1 A selection of commercially available trihaloethene derivatives. | ||
Although trihaloethenes are generally available from commercial sources, iodine containing derivatives are far less common. Scheme 1 outlines the four approaches involved in the stereoselective synthesis of differentially substituted trihaloethenes. The synthesis of 1,1-difluoro-2-iodoethene (12) is based on the addition of iodinemonochloride and subsequent elimination of hydrogen chloride (Scheme 1a).22 This reaction could be conducted on a two mole scale without noticeable loss of yield. Burton published a selective synthesis of (Z)-1,2-difluoro-1-iodoethene (15) (Scheme 1b).23 In the first two steps, silylation via chlorine–lithium exchange and reduction with lithium aluminium hydride afforded 14 in a (Z):(E) ratio of 95:5. Subsequent deprotonation and iodination followed by protodesilylation yielded isomerically pure 15. The enrichment of the (Z)-isomer was explained due to decomposition of the lithiated (E)-isomer via elimination of lithium fluoride. In 2007, the group of Castanet reported the synthesis of (Z)-1,2-dichloro-1-iodoethene (16) from TCE (1) (Scheme 1c).24 Since 1 is used as the solvent and the reaction proceeds via the intermediacy of the highly explosive dichloroacetylene (DCA, 41), only substoichiometric quantities of base were used in order to avoid accumulation of large amounts of 41. (Z)-1-Bromo-1,2-dichloroethene (18) was prepared in 1966 by Köbrich and Flory by lithiation of (E)-1,2-dichloroethene (17), followed by treatment with bromine (Scheme 1d).25
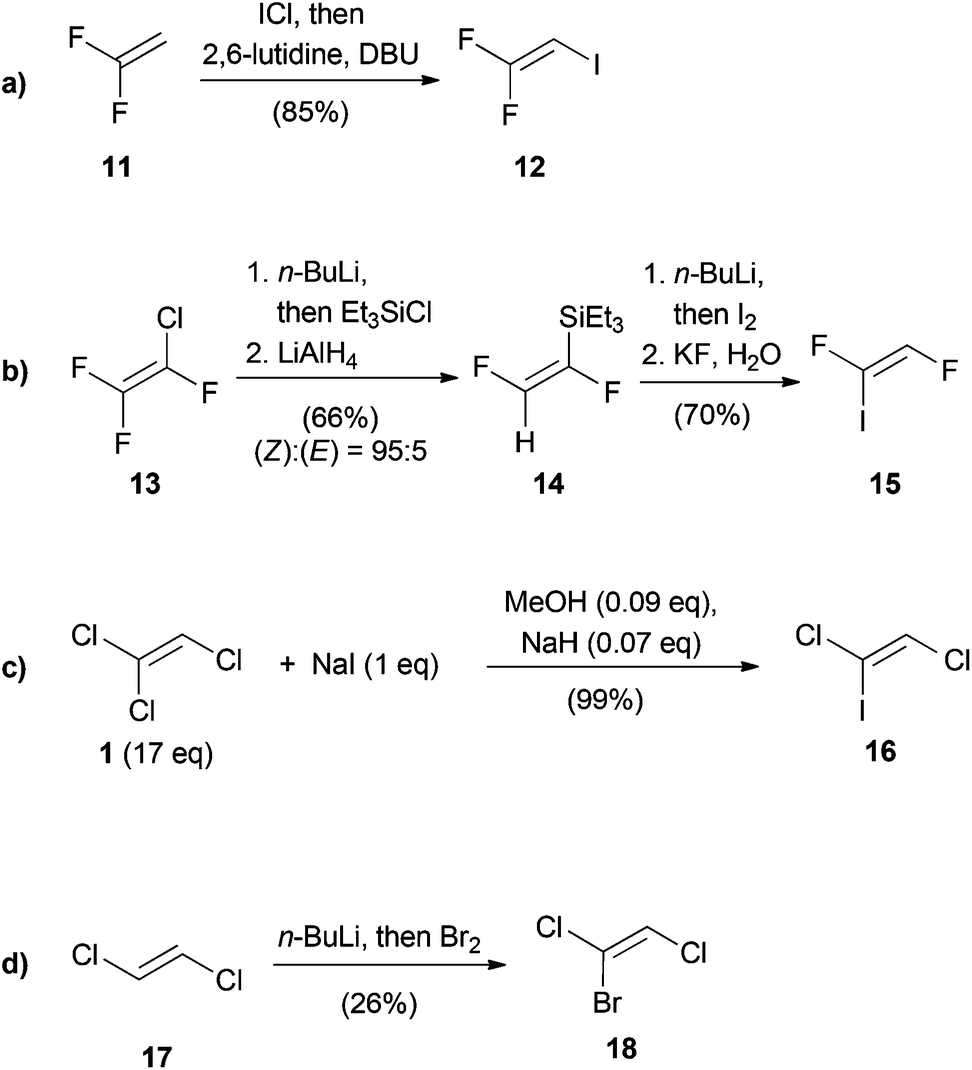 | ||
| Scheme 1 Synthesis of the trihaloethenes 12, 15, 16 and 18. | ||
The following sections will focus on the chemistry of trihaloethenes, in particular TCE (1). Their functionalization via addition, elimination and transition metal-catalyzed reactions is presented, as well as their application in the synthesis of natural products.
2. Functionalization of trihaloethenes via addition and elimination reactions
Addition reactions
Trihaloethenes are very versatile reagents for nucleophilic or electrophilic addition reactions. Additionally, the increased acidity of the vinylic proton (pKa (1) = 27–29)26 enables the simple use of lithiated trihaloethenes as valuable nucleophiles.The Humphrey group reported an addition reaction to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond as an efficient method to synthesize halogenated aryl and heteroaryl ethers.27 A variety of phenols and 3-hydroxypyridines underwent nucleophilic addition to 2-bromo-1,1-difluoroethene (4) under basic conditions (Scheme 2). A small amount of water was a necessary additive to immediately protonate the initially formed anionic adduct and therefore prevent elimination of fluoride. In all reactions, less than 5% elimination was observed, except in the case of 19h, where 14% elimination took place. In this instance, the authors suggested that the proton transfer might be hindered due to the presence of the sterically encumbered isopropoxy group.
C double bond as an efficient method to synthesize halogenated aryl and heteroaryl ethers.27 A variety of phenols and 3-hydroxypyridines underwent nucleophilic addition to 2-bromo-1,1-difluoroethene (4) under basic conditions (Scheme 2). A small amount of water was a necessary additive to immediately protonate the initially formed anionic adduct and therefore prevent elimination of fluoride. In all reactions, less than 5% elimination was observed, except in the case of 19h, where 14% elimination took place. In this instance, the authors suggested that the proton transfer might be hindered due to the presence of the sterically encumbered isopropoxy group.
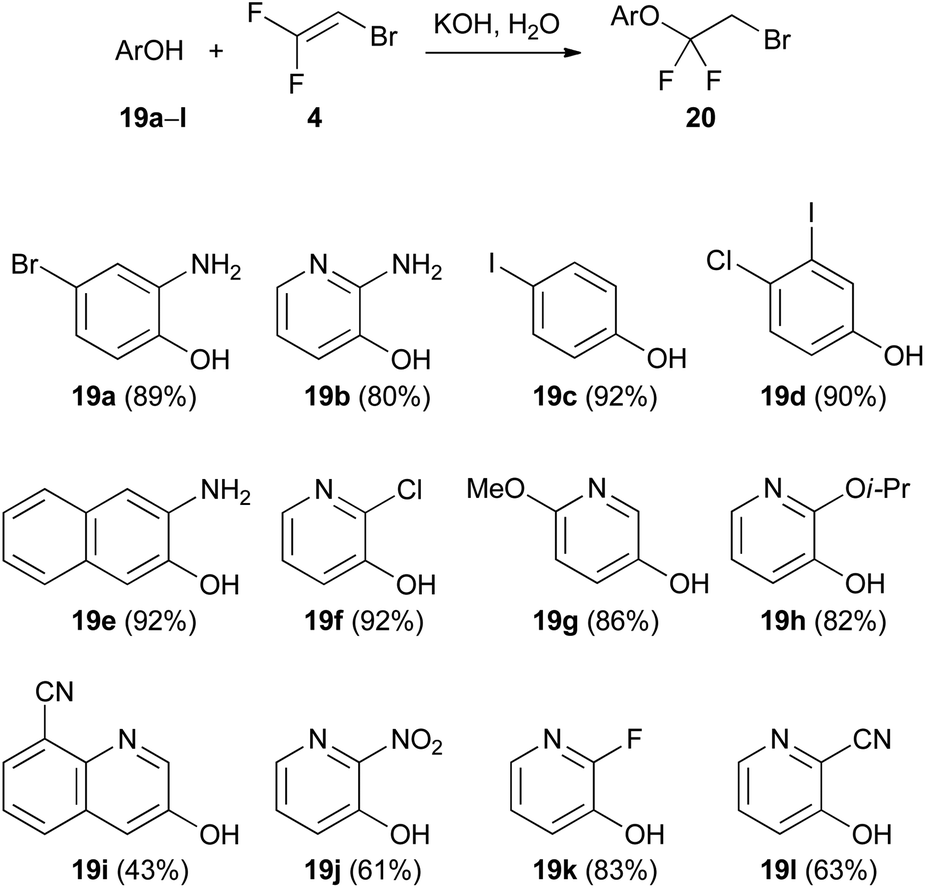 | ||
| Scheme 2 Addition reaction of hydroxy arenes and heteroarenes 19a–l to 2-bromo-1,1-difluoroethene (4). | ||
Despite having three electron withdrawing chlorine substituents, TCE (1) is capable of nucleophilic addition to formaldehyde (21) under strongly acidic conditions (Scheme 3). This reaction was first reported by Prins in 1932, who claimed that the final product was 3,3′-oxybis(2-chloropropanoic acid).28 However, in subsequent studies29 the product was identified as 2-chloroacrylic acid (23). The putative intermediate 22 could not be isolated due to spontaneous dehydration under the reaction conditions.30
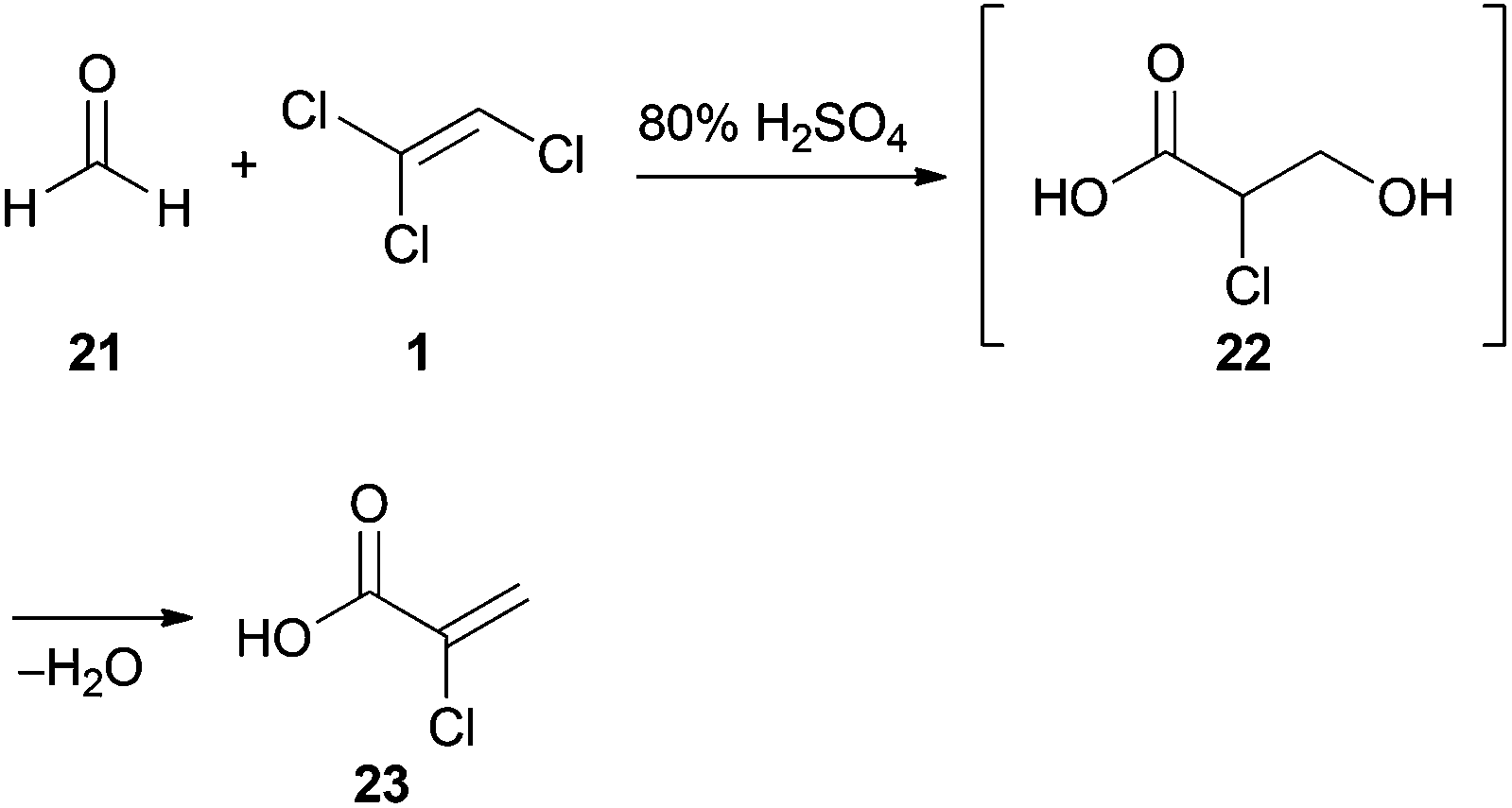 | ||
| Scheme 3 Prins reaction of 1 with formaldehyde (1) under strongly acidic conditions. | ||
In 1966, Köbrich and Flory reported the reaction of (Z)-1-bromo-1,2-dichloroethene (18) with n-butyllithium and carbon dioxide (Scheme 4).25 Surprisingly, depending on the reaction solvent, the differently lithiated products 24 or 25 were formed in situ instead of the expected elimination products. While 24 is the product of vinylic deprotonation, halogen–lithium exchange leads to the formation of 25. They proposed that deprotonation is quicker than the halogen–lithium exchange in the more polar and strongly coordinating solvent tetrahydrofuran.25,31 Based on this discovery, various trihaloethenes could be metalated and used as nucleophiles in addition reactions with carbon dioxide to form the corresponding acids.
 | ||
| Scheme 4 Solvent effects on the regioselective lithiation of 18. | ||
Similarly, Köbrich and Flory metalated 1 with n-butyllithium and treated the so generated vinyl lithium intermediate 26 with carbon dioxide to afford trichloroacrylic acid (27) (Scheme 5).25
 | ||
| Scheme 5 Carboxylation of 1 with carbon dioxide. | ||
Based on the results of Köbrich and Flory, Tarrant prepared fluorine-containing vinyllithium reagents using 28. Treatment with a variety of carbonyl compounds 29 afforded the corresponding 1,2-addition products 30 (Scheme 6a).32
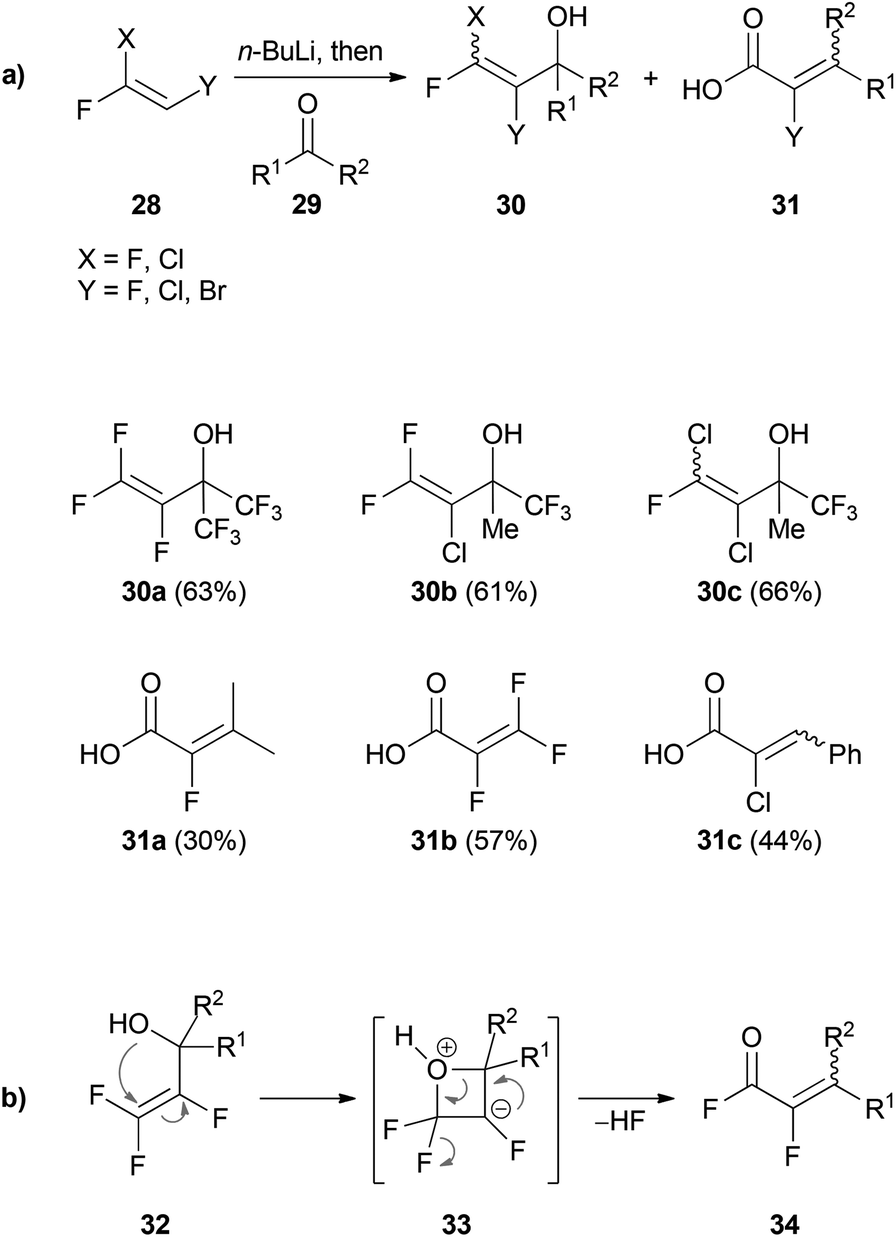 | ||
| Scheme 6 (a) 1,2-Addition of trihalovinyllithium reagents to carbonyl compounds 29. (b) Reaction mechanism for the 1,3-rearrangement of 32. | ||
In the cases where the carbonyl compounds 29 did not have electron withdrawing CF3-groups, subsequent rearrangement of the intermediate alcohols to 31 was usually observed. The postulated mechanism of the 1,3-rearrangement is illustrated in Scheme 6b. Attack of the hydroxyl group at the vinylic gem-difluoro group of 32 leads to formation of the oxetane intermediate 33. Subsequent elimination of hydrogen fluoride gives rise to the acid fluoride 34 which is then hydrolyzed to give the acid 31.
Addition–elimination reactions
Reactions wherein a nucleophilic addition to the trihaloethene followed by elimination of a halide anion takes place are designated as addition–elimination reactions. A useful application of this concept was demonstrated in 2000 by Tellier for the stereoselective synthesis of halogenated carboxylic acids (Scheme 7).33 Various allylic alcohols 35 were treated with potassium hydride to form the corresponding alkoxides, which then reacted with 2-bromo-1,1-difluoroethene (4) at −90 °C to selectively form the E-enol ether 37. At temperatures around −30 °C, 37 underwent a Claisen rearrangement to give the acid fluoride 38, which afforded the anti substituted acid 36 upon aqueous workup.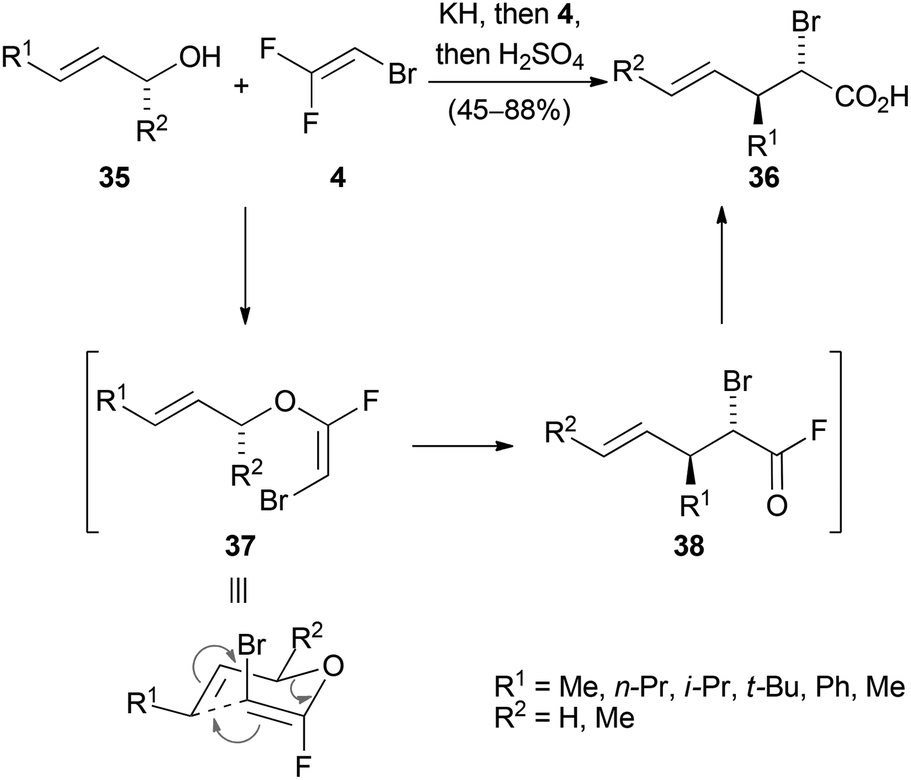 | ||
| Scheme 7 Claisen rearrangements of halogenated allyl vinyl ethers. | ||
Elimination–addition reactions
These transformations, in some cases referred to as condensation34 or substitution30,35 reactions, occur readily with TCE (1). Seminal studies concerning their mechanism were performed by Kende.34 Based on these findings, Hultin described a detailed mechanism using phenols as nucleophiles (Scheme 8).35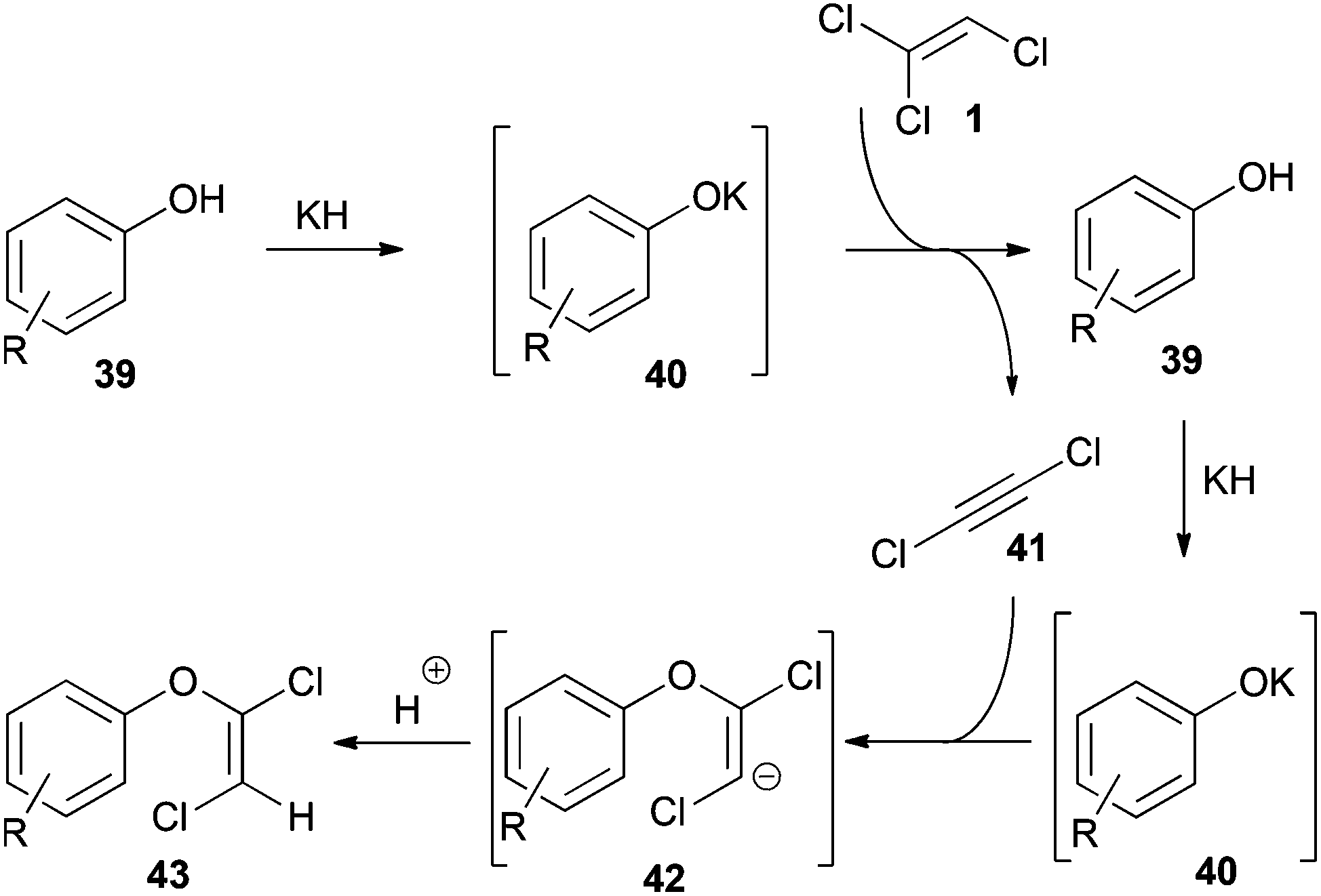 | ||
| Scheme 8 Mechanism of the elimination–addition reaction of 1 using potassium phenolates 40. | ||
In the first step DCA (41) is formed by deprotonation of 1 with the potassium phenolate 40. Regeneration of 40 by a second equivalent of base then initiates the addition reaction to the in situ generated 41. Finally, protonation of the vinyl anion 42 affords the 1,2-dichlorovinyl ether 43. The addition step proceeds highly stereoselective and provides the E-configured vinyl ether as the main product.35
The dehydrochlorination of TCE (1) is a very common reaction that was also studied in more detail.30 The β-elimination generally occurs by treatment of 1 with a strong base and leads to the formation of DCA (41).36 In a procedure reported in 1961, a gaseous mixture of 1 in diethyl ether was passed through pyrex tubes containing a potassium hydroxide/calcium oxide mixture.37 Two other reports describe the synthesis of 41via phase-transfer catalysis (PTC).36d,38 While Kende synthesized 41 by deprotonating 1 with lithium bis(trimethylsilyl)amide,39 Greene employed potassium hydride and a catalytic amount of methanol for this transformation (Scheme 9).36b
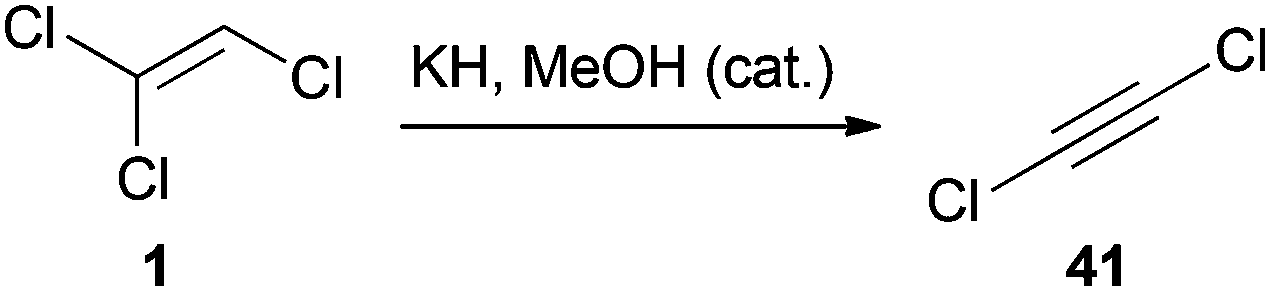 | ||
| Scheme 9 Synthesis of DCA (41) from 1. | ||
Preparing 41 as a solution in diethyl ether forms a rather stable diethyl ether–DCA (41) complex and proved to be a practical method for the handling of this reagent, as pure 41 is toxic and highly explosive.30,56
Kende employed an elimination–addition reaction with in situ generated 41 to prepare various dichlorovinyl ketones in up to 64% yield (Scheme 10a).34 The same conditions were investigated for the reaction of 49 with 1,2-dichloro-1-fluoroethene (9). Although 9 was used as a 1:1 mixture of (E) and (Z)-isomers, only the (E)-isomer 50 was observed as the product (Scheme 10b). This result implies the formation of chlorofluoroacetylene as an intermediate, similar to the mechanism depicted in Scheme 8.
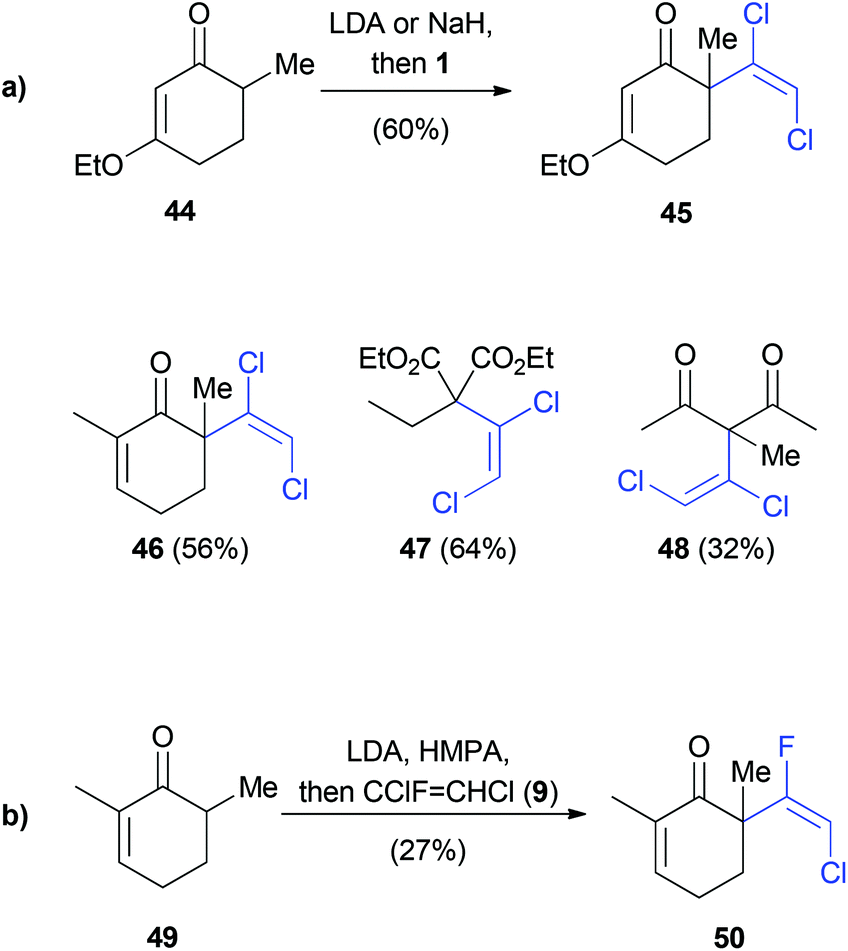 | ||
| Scheme 10 α-Vinylation of ketones. | ||
Greene reported a method based on a similar protocol (Scheme 11).40 After the treatment of alcohols 51 with potassium hydride and TCE (1) to form the intermediate dichlorovinyl ethers, addition of excess n-butyllithium gave the corresponding chloro ynol ethers. Under the reaction conditions, a subsequent halogen–lithium exchange took place and the generated lithium acetylide is then either protonated or alkylated to yield the ynol ether 52.
 | ||
| Scheme 11 Preparation of ynol ethers. | ||
In 1995, Greene expanded the one-pot methodology for the synthesis of ynol ethers to the preparation of ynethiol ethers 58 by implementing the corresponding thiols 57 (Scheme 12).41 Primary, secondary and tertiary thiols served as substrates in this reaction and the obtained yields ranged from 69–98%.
 | ||
| Scheme 12 Synthesis of ynethiol ethers. | ||
Pielichowski reported the reaction of TCE (1) with secondary amines 59 to obtain glycinamides 60 (Scheme 13a).38,42 The proposed reaction mechanism is depicted in Scheme 13b. DCA (41) is formed by deprotonation with sodium hydroxide under PTC conditions. The secondary amine 59 adds to 41 and the resulting enamine 61 undergoes a further elimination to the ynamide 62. Addition of water to 63 and replacement of the α-chloro ketone in 64 with the second equivalent of amine leads to the glycinamide 60.
 | ||
| Scheme 13 (a) Synthesis of substituted glycinamides 60. (b) Proposed mechanism. | ||
Radical addition reactions
Trihaloethenes can not only undergo nucleophilic addition reactions but also radical addition reactions (Scheme 14).43 Iron(II) sulfate reduces the diazonium salt 65 to generate an aryl radical, which undergoes a 5-exo-trig cyclization followed by addition to 1. Subsequent elimination of chlorine leads to 66 in 58% yield. | ||
| Scheme 14 Radical cyclization and addition of diazionium salt 65 to 1. | ||
A related reaction reported by the Sandford group relies on the radical hydroalkylation of fluorinated trihaloethenes with cyclopentane (67a) and cyclohexane (67b) (Scheme 15).21 The alkyl radicals, generated by γ-ray irradiation or treatment with di-tert-butyl peroxide, regioselectively substitute the gem-difluoro carbon atom. The authors suggest that the regioselectivity is governed by the little sterical hindrance and the increased electrophilicity of this position.
 | ||
| Scheme 15 Free radical addition of trihaloethenes 4 and 6. | ||
3. Cycloadditions
[4 + 2] cycloadditions
Trihaloethenes have also found application as dienophiles in cycloaddition reactions. Chambers reported Diels–Alder reactions between trifluoroethene (3) and various substituted furan derivatives 69 (Scheme 16).44 The reactions were carried out in an autoclave at 200 °C under autogenous pressure to yield fluorinated oxabicyclo[2.2.1]hept-2-enes 70. | ||
| Scheme 16 [4 + 2] Cycloaddition with TrFE (3). | ||
Another example of a [4 + 2] cycloaddition was published by Borsato (Scheme 17).45 They synthesized 3-chloronorbornenone (76) via a three-step synthesis starting with a Diels–Alder reaction between TCE (1) and cyclopentadiene. The formed trichloronorbornene 72 was converted to the dichloronorbornadiene 73 under basic conditions and subsequently desymmetrized with the chiral auxiliary (−)-ephedrine (74). Hydrolysis with pyridinium p-toluenesulfonate (PPTS) then afforded bicyclic ketone 76.
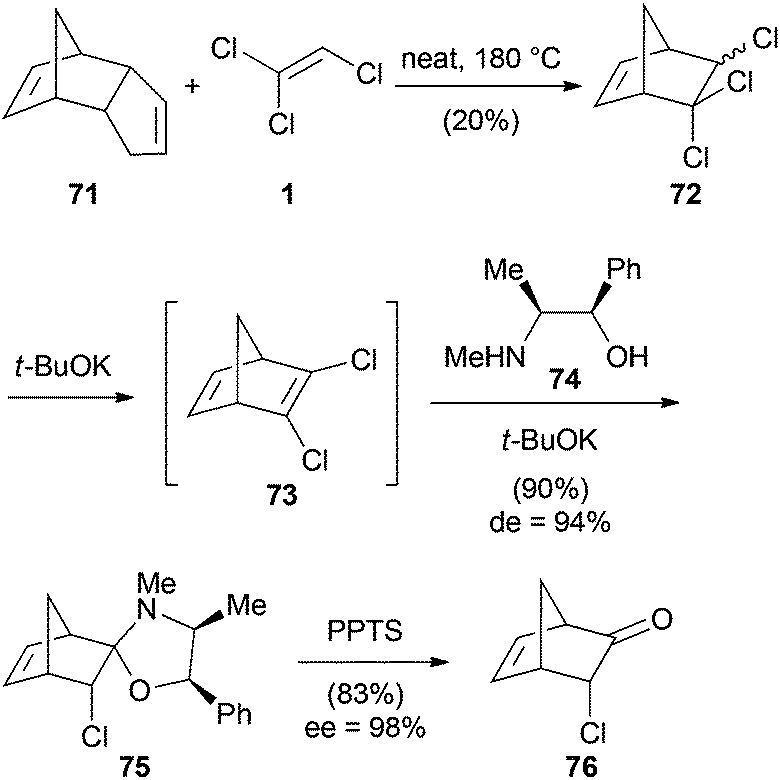 | ||
| Scheme 17 [4 + 2] Cycloaddition with TCE (1). | ||
[2 + 2] cycloaddition
A photochemical [2 + 2] cycloaddition reaction with TCE (1) was reported by Tobe (Scheme 18).46 The bicyclic enone 77 was dissolved in 1 and irradiated with a 500 W high-pressure mercury lamp. The following elimination of hydrochloric acid under basic conditions allowed isolation of the cyclobutene 78. | ||
| Scheme 18 [2 + 2] Cycloaddition of an enone with 1. | ||
1,3-Dipolar cycloaddition
Mani and Sales discovered an intramolecular 1,3-dipolar cycloaddition one-pot protocol for the concise synthesis of benzofuropyrazoles (Scheme 19).47 Starting from salicylaldehyde (79), the corresponding dichlorovinyl ether was generated and treated with benzenesulfonyl hydrazide to form the corresponding hydrazine. Raising the temperature to 50 °C afforded aryldiazomethane 80, which spontaneously cyclized to form chloropyrazole 81 in 55% overall yield. The product could be further functionalized by Buchwald–Hartwig or Suzuki–Miyaura cross-coupling reactions.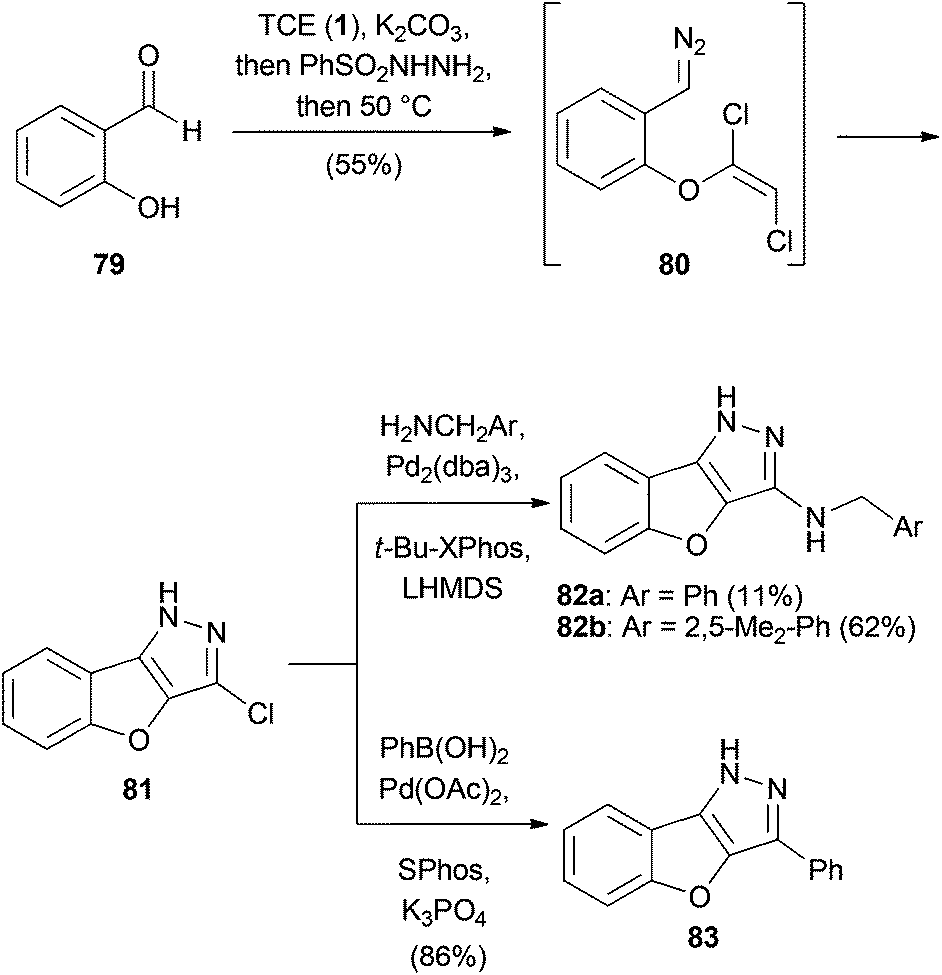 | ||
| Scheme 19 A one-pot protocol for the synthesis of benzofuropyrazoles. | ||
4. Transition metal-catalyzed reactions
Since the reactivities of the three C–Cl bonds of TCE (1) differ significantly from each other, it can be used as versatile reagent for cross-coupling reactions. Hultin reported detailed studies concerning the different possibilities to sequentially and selectively functionalize 1 using traditional palladium(0)-catalyzed transformations such as Suzuki–Miyaura, Negishi, and Sonogashira cross-coupling reactions (Fig. 2).48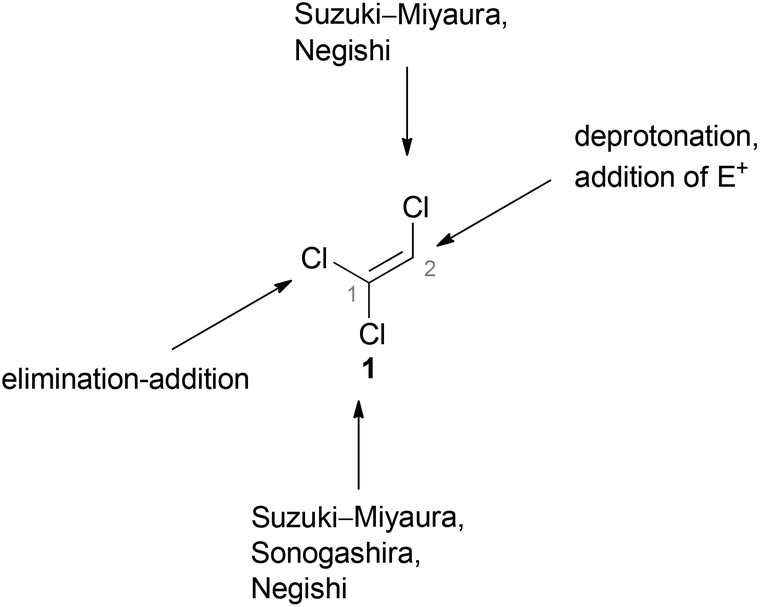 | ||
| Fig. 2 Overview of the different functionalization possibilities of 1. | ||
Hultin exploited the unique reactivity of 1 which enabled the preparation of highly substituted ethene derivatives in a controlled fashion (Scheme 20).48 In each case, the first step involves formation of a (E)-1,2-dichlorovinyl ether 84 by an elimination–addition reaction. As depicted in route A, deprotonation of the C(2)–H group in 84 followed by subsequent addition of an electrophile allows the preparation of substituted 1,2-dichlorovinyl ether 85. In route B, the C(1)–Cl group of 84 is able to participate in cross-coupling reactions just as the C(2)–Cl group in the ensuing step (route C). The trisubstituted alkene 87 can be obtained via route C. Route D starts from 86 as well, but the following step involves the deprotonation of C(2)–H and replacement with an electrophile. A further cross-coupling reaction of the C(2)–Cl group of 88 then gives rise to the tetrasubstituted alkene 89.
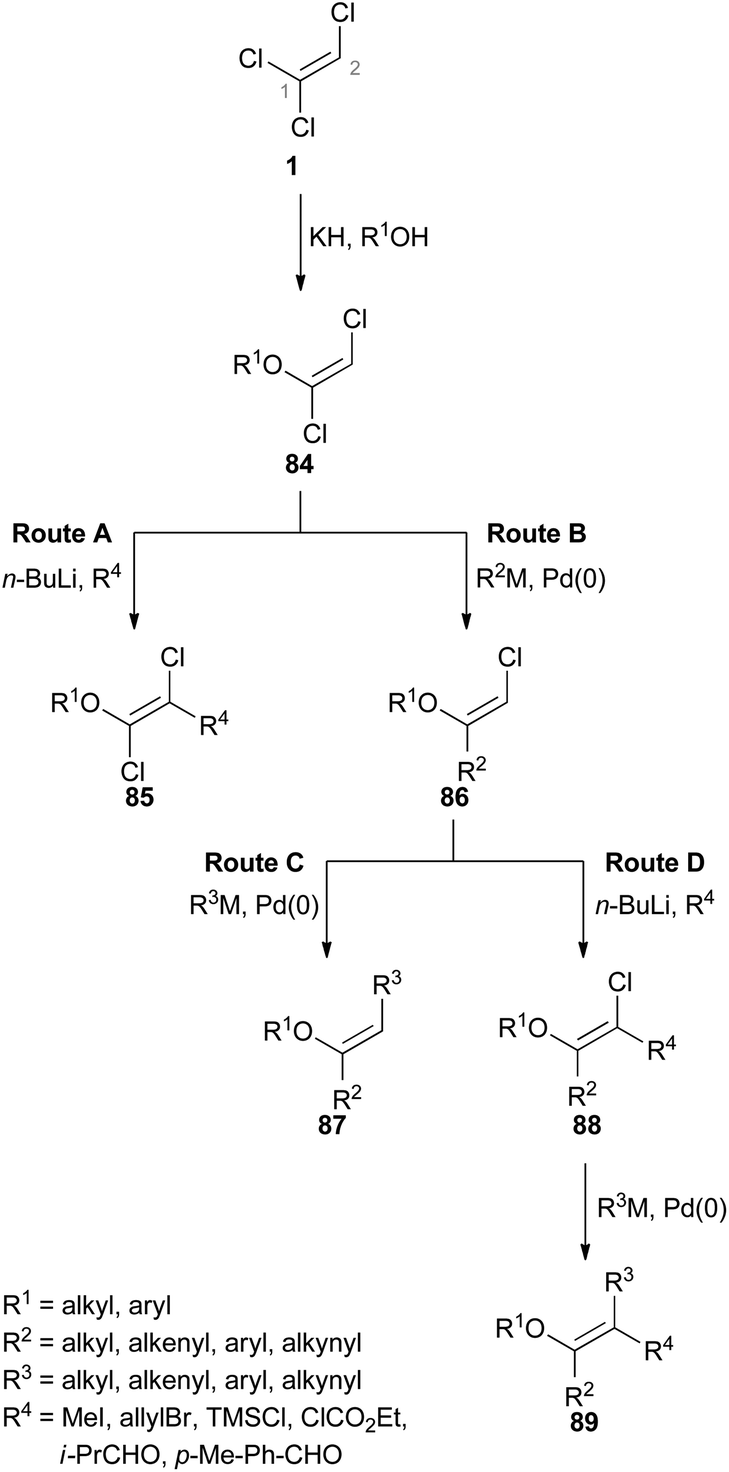 | ||
| Scheme 20 Reaction sequences for the functionalization of 1. | ||
However, there are limitations to the reaction pathways depicted in route A and C. While Suzuki–Miyaura cross coupling reactions at the C(1)–Cl group of product 85 (route A) were not stereoselective, Sonogashira cross-couplings proved to be unsuccessful at all. In route C, deprotonation of the C(2)–H group of 87 was problematic and generation of a tetrasubstituted alkene was not possible via this route.48
Suzuki–Miyaura coupling
Hultin and Geary35,49 expanded the Suzuki–Miyaura cross-coupling reactions to include 1,2-dichlorovinylaryl ethers 90. The latter were derived from symmetrical phenols and 1, and enabled the synthesis of 2-substituted benzofurans 91a,b (Scheme 21).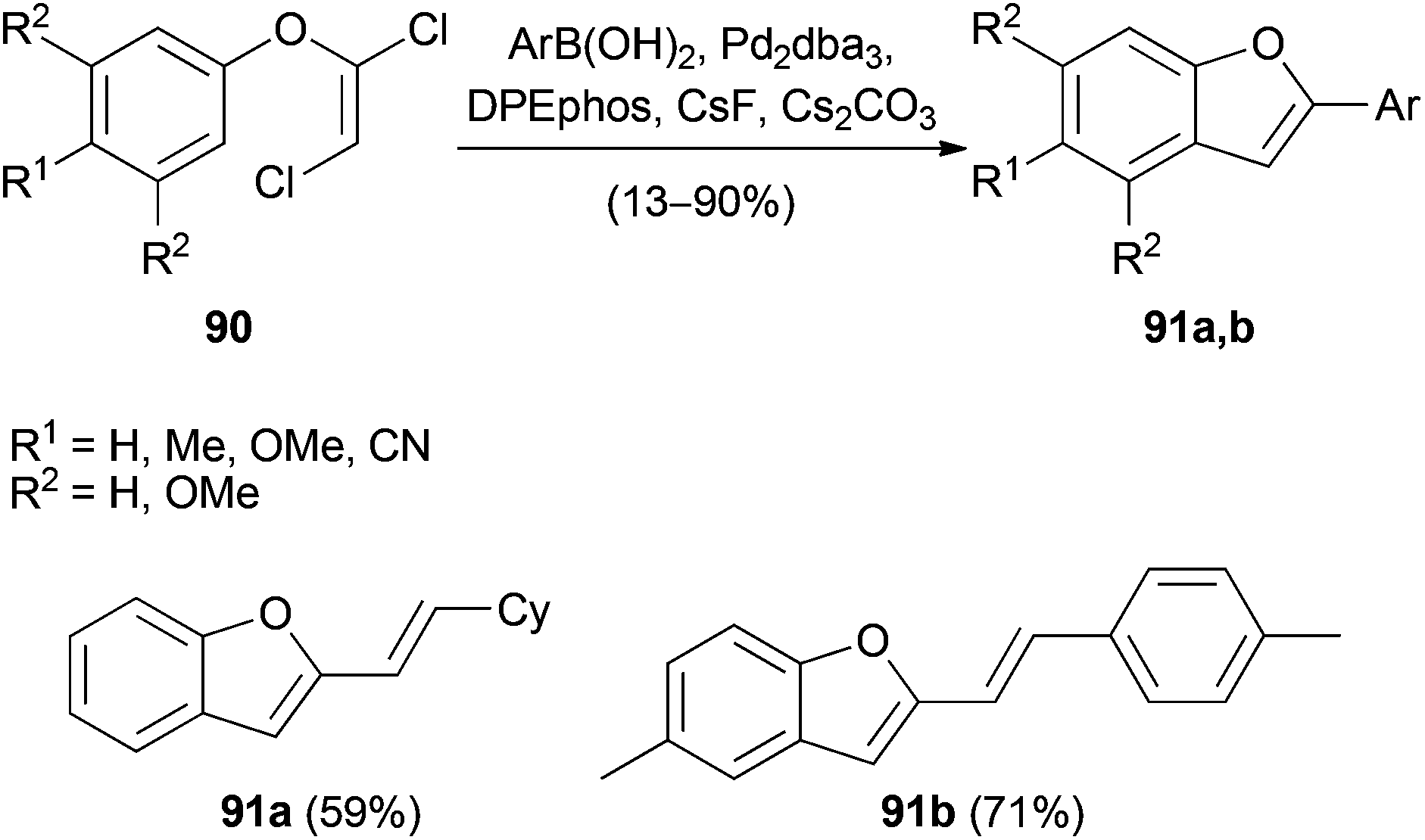 | ||
| Scheme 21 One-pot preparation of benzofurans from symmetrical phenols. | ||
The cross-coupling occurred selectively at the C(1)–Cl group of the electrophile and gave a single regio- and stereoisomer in all cases. This is due to the oxidative addition of palladium to the most electrophilic carbon atom. The authors also observed spontaneous cyclization to the benzofuran upon prolonged heating after completion of the Suzuki coupling. However, if the cross-coupling was incomplete, the subsequent cyclization did not occur and thus limited the utility of the one-pot protocol. Hultin and Geary further employed this method for the synthesis of indoles with substituents in C(2)-position. However, yields were generally lower for these substrates (29–47% yield).49
In further experiments it was demonstrated that 1,2-dichlorovinyl ethers 92a, derived from unsymmetrical electron rich phenols, afforded substituted benzofurans 93a as single regioisomers (Scheme 22a).35 However, with electron deficient phenol derivatives 92b, a mixture of the regioisomers 93b and 94 was obtained (Scheme 22b).
 | ||
| Scheme 22 Synthesis of benzofurans from unsymmetrical phenols. | ||
After further experimentation, the authors suggested four mechanistic possibilities: electrophilic aromatic substitution, σ-bond metathesis and two different assisted intermolecular palladations. Finally, the data were found to be consistent with a C–H metathesis as well as with an assisted palladation pathway. However, further discrimination was not possible so far.
Stereoselective Suzuki–Miyaura cross coupling reactions are also possible with TCE (1) itself. Matsuda reported the synthesis of (2,2-dichlorovinyl)arenes 95 by a rhodium(I)-catalyzed cross-coupling reaction of 1 with arylboronic esters (Scheme 23).50 The reaction occurred selectively at the C(2)–Cl bond and a variety of β,β-dichlorostyrenes were synthesized.
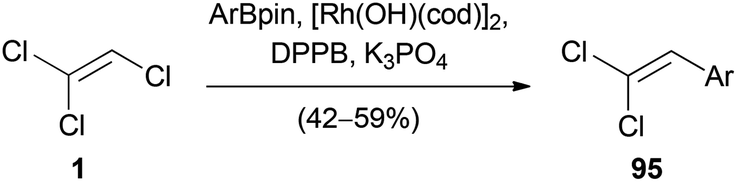 | ||
| Scheme 23 Rhodium(I)-catalyzed Suzuki–Miyaura cross-coupling with TCE (1). | ||
The observed selectivity differed significantly from the analogous rhodium(I)-catalyzed reaction using TBE (2), since a 3:1 mixture of 2-(2,2-dibromovinyl)-naphthalene and (Z)-(1,2-dibromovinyl)naphthalene was obtained in low yield.50
An alternative Suzuki–Miyaura coupling with TBE (2) using a palladium(0) catalyst was reported by the group of Organ. For this reaction, the (Z)-isomer of 1,2-dibromovinyl benzene (96) was formed in good yield (Scheme 24).51 Unfortunately, all attempts to further diversify bromide 96 by cross-coupling reactions, even under mild Negishi conditions, failed. Instead, elimination and homocoupling resulted in the formation of 97.
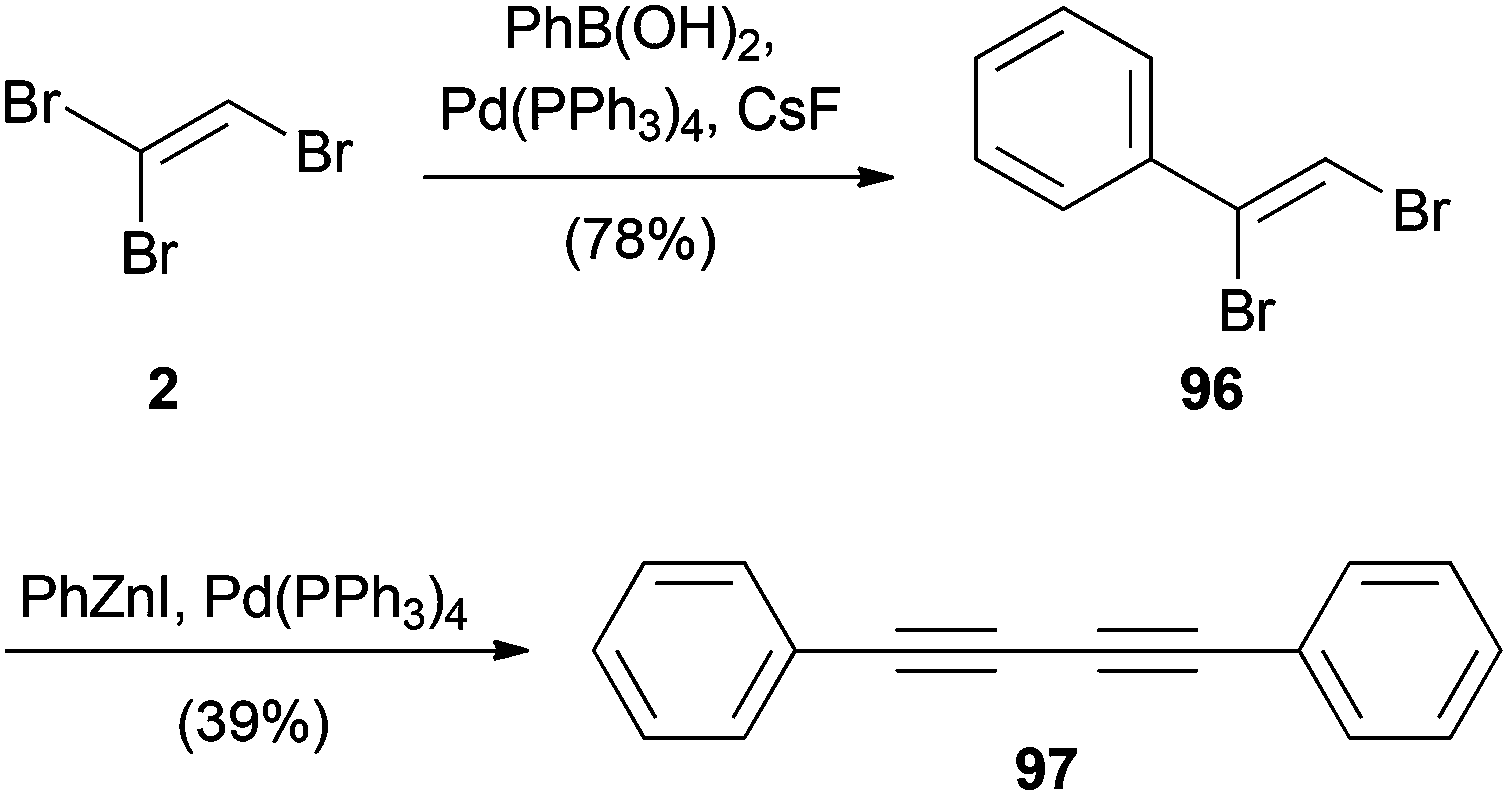 | ||
| Scheme 24 Suzuki–Miyaura cross-coupling with TBE (2) and subsequent homocoupling under Negishi cross-coupling conditions. | ||
Negishi coupling
Burton and Liu reported the stereoselective preparation of (Z)-1,2-difluoro-1-iodoethane (15), which can undergo stereospecific Negishi cross-coupling reactions to obtain (E)-1,2-difluorostyrene derivatives 99 (Scheme 25a).23 The metalation of 15 with activated zinc dust afforded exclusively the (E)-isomer 98. The best yields were obtained by using N,N-dimethylacetamide, while the formation of several side products was observed when the reaction was conducted in N,N-dimethylformamide. By using substoichiometric amounts of copper(I) bromide as an additive for the Negishi cross-coupling, the reaction rate could be increased and full conversion was achieved within less than six hours. In an analogous reaction reported by Burton 1,1-difluoro-2-iodoethene (12) was used to prepare the metalated species 100 (Scheme 25b).52 Palladium(0)-catalyzed coupling with various substituted aryl halides gave the corresponding 2,2-difluorostyrenes 101 in up to 92% yield without the need for an additional copper(I) additive. | ||
| Scheme 25 Negishi cross-coupling with 15 and 12. | ||
Another Negishi coupling was reported by Lentz and co-workers in 2010 (Scheme 26).22 The organozinc compound was prepared from 12 using activated zinc powder and gave rise to the homocoupled product 1,1,4,4-tetrafluorobuta-1,3-diene (102), which was brominated in a 1,4-fashion to yield 103. Intermediate 103 was then converted to tetrafluorobutatriene (104) via a double elimination reaction using potassium hydroxide.
 | ||
| Scheme 26 Negishi cross-coupling with 1,1-difluoro-2-iodoethene (12) and formation of tetrafluorobutatriene (104). | ||
Stille coupling
Lentz also reported a palladium(II)-catalyzed Stille cross-coupling reaction of 12 with tributyl(ethynyl)stannane (105) (Scheme 27).22 Attempts to convert product 106 to the corresponding butatriene 107 using UV-irradiation were unsuccessful. | ||
| Scheme 27 Stille cross-coupling with 12. | ||
Heck reactions
To the best of our knowledge, only one report is detailing a Heck reaction using a trihaloethene. As shown in Scheme 28, TrFE (3) was treated with iodobenzene, triethylamine and a catalytic amount of palladium(II) acetate to obtain (Z)-(1,2-difluorovinyl)benzene 108 in 39% yield.53 The other reaction products 109 and 110 were also formed in low yields. The formation of 108 is consistent with a β-fluoride elimination of the palladium(II) intermediate. To support the mechanistic proposal, which involves attack of the arylpalladium(II) intermediate at the higher substituted carbon atom, MNDO-calculations (Modified Neglect of Diatomic Overlap) were conducted. The calculated net atomic charges of both C-atoms differ considerably (C(1) = +0.28; C(2) = +0.09), which indicates a charge controlled insertion reaction to 3. | ||
| Scheme 28 Heck reaction with TrFE (3). | ||
Pd-catalyzed CO insertion
A palladium-catalyzed carboalkoxylation of (Z)-1,2-difluoro-1-iodoethene (15) was published by Burton (Scheme 29).54 Based on the modification of a former procedure of Burton and Wesolowski,55 ethyl (E)-2,3-difluoroacrylate (111) could be prepared in 63% yield.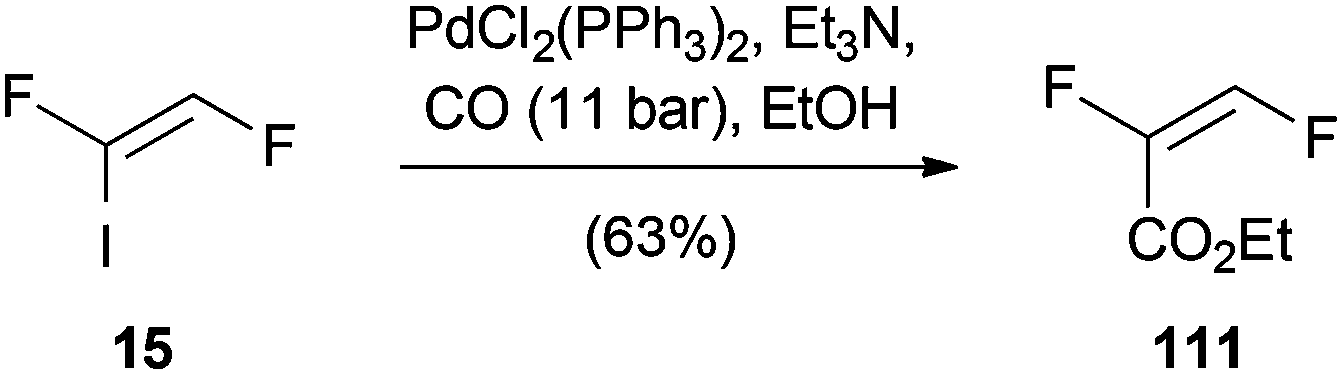 | ||
| Scheme 29 Palladium(0)-catalyzed carboalkoxylation of 15. | ||
A very similar procedure was used by Castanet for the palladium(0)-catalyzed alkoxycarbonylation of (Z)-1,2-dichloro-1-iodoethene (16) (Scheme 30).24 It was not possible to obtain the reaction product 112 by direct methoxycarbonylation of TCE (1). Therefore, the mono-iodo analogue 16 was synthesized to increase the rate of oxidative addition. In this manner, 112 could be obtained in moderate yield.
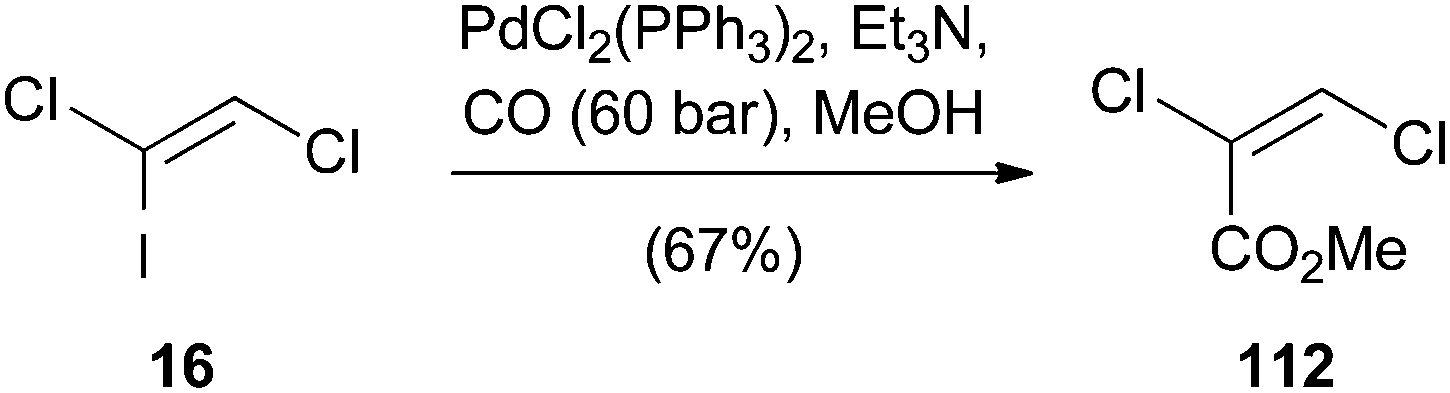 | ||
| Scheme 30 Palladium-catalyzed methoxycarbonylation of 16. | ||
Kumada coupling
The palladium or nickel-catalyzed reaction of a Grignard reagent with 1 was reported in 1985.56 Under mild conditions, 1 reacts with primary and secondary alkyl magnesium chlorides to yield 1,1-dichloroalkenes 113 in up to 81% yield (Scheme 31). The proposed mechanism, oxidative addition of the transition metal to 1, transmetalation with the Grignard reagent and reductive elimination, is consistent with the general mechanism of Kumada cross-coupling reactions.57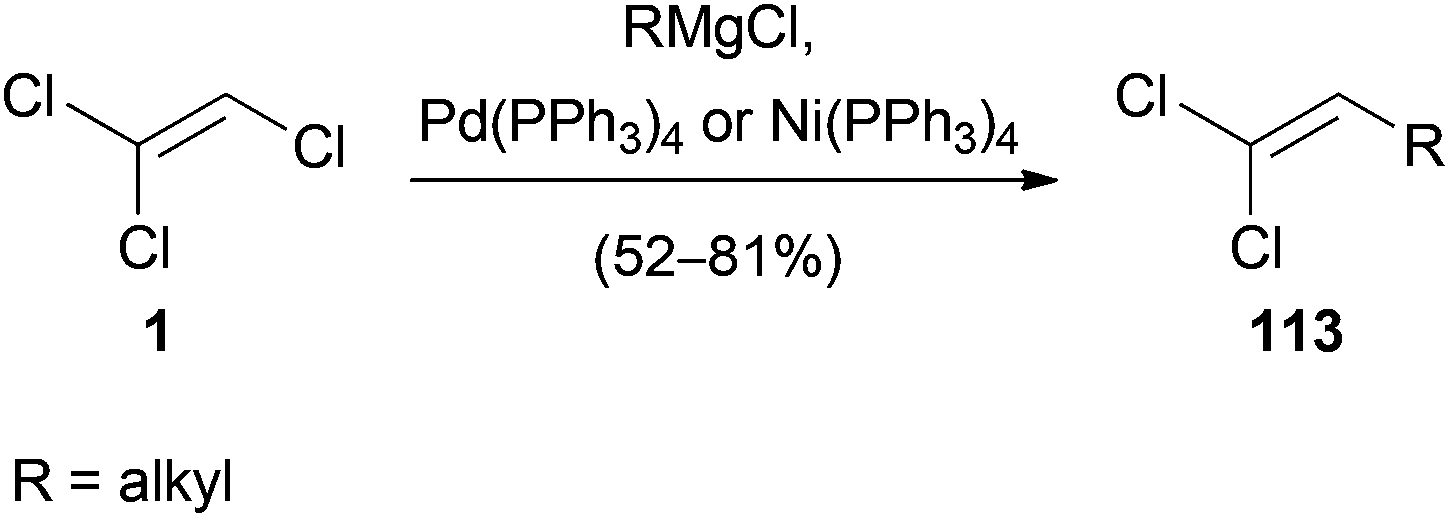 | ||
| Scheme 31 Palladium- and nickel-catalyzed coupling of Grignard reagents with 1. | ||
Another example of a palladium(0)-catalyzed coupling reaction of 1 with Grignard reagents was published in 1987 by Minato and Tamao (Scheme 32).58
 | ||
| Scheme 32 Regioselective palladium(0)-catalyzed Kumada coupling. | ||
However, in contrast to the reaction depicted in Scheme 31, the arylation occurred at the C(1)–Cl group. This is remarkable, since former studies document that the oxidative addition generally takes place at the C(2)–Cl group.59 It can be hypothesized that the regioselectivity of the reaction is dependent on the type of catalyst, but no explanation was given by the authors.9a
5. Application in natural product synthesis
The previously discussed reactions and methodologies have also been used for the synthesis of more complex molecules. In the following section, the synthesis of natural products involving transformations with trihaloethenes will be disclosed. Clark and co-workers conducted studies for the synthesis of gambieric acids A–D (Scheme 33).60 For the synthesis of the F–J fragment 117, 1 was employed to prepare enol ether 116. In the first step, the 1,2-dichlorovinylether was formed which was treated with n-butyllithium to afford the corresponding ynol ether. Reduction with Lindlar catalyst then gave rise to 116. The latter compound served as the substrate to simultaneously construct the six- and nine-membered cyclic ether 117 by ring-closing metathesis using Grubbs second generation catalyst. | ||
| Scheme 33 Synthesis of the F–J fragment of the gambieric acids (117). | ||
Poisson's total syntheses of (−)-swainsonine (125) and (+)-6-epicastanospermine (126) started with the preparation of the chiral 1,2-dichloroenol ether 119 (Scheme 34).61 Treatment of 119 with two equivalents of n-butyllithium gave rise to the corresponding acetylide, which was treated with allyl iodide to afford the ynol ether 120. After selective reduction of the alkine moiety to the Z-double bond using DIBAL-H, a [2 + 2] cycloaddition with in situ generated dichloroketene took place, yielding cyclobutanone 121. Tamura's reagent (122) was then used for the Beckmann ring expansion to yield a dichlorolactam, which was dechlorinated with zinc–copper couple in acidic medium. The so formed pyrrolidinone 123 was converted to the indolizidinone 124 in seven further steps. (−)-Swainsonine (125) and (+)-6-epicastanospermine (126) were prepared in eight and four steps, respectively, from this common indolizidinone precursor.
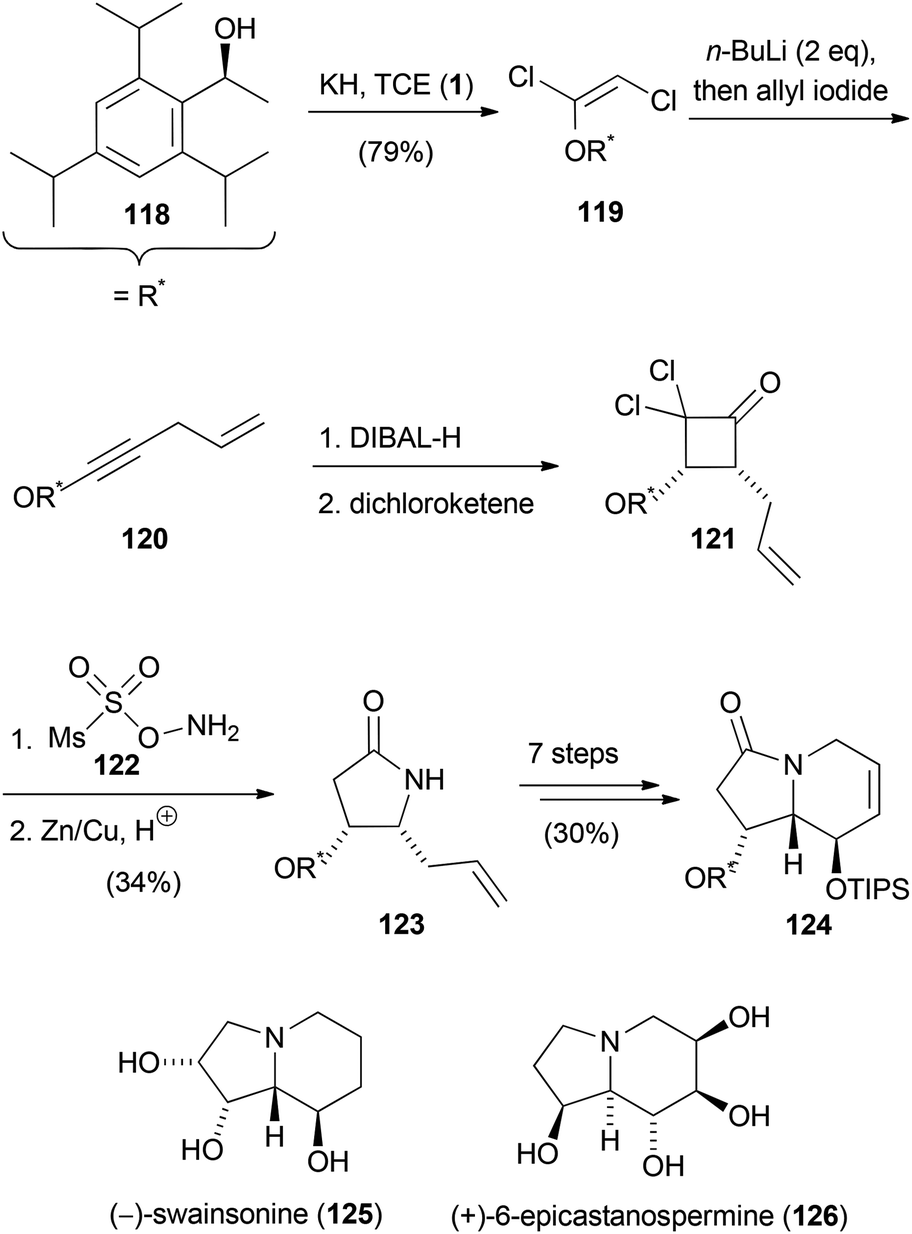 | ||
| Scheme 34 Synthesis of an intermediate for the synthesis of (−)-swainsonine (125) and (+)-6-epicastanospermine (126). | ||
In the synthesis of the hydroazulene core 136 of guanacastepene A (137) by Tius, TBE (2) was used as the starting material (Scheme 35).62 Reagent 2 was converted to 1-bromo-2-morpholinoacetylene (128) via an elimination–addition–elimination sequence. Nucleophilic addition to isobutyraldehyde (129) formed the zwitterionic ketene intermediate 130 which presumably rearranges via oxetane 131 to produce α-bromoamide 132 in up to 73% yield. In the following reaction, 132 was treated with a slight excess of allenylithium 133. The in situ formed addition product 134 underwent an allenylithium/vinyl amide cyclopentannelation reaction (Nazarov cyclization) to afford the highly substituted cyclopentenone 135, which was converted to the hydroazulene core 136 in ten steps.
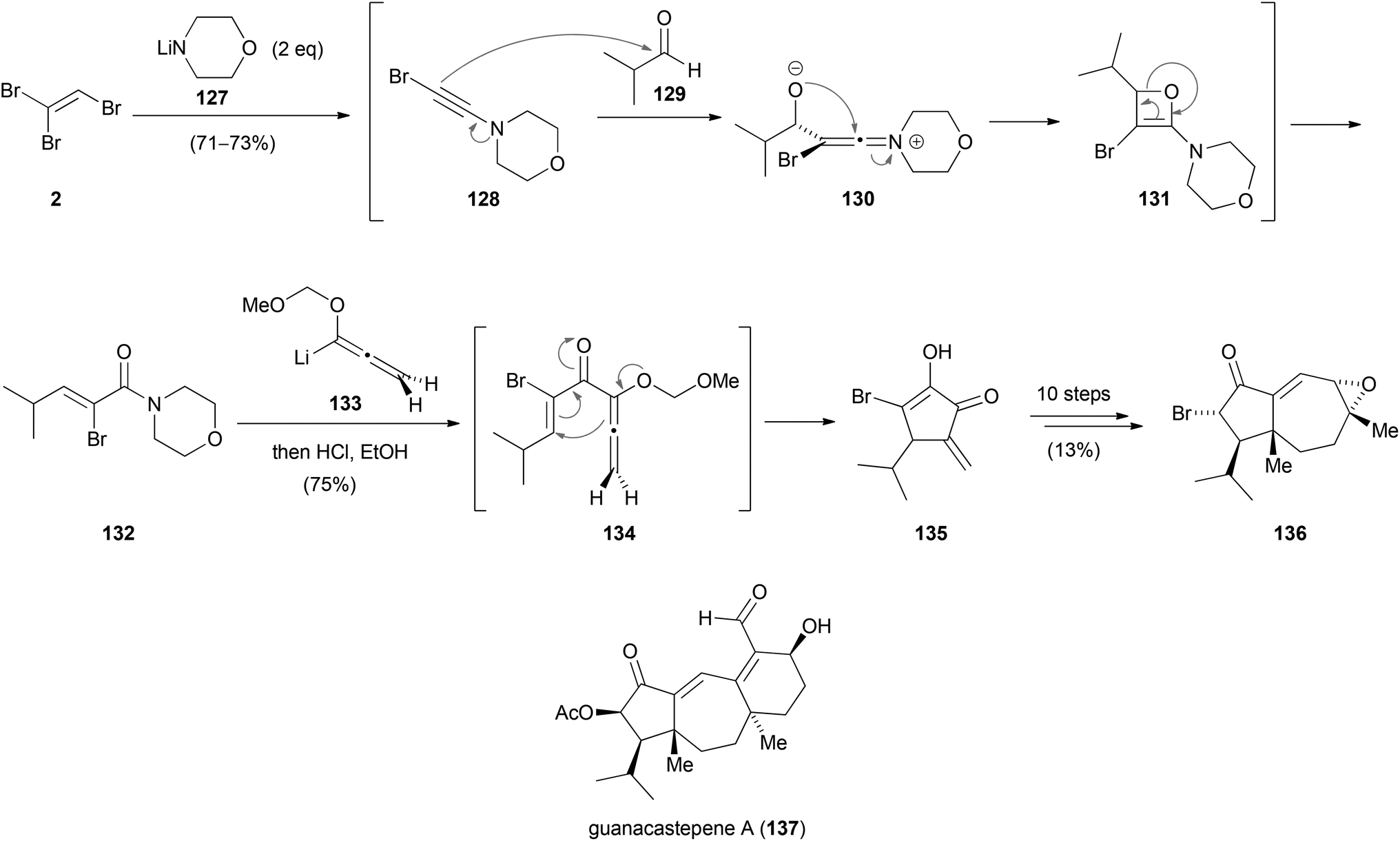 | ||
| Scheme 35 Synthesis of the hydroazulene core 136 of the natural product guanacastepene A (137). | ||
7. Conclusions
In this review we outlined the chemistry of trihaloethenes in general and of TCE (1) in particular. The outstanding versatility of all these reagents was demonstrated for selected reactions. Trihaloethenes combine the properties of alkenes and halogenated compounds and can therefore participate in addition and elimination reactions or combinations thereof, as well as in transition metal-catalyzed reactions. Despite their versatile reactivity, trihaloethenes are only sparsely applied in the synthesis of natural products and their main application is the formation of vinyl or acetylenic ethers. By this review, we hope to turn more attention to this fascinating class of molecules and to extend their synthetic applications.Acknowledgements
We gratefully acknowledge financial support from the FCI (Sachkostenzuschuss to T. M.) and the DFG (Emmy-Noether Program to T. M.). We thank MSc Cedric L. Hugelshofer and PhD Bryan Matsuura for helpful discussions during the preparation of this manuscript.Notes and references
- E. Fischer, Jenaische Zeitschrift für Medizin und Naturwiss., 1864, 1, 123 Search PubMed.
- E.-L. Dreher, T. R. Torkelson and K. K. Beutel, Ullmann's Encycl. Ind. Chem, 2015 Search PubMed.
- TSCA Work Plan Chemical Risk Assessment Trichloroethylene: Degreasing, Spot Cleaning and Arts & Crafts Uses, U.S. Environmental Protection Agency, Washington, D.C., 2014 Search PubMed.
- (a) Organofluorine Chemistry - Principles and Commercial Applications, ed. R. E. Banks, B. E. Smart and J. C. Tatlow, Plenum Press, New York and London, 1994 Search PubMed; (b) R. L. Powell, in Fluorine chemistry at the millennium, ed. R. E. Banks, Elsevier Ltd, 1st edn, 2000, pp. 339 Search PubMed; (c) J. D. Scott and R. A. Steven, US5744658A, 1998 Search PubMed; (d) J. D. Scott and R. A. Steven, US5395996A, 1995 Search PubMed; (e) J. D. Scott and R. A. Steven, US5243107A, 1993 Search PubMed.
- (a) A. Bale, S. Barone, R. Brown, J. C. Caldwell, C. Chen, W. A. Chiu, G. Cooper, G. Dannan, M. Evans, J. Fox, K. Z. Guyton, M. R. Gwinn, J. Jinot, N. Keshava, J. Lipscomb, S. Makris, M. Okino, F. Power, J. Schaum and C. Siegel Scott, Toxicological Review of Trichloroethylene, U.S. Environmental Protection Agency, Washington, D.C., 2011 Search PubMed; (b) 1 Trichloroethylene (CASRN 79-01-6), U.S. Environmental Protection Agency, Washington, D.C, 2011 Search PubMed.
- Trichloroethylene - Legislation, Markets and Uses, ECSA - European Chlorinated Solvent Association Search PubMed.
- B. Schmidt, D. K. Ehlert and H. A. Braun, Tetrahedron Lett., 2004, 45, 1751 CrossRef CAS.
- (a) Y. K. Kang, P. Deria, P. J. Carroll and M. J. Therien, Org. Lett., 2008, 10, 1341 CrossRef CAS PubMed; (b) S. Ijadi-Maghsoodi, Y. Pang and T. J. Barton, J. Polym. Sci., Part A: Polym. Chem., 1990, 28, 955 CrossRef CAS.
- (a) J. Anthony, A. M. Boldi, Y. Rubin, M. Hobi, V. Gramlich, C. B. Knobler, P. Seiler and F. Diederich, Helv. Chim. Acta, 1995, 78, 13 CrossRef CAS; (b) T. Nagai, T. Shibanuma, S. Ogoshi and M. Ohashi, US2012/0330072A1, 2012 Search PubMed.
- (a) A. C. W. Lennox, Justus Liebigs Ann. Chem., 1862, 122, 122 CrossRef; (b) A. Sabanejeff, Justus Liebigs Ann. Chem., 1875, 178, 109 CrossRef.
- M. Mai, S. Ke, P. Lin and X. Zeng, J. Nanomater., 2015, 2015, 1 CrossRef.
- (a) GB 913040A, 1962 Search PubMed; (b) H. E. Bellis, US3232889A, 1966 Search PubMed.
- P. Schmittinger, T. Florkiewicz, L. C. Curlin, B. Lücke, R. Scannell, T. Navin, E. Zelfel and R. Bartsch, Ullmann's Encycl. Ind. Chem, 2015 Search PubMed.
- (a) M. Heisler, W. Gabler, E. Pichl and R. Straßer, 3804265A1, 1989; (b) L. N. Ito, A. Harley, M. Holbrook, D. D. Smith, C. B. Murchison and M. D. Cisneros, WO94/07824, 1994 Search PubMed; (c) L. N. Ito, C. B. Murchison, M. T. Holbrook, A. Harley and D. D. Smith, US5476979 A1, 1995 Search PubMed.
- R. Ohnishi, I. Suzuki and M. Ichikawa, Chem. Lett., 1991, 841 CrossRef CAS.
- E. R. Wagoner, J. L. Hayes, J. A. Karty and D. G. Peters, J. Electroanal. Chem., 2012, 676, 6 CrossRef CAS.
- (a) B.-C. Meng, Z.-Y. Sun, J.-P. Ma, G.-P. Cao and W.-K. Yuan, Catal. Lett., 2010, 138, 68 CrossRef CAS; (b) S. Millefanti, V. Tortelli and G. Marchionni, WO2012/000853A1, 2011 Search PubMed.
- (a) W. Jia, Q. Wu, X. Lang, C. Hu, G. Zhao, J. Li and Z. Zhu, Catal. Lett., 2015, 145, 654 CrossRef CAS; (b) W. Jia, M. Liu, X. Lang, C. Hu, J. Li and Z. Zhu, Catal. Sci. Technol., 2015, 5, 3103 RSC.
- R. B. Bayatyan, B. E. Bayatyan and L. A. Saakyan, Russ. J. Appl. Chem., 2006, 79, 1849 CrossRef CAS.
- G. K. Kostov, L. Sauguet, B. Ameduri, H. Kaspar, T. Zipplies and K. Hintzer, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 3964 CrossRef CAS.
- J. A. Cooper, E. Copin and G. Sandford, J. Fluorine Chem., 2002, 115, 83 CrossRef CAS.
- C. Ehm, F. A. Akkerman and D. Lentz, J. Fluorine Chem., 2010, 131, 1173 CrossRef CAS.
- Q. Liu and D. J. Burton, J. Fluorine Chem., 2011, 132, 78 CrossRef CAS.
- S. Pellegrini, Y. Castanet and A. Mortreux, J. Mol. Catal. A: Chem., 2007, 277, 21 CrossRef CAS.
- G. Köbrich and K. Flory, Chem. Ber., 1966, 99, 1773 CrossRef.
- M. Gabričević, G. Lente and I. Fábián, J. Phys. Chem. A, 2015, 119, 12627 CrossRef PubMed.
- E. Yang, M. R. Reese and J. M. Humphrey, Org. Lett., 2012, 14, 3944 CrossRef CAS PubMed.
- H. J. Prins, Recl. des Trav. Chim. des Pays-Bas, 1932, 51, 469 CrossRef CAS.
- (a) J. W. C. Crawford and N. McLeish, US2233835, 1941 Search PubMed; (b) J. J. Gajewski, K. B. Peterson, J. R. Kagel and Y. C. J. Huang, J. Am. Chem. Soc., 1987, 111, 9078 CrossRef.
- R. V. Kaberdin and V. I. Potkin, Russ. Chem. Rev., 1994, 63, 641 CrossRef.
- G. Köbrich, A. Akhtar, F. Ansari, W. E. Breckoff, H. Büttner, W. Drischel, R. H. Fischer, K. Flory, H. Fröhlich, W. Goyert, H. Heinemann, I. Hornke, H. R. Merkle, H. Trapp and W. Zündorf, Angew. Chem., Int. Ed., 1967, 6, 41 CrossRef.
- (a) P. Tarrant, P. Johncock and J. Savory, J. Org. Chem., 1963, 28, 839 CrossRef CAS; (b) F. G. Drakesmith, R. D. Richardson, O. J. Stewart and P. Tarrant, J. Org. Chem., 1968, 33, 286 CrossRef CAS.
- F. Tellier, M. Audouin, M. Baudry and R. Sauvetre, Eur. J. Org. Chem., 2000, 1933 CrossRef CAS.
- A. Kende, P. Fludzinski, J. Hill, W. Swenson and J. Clardy, J. Am. Chem. Soc., 1984, 106, 3551 CrossRef CAS.
- L. M. Geary and P. G. Hultin, Eur. J. Org. Chem., 2010, 5563 CrossRef CAS.
- (a) P. Ruggli and I. Marszak, Helv. Chim. Acta, 1928, 11, 180 CrossRef CAS; (b) J.-N. Denis, A. Moyano and A. E. Greene, J. Org. Chem., 1987, 52, 3461 CrossRef CAS; (c) E. Kloster-Jenson, Tetrahedron, 1971, 27, 33 CrossRef; (d) A. H. Gierczak and A. Jonczyk, Tetrahedron, 2000, 56, 6083 CrossRef.
- J. H. Wotiz, F. Huba and R. Vendeley, J. Org. Chem., 1961, 26, 1626 CrossRef CAS.
- J. Pielichowsky and R. Popielarz, Synthesis, 1984, 433 CrossRef.
- A. S. Kende and P. Fludzinski, Synthesis, 1982, 455 CrossRef CAS.
- A. Moyano, F. Charbonnier and A. E. Greene, J. Org. Chem., 1987, 52, 2919 CrossRef CAS.
- P. Nebois, N. Kann and A. E. Greene, J. Org. Chem., 1995, 60, 7690 CrossRef CAS.
- J. Pielichowski and R. Popielarz, Tetrahedron, 1984, 40, 2671 CrossRef CAS.
- H. Jasch, Y. Landais and M. R. Heinrich, Chem. – Eur. J., 2013, 19, 8411 CrossRef CAS PubMed.
- R. D. Chambers, A. F. Gilbert and R. L. Powell, J. Fluorine Chem., 2000, 104, 233 CrossRef CAS.
- G. Borsato, A. Linden, O. De Lucchi, V. Lucchini, D. Wolstenholme and A. Zambon, J. Org. Chem., 2007, 72, 4272 CrossRef CAS PubMed.
- Y. Tobe, T. Fujii, H. Matsumoto, K. Tsumuraya, D. Noguchi, N. Nakagawa, M. Sonoda, K. Naemura, Y. Achiba and T. Wakabayashi, J. Am. Chem. Soc., 2000, 122, 1762 CrossRef CAS.
- Z. S. Sales and N. S. Mani, J. Org. Chem., 2009, 74, 891 CrossRef CAS PubMed.
- L. M. Geary and P. G. Hultin, J. Org. Chem., 2010, 75, 6354 CrossRef CAS PubMed.
- L. M. Geary and P. G. Hultin, Org. Lett., 2009, 11, 5478 CrossRef CAS PubMed.
- T. Matsuda, K. Suzuki and N. Miura, Adv. Synth. Catal., 2013, 355, 3396 CrossRef CAS.
- M. G. Organ, H. Ghasemi and C. Valente, Tetrahedron, 2004, 60, 9453 CrossRef CAS.
- B. V. Nguyen and D. J. Burton, J. Org. Chem., 1997, 62, 7758 CrossRef CAS.
- (a) W. Heitz and A. Knebelkamp, Makromol. Chem., Rapid Commun., 1991, 12, 69 CrossRef CAS; (b) W. Heitz and A. Knebelkamp, US5313001A1, 1994 Search PubMed.
- Y. Wang, L. Lu and D. J. Burton, J. Org. Chem., 2005, 70, 10743 CrossRef CAS PubMed.
- C. A. Wesolowski and D. J. Burton, Tetrahedron Lett., 1999, 40, 2243 CrossRef CAS.
- V. Ratovelmanana, G. Linstrumelle and J.-F. Normant, Tetrahedron Lett., 1985, 26, 2575 CrossRef.
- L. Kürti and B. Czako, in Strategic Applications of Named Reactions in Organic Synthesis, Elsevier Academic Press, 2005 Search PubMed.
- A. Minato, K. Suzuki and K. Tamao, J. Am. Chem. Soc., 1987, 109, 1257 CrossRef CAS.
- (a) I. Moritani, Y. Fujiwara and S. Danno, J. Organomet. Chem., 1971, 27, 279 CrossRef CAS; (b) P. Fitton and J. E. McKeon, Chem. Commun., 1968, 4 RSC.
- J. S. Clark, M. C. Kimber, J. Robertson, C. S. P. McErlean and C. Wilson, Angew. Chem., Int. Ed., 2005, 44, 6157 CrossRef CAS PubMed.
- J. Ceccon, A. E. Greene and J. F. Poisson, Org. Lett., 2006, 8, 4739 CrossRef CAS PubMed.
- A. Nakazaki, U. Sharma and M. A. Tius, Org. Lett., 2002, 4, 3363 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |