
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructure–activity correlation in transfection promoted by pyridinium cationic lipids†
P.
Parvizi-Bahktar
a,
J.
Mendez-Campos
a,
L.
Raju
b,
N. A.
Khalique
b,
E.
Jubeli
bc,
H.
Larsen
d,
D.
Nicholson
e,
M. D.
Pungente
 f and
T. M.
Fyles
*a
f and
T. M.
Fyles
*a
aDepartment of Chemistry, University of Victoria, P.O. Box 3065, Stn CSC, Victoria, BC V8W 3V6, Canada. E-mail: tmf@uvic.ca
bResearch Division, Weill Cornell Medicine in Qatar, Education City, P.O. Box 24144, Doha, Qatar
cUniversité Paris-Sud, EA 401, IFR 141, Faculté de Pharmacie, F-92296 Châtenay Malabry, France
dDepartment of Physics, University of Stavanger, 4036 Stavanger, Norway
eDepartment of Chemistry, Norwegian University of Science and Technology, 7491 Trondheim, Norway
fPremedical Unit, Weill Cornell Medicine in Qatar, Education City, P.O. Box 24144, Doha, Qatar
First published on 15th February 2016
Abstract
The efficiency of the transfection of a plasmid DNA encoding a galactosidase promoted by a series of pyridinium lipids in mixtures with other cationic lipids and neutral lipids was assessed in CHO-K1 cells. We identify key molecular parameters of the lipids in the mixture – clog![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P, lipid length, partial molar volume – to predict the morphology of the lipid-DNA lipoplex and then correlate these same parameters with transfection efficiency in an in vitro assay. We define a Transfection Index that provides a linear correlation with normalized transfection efficiency over a series of 90 different lipoplex compositions. We also explore the influence of the same set of molecular parameters on the cytotoxicity of the formulations.
P, lipid length, partial molar volume – to predict the morphology of the lipid-DNA lipoplex and then correlate these same parameters with transfection efficiency in an in vitro assay. We define a Transfection Index that provides a linear correlation with normalized transfection efficiency over a series of 90 different lipoplex compositions. We also explore the influence of the same set of molecular parameters on the cytotoxicity of the formulations.
Transfection of nucleic acid into cells is an essential in vitro biochemical tool and holds the promise for future genetic therapies, either based upon incorporation and translation of inserted DNA, or via gene silencing by RNA interference.1–3 Although conceptually simple, several decades of experience have demonstrated that implementation of transfection is very challenging.4 A key bottleneck has been the lack of effective delivery methods that protect the nucleic acid cargo from destruction in the extra-cellular environments encountered, recognize the target cell, then penetrate and release the cargo efficiently.5–8 Not only is effective delivery required, the delivery agents must have low cytotoxicity and immunogenicity. Viral vectors can be very efficient agents based on their evolved supramolecular structures that are optimized to respond to the changing requirements from tight initial packaging through to insertion and to dispersion of the cargo within the cell. Such vectors are however immunogenic and thus have constrained scope for general use in a course of treatments.1,2
Non-viral nucleic acid delivery via lipid–nucleic acid complexes (lipoplexes) is significantly less effective compared to viral delivery but the low immunogenic potential and generally moderate toxicity of the active agents make it a very appealing alternative.5,9,10 Since the first report by Felgner et al.11 in 1987 describing the use of the cationic lipid N-[1-(2,3-dioleyloxy)propyl]-N,N,N-trimethylammonium (DOTMA) as a vehicle for nucleic acid delivery, the following decades brought modifications and refinements mainly to lipid architectures and particle formulations with the hope of vastly improving gene transfer efficiencies.9,10,12,13 The majority of structural modifications to the lipid architecture focused on features having a significant impact on particle integrity and packing morphology, specifically the lipid hydrophobic domain,14,15 the functional nature of the charged head group,16–18 and the type of chemical bond or linkage through which these groups are connected.19–22 Although a few general trends emerged such as the advantage of using a double-chain hydrophobic domain23 and the replacement of saturated with monounsaturated chains,13 it is difficult to correlate cationic lipid structural details with transfection efficacy using simple “golden rules” and the strategies to improve performance are still mainly empirical.
Typically the anionic nucleic acid is rendered hydrophobic via electrostatic complex formation with cationic amphiphiles or polymers which, in the presence of neutral co-lipids, form into particles, liposomes (vesicles) or other aggregates. The packaged nucleic acid is protected within a hydrophobic environment, the particles themselves are not initially recognized by the immune system, and the individual components are potentially of low toxicity24 and can be manufactured to high purity with conventional GMP.9 The functional supramolecular sophistication of these systems is not very evolved and there is still considerable uncertainty over which supramolecular organizations or properties hold the keys to highly effective transfection.9 Supramolecular structure and assembly follows from the molecular properties of the components so is potentially amenable to optimization through the structures of the components. Our focus here is on low molecular weight cationic lipids and neutral co-lipids. Our premise is that the molecular properties of these small molecules will control supramolecular organization and function and thus will control transfection efficiency.
Which molecular and supramolecular properties are known to play a role in transfection by cationic lipids? Extra-cellular stability is obviously important; the particle must resist both enzymatic and thermal degradation (aggregation, decomposition), and in vivo must resist opsonation (labelling for destruction). This is usually accomplished through a surface coating of lipid-anchored polyethylene glycol (“pegylation”) that provides a steric cushion that limits the interactions with proteins in the extra-cellular medium,25,26 although cationic lipid head group optimization can also produce serum-resistant formulations.27 Physical factors such as particle size and surface charge density are clearly important at the level of lipoplex transport and uptake in vivo.7,8,28–32 Cellular uptake of larger particles in vitro occurs readily.33–35 Cells are diverse and utilize parallel uptake pathways each having particular triggering events,28 but the basic events from the particle perspective are similar between pathways; the particle is recognized for uptake, is pulled into the cell, and is initially held within an endosome or similar compartment within the cell. These stages can handle particles up to 400–500 nm in diameter,28 although in vivo applications likely require particles smaller than 125 nm (ref. 36) to avoid filtering by the spleen. The initial recognition of the particle may depend on the particle lipid phase morphology as well.8,37
Lipid phase behaviour plays a key role in the release of the nucleic acid cargo from within the endosome.8,37–42 Release is clearly correlated with the propensity to form inverted lipid phases, either hexagonal or cubic. This propensity is proposed to facilitate migration of lipids to the endosomal membrane, to reorganize the lipids locally to an inverted phase leading to rupture of the lamellar bilayer that contains the particle and its cargo.40,42,43 This key phase behaviour is temperature-dependent, and transfection is optimal when the inverted phase transition is close to the temperature of transfection (typically 37 °C).41,42 Lipid phase morphology is directly related to lipid structure through the shape or packing parameter S = V/(a0 × lc); where V is the equilibrium hydrocarbon chain volume, a0 is the equilibrium head group area at the polar-non-polar interface, and lc is the equilibrium hydrocarbon chain length.44,45 Pure lipids with ∼0.7 < S < 1.0 form lamellar phases; S > 1.0 leads to inverted phases. Lipid mixtures behave as pure lipids and have a shape parameter Smix given as the molar weighted average of the S values of the components.46
A common structural variable in synthesis-based structure–activity studies of cationic lipids is the hydrocarbon chain (length, degree of unsaturation).10,13 This parameter will influence S to a limited extent but plays a more significant role in varying the hydrophobicity and partitioning behaviour of the lipid. Quantitative structure–activity explorations47,48 of transfection efficiency include partitioning using a calculated logP term (clogP) based on additive structural increments. The rationale is that a more hydrophobic lipid will increase the structural stability of the initial particle, but at some point will inhibit cargo release leading to a bell-shaped dependence48 usually described as hydrophilic–lipophilic balance.49
We previously reported the development of a Transfection Index (TI) that was based upon S, clogP, and the geometric constraints of packing lipids and DNA into a lipoplex having overall hexagonal (HII) morphology.50 The goal was to develop a predictive tool based upon additive molecular structural parameters, ideally without recourse to any adjustable parameters. To the extent that we uncovered a linear correlation between TI and transfection efficiency by a limited number of previously reported pyridinium diesters51 in a range of formulations, we were encouraged that the structural parameters selected and the overall approach had some potential. However, we need to evaluate a broader set of compounds and formulations to establish if the previous correlation was adventitious.
We report here transfection, cell viability, and structural experiments for a larger set of pyridinium diesters, including binary and ternary mixtures of hydrocarbon chains. This dataset can be correlated using a modified TI that takes into account the potential role lipid phase behaviour plays in transfection. We also explore the correlation of formulation and structural parameters on cytotoxicity.
Results and discussion
Synthetic methyl pyridinium diesters are a known class of compound with previously published syntheses.50–53 Our syntheses are summarized in Scheme 1. Throughout we use a standard naming system in which we list the hydrocarbon components by chain length; di16:0 is the amphiphile with two saturated C16 chains. Other groups are C16:1 (15-hexadecen-1-yl), C18:1 (oleyl), and the two branched hydrocarbons given in Scheme 1.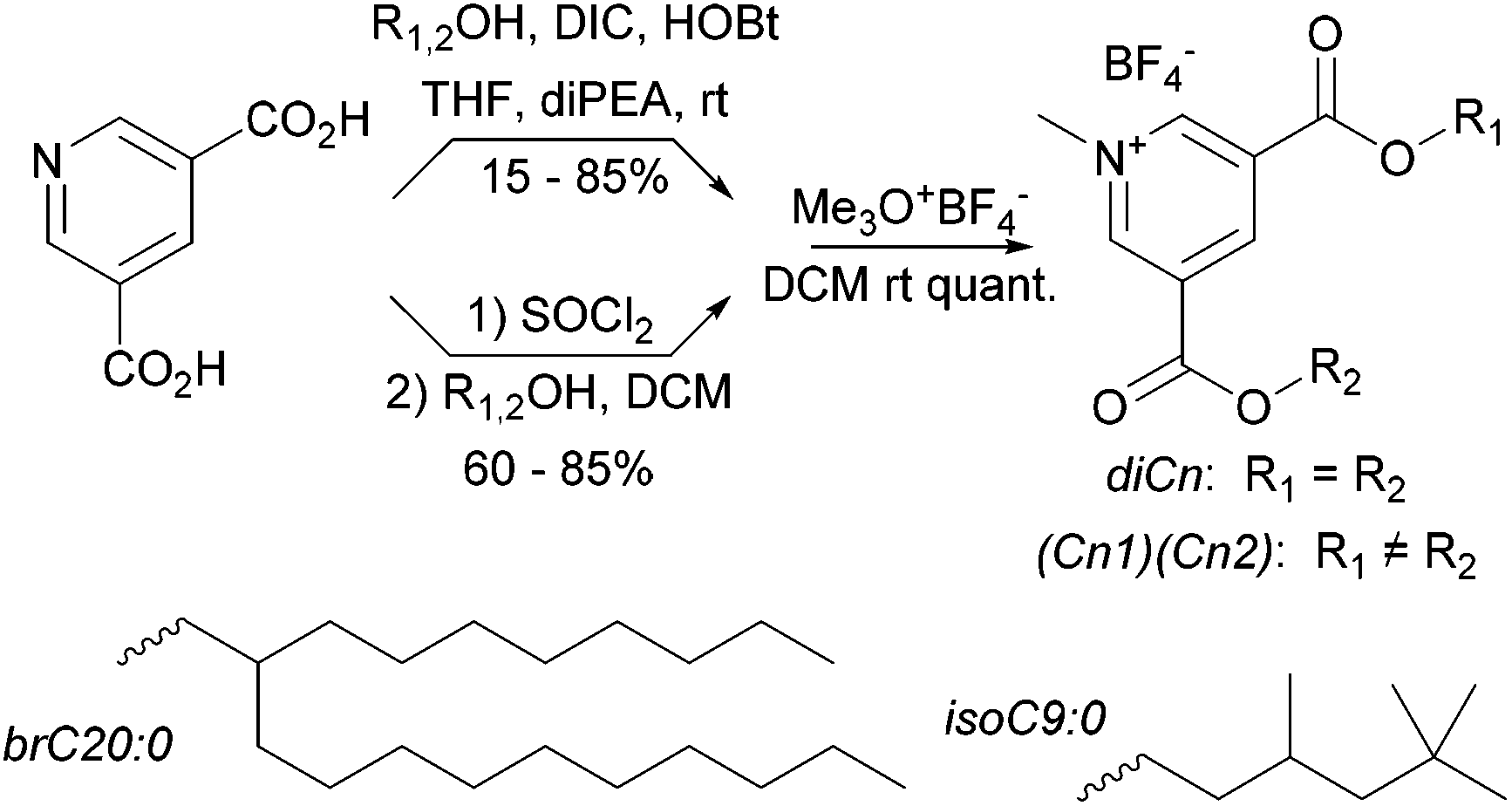 | ||
| Scheme 1 Synthesis of methyl pyridinium diesters. | ||
The yields of the intermediate diester pyridines are variable, primarily due to chromatographic losses. The diester pyridines are very weakly basic54 and difficult to directly N-alkylate. In our hands, alkylating agents that produce nucleophilic counterions, e.g. methyl iodide, also produce a small amount of addition product (N-alkyl-2-X-dihydropyridines). To circumvent this problem we use the forcing trimethyloxonium tetrafluoroborate for the methylation step which occurs quantitatively.
Below we explore binary blends of symmetrical diesters as a means to vary S and clogP. We were interested as well in mixed chain species as a comparison. A statistical synthesis with mixed starting alcohols e.g. oleyl alcohol and HO-brC20:0, produces a ternary mixture (diC18:1, dibrC20:0, and (C18:1)(brC20:0)). Electrospray MS, either of the protonated pyridines or of the methyl pyridiniums, shows only single molecular ions whose intensity ratios relative to a fixed internal standard (tetrapentylammonium) are independent of total concentration over a ten-fold concentration range. We take this as evidence that matrix suppression is negligible in this system and that the intensity ratios directly reflect the composition of the samples. In the example above an initial 1:1 ratio of alcohols produced a 16:28:56 mixture of diC18:1, dibrC20:0, and (C18:1)(brC20:0). Details of the synthetic and analytical procedures and compound characterization data are available in the ESI.†
Lipoplex formulation
Pyridinium lipids were mixed with the cationic lipid EPC (1,2-dimyristoyl-sn-glycero-3-ethylphosphocholine) in a 1:1 mole ratio and these mixed cationic lipids were co-formulated with a neutral co-lipid (cholesterol or DOPE 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine) in a 3:2 mole ratio. The hydration method of liposome formation results in high variability of size and dispersity. This PDI is expected for liposomes prepared without extrusion and without added surfactant. In some experiments binary and tertiary mixtures of pyridinium compounds were formulated with EPC and a co-lipid keeping the overall molar ratio of the cationic to neutral lipid 3:2. A dried mixed lipid film was hydrated and dispersed by sonication to produce a polydisperse liposome dispersion as assessed by dynamic light scattering; average diameter ranged from 0.2–2 μm with 0.2 < PDI < 0.9).
In some experiments, it was obvious that the more hydrophobic pyridinium lipids (diC20:0 and dibrC20:0) were not entirely incorporated into the lipid dispersion. Electrospray-mass spectrometry (ESI-MS) analysis established that the ratio of dibrC20:0:EPC in an aqueous dispersion is significantly decreased from the formulated 1:1 ratio. Data are available in the ESI.†
Aliquots of the liposome dispersion were added to plasmid DNA at molar charge ratios (CR; pyridinium N: phosphate P) of 0.5, 1.5, 3, 5 and 10. Dynamic light scattering analysis showed general increase in average diameter from liposome to lipoplex (0.3–10 μm) that is dependent on the charge ratio. A similar trend of increased lipoplex size compared to liposome is reported as due to fusion in the buffer solution, electrostatic adhesion of neighbouring liposomes and/or from lipoplex aggregation.55,56 In the highly diluted samples used in this study, DLS measurements are influenced by the presence of a small number of large aggregates. The largest particles are associated with intermediate values of CR (1.5–5) in most cases. The polydispersity of all formulations is high (0.2 < PDI < 0.9); the polydispersity of the majority of formulations were lower than those of the corresponding liposomes. Previous studies of in vitro transfection revealed that large lipoplexes of the sizes observed here are effective in transfection.33–35 The hydration method of lipoplex formation results in high variability in size and dispersity. No relation could be found between lipoplex size and either transfection efficiency or cell viability, but it is likely that some of the observed variation arises from this source. Particle sizing data are available in Table S1 in the ESI.†
Lipid phase morphology
The lipid phase morphology of the lipoplexes was assessed by synchrotron-based small-angle X-ray diffraction (SAXD; Table 1; additional data are given in Table S2 of the ESI†). In previous work there was a good correlation between the calculated Smix and the lipid phase. This correlation remains acceptable for pyridinium lipid mixtures of intermediate chain lengths but fails for the longest and shortest members of the series. As noted above, the hydrophobic compounds may not be well incorporated into the dispersions which would reduce Smix. Of course, the methods used to compute Smix may be flawed; this issue is discussed in a subsequent section. Lipid phase morphology is not correlated with the size or dispersity of the resulting lipoplexes. This suggests that the observed large particle size was due to inter-lipoplex interactions rather than to differences in lipid packing within the lipid phase.| Pyridinium lipida | Co-lipid | Charge ratio (N:P) |
Lipid phase | Lattice parameter (δ, Å) | S mix |
|---|---|---|---|---|---|
|
a Lipoplexes formulated with a 3:2 ratio of cationic lipid:co-lipid. Pyridinium lipids were mixed 1:1 with EPC. For single valued lattice parameters a typical uncertainty of ±1 Å may be associated. Owing to the broad diffraction peaks produced by some of the samples, an interval is given encompassing the distribution of possible lattice parameters.
|
|||||
| diC12:0 | DOPE | 1.5, 3.0 | Hexagonal | 63 | 1.09 |
| diC14:0 | DOPE | 1.5, 3.0 | Hexagonal | 69 | 1.09 |
| diC16:0 | DOPE | 1.5 | Hexagonal | 68 | 1.09 |
| diC18:0 | DOPE | 1.5 | Hexagonal | 76–84 | 1.09 |
| diC18:1 | DOPE | 1.5 | Hexagonal | 68 | 1.10 |
| diC12:0 | Cholesterol | 1.5, 3.0 | Hexagonal | 80, 84 | 1.08 |
| diC14:0 | Cholesterol | 3.0 | Hexagonal | 84 | 1.08 |
| diC16:0 | Cholesterol | 1.5 | Hexagonal | 82 | 1.09 |
| diC18:0 | Cholesterol | 3.0 | Lamellar | 70 | 1.09 |
| diC18:1 | Cholesterol | 1.5 | Hexagonal | 82 | 1.10 |
| diC9:0 | DOPE | 1.5 | Lamellar | 63 | 1.08 |
| diisoC9:0 | DOPE | 1.5 | Lamellar | 68 | 1.23 |
| diC20:0 | DOPE | 3.0 | Hexagonal | 64 | 1.10 |
| dibrC20:0 | DOPE | 3.0 | Hexagonal | 68 | 1.38 |
| diC9:0 | Cholesterol | 1.5 | Lamellar | 68 | 1.08 |
| diisoC9:0 | Cholesterol | 1.5 | Lamellar | 68–85 | 1.22 |
| diC20:0 | Cholesterol | 3.0 | Lamellar | 71 | 1.09 |
| dibrC20:0 | Cholesterol | 3.0 | Lamellar | 70 | 1.37 |
Lipoplex packaging of DNA
Excluding exploratory experiments, 123 unique formulations of pyridinium lipid(s), EPC, and a neutral co-lipid (cholesterol or DOPE) were assessed for their potential to encapsulate a DNA plasmid (pDNA). Effective DNA packaging is associated with the ability of the lipoplex to neutralize the DNA charge and therefore retard the electrophoretic migration of pDNA in a gel. Fig. 1A gives a representative example; full data are available pages 11–17 in the ESI.† EPC: cholesterol and EPC:DOPE lipoplexes are effective at CR 5 and greater with CR 3 showing a mixture of free pDNA and DNA in an intact lipoplex. A majority of the formulations containing pyridinium lipids show similar gel retardation of DNA at CR 3 and above without any marked differentiation between cholesterol or DOPE as co-lipid.
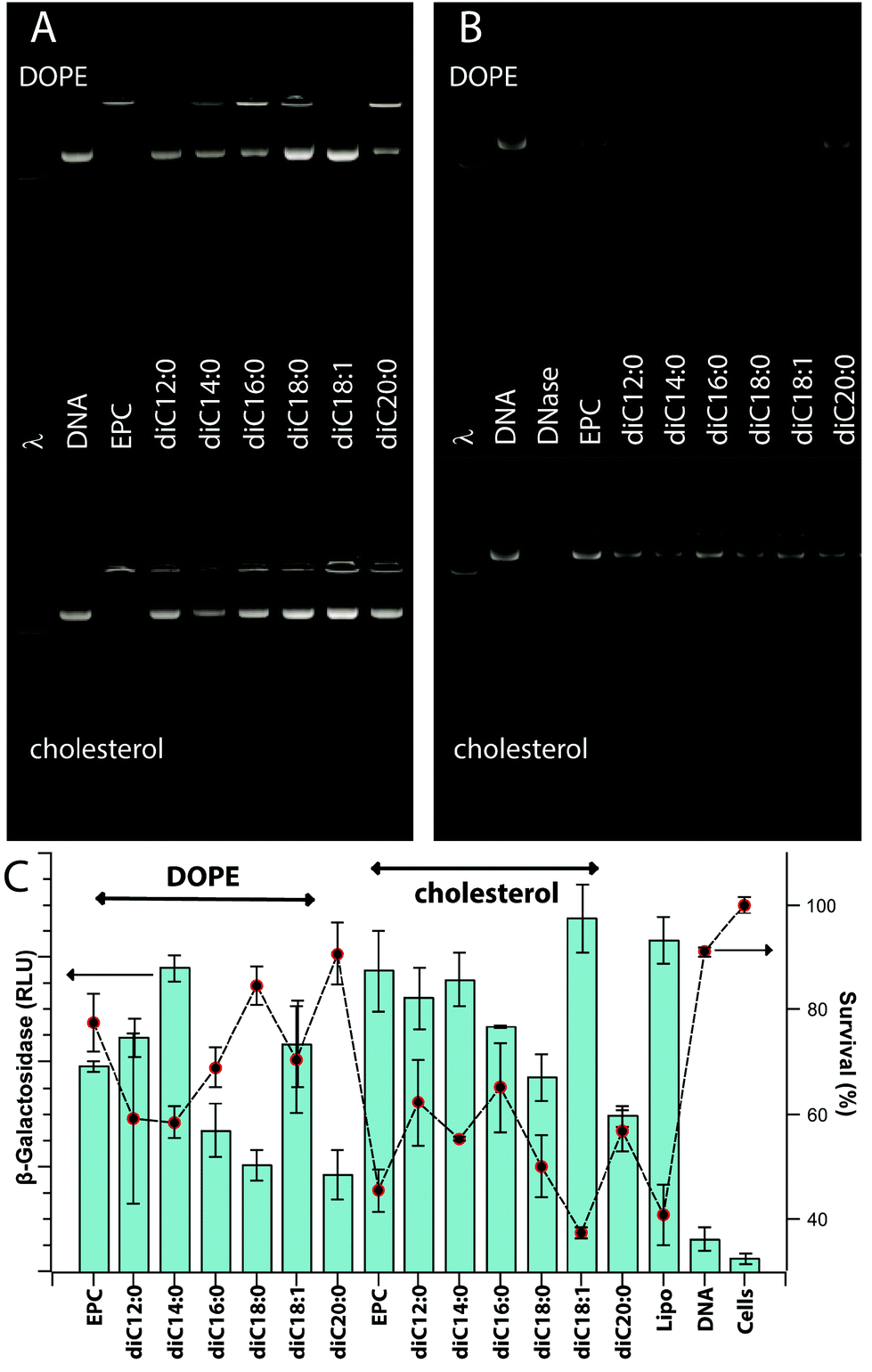 | ||
| Fig. 1 Representative data for gel retardation of formulated lipoplexes (panel A), DNase protection of encapsulated pDNA (panel B), transfection efficiency as luminescence readings of β-galactosidase activity (panel C; bars) and cell viability (panel C; line + symbol). Lipoplexes were formulated from the homologous series of pyridinium lipids diC12:0 to diC20:0/EPC/co-lipid at a mol ratio of 1.5:1.5:2 with pDNA at CR = 3. Lipofectamine2000™ (Lipo) as a positive control, and plasmid DNA alone and CHO-K1 cells alone as negative controls are given. Charts show n = 9; mean ± SD). | ||
Direct protection of encapsulated pDNA from hydrolysis by DNase I was also assessed. Fig. 1B gives a representative example; full data are available on pages 18–23 in the ESI.† EPC:cholesterol and EPC:DOPE afford some level of protection at CR 1.5 and above and again, the majority of pyridinium-containing formulations behave similarly. Notable exceptions include diC9:0 and diisoC9:0 which show only low levels of DNA protection at CR 3 and above, and diC20:0 and dibrC20:0 which are protective only at CR 5 and above. Some formulations based on mixtures and mixed chain species diC18:1, dibrC20:0, and (C18:1)(brC20:0) show a maximal protection at intermediate 1.5 < CR < 5 with CR 10 producing a low level of protection. This issue is discussed in a subsequent section. Data for these experiments are available in the Biological data – Figures section of the ESI.†
Transfection efficiency
Transfection efficiencies of pyridinium lipid containing formulations ranged from 2 to 120% relative efficiency compared to Lipofectamine2000™. Fig. 1C gives a representative example; bar chart data (pp. 24–30) and numerical data (Table S3†) are available in the ESI.† For a majority of relatively active formulations, clear maxima were observed for CR 3 or 5 relative to formulations of the same components at lower or higher CR. Among the least active pyridinium compounds were diC9:0 and diC20:0 especially for formulations of CR 3 or lower. Both these compounds did produce intermediate levels of transfection at CR 10.Transfection experiments with controls and replicates limit the number of independent formulations that can be assessed in parallel within a single experiment to about thirty, or six compounds at five levels of CR. Independent day replicates gave the same relative transfection efficiencies, but direct merging of data from different days required the use of a normalized transfection efficiency (TEnorm) defined as the average transfection efficiency (n = 6) relative to the maximum and minimum levels of transfection observed in the experiment. The maximum was usually the Lipofectamine2000™ result and the minimum was the cells alone without added DNA or lipids. The values of TEnorm from comparable experiments conducted months apart differed by less than 15%. This is comparable to the overall relative standard error for different conditions within a single experiment which indicates that the normalization process used does not introduce significant bias at the same time as it allows comparisons between experiments conducted over many months. The numerical transfection data (raw and normalized) are available in the ESI.†
Cell viability
Cationic lipids – pyridinium salts, EPC – show a degree of dose-dependent toxicity to CHO-K1 cells. Cell viability was assessed using an assay based on the mitochondrial reduction, by living cells, of a tetrazolium salt to a coloured formazan. Values are reported as a percent of the value for untreated cells and range from 5 to 100 ± 5%. Fig. 1C gives a representative example; bar chart data (pp. 24–30) and numerical data (Table S3) are available in the ESI.† Some formulations that were active for transfection (TEnorm > 0.75) were relatively non-toxic (viability > 80%); Lipofectamine2000™ was significantly toxic in all experiments (15% < viability < 30%). Numerical data are available in the ESI.†Molecular parameters
The general procedures used to estimate the required molecular parameters for development of the transfection index are unchanged from our previous report,50 with a few minor refinements for consistency. The earlier work relied upon a mixture of additive values together with “consensus” experimental data for cholesterol.46 More recent experimental and computational work has shown that the equilibrium area of cholesterol strongly depends upon the composition of the lipid phase in which it is located;57–59 the length and volume are relatively unaffected, but the cholesterol position in the phase leads to considerably variable area projected at the head-group plane. We therefore looked at the parameters for cholesterol in more detail.Our earlier approach results in a mixture of meanings for the terms involved. As noted above, V, a0, and lc are the equilibrium tail volume, head group area, and critical chain length of the lipid. These are experimental parameters and do not directly reflect a physical volume, area, or length.60 By use of an additive model, we implicitly assume they do take a physical meaning. The success of the previous predictions depends in part on the cancellation of errors between the three terms in the derived parameter S.50 We wish to continue to treat the additive approach as if it generated physical parameters, so we require estimates for cholesterol based upon the same approach, recognizing that we freeze what we know to be a variable quantity – the cholesterol head group area. The length of cholesterol is certainly 17.5 Å either directly from experiment or by bond length additive methods. We note that the idea of “head-group” either in its equilibrium sense or a physical sense relates to an incompressible area below which steric repulsions rapidly increase the potential energy.44 For cholesterol this is the span C-2 to C-6. Using this region as head group we derived a value of a0 = 27.5 Å2 and V = 545 Å3. Together these give a value of S = 1.14, in reasonable agreement with the previous value of 1.20.
Our previous approach ignored the counterion of the cationic lipid in the determination of the head group area. A closer reading of the background to additive partial molar volumes suggested the parameters applied only to complete salts.61 We have incorporated a chloride ion in the current model. A table of the molecular parameters utilized is given in Table S4 in the ESI.†
Development of Transfection Index
We previously reported a Transfection Index (TIPSV) based on the product of a partition term, a shape term and a volume filling term50 given by the following equations:| partition term = clogPmix/|clogP+ − clogP0| | (1) |
| shape term = |S+ − 1| | (2) |
| volume term = (nexp/nlat) − (nexp/ncyl); nexp < nlat | (3a) |
| volume term = 1 − (nexp/ncyl); nexp ≥ nlat | (3b) |
The clogP terms of eqn (1) are computed for the mixture of lipids, the mixture of cationic lipids, and the mixture of neutral lipids with subscripts mix, +, and 0 respectively. We note a problem with this term that allows it to approach ∞ as the difference in the denominator approaches 0; we resolve this issue below. Eqn (2) focussed solely on the computed shape parameter of the cationic lipid. The volume filling term (eqn (3a) and (3b)) is based on the number of lipids per DNA base pair (n) required to fulfil the volumetric requirements of a hexagonal lattice of lipid-coated DNA and of a cylinder of bilayer-coated DNA (subscripts lat and cyl respectively) relative to the experimentally set number of lipids per base pair (subscript exp).
Despite the adequate correlation that TIPSV provided for our initial dataset, that result was clearly adventitious as illustrated in Fig. 2A for a “training” subset of pyridinium lipids of varying linear hydrocarbon chain length diC12:0 to diC20:0 at a CR = 3 with two different neutral lipids. A closer look at the data suggested that there was an optimum chain length at di14:0 (Fig. 2B). Addition of data for diC18:1 and dibrC20:0 shows that the variation in transfection cannot be related to partition; diC20:0 and dibrC20:0 have closely similar clogP but differ significantly in activity. The parameter of merit is length of the longest carbon chain, although in itself this is incomplete as diC18:0 is much less active that di18:1.
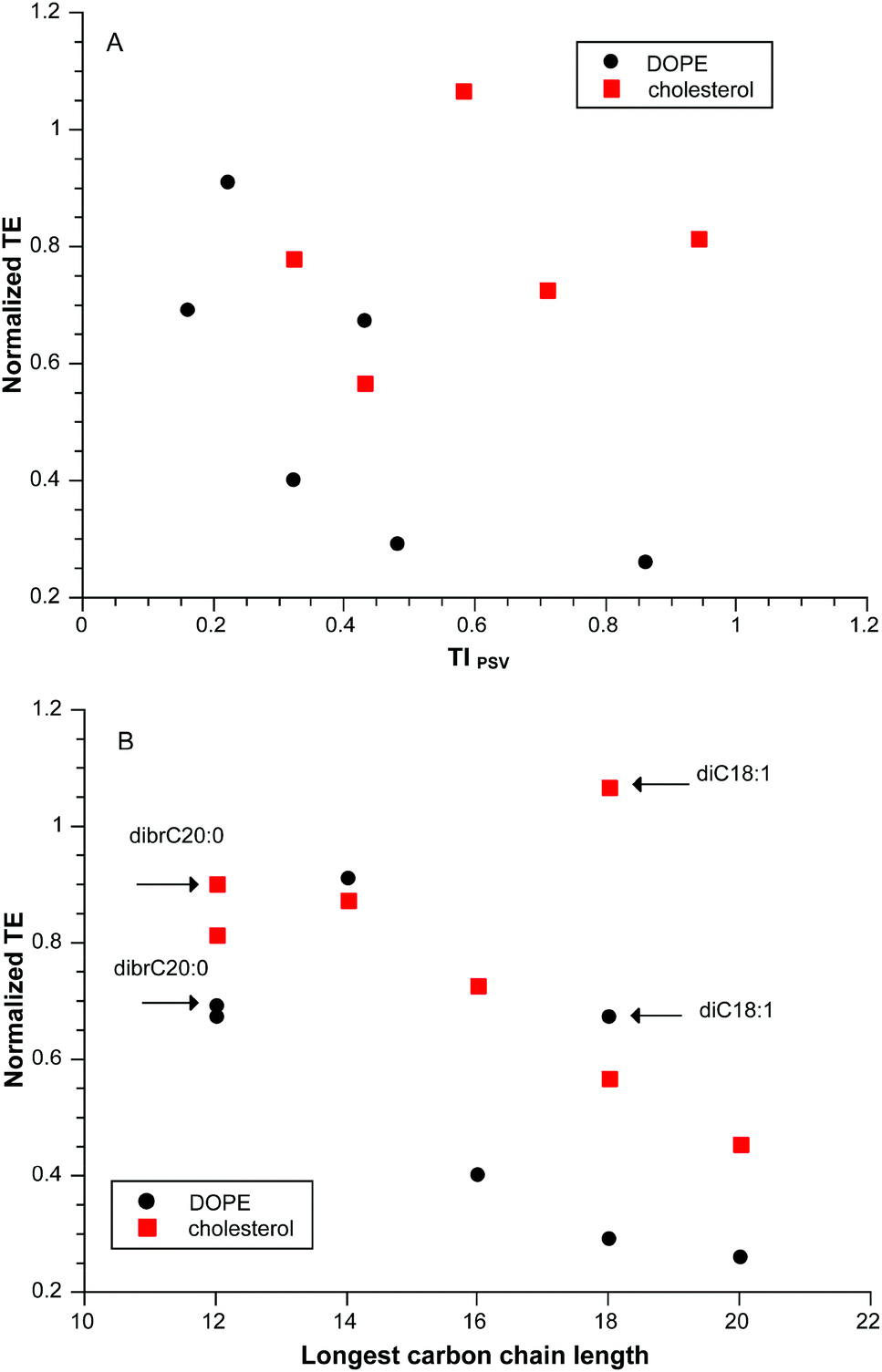 | ||
| Fig. 2 Panel A Normalized transfection efficiency as a function of computed TIPSV for diCn:0 (n = 12, 14, 16, 18, 20) formulated with EPC and neutral lipids DOPE or cholesterol at CR = 3; panel B: normalized transfection efficiency as a function of longest carbon chain length for the same data as panel A plus additional values for diC18:1 and dibrC20:0. | ||
The problem with the partition term was noted above so we explored alternative expressions. Eqn (4) is one transformation of the same terms used previously and can reach a maximum value of 1, similar to the volume term of eqn (3a) and (3b).
| partition term = 1/(clogPmix − |clogP+ − clogPmix|) | (4) |
This change made no change to the fortunate correlations observed previously, or to the lack of correlation in Fig. 2A. We note as well that the shape term (eqn (2)) is not an independent term as it contains the same geometric parameters of volume, length and area as are used in the volume filling term (eqn (3a) and (3b)). However, these are technical refinements that do not address the core issues.
Hydrocarbon chain length directly influences two physical properties of lipid mixtures. One is the melting behaviour of the pure lipid components which scales with carbon number for a range of head groups and lipid structures.62 Another parameter is the miscibility of lipid mixtures which is directly related to chain-length mismatch in simple cases.63 Previous studies that have concurrently investigated the relationship between lipid phase behaviour and transfection have shown that the melting temperature of a binary mixture of EPC with saturated C14 and C16 chains plays a critical role in transfection efficiency.13,42,64 The most effective compositions are those where the melting temperature is close to the temperature of the transfection experiment (37 °C). Lipid mobility is enhanced at the melting temperature via the inevitable formation of defects as the structured phase melts.62
Our ternary lipid mixtures are unlikely to have simple phase behaviours and it is impossible to predict their true nature. However, it is likely that as the chain length of the pyridinium component increases, there will be an increase in the “melting” temperatures of the mixture or a lipid redistribution between coexisting phases that may be present. If there are fluid phases present, then variation in chain length will have little effect on transfection. However, above some threshold value of pyridinium chain length the melting temperature may shift to higher than the experimental temperature with an increasingly negative effect on transfection efficiency due to decreased lipid mobility.
We attempt to capture this in a melting term (eqn (5a)–(5c)):
| melting term = 1/(Cn+ − Cnmix); Cn+ ≥ Cnmix | (5a) |
| melting term = 1; Cn+ < Cnmix | (5b) |
| melting term = 1; |1/(Cn+ − Cnmix| > 1 | (5c) |
The focus is on Cn, the number of independent rotors in the longest continuous chain of either the pyridinium lipid (subscript +) or the lipid mixture (subscript mix; determined as the molar weighted average of the Cn of the components present). This approach follows directly from the correlations derived from both melting and mixing of pure lipids and binary mixtures.62,63 This term will vary only under the conditions of eqn (5a) to diminish the overall TI under conditions where the pyridinium lipid significantly pushes the mixture towards higher melting temperature. If the pyridinium chain length is shorter than the molar weighted average of the mixture then the pyridinium lipid will play a melting temperature reducing role in the overall phase behaviour and the term will take its maximum value (eqn (5b)). Similarly, if the pyridinium is roughly comparable to the mixture and the denominator of (5a) becomes large, eqn (5c) ensures the term does not exceed the maximum of 1.
For linear hydrocarbon chains Cn is the total carbon length less the two end groups as this defines the number of independent rotors. For an oleyl chain (18:1) this value is 8 as previously reported.62,63 Cholesterol behaves less as a chain and more as a diluent to reduce the cooperative length of the phase transition;62,63 we assume it behaves as if it were a diluent of the same overall length as a C16 hydrocarbon chain as these two species have very similar value of lc (17.4 Å) and thus the same value of Cn ( = 14).
We can therefore write an expression for TIPVM as a product of there terms:
| TIPVM = partition term × shape term × melting term |
The required terms are eqn (1), (3a)–(3b) and (5a)–(5c) respectively. This produces values between 0 and 0.1 for the range of clogP values in the experiments so we multiply by ten to get a convenient scaling. Does this approach work any better than in Fig. 2A? The results for the same dataset are given in Fig. 3. Visually the correlation is more persuasive. Statistically we fit a line to the data using the known standard errors in the normalized transfection efficiencies to give the results shown with an r2 = 0.89. There is obvious scatter but there is a basis for deeper exploration of TIPVM beyond the initial 14 formulations of the “training” subset in Fig. 3.
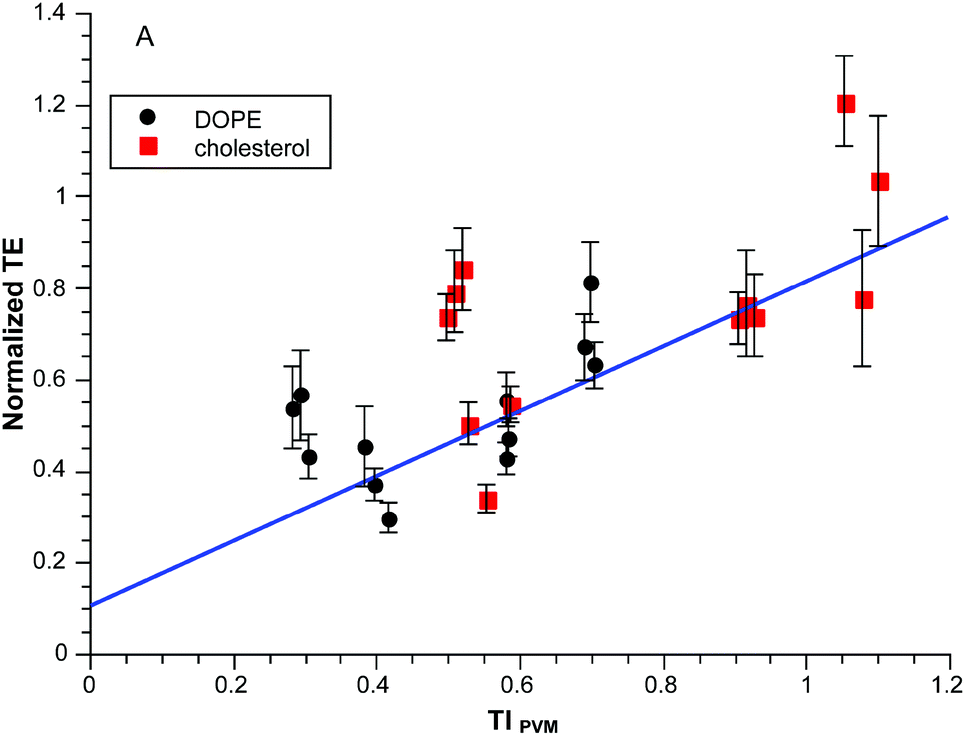 | ||
| Fig. 3 Normalized transfection efficiency as a function of TIPVM. The experimental data is the same as Fig. 2. | ||
Although TIPVM has removed the lipid shape parameter as an explicit term, pyridinium lipid shape is presumed to be an important factor at the point of lipoplex release of cargo within the cell.13,40 The range of S for the series of compounds correlated in Fig. 3 is quite limited (S = 1.21 ± 0.02) resulting in a narrow range of values in the transfection experiments (Smix = 1.09 ± 0.01); we require compounds with high values of S as a means to probe TIPVM more extensively. The bis-isononyl ester diisoC9:0 (S = 1.67) proved to be quite cytotoxic (see below), but dibrC20:0 (S = 2.16) had acceptably low toxicity and at high CR was an active transfection agent. As noted above, the lipid films and initial liposome dispersions of the dibrC20:0 resulted in lipid fractionation. The lamellar phases observed by SAXD for samples with high Smix (Table 1) are consistent with this type of fractionation.
Fortunately, binary blends of diC18:1 and dibrC20:0 were apparently jointly taken into the liposomes so it was possible to explore the effect of S using mixtures of these two components. For example, a 1:1 blend of these components produces an effective S = 1.70 which in turn produces Smix = 1.24 when mixed with EPC and a co-lipid. Fig. 4A shows normalized TE as a function of TIPVM for binary blends with values of Smix ranging from 1.24–1.33. The correlation is similar (r2 = 0.92) to that of Fig. 3.
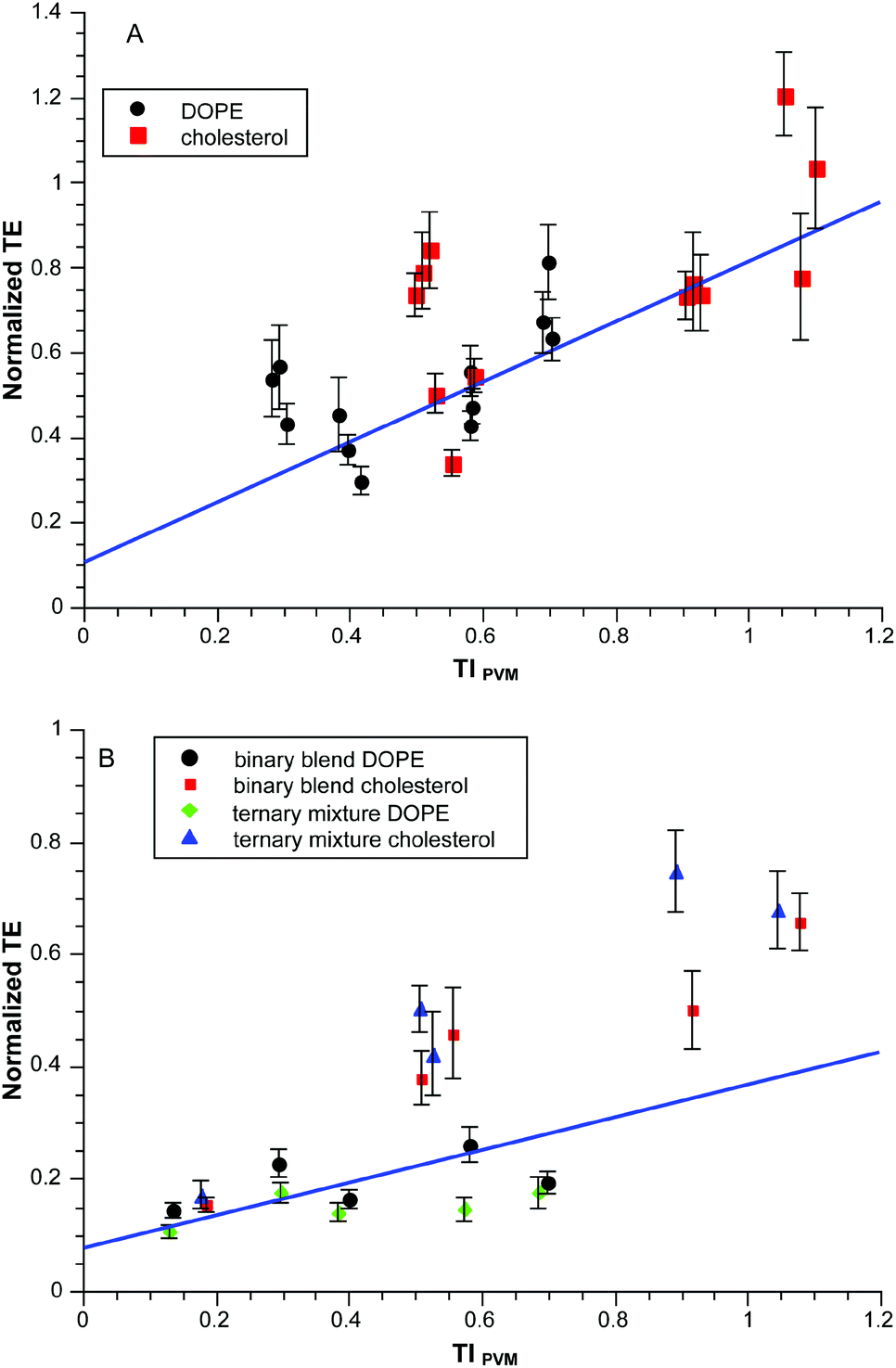 | ||
| Fig. 4 Normalized transfection efficiency as a function of TIPVM. A: binary blends of diC18:1 and dibrC20:0 in EPC:co-lipid to produce a range of values of Smix CR = 3.0; B: binary blends of diC18:1 and dibrC20:0 and a ternary mixture of C18:1: dibrC20:0: (C18:1)(brC20:0) to produce Smix = 1.28, CR = 0.5, 1.5, 3, 5, 10. | ||
As a further check we directly prepared ternary mixtures from a 1:1 mixture of the starting alcohols. ESI MS analysis of the product mixture gave the composition 16:28:56 mol% diC18:1, dibrC20:0 and the mixed chain species (C18:1)(brC20:0) with an effective S = 1.70, which in turn produced lipoplexes with Smix = 1.28. A binary blend of diC18:1 and dibrC20:0 in a ratio of 32:68 produces the same final Smix. Fig. 4B shows normalized TE as a function of TIPVM for these ternary mixtures and binary blends, all having the same value of Smix. The correlation in this case is poorer than previously (r2 = 0.70) and appears to be dependent upon the neutral co-lipid. For a given co-lipid the correlations are comparable to one another and the previous datasets (DOPE r2 = 0.86; cholesterol r2 = 0.77). It is possible that some differential lipid fractionation occurred as these systems do involve dibrC20:0.
The full dataset consisted of 123 independent conditions conducted in independently duplicated triplicates (see ESI†). Some conditions were quite toxic to the cells leaving 90 conditions where cell survival exceeded 20% (50 with DOPE; 40 with cholesterol as co-lipid). The selection of 20% survivorship is both arbitrary and quite low. It is largely guided by two factors: (1) Lipofectamine2000™ is quite toxic in this cell line and in some experiments cell viability in the Lipofectamine2000™ treatment is about this level; (2) We are interested in capturing the full effect of structural variables on transfection so we need to consider as wide a range of compounds as possible, including those with toxicity that precludes any subsequent practical use.
The correlation of the full dataset with TIPVM weighted by the known standard errors in normalized TE is given in Fig. 5. The overall r2 for this weighted data is 0.7; there is no difference between the cholesterol and DOPE subsets considered separately. The scatter is obvious in the dataset but does indicate an underlying correlation with TIPVM which in turn is based upon a handful of parameters which are not adjusted.
 | ||
| Fig. 5 Normalized transfection efficiency as a function of TIPVM for all available data on pyridinium lipids with EPC and co-lipid. | ||
Cytotoxicty
As noted above, some formulations were significantly toxic to the cells and in some cases the extent of transfection appeared to increase as the formulation becomes more toxic. We therefore explored the dataset to uncover factors that affect the survival of the cells. Fig. 6A shows the cell survival as a function of charge ratio for several systems in a semi-log plot; this is a dose–response curve as the amount of DNA is constant and the amount of cationic lipid scales directly. The correlation of log(survival%) with CR is acceptable (r2 > 0.9) in all cases except for the toxic diisoC9:0 compound. The isomeric pyridinium diC9:0 is relatively less toxic and similar to longer chain pyridinium compounds. The pyridinium compounds as a group are less toxic than EPC alone.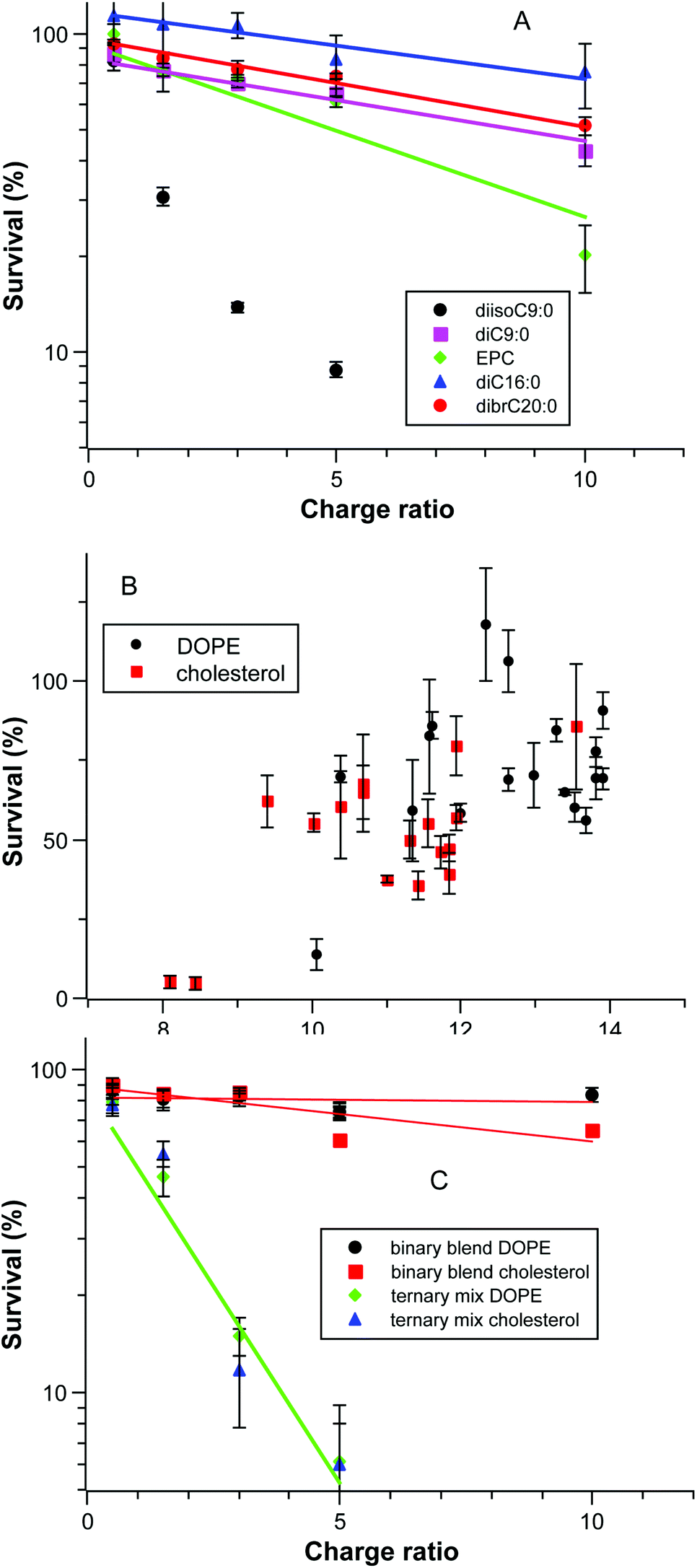 | ||
| Fig. 6 Relationship of cell survival to transfection mixture parameters. (A) Selected data of log(cell survival) as a function of charge ratio of the lipoplex; (B) Cell survival as a function of the clogPmix of the lipid mixture (C) log(cell survival) as a function of charge ratio for binary blends of diC18:1 and dibrC20:0 and a ternary mixture of diC18:1: dibrC20:0: (C18:1)(brC20:0) each having Smix = 1.28. | ||
A second factor controlling the toxicity of a formulation is the lipophilic character of lipid mixture (Fig. 6B). Cholesterol has a substantially lower clogP (9.9) than DOPE (14.8) so the clogPmix of a given pyridinium lipid in a mixture has an offset that is dependent upon the co-lipid; the offset is 1.9 at the ratio of cationic to neutral lipids used. Although there appears to be a general increase in cell survival as clogPmix increases, the correlation is poor and other factors must be important. One of those factors appears in the comparison of the toxicity of binary blends and ternary mixtures of diC18:1, dibrC20:0, and (C18:1)(brC20:0) as given in Fig. 6C; clogPmix alone is insufficient to rationalize the observed toxicity. Fig. 6C and 4B are derived from the same experiments, the Smix is fixed at 1.28, and the clogPmix values for a given co-lipid are the same (DOPE 13.3; cholesterol 11.4). Even with close similarities the two binary blend sample series are much less toxic than the two ternary mixture series. Note as well that this is independent of the relative transfection efficiencies as given in Fig. 4B; transfection and toxicity are uncorrelated. The key difference between the binary blended samples and the ternary mixture is the presence of (18:1)(br20:0) in the latter. The toxicity evident in Fig. 6C must be due to this lipophilic component.
The two most toxic pyridinium compounds uncovered are diisoC9:0 and (18:1)(brC20:0). They lie at the extremes of the range of clogP for the set of compounds investigated but have high and similar S values (1.67 and 1.60 respectively). They are also the least “lipid-like” compounds in this study (branching in the chains; chain-length mis-match). It is possible that neither of these compounds is well-tolerated in cell membranes and as a result these compounds are more mobile in the cytoplasm and can reach cellular targets more easily than less mobile components.
The experimental work in this study is based on a 1:1 ratio of a pyridinium compound and EPC. To further extract the toxic effect of the pyridinum compound from the toxic effect of EPC this ratio would have to vary. More detailed conclusions on the structural factors that control the toxicity of a given pyridinum compound await that dataset.
Conclusions
The goal of this study was to examine our earlier proposal of a transfection index based upon partitioning, lipid shape, and lattice filling considerations. It is clear that the previous correlation was adventitious and we here developed a modification built on the same concepts which does a better job on a much larger dataset. To the extent that we have succeeded we have confirmed that the assembly of lipoplexes is under the structural control of the components. We also examined potential factors that influence the cytotoxicity of the mixtures utilized.It is doubtful that we have identified all the key components even within this relatively limited system as we have restricted ourselves to pyridinium-based cationic lipids. There is ample evidence that the overall charge on the lipoplex plays a significant role in transfection.27,29,32 We have been able to ignore this factor as the pyridinium lipids have fixed charges and are mixed in fixed proportions to other fixed-charge species (EPC, DNA) to produce – we assume – lipoplexes with approximately constant surface charge. The same type of study could examine the issue of charge as a contributing term using ionisable lipids (polyamines) in conjunction with related quaternized ammonium compounds.
Another as yet unexplored dimension is the influence of cell type on transfection with defined lipoplexes. Commercial transfection kits promise a broad spectrum of activity. Is this a consequence of a general structure-derived source of activity as our correlation here suggests? The same question arises with respect to the nature of the nucleic acids.
In the end, the robust packaging of the cargo is the key issue. We are a long way from packaging that is too robust based on supramolecular assembly from lipidic components. As noted above, we have relatively poor control over the particle size and dispersity with the preparative method utilized. It is possible that some of the scatter between replicates and some of the deviations from the overall trend are simply due to variations in the physical parameters of the assemblies. Microfluidic mixing is an emerging technology for the formation of lipid-nucleic acid nanoparticles for siRNA gene silencing.65,66 Such techniques offer considerable control over particle size and are an important next step in establishing that the correlations reported here are themselves not simply adventitious.
Experimental methods
The synthetic procedures generally follow previous reports;50 details for preparations and characterizations are given in the ESI.† The biochemical methods were the previously published methods;50 data on gel retardation, DNase protection, transfection and viability assays are in the ESI.† The SAXD data were acquired using previously published protocols and analysed using the same methodology;50 the findings are reported in Table 1. The molecular parameters were calculated by fragment additive methods as previously described.50 A table of molecular parameters utilized is given in the ESI† along with the full dataset from which the graphs of the paper were derived. Linear correlations with uncertainty in the transfection efficiency were computed using the linest package in Python.Acknowledgements
This work was made possible by a grant from the Qatar National Priorities Research Program, award NPRP08-705-3-144. The contents are solely the responsibility of the authors and do not necessarily represent the official views of the Qatar National Research Fund. Synchrotron beam time on the BioSAXS beam line (BM29) was provided by the European Synchrotron Radiation Facility in Grenoble, France. On-going support from the National Science and Engineering Research Council of Canada is gratefully acknowledged.References
- F. Mingozzi and K. A. High, Nat. Rev. Genet., 2011, 12, 341–355 CrossRef CAS PubMed.
- M. A. Kay, Nat. Rev. Genet., 2011, 12, 316–328 CrossRef CAS PubMed.
- B. L. Davidson and P. B. McCray, Nat. Rev. Genet., 2011, 12, 329–340 CrossRef CAS PubMed.
- T. Wirth, N. Parker and S. Yla-Herttuala, Gene, 2013, 525, 162–169 CrossRef CAS PubMed.
- H. Yin, R. L. Kanasty, A. A. Eltoukhy, A. J. Vegas, J. R. Dorkin and D. G. Anderson, Nat. Rev. Genet., 2014, 15, 541–555 CrossRef CAS PubMed.
- R. Kanasty, J. R. Dorkin, A. Vegas and D. Anderson, Nat. Mater., 2013, 12, 967–977 CrossRef CAS PubMed.
- T. M. Allen and P. R. Cullis, Adv. Drug Delivery Rev., 2013, 65, 36–48 CrossRef CAS PubMed.
- I. S. Zuhorn, J. Engberts and D. Hoekstra, Eur. Biophys. J. Biophys. Lett., 2007, 36, 349–362 CrossRef CAS PubMed.
- B. Draghici and M. A. Ilies, J. Med. Chem., 2015, 58, 4091–4130 CrossRef CAS PubMed.
- M. A. Mintzer and E. E. Simanek, Chem. Rev., 2009, 109, 259–302 CrossRef CAS PubMed.
- P. L. Felgner, T. R. Gadek, M. Holm, R. Roman, H. W. Chan, M. Wenz, J. P. Northrop, G. M. Ringold and M. Danielsen, Proc. Natl. Acad. Sci. U. S. A., 1987, 84, 7413–7417 CrossRef CAS.
- W. Li and F. C. J. Szoka, Pharm. Res., 2007, 24, 438–449 CrossRef CAS PubMed.
- R. Koynova and B. Tenchov, in Nucleic Acid Transfection, ed. W. Bielke and C. Erbacher, Springer-Verlag Berlin, Berlin, 2010, vol. 296, pp. 51–93 Search PubMed.
- R. Koynova, B. Tenchov, L. Wang and R. C. MacDonald, Mol. Pharmaceutics, 2009, 6, 951–958 CrossRef CAS PubMed.
- M. Dittrich, M. Heinze, C. Wölk, S. S. Funari, B. Dobner, H. Möhwald and G. Brezesinski, ChemPhysChem, 2011, 12, 2328–2337 CrossRef CAS PubMed.
- D. A. Medvedeva, M. A. Maslov, R. N. Serikov, N. G. Morozova, G. A. Serebrenikova, D. V. Sheglov, A. V. Latyshev, V. V. Vlassov and M. A. Zenkova, J. Med. Chem., 2009, 52, 6558–6568 CrossRef CAS PubMed.
- D. Zhi, S. Zhang, S. Cui, Y. Zhao, Y. Wang and D. Zhao, Bioconjugate Chem., 2013, 24, 487–519 CrossRef CAS PubMed.
- R. Sheng, T. Luo, H. Li, J. Sun, Z. Wang and A. Cao, Bioorg. Med. Chem., 2013, 21, 6366–6377 CrossRef CAS PubMed.
- K. Kostarelos and A. D. Miller, Chem. Soc. Rev., 2005, 34, 970–994 RSC.
- T. Montier, T. Benvegnu, P.-A. Jaffres, J.-J. Yaouanc and P. Lehn, Curr. Gene Ther., 2008, 8, 296–312 CrossRef CAS PubMed.
- D. A. Balazs and W. Godbey, J. Drug Delivery, 2011, 2011 Search PubMed.
- A. Pensado, B. Seijo and A. Sanchez, Expert Opin. Drug Delivery, 2014, 11, 1721–1731 CrossRef CAS PubMed.
- E. A. Ivanova, M. A. Maslov, T. O. Kabilova, P. A. Puchkov, A. S. Alekseeva, I. A. Boldyrev, V. V. Vlassov, G. A. Serebrennikova, N. G. Morozova and M. A. Zenkova, Org. Biomol. Chem., 2013, 11, 7164–7178 CAS.
- H. Lv, S. Zhang, B. Wang, S. Cui and J. Yan, J. Controlled Release, 2006, 114, 100–109 CrossRef CAS PubMed.
- R. Gref, A. Domb, P. Quellec, T. Blunk, R. H. Müller, J. M. Verbavatz and R. Langer, Adv. Drug Delivery Rev., 2012, 64(Supplement), 316–326 CrossRef.
- K. Buyens, S. C. De Smedt, K. Braeckmans, J. Demeester, L. Peeters, L. A. van Grunsven, X. de Mollerat du Jeu, R. Sawant, V. Torchilin, K. Farkasova, M. Ogris and N. N. Sanders, J. Controlled Release, 2012, 158, 362–370 CrossRef CAS PubMed.
- C. L. Chan, K. K. Ewert, R. N. Majzoub, Y. K. Hwu, K. S. Liang, C. Leal and C. R. Safinya, J. Gene Med., 2014, 16, 84–96 CrossRef CAS PubMed.
- Z. U. Rehman, I. S. Zuhorn and D. Hoekstra, J. Controlled Release, 2013, 166, 46–56 CrossRef PubMed.
- M. Jayaraman, S. M. Ansell, B. L. Mui, Y. K. Tam, J. Chen, X. Du, D. Butler, L. Eltepu, S. Matsuda, J. K. Narayanannair, K. G. Rajeev, I. M. Hafez, A. Akinc, M. A. Maier, M. A. Tracy, P. R. Cullis, T. D. Madden, M. Manoharan and M. J. Hope, Angew. Chem., Int. Ed., 2012, 51, 8529–8533 CrossRef CAS PubMed.
- K. K. Ewert, A. Zidovska, A. Ahmad, N. F. Bouxsein, H. M. Evans, C. S. McAllister, C. E. Samuel and C. R. Safinya, in Nucleic Acid Transfection, ed. W. Bielke and C. Erbacher, 2010, vol. 296, pp. 191–226 Search PubMed.
- A. Zidovska, H. M. Evans, A. Ahmad, K. K. Ewert and C. R. Safinya, J. Phys. Chem. B, 2009, 113, 5208–5216 CrossRef CAS PubMed.
- A. Ahmad, H. M. Evans, K. Ewert, C. X. George, C. E. Samuel and C. R. Safinya, J. Gene Med., 2005, 7, 739–748 CrossRef CAS PubMed.
- M. Stanulla, E. Schaeffeler and M. Schwab, in Genomics and Pharmacogenomics in Anticancer Drug Development and Clinical Response, ed. F. Innocenti, Humana Press, 2009, ch. 11, pp. 173–201, DOI:10.1007/978-1-60327-088-5_11.
- S. Sahasranaman, D. Howard and S. Roy, Eur. J. Clin. Pharmacol., 2008, 64, 753–767 CrossRef CAS PubMed.
- P. Karran and N. Attard, Nat. Rev. Cancer, 2008, 8, 24–36 CrossRef CAS PubMed.
- G. Basha, T. I. Novobrantseva, N. Rosin, Y. Y. C. Tam, I. M. Hafez, M. K. Wong, T. Sugo, V. M. Ruda, J. Qin, B. Klebanov, M. Ciufolini, A. Akinc, Y. K. Tam, M. J. Hope and P. R. Cullis, Mol. Ther., 2011, 19, 2186–2200 CrossRef CAS PubMed.
- B. C. Ma, S. B. Zhang, H. M. Jiang, B. D. Zhao and H. T. Lv, J. Controlled Release, 2007, 123, 184–194 CrossRef CAS PubMed.
- I. M. Hafez, N. Maurer and P. R. Cullis, Gene Ther., 2001, 8, 1188–1196 CrossRef CAS PubMed.
- J. Šmisterová, A. Wagenaar, M. C. A. Stuart, E. Polushkin, G. ten Brinke, R. Hulst, J. B. F. N. Engberts and D. Hoekstra, J. Biol. Chem., 2001, 276, 47615–47622 CrossRef PubMed.
- R. Koynova, L. Wang and R. C. MacDonald, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 14373–14378 CrossRef CAS PubMed.
- B. G. Tenchov, L. Wang, R. Koynova and R. C. MacDonald, Biochim. Biophys. Acta, 2008, 1778, 2405–2412 CrossRef CAS PubMed.
- R. Koynova and B. Tenchov, Soft Matter, 2009, 5, 3187–3200 RSC.
- C. Leal, K. K. Ewert, R. S. Shirazi, N. F. Bouxsein and C. R. Safinya, Langmuir, 2011, 27, 7691–7697 CrossRef CAS PubMed.
- J. Israelachvili, Intermolecular and Surface Forces, Academic Press, London, Second edn., 1992 Search PubMed.
- J. N. Israelachvili, D. J. Mitchell and B. W. Ninham, J. Chem. Soc., Faraday Trans. 2, 1976, 72, 1525–1568 RSC.
- V. V. Kumar, Proc. Natl. Acad. Sci. U. S. A., 1991, 88, 444–448 CrossRef CAS.
- J. A. Gruneich and S. L. Diamond, J. Gene Med., 2007, 9, 381–391 CrossRef CAS PubMed.
- R. W. Horobin and V. Weissig, J. Gene Med., 2005, 7, 1023–1034 CrossRef CAS PubMed.
- S. P. Jones, N. P. Gabrielson, C.-H. Wong, H.-F. Chow, D. W. Pack, P. Posocco, M. Fermeglia, S. Pricl and D. K. Smith, Mol. Pharm., 2011, 8, 416–429 CrossRef CAS PubMed.
- P. Parvizi, E. Jubeli, L. Raju, N. A. Khalique, A. Almeer, H. Allam, M. A. Manaa, H. Larsen, D. Nicholson, M. D. Pungente and T. M. Fyles, Int. J. Pharm., 2014, 461, 145–156 CrossRef CAS PubMed.
- D. Pijper, E. Bulten, J. Smisterova, A. Wagenaar, D. Hoekstra, J. Engberts and R. Hulst, Eur. J. Org. Chem., 2003, 4406–4412 CrossRef CAS.
- E. J. R. Sudholter, J. B. F. N. Engberts and W. H. De Jeu, J. Phys. Chem., 1982, 86, 1908–1913 CrossRef CAS.
- A. A. P. Meekel, A. Wagenaar, J. Smisterova, J. E. Kroeze, P. Haadsma, B. Bosgraaf, M. C. A. Stuart, A. Brisson, M. H. J. Ruiters, D. Hoekstra and J. Engberts, Eur. J. Org. Chem., 2000, 665–673 CrossRef CAS.
- M. A. Ilies, T. V. Sommers, L. C. He, A. Kizewski and V. D. Sharma, in Amphiphiles: Molecular Assembly and Applications, American Chemical Society, 2011, vol. 1070, ch. 2, pp. 23–38 Search PubMed.
- B. Kedika and S. V. Patri, J. Med. Chem., 2011, 54, 548–561 CrossRef CAS PubMed.
- T. A. Balbino, A. A. M. Gasperini, C. L. P. Oliveira, A. R. Azzoni, L. P. Cavalcanti and L. G. de La Torre, Langmuir, 2012, 28, 11535–11545 CrossRef CAS PubMed.
- E. Falck, M. Patra, M. Karttunen, M. T. Hyvönen and I. Vattulainen, Biophys. J., 2004, 87, 1076–1091 CrossRef CAS PubMed.
- S. A. Pandit, S. W. Chiu, E. Jakobsson, A. Grama and H. L. Scott, Langmuir, 2008, 24, 6858–6865 CrossRef CAS PubMed.
- C. Hofsass, E. Lindahl and O. Edholm, Biophys. J., 2003, 84, 2192–2206 CrossRef CAS PubMed.
- R. Nagarajan, Langmuir, 2002, 18, 31–38 CrossRef CAS.
- H. Durchschlag and P. Zipper, in Ultracentrifugation, ed. M. D. Lechner, Dr Dietrich Steinkopff Verlag, Berlin 33, 1994, vol. 94, pp. 20–39 Search PubMed.
- T. Heimberg, Thermal Biophysics of Membranes, Wiley-VCH, Weinheim, 2007 Search PubMed.
- D. Marsh, Biochim. Biophys. Acta, 2010, 1798, 40–51 CrossRef CAS PubMed.
- R. Koynova, L. Wang and R. C. MacDonald, J. Phys. Chem. B, 2007, 111, 7786–7795 CrossRef CAS PubMed.
- N. M. Belliveau, J. Huft, P. J. C. Lin, S. Chen, A. K. K. Leung, T. J. Leaver, A. W. Wild, J. B. Lee, R. J. Taylor, Y. K. Tam, C. L. Hansen and P. R. Cullis, Mol. Ther. - Nuclic Acids, 2012, 1, e37 CrossRef PubMed.
- A. K. K. Leung, I. M. Hafez, S. Baoukina, N. M. Belliveau, I. V. Zhigaltsev, E. Afshinmanesh, D. P. Tieleman, C. L. Hansen, M. J. Hope and P. R. Cullis, J. Phys. Chem. C, 2012, 116, 18440–18450 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic methods and compound characterization; biological methods and data. See DOI: 10.1039/c6ob00041j |
| This journal is © The Royal Society of Chemistry 2016 |