
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis, analysis and biological evaluation of novel indolquinonecryptolepine analogues as potential anti-tumour agents†
A.
Le Gresley
*,
V.
Gudivaka
,
S.
Carrington
,
A.
Sinclair
and
J. E.
Brown
School of Life, Chemical and Pharmaceutical Sciences, SEC Faculty, Kingston University, Kingston-upon-Thames, KT1 2EE, UK. E-mail: a.legresley@kingston.ac.uk
First published on 12th February 2016
Abstract
A small library of cryptolepine analogues were synthesised incorporating halogens and/or nitrogen containing side chains to optimise their interaction with the sugar–phosphate backbone of DNA to give improved binding, interfering with topoisomerase II hence enhancing cytotoxicity. Cell viability, DNA binding and Topoisomerase II inhibition is discussed for these compounds. Fluorescence microscopy was used to investigate the uptake of the synthesised cryptolepines into the nucleus. We report the synthesis and anti-cancer biological evaluation of nine novel cryptolepine analogues, which have greater cytotoxicity than the parent compound and are important lead compounds in the development of novel potent and selective indoloquinone anti-neoplastic agents.
Introduction
Cryptolepine (1) has been known for some years to possess activity as an anticancer agent not only by intercalating into DNA but also by inhibiting the topoisomerase II. This has led to a number of derivatives being synthesised incorporating halogens at various ring positions.1–6 Comparison of anti-cancer activity of cryptolepines with chloro, bromo and iodo atoms at the 11 position showed considerable variation, with 11-bromo showing an improved cytotoxicity profile and the 11-chlorinated compound showing a reduction in the activity compared to the parent compound in the tested cell lines. Of those synthesized, 7,11-dichlorocryptolepine (2) showed the highest cytotoxic activity against MAC15a (colon adenocarcinoma) cells with an IC50 value of 14.4 ± 2.8 μM (after 1 hour exposure).7,8Whilst the literature is generally in agreement that cryptolepines are an interesting target for anti-cancer research, the mode of action is still not that well understood.9 For example, it is observed that the cytotoxicity of cryptolepine is linked to its ability to inhibit the topoisomerase II enzyme so it would be expected that bromo and iodo compounds would show greater topoisomerase II inhibition owing to their increased lipophilicity. Topoisomerase II assay results of these 11-halogenated compounds showed they are responsible for greater inhibition compared to the parent molecule 1, but surprisingly the 11-chloro derivative showed the highest topoisomerase II inhibition despite its lower in vitrocyototoxicity.10–12 It was hyptothesised that this could be due to the changes in drug–DNA interactions or drug uptake based on the structure and size of the molecule.13
Materials and methods
Synthetic chemistry
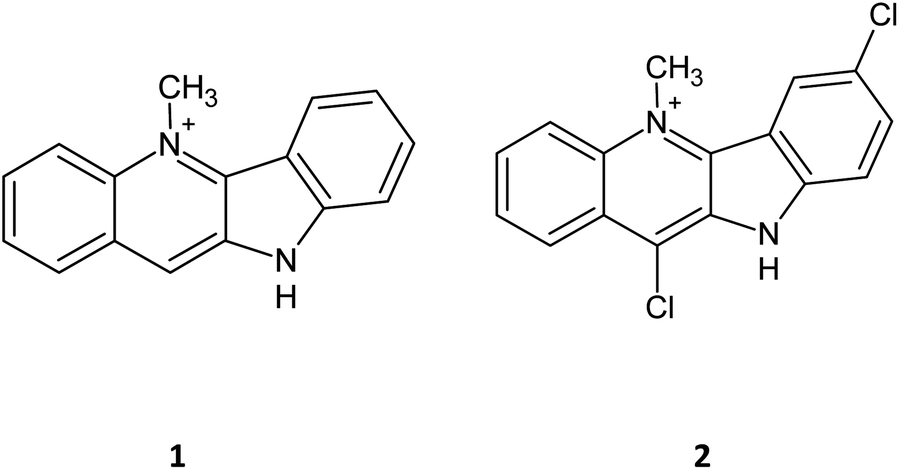
The synthetic techniques used in the synthesis of the chosen targets are reasonably well known and details of key coupling and quaternisation reactions are given in more detail under general procedures. Full spectroscopic data and detailed methodology is provided in the ESI.†Scheme 1 indicates the varied routes taken to achieve the synthesis of a series of amido, amino and halo-derivatised cryptolepines.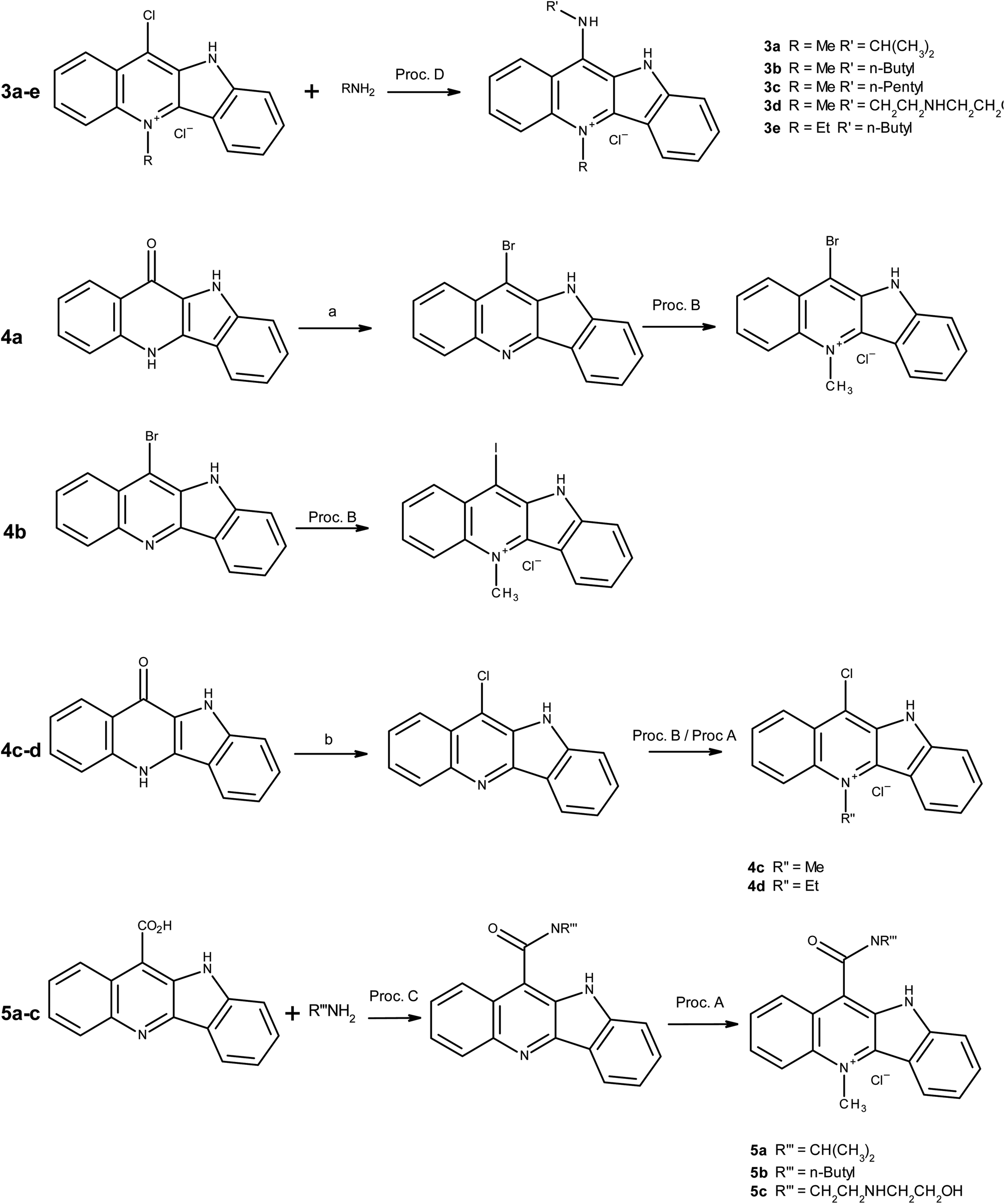 | ||
| Scheme 1 Synthetic route for the cryptolepine derivatives. (a) POBr3 PBr3, 16 h, reflux (b) POCl3, PCl5, 4 h reflux. General procedures are given as Proc. A–D and are discussed in more depth in the methods section: (A) methyl/ethyl triflate, 34 h, RT (B) MeI, sulfolane 16 h RT (C) COMU, Et3N, DCM 16 h RT (D) EtOAC, amine, 16 h, reflux. | ||
Instrumentation
NMR spectra were recorded on Bruker Avance III 400 two-channel FT-NMR spectrometer (AV400). It is equipped with a 5 mm BBFO+ probe (broadband multinuclear, auto-tune, with Z-gradients). Spectra were recorded using TopSpin 3.0 pl4, © 2010 Brüker BioSpin, control and processing software and Icon NMR 4.5.1 Build 51 for TopSpin 3.0 automation software. Mass spectra of compounds were obtained using a Waters LCT-TOF-MS instrument using ESI selective mass detector and Agilent 5973 GC-MS. High resolution mass spectra of compounds were obtained from Medac labs, UK (BS EN ISO9001:2008) using Waters LCT Premier (ES-ToF)/Acquity i-Class mass spectrometer. Infrared spectra were recorded on Thermo Scientific™ Nicolet™ iS™5 FT-IR Spectrometer. Melting points were measured using Gallenkamp digital melting-point apparatus 5A 6797. Up to 350 °C (division: 0.1 °C. 220 V) unless otherwise stated, all the chemicals used in the synthesis were purchased from Sigma Aldrich, UK.General procedures
Specific details of intermediate synthesis and spectroscopic data are found in ESI.† The below general procedures outline the final synthetic steps for the cryptolepine derivatives as indicated in Table 1.| Compound | Yield (%) |
|---|---|
| a Denotes the ethyl quaternary ammonium salt rather than methyl, which is the case for the rest. Details of the synthetic methodology and supporting spectroscopic data are available in ESI. b Denotes the synthesis compounds first reported in this paper. | |
| 1 | N/A |
| 3a | 61b |
| 3b | 68b |
| 3c | 67b |
| 3d | 23b |
| 3e | 35a,b |
| 4a | 11 |
| 4b | 65 |
| 4c | 56 |
| 4d | 74a |
| 5a | 83b |
| 5b | 91b |
| 5c | 12b |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :methanol:NH4OH (5:1:0.1) as the eluent. Finally the free base was converted to its hydrochloride salt by neutralising with 0.1 M methanolic hydrochloric acid (5 ml) and then solution was evaporated in vacuo to give the desired N5-methyl or N5-ethyl cryptolepine derivative, as a yellow coloured solid.
:methanol:NH4OH (5:1:0.1) as the eluent. Finally the free base was converted to its hydrochloride salt by neutralising with 0.1 M methanolic hydrochloric acid (5 ml) and then solution was evaporated in vacuo to give the desired N5-methyl or N5-ethyl cryptolepine derivative, as a yellow coloured solid.
The reaction mixture was added to ethyl acetate (100 ml) and extracted with aqueous 1 M HCl solution (2 × 30 ml) and aqueous 1 M sodium bicarbonate solution (2 × 30 ml). Then the organic layers were collected and dried using magnesium sulphate and evaporated in vacuo to give the crude quindoline amide, which was purified by column chromatography over silica gel using ethyl acetate:hexane (1:1) and dried in vacuo to give desired alkylamidoquindoline.
Biological methods
000 U per mL penicillin, 10 mg per mL streptomycin), L-glutamine (200 mM), Trypsin, Trypan blue solution, MTT, Doxorubicin, Dimethyl sulfoxide (DMSO) (biological grade) were obtained from Sigma Aldrich, UK. 96 well culture plates, T75 ml flasks, and pipettes were purchased from Fischer Scientific, UK. Plate reader used to scan the 96 well plates was Lab-tech LT-4000. The Incubator used to grow the cells was from Thermo Scientific, UK. The human cell lines, MCF-7, A549 and DLD-1 cells were kindly provided by the Institute of Cancer Therapeutics, Bradford University, Bradford, UK.
000 U per mL penicillin, 10 mg per mL streptomycin and 200 mM L-glutamine. Cells were grown in 95% humidified air mixed with 5% carbon dioxide at a fixed temperature of 37 °C. Exhausted growth medium was replaced with fresh growth medium every two days. When the cells reached 90% confluence, they were sub-cultured. Firstly, in a laminar flow chamber, the media was removed and the cell were washed with 15 ml of PBS and then 2.5 ml of trypsin-EDTA solution was added then incubated for 5–7 min to allow the cell splitting to takes place. Then cells were observed under a light microscope (20×) to check if they were detached. If needed they were tapped gently to detach from each other. Fresh growth medium (10 ml) was added and the suspension was aspirated using a pasture pipette. For further culturing 2 ml of that suspension was taken and added to 20 ml of fresh growth medium in a T75 flask and incubated.
000, 32000 and 64000) at a final volume of 200 μL and incubated for 96 hours. Two-hundred microliters of the growth medium without cells served as the blank for the assay. Following incubation, the medium was removed and 25 μL of the MTT reagent (concentration) was added to each well and incubated for 3 hours. The purple formazan crystals were solubilised with 100 μL of DMSO and gentle shaking. Plates were then left in the dark for an hour and were then scanned at 570 nm using a plate reader. A graph was plotted with the average absorbance on the Y-axis and cell count on the X-axis. This was replicated thrice on the same plate in three rows.
After MTT treatment purple coloured crystals were observed under the microscope then medium from each well was carefully removed and 100 μL of DMSO were added to dissolve the precipitate and the plate was left in the dark for an hour. The obtained raw data was then analysed using MS-Excel (2010) software and the IC50 values were calculated using the GraphPad Prism software (version 6.0).
:drugs:cryptolepine and its analogues Eppendorf tubes, Micro pipettes and pipette tips, A grade glassware. 1% Agarose gels were prepared (0.75 g agarose in 75 mL tris buffer) and ethidium bromide (0.5 μg mL−1) added to the solution. The solution was allowed to solidify at room temperature for an hour prior to loading. Running Buffer (TAE buffer): 50× concentration: tris base (242 g), glacial acetic acid (57.1 mL) and of 0.5 M EDTA (100 mL). Diluted to 1× cryptolepine and its analogues were initially dissolved in DMSO and then diluted to reach the desired concentration. Serial dilutions were carried out to produce a concentration range of 100 μM to 0.01 μM. The maximum DMSO concentration did not exceed 0.1% v/v. Buffer (2 μL complete reaction buffer = buffer A + buffer B in 1:1 ratio), kDNA (0.25 μg mL−1, 1 μL), bovine serum albumin (30 μg mL−1, 1 μL), distilled water (to make up to 20 μL), drug to be tested in desired concentration (1 μL) were pipetted out to a Eppendorf tube and incubated for 30 minutes at 37 °C.
The reaction was then stopped by adding 4 μL of stopping buffer and 2 μL of protein kinase. The mixture was then incubated for digestion for 30 minutes. The reaction products were then loaded on to the ethidium bromide-agarose gel and electrophoresis was carried out at 65 V for 2.5 hours in TAE buffer. After the separation, the gel was visualised under UV illumination.
Cells were grown in a 96 well plate for 24 hours in phenol red-free RPMI medium at a seeding density of 20000 cells per well. Seven rows of 3 columns (7 × 3 = 21 wells) were loaded with cell suspension for incubation. Twenty four hours post incubation, media from the first row of the cells was removed and loaded with 100 μL of drug solution (solvent = media) with a concentration of 10 μM and the plate was placed back in to the incubator. After 30 min. the plate was removed from the incubator and the media from the second row was removed and loaded with 100 μL of drug solution (10 μM). The same procedure was repeated for the remainder of the rows every 30 minutes, leaving the last row with no treatment (blank). After the treatments were complete, the drug solutions were gently removed and 100 μL of DMSO was added to the wells and shaken gently for 30 minutes. The positive control wells were filled with 100 μL of 10 μM drug solution.
Cells were lysed using Triton X-100 (a non-ionic surfactant which increases the cell permeation) and the drug was extracted using methanol. The plate was then scanned using a fluorescence plate reader measuring the intensity of the fluorescence from each well (25 scans for each well). The 8th was filled with 10 μM drug solution, which served as a positive control for the experiment. Using a positive control (100% drug solution), data was analysed and compared to generate the report of percentage drug uploaded into the cells over the period of three hours.
000 nm min−1, radiation source is Xenon flash lamp with room light immunity) with carry eclipse spectrophotometric software. Sodium cacodylate was purchased from Sigma Aldrich, UK. Made using 0.2 M sodium cacodylate and 0.2 M HCl thiazole orange was purchased from Sigma Aldrich, UK. Cacodylate buffer was made by dissolving sodium cacodylate crystals (42.8 g) in distilled water (1 L) and the pH was adjusted to 7.3 using aqueous HCl solution (0.2 M). The G-quadruplexes were made by heating the 22AG nucleotide in the cacodylate buffer (10 mM, pH 7.3), 100 mM KCl at 90 °C for five minutes and then the solution was cooled in ice to form G-quadruplexes by intermolecular folding. This duplex nucleotide sequence was made by heating DS26 in the cacodylate buffer (10 mM, pH 7.3), 100 mM KCl at 90 °C for five minutes and then slowly cooling over a period of 6 hours. Once these DNA solutions were made, the concentrations were measured using the UV-VIS spectrophotometer at 260 nm using their extinction coefficient values.
Cacodylate buffer containing 100 mM of KCl was used for all the dilutions to get desired concentrations. In a 3 mL capacity cuvette, 1 mL of thiaozole orange solution and 1 mL of DS26 solution were pipetted and then the volume was adjusted to 3 mL using the blank buffer. The fluorescence was measured using the fluorimeter. This served as a control for the titration. For the ligand binding experiments, first 1 mL of thiozole orange and 1 mL of DS26 solution were put into a cuvette and then the total concentration of ligand was made to 0.375 μM by adding the desired ligand and the volume was made up using the cacodylate buffer. The solution was allowed to equilibrate for 3 min and then the fluorescence intensity was measured.
The same was repeated for each fluorometric titration by increasing the concentration of ligand to 0.75 μM, 1.5 μM, 3.0 μM, 4.5 μM, 7.5 μM in the cell and keeping the total volume of the cell constant at 3 mL. The same procedure was carried out for the G-quadruplex sequence with the only difference being the concentration of thiazole orange used which was compared to DS26. The reason for this is that quadruplex sequence has a smaller number of binding sites compared to the duplex sequence of DS26. During the titration, depending on the activity of the ligand, a gradual fall in the fluorescence was seen as the concentration of the ligand increased.
All the concentrations were recorded using the Carry Eclipse software and the integral graphs were plotted for each ligand concentration measured. The decrease in percentage of fluorescence intensity was measured for each ligand concentration and hence the binding efficiency of the ligand was interpreted.
Results – synthesis
In addition to probing the halocryptolepine activity, alkylamino side chains were attached to increase lipophilicity and improve overall drug uptake. For the same reason alkylamino side chains with different lengths were selected to see if there is any correlation between side chain length and the drug uptake in the cell lines in vitro.11-Chlorocryptolepine served as the key intermediate to make the derivatives with amino linkages at 11-position via nucleophilic substitution (see general procedure D). In the derivatives with amino linkages, it is unlikely that the secondary amine present on the side chain can be protonated at physiological pH but it is possible that the positive charge arising from the pyridinium nitrogen (Fig. 1) can resonate to transfer the positive charge to the side chain. This structure can potentially better interact with the negatively charged phosphate backbone of DNA which should ultimately lead to improved binding interactions.
 | ||
| Fig. 1 Possible resonance structure for 11-aminocryptolepine derivatives. | ||
Unlike derivatives with amino linkages, in the derivatives with amido linkages, the positive charge will not be able to move to the aromatic ring (Fig. 1). So these compounds were made to see how having an amide bond between side chains and the aromatic system of cryptolepine will affect the cytotoxicity and the in vitro drug uptake in cancer cells.
Results – in vitro and biophysical testing
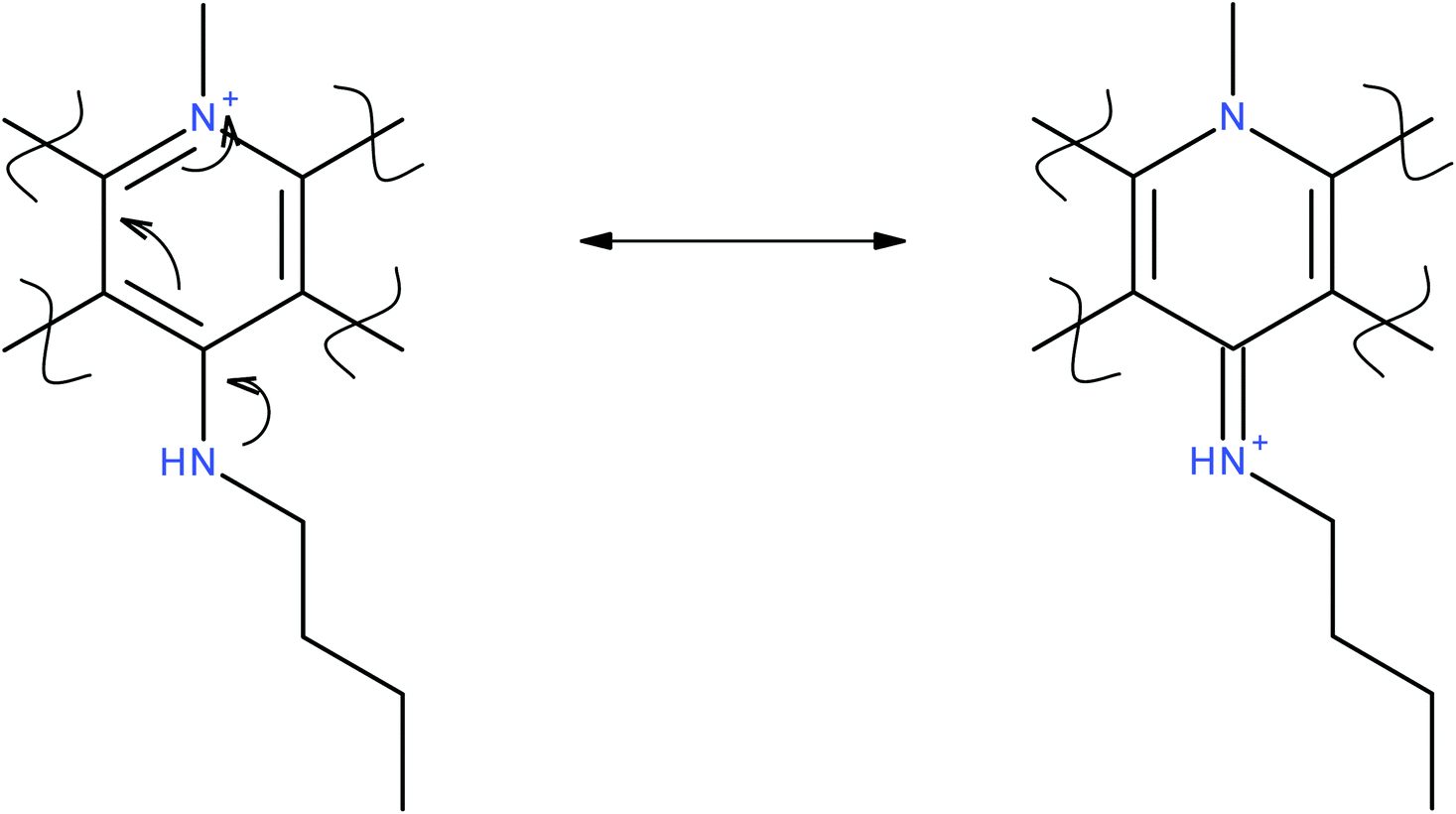
Three different cancer cell lines: MCF-7 (breast cancer) A549 (lung cancer) DLD-1 (colon cancer) were used to test the anticancer activity of the synthesised cryptolepine analogues. In addition to ease of culture and laboratory maintenance, choosing these cell lines make it possible to compare the results with previous literature.14a–c All three cell lines incubated with cryptolepine 1 for 96 hours, showed anti-proliferative activity. The greatest activity was against A459 cells with an IC50 value of 0.47 ± 0.12 μM compared to MCF-7 and DLD-1 cells on which it gave IC50 values of 5.9 ± 0.69 μM and 1.25 ± 0.77 μM respectively (Table 2), which are in agreement with similar experiments carried out by Gokcek et al. on A-549 and DLD-1 cells and reported IC50 values of 0.55 ± 0.05 μM and 1.44 ± 0.002 μM.15 From the dose response curve it is observed that no significant activity was seen until a 0.10 μM concentration of cryptolepine was applied. After that there was a gradual downward trend until 1.00 μM. Once the concentration reached 10 μM, the percentage cell survival was essentially zero.| Compound | A549 cells IC50 (μM) | MCF-7 cells IC50 (μM) | DLD-1 cells IC50 (μM) | Topoisomerase II inhibition MIC (μM) | DS26 binding (Kd) | 22AG binding (Kd) |
|---|---|---|---|---|---|---|
| 1 | 0.47 ± 0.12 | 5.90 ± 0.69 | 1.25 ± 0.77 | 0.5 | 1.62 ± 0.62 | 2.45 ± 0.36 |
| 3a | 20.38 ± 2.01 | 17.92 ± 2.31 | 37.05 ± 2.36 | — | 2.51 ± 0.33 | 39.81 ± 5.17 |
| 3b | 1.05 ± 0.15 | 0.63 ± 0.51 | 2.85 ± 0.87 | 0.25 | 0.79 ± 0.37 | 5.01 ± 1.85 |
| 3c | 0.83 ± 0.28 | 4.253 ± 1.88 | 2.4 ± 0.96 | — | 1.26 ± 0.26 | 6.31 ± 3.15 |
| 3d | 10.41 ± 0.75 | 6.77 ± 2.03 | 12.23 ± 1.20 | — | 2.51 ± 0.58 | 3.98 ± 1.53 |
| 3e | 12.58 ± 2.12 | 43.59 ± 4.72 | 26.73 ± 3.92 | 1.55 ± 0.55 | 26.92 ± 6.96 | |
| 4a | 5.16 ± 1.25 | 2.15 ± 0.41 | 3.35 ± 1.62 | — | — | — |
| 4b | 0.28 ± 0.1 | 0.1 ± 0.02 | 0.34 ± 0.05 | 0.10 ± 0.06 | 0.32 ± 0.10 | |
| 4c | 42.83 ± 3.74 | 62.87 ± 2.71 | 81.58 ± 2.61 | 0.25 | 0.40 ± 0.20 | 0.63 ± 0.02 |
| 4d | 15.24 ± 2.20 | 21.00 ± 3.62 | 31.84 ± 3.86 | — | — | — |
| 5a | 42.64 ± 3.17 | 31.72 ± 2.56 | 75.01 ± 2.92 | >100 | — | — |
| 5b | 21.41 ± 1.76 | 7.26 ± 1.24 | 4.14 ± 0.89 | — | 2.00 ± 0.74 | 3.55 ± 1.04 |
| 5c | 12.38 ± 2.16 | 9.15 ± 1.56 | 15.21 ± 3.23 | — | — | — |
Cell viability
After exposing cells to 1 μM or higher It was observed under the microscope that cells started changing morphology. As the concentration increased, more cells were lysed and more cell debris was observed. Compounds 3b and 3c showed a higher level of activity compared to 3a suggesting that having an alkylamino side chain is structurally significant. In particular, compound 3b showed 10 times greater activity than the parent compound 1 with an IC50 value of 0.63 ± 0.51 μM. Interestingly 3c derivative showed a greater activity than 3b for A549 and DLD-1 cells with IC50 values of 0.83 ± 0.28 μM and 2.4 ± 0.96 respectively (Fig. 2). | ||
| Fig. 2 Dose–response curve for 1 against MCF 7 (-○-), A549 (□) and DLD-1 (△) cancer cell lines. | ||
Unsurprisingly, these results indicate the dependence of the activity in the specific cell line being tested, however the results of testing against cancer cell lines suggest that alkylamino amino side chains may be useful in forming electrostatic interactions with the negatively charged DNA backbone.16,17 Addition of a hydrophilic motif as in the case of 3d failed to improve upon the anticancer activity, however it was shown to still be superior to the more lipophilic 3b derivative.
The derivative 3d is actually similar to mitoxantrone and showed greatest activity on the breast cancer cells with an IC50 value of 6.77 ± 2.03 μM. This selectivity was also observed when Von Hoff and his team tested mitoxantrone on 20 different tumour cell lines, where greatest activity against breast cancer (MCF-7) cells compared to DLD-1 cells was observed.18a,19 The anti-proliferative activity shown by 4b was most potent, with 4a showing the second highest activity among all the halogenated compounds tested. The 11 halogenated analogues followed a downward trend of activity from 11-iodo to the 11-chloro derivative indicating the importance of a lipophilic group in the 11 position. The importance of halogen interactions has been reported by Sarwar et al. where the potential for encapsulation augments the role of halogen interactions, which are negligible in comparison with the bulk solvent interactions.18b
This van der Waals interaction between iodine and amino hydrogens is important (3.43 ± 0.33 A) for optimal binding and selectivity of drug towards that particular DNA sequence.18a A gradual decrease in the DNA-binding has been similarly observed from iodo to fluoro analogues of Calicheamicine. This could mean that the presence of Iodine atom at 11-position may have been involved in similar molecular interactions which were contributing for the DNA binding efficiency of 4b.
Derivatives 5a–5c showed comparatively less activity than the other sets. Amongst the amido-derivatives, 5c showed the highest activity whereas the 5a derivative showed lowest activity. Regardless of whether it is an amino derivative and an amido derivative, the isopropyl group reduced the activity significantly, possibly due to the steric hindrance imparted by the isopropyl functional group.
Amongst all the molecules synthesised, derivatives with 11-alkylamino linkages and 11-halogenated compounds showed comparatively greater activity than compounds having 11-amido side chains.
Topoisomerase II inhibition studies
Cryptolepine has been tested for topoisomerase II activity in the past using both of the methods outlines above and it has been proposed that cryptolepine acts as a topoisomerase II poison.20,21 A random derivative was selected from each set for Topoisomerase II testing. When cryptolepine 1 was tested for its topoisomerase II activity it gave a minimum inhibitory concentration value (MIC) of 0.5 μM. The same result was reported by Bonjean and his team on supercoiled plasmid DNA obtained from Escherichia coli.10Cryptolepine 3b showed the highest topoisomerase II inhibition of the compounds tested and also the highest overall cytotoxicity. This continues to support the link between the two. The analogue 5a gave much lower topoisomerase II activity than the parent compound; however the 4c derivative failed to confirm this correlation between Topoisomerase II inhibition and cytotoxicity. The topoisomerase II activities obtained from both compounds 3b and 4c are significantly higher than that of the parent compound.
It is possible that the lack of correlation between Topoisomerase II inhibition and cytotoxicity may be due to differences in the pharmacokinetics profile of the compounds in so drug uptake was investigated.
Fluorescence microscopy
Fluorescence microscopy was used to investigate whether the derivatives were reaching the nucleus by crossing the cellular membrane and nuclear envelope. For this experiment A549 cells were selected by observing the morphology under a microscope. Though the cultured cells are in monolayers, MCF-7 and DLD-1 cells tend to form clusters in the culture plate whereas A549 cells are evenly spread. Out of the compounds scanned, 4a–4c and 3b emitted high enough fluorescence intensity to facilitate fluorescence microscopy. Derivative 3b was selected as one of the most active and 4c was included with view to identifying the reason for its low cytotoxicity despite high topoisomerase II activity. Fluorescence microscopic pictures were taken for both DAPI and the cryptolepine derivatives by exciting them at their respective excitation wavelengths and then overlaying them to identify co-localisation.22,23Co-localisation of DAPI and 3b in the nucleus of A549 cells indicated that although 3b was spread all over the cell, high concentrations are localised at nucleus, which was clearly seen in the pictures as more bright fluorescence was arising from the nucleus of the cell which is co-localised with DAPI (Fig. 3).
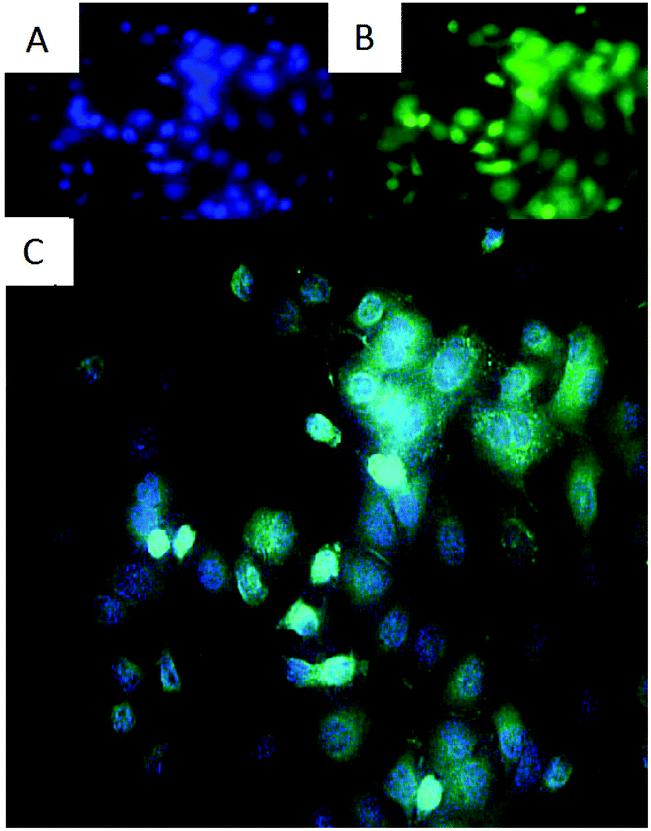 | ||
| Fig. 3 Showing co-localisation of DAPI and drug in the nucleus of A549 cells. Magnification: 40×, green: 3b (A), blue: DAPI nuclear dye (B), overlay of both 3b and DAPI (C). | ||
The same was observed with 4c but the fluorescence intensity seemed to be lower than 3b (Fig. 4). This is supported by similar experiments conducted by Redshaw et al. to see the cellular localisation of fluorescent zinc complexes using DAPI as a counterstain.24
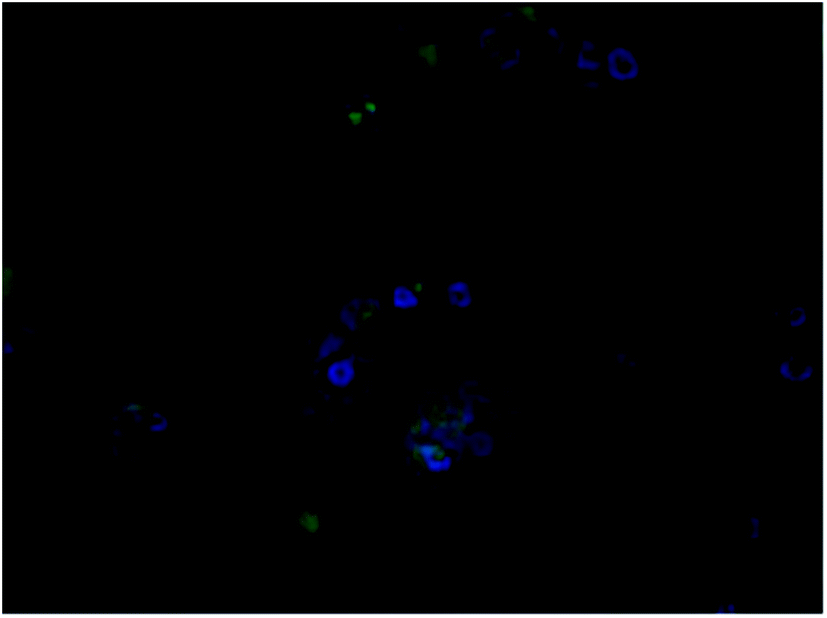 | ||
| Fig. 4 Showing co-localisation DAPI and drug in the nucleus of A549 cells. Magnification: 40×, green: 11-chloro cryptolepine, blue: DAPI nuclear dye, overlay of both drug and DAPI. | ||
These drug localisation experiments only gave information about whether the drug is present in the nucleus or not. By measuring the amount of drug taken up by the cells after treatment for particular time will give quantitative information about the rate of drug uptake in the cells.
In vitro drug uptake studies
To carry out the drug uptake studies A549 cells were selected because, after drug treatment, they make evenly spread monolayers. MCF-7 and DLD-1 cells tend to form clusters in the culture plates this was observed under microscope while performing cytotoxicity assays. Based on the observed lack of cell expansion the images suggest apoptosis as the pathway and this is supported by the literature.25Drug uptake was determined using the method of Kunwar et al.26 Fluorescence intensity was measured and compared using a positive control of the same drug solution and result was expressed as percentage drug uptake. Cryptolepine derivatives from each set were chosen at random. The amount of fluorescence exhibited by each drug can be different, so the same drug was taken as positive control (without any cells being present – 100%) percentage drug uptake was calculated. This percentage drug uptake was directly proportional to the amount of drug present in the cells.
Looking at the in vitro drug uptake results, 3b was taken up into the cells more quickly than any other compounds tested reaching 40% within first 30 min after drug treatment, then it continued to rise up to 60% in 1.5 hours. After 1.5 hours percentage drug uptake started to decrease reaching to a 20% at 3 hours this could be due the oozing of cellular contents along with drug from ruptured cell membranes of dying cells. This high rapid percentage drug uptake in to the cells may be the reason for high activity of 3b (Fig. 5).
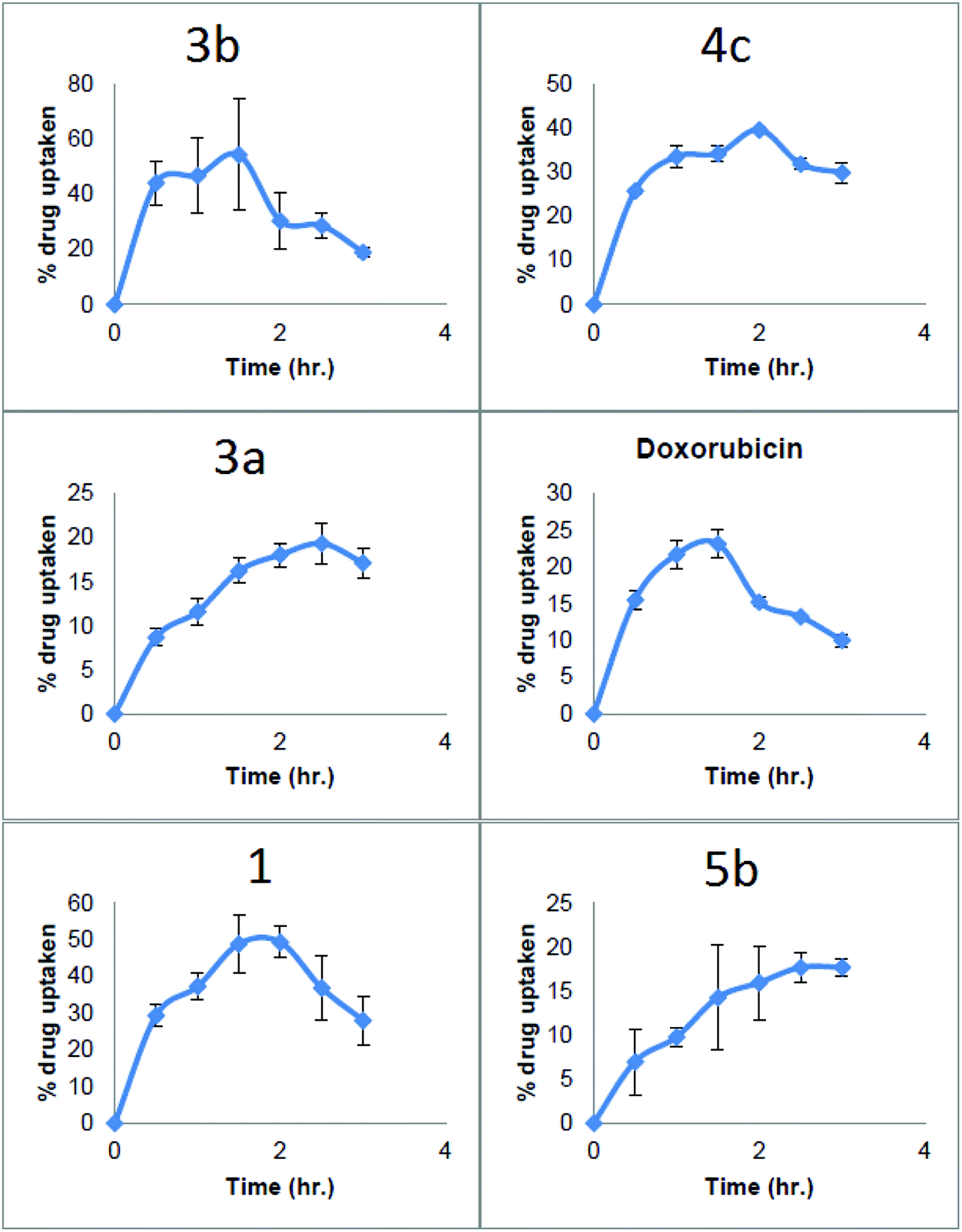 | ||
| Fig. 5 Showing the rate of drug uptake of a range of cryptolepine derivatives in A549 cells. | ||
In the case of 4c, the percentage drug uptake only reached 25% in the first 30 minutes and then increased to 40% after 2 hours then it started to decline and reached about 30% after three hours. This lower uptake rate might be the reason behind the lower cytotoxicity of 4c. Another important issue in the drug uptake profile of this compound was that the intracellular concentration of 4c did not fall as quickly as 3b. This may be due to lower cytotoxicity of the 4c.
11-Isopropylaminocryptolepine 3a gave a poor uptake profile with a percentage uptake of 8% in the first thirty minutes which is 6 times lower compared to 3b and approximately 4 times lower than the parent compound 1. The maximum drug uptake for 3a of 18% was seen at 2.5 hours which is 3 times lower than 3b.
Drug–DNA binding
Since cryptolepines have the potential to act as an anticancer agent by intercalating into DNA, it was felt prudent to investigate binding efficiencies to see whether the novel derivatives showed improved binding with respect to 1. The aim was also to better understand the mode of action to address the counterintuitive relationship between for example, the high topoisomerase II activity, yet low cytotoxicity for 4c.Fluorescence displacement assay was selected as method of choice for the current project for its robustness and the fact that we could use different types of DNA sequences (duplex, quadruplex) to test a group of random cryptolepine derivatives covering the different classes synthesised.
Using this fluorimetric titration assay, cryptolepine derivatives were tested for their selectivity and efficiency of DNA binding. This assay was done using two types of DNA sequences; one was a duplex-forming DNA (DS26) and the other was quadruplex-forming DNA (22AG).
22AG is an oligonucleotide which is similar to the human telomeric sequence. It is a repeat of the [5′-AG3(T2AG3)3-3′] which forms G quadruplex DNA. DS26 is a self-complementary DNA sequence 5′-CA2TCG2ATCGA2T2CGATC2GAT2G-3′] which form a duplex DNA.24 The gradual decrease in the fluorescence arising from displacement of thiazole orange from quadruplex and duplex DNA is used to profile the binding upon gradual increase of the ligand concentration.
Reference to Table 2 shows that the 11-halocryptolepines 4b & 4c had the best DNA binding overall with slightly more selectivity towards duplex DNA over G-quadruplex DNA. The 11-iodo compound 4b showed the highest DNA binding constant in both duplex and quadruplex sequences. This reinforces the cytotoxicity data as high cytotoxicity was shown by the 11-iodo compound 4b. Interestingly the 11-chloro derivative 4c showed better DNA binding efficiency than parent molecule despite having a poorer cytotoxicity profile.
The 11-alkylamino derivatives 3a–3e showed better DNA binding than 1, especially the molecules 3b and 3c with the 11-butylamino and pentylamino side chains. The same pattern of DNA binding for this class of cryptolepine derivatives was observed for the cytotoxicity data. The 11-isopropylamino derivative 3a showed binding efficiency to G-quadruplex which is 16 times lower than its duplex DNA affinity. When compared to 1, 3b and 3c derivatives showed higher binding efficiency to duplex DNA, implying that the compounds with 11-alkylamino side chains prefer duplex DNA over quadruplex DNA.
Even though derivative 3d with the aminoalcohol side chain did not show particularly strong binding to duplex DNA, its quadruplex binding efficiency is considerably higher than derivatives 3a–3c. More importantly there is not much selectivity of 3d for duplex or quadruplex DNA.
Derivative 5b was selected from the 11-alkylamido class as it showed higher cytotoxicity compared to the other 11-alkyl amido derivatives. It showed less binding efficiency towards both duplex and quadruplex DNAs when compared to 1 but it showed better quadruplex binding than the 11-alkylamino derivatives implying that it prefers G-quadruplex structures than duplexes. Overall cryptolepines appear to bind preferentially to duplex DNA over G-quadruplex DNA, this may be because of its selectivity towards GC rich sequences present in the duplex DNA.27
Conclusions
The purpose of this study was to attach halogens (Cl, Br and I) and amino/amido-alkyl side chains at the 11-position of cryptolepine and then investigate these molecules for anti-cancer activity using established in vitro cytotoxicity assay and biophysical studies including DNA binding assays, topoisomerase II assays, fluorescence microscopy and quantitative drug uptake studies.Of the novel derivatives synthesised the 11-aminoalkyl and 11-halo cryptolepines (3a–3d & 4a–4c resp.) showed greater activity than those with 11-amido side chains (5a–5c). Overall, the 11-iodo 4b and the 11-butylamino 3b derivative showed the highest activity (Table 2). The reason for the enhanced cytotoxicity of 3b could be the lipophilic alkyl side chain which may facilitate faster drug-uptake by the cells.
Topoisomerase II results reinforced the link between the cytotoxicity and topoisomerase II inhibition. Surprisingly, despite its lower cytotoxicity, the 11-chloro derivative 4c showed the highest topoisomerase II activity with an MIC value of 0.25 μM. When analysing its drug uptake profile, the percentage drug uptake was lower than both the 11-butylamino derivative 3b and parent cryptolepine 1, reaching an intracellular concentration of only 25% in the first 30 minutes. In contrast to this despite its lower topoisomerase II activity, 11-iodocryptolepine 4b (MIC = 0.5 μM) gave a higher cytotoxicity value compared to the 11-chlorocryptolepine.
Fluorescence microscopic images showed that, although 3b was dispersed throughout the cell, high concentrations were localised in the nucleus. Despite this also being observed in 11-chlorocryptolepine 4c, the amount of drug localised in the nucleus is dramatically lower than for 3b. This indicates not only that a smaller amount of the 4c is being taken up by the cells but also that it is not reaching the nucleus effectively. We have also shown that 5-ethyl cryptolepines (3e & 4d) have substantially lower anti-cancer activity that 5-methyl derivatives and make unlikely lead compounds.
We report the synthesis, characterisation and in vitro testing of two new classes of cryptolepine derivatives and have considered their interaction with topoisomerase II and as DNA intercalators. In addition, we highlight that whilst topoisomerase II inhibition is a potentially important mode of action for cryptolepine anti-cancer activity, the direct intercalation of duplex DNA as well as cell permeability significantly influence the anti-cancer profile of any such derivative. This is important in deciding what potential anticancer motifs derivatives of this class of molecule will require to enhance activity and selectivity. Our group is currently working on the combination of halogenations and substitution of the crypolepine with amine bearing moieties.
In conclusion, this work has shown that novel amino and halogenated cryptolepine analogues have greater in vitro cytotoxicity than the parent compound and are important lead compounds in the development of novel potent and selective indoloquinone anti-neoplastic agents. Further synthesis of derivatives which incorporate the amino and halogen groups may yet result in enhanced activity and potential selectivity for G-quadruplex DNA, leading to indirect telomerase inhibition, which are currently being investigated.
Acknowledgements
The authors would like to thank Kingston University for the provision of support for Venkatesh Gudivika and Dr Jean-Marie Peron for excellent support in NMR interpretation. A special thanks go to Dr Eleanor Polycarpou for proof reading and useful discussions on the biological aspects.Notes and references
- J. N. Lisgarten, M. Coll, J. Portugal, C. W. Wright and J. Aymami, Nat. Struct. Biol., 2002, 9, 57 CrossRef PubMed.
- C. W. Wright, J. Addae-Kyereme, A. G. Breen, J. E. Brown, M. F. Cox, S. L. Croft, Y. Gokcek, H. Kendrick, R. M. Phillips and P. L. Pollet, J. Med. Chem., 2001, 44, 3187 CrossRef CAS PubMed.
- O. Onyeibor, S. L. Croft, H. Dodson, M. Feiz-Haddad, H. Kendrick, N. J. Millington, S. Parapini, R. M. Phillips, S. Seville, S. D. Shnyder, S. Taramelli and C. W. Wright, J. Med. Chem., 2005, 48, 2701 CrossRef CAS PubMed.
- L. G. Mardenborough, X. Y. Zhu, P. C. Fan, M. R. Jacob, S. I. Khan, L. A. Walker and S. Y. Ablordeppey, Bioorg. Med. Chem., 2005, 13, 3955 CrossRef CAS PubMed.
- S. Seville, O. Onyeibor, M. Feiz-haddad, N. Karodia, R. M. Phillips and C. W. Wright, J. Pharm. Pharmacol., 2003, 55, S.32 Search PubMed.
- P. Jerrum, C. W. Wright, A. Jewell, J. E. Brown and S. Carrington, J. Pharm. Pharmacol., 2007, 59, A2 Search PubMed.
- P. Jerrum, C. W. Wright, R. M. Phillips, S. R. Gouni, J. E. Brown and S. Carrington, J. Pharm. Pharmacol., 2006, 58, A59 Search PubMed.
- S. R. Gouni, S. Carrington and C. W. Wright, J. Heterocycl. Chem., 2006, 43, 171 CrossRef CAS.
- J. Lavrado, R. Moreira and A. Paulo, Curr. Med. Chem., 2010, 17, 2348 CrossRef CAS PubMed.
- K. Bonjean, M. C. De Pauw-Gillet, M. P. Defresne, P. Colson, C. Houssier, L. Dassonneville, C. Bailly, R. Greimers, C. Wright, J. Quetin-Leclercq, M. Tits and L. Angenot, Biochemistry, 1998, 37, 5136 CrossRef CAS PubMed.
- L. Dassonneville, K. Bonjean, M. C. De Pauw-Gillet, P. Colson, C. Houssier, J. Quetin-Leclercq, L. Angenot and C. Bailly, Biochemistry, 1999, 38, 7719 CrossRef CAS PubMed.
- W. Luniewski, J. Wietrzyk, J. Godlewska, M. Switalska, M. Piskozub, W. Pec-zynska-Czoch and L. Kaczmarek, Bioorg. Med. Chem. Lett., 2012, 22, 6103 CrossRef CAS PubMed.
- L. Guittat, P. Alberti, F. Rosu, S. Van Miert, E. Thetiot, L. Pieters, V. Gabeli-ca, E. De Pauw, A. Ottaviani, J. F. Riou and J. L. Mergny, Biochimie, 2003, 85, 535 CrossRef CAS PubMed.
- (a) O. A. Olajide, E. H. Heiss, D. Schachner, C. W. Wright, A. M. Vollmar and V. M. Dirsch, Bioorg. Med. Chem., 2007, 15, 43–49 CrossRef CAS PubMed; (b) D. Laryea, A. Isaksson, C. W. Wright, R. Larsson and P. Nygren, Invest. New Drugs, 2009, 27, 402–411 CrossRef CAS PubMed; (c) H. Zhu and N. J. Gooderham, Toxicol. Sci., 2006, 91, 132–139 CrossRef CAS PubMed.
- Y. Gokcek, P. Pollet, C. W. Wright and J. E. Brown, J. Pharm. Pharmacol., 2000, 52, 109 Search PubMed.
- C. Avendaño and J. C. Menendez, Medicinal chemistry of anticancer drugs, Elsevier, 2008 Search PubMed.
- D. S. Alberts, Y. M. Peng, G. T. Bowden, W. S. Dalton and C. Mackel, Invest. New Drugs, 1985, 3, 101 CAS.
- (a) D. D. Von Hoff, C. A. Coltman and B. Forseth, Cancer Res., 1981, 41, 1853 CAS; (b) M. Sarwar, D. Ajami, G. Theodorakopoulos, I. Petsalakis and J. Rebek Jr., J. Am. Chem. Soc., 2013, 135, 13672 CrossRef CAS PubMed.
- T. Li, Z. Zeng, V. A. Estevez, K. U. Baldenius, K. Nicolaou and G. F. Joyce, J. Am. Chem. Soc., 1994, 116, 3709 CrossRef CAS.
- L. Dassonneville, A. Lansiaux, A. Wattelet, N. Wattez, C. Mahieu, S. Van Miert, L. Pieters and C. Bailly, Eur. J. Pharmacol., 2000, 409, 9 CrossRef CAS PubMed.
- C. Bailly, W. Laine, B. Baldeyrou, M. C. De Pauw-Gillet, P. Colson, C. Houssier, K. Cimanga, S. Van Miert, A. J. Vlietinck and L. Pieters, Anti-Cancer Drug Des., 2000, 15, 191 CAS.
- A. M. Burger, T. C. Jenkins, J. A. Double and M. C. Bibby, Br. J. Cancer, 1999, 81, 367 CrossRef CAS PubMed.
- C. Molenaar, J. Teuben, R. J. Heetebrij, H. J. Tanke and J. Reedijk, J. Biol. Inorg. Chem., 2000, 5, 655 CrossRef CAS PubMed.
- C. Redshaw, M. R. Elsegood, J. W. Frese, S. Ashby, Y. Chao and A. Mueller, Chem. Commun., 2012, 48, 6627 RSC.
- M. Cheng, Y. Yang, Y. Wong, T. Chen, C. Lee, C. Yang, S. Chen, I. Yang, Y. Yang, H. Huang, C. Yang, M. Huang and H. Chiu, Anti-Cancer Drugs, 2012, 23, 191 CrossRef CAS PubMed.
- A. Kunwar, A. Barik, B. Mishra, K. Rathinasamy, R. Pandey and K. I. Priyadarsini, Biochim. Biophys. Acta, 2008, 1780, 673 CrossRef CAS PubMed.
- D. Monchaud, C. Allain, H. Bertrand, N. Smargiasso, F. Rosu, V. Gabelica, A. De Cian, J. Mergny and M. Teulade-Fichou, Biochimie, 2008, 90, 1207 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic details and spectroscopic data. See DOI: 10.1039/c5ob02408k |
| This journal is © The Royal Society of Chemistry 2016 |