
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis, structure and pyrolysis of stabilised phosphonium ylides containing saturated oxygen heterocycles†
R. Alan
Aitken
*a,
Nazira
Karodia‡
*b,
Hollie B.
McCarron
a,
Cécile
Rouxel
a,
Nina
Sahabo
b and
Alexandra M. Z.
Slawin
a
aEaStCHEM School of Chemistry, University of St Andrews, St Andrews, Fife, KY16 9ST, UK. E-mail: raa@st-and.ac.uk
bSchool of Life Sciences, University of Bradford, Bradford, W Yorkshire, BD7 1DP, UK
First published on 5th January 2016
Abstract
A range of twelve stabilised phosphonium ylides containing tetrahydrofuran, tetrahydropyran or 2,2-dimethyl-1,3-dioxolane rings have been prepared and fully characterised, including one X-ray structure determination of each type. The X-ray structures confirm the P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and CO functions to be syn and all the compounds undergo thermal extrusion of Ph3PO to give the corresponding alkynes. In some cases there is also competing loss of Ph3P to give different carbene-derived products and evidence has been obtained for the generation of 2-phenyloxete in this way. Raising the pyrolysis temperature leads in several cases to new secondary reactions of the alkyne products involving a sequence of alkyne to vinylidene isomerisation, intramolecular CH insertion, and retro Diels Alder reaction.
C and CO functions to be syn and all the compounds undergo thermal extrusion of Ph3PO to give the corresponding alkynes. In some cases there is also competing loss of Ph3P to give different carbene-derived products and evidence has been obtained for the generation of 2-phenyloxete in this way. Raising the pyrolysis temperature leads in several cases to new secondary reactions of the alkyne products involving a sequence of alkyne to vinylidene isomerisation, intramolecular CH insertion, and retro Diels Alder reaction.
Introduction
Thermal extrusion of Ph3PO from suitably substituted β-oxophosphonium ylides in the so-called “intramolecular Wittig reaction” is a versatile method of alkyne synthesis.1 The ylides are readily prepared in a few simple steps from alkyl halides and acid chlorides as stable crystalline solids, and overall this represents an unusual way to construct the alkyne triple bond (Scheme 1). The process was first described in 1959 for R1 = R2 = Ph where simply heating the ylide at 300 °C resulted in the desired elimination of Ph3PO to give diphenylacetylene in 59% yield.2 Shortly thereafter a convenient synthesis of acetylenic esters by pyrolysis of the ylides with R1 = CO2Me was reported.3 Over the next 25 years many further examples appeared, but using conventional pyrolysis the scope was limited to examples where R1 was an electron withdrawing group.4 This restriction was overcome by the use of flash vacuum pyrolysis (FVP) which allowed synthesis of purely aliphatic and terminal alkynes from ylides with R1 = alkyl or H.5 Using this method, a wide range of functionalised alkynes have been prepared and typical examples include the synthesis of fused ring heterocycles via cascade cyclisation6,7 and chiral amino acid-derived alkynes.8 FVP is a technique of increasing importance in the synthesis of heterocyclic compounds,9 but has not so far seen any significant application in carbohydrate chemistry. In fact the use of FVP in carbohydrate chemistry is effectively limited to a single example: the pyrolytic elimination of acetic acid to form a derivative of zanamivir (Relenza).10 In this paper we describe the preparation and pyrolysis behaviour of a series of ylides containing tetrahydrofuran, tetrahydropyran and 2,2-dimethyl-1,3-dioxolane rings to check the compatibility of these ring systems common in carbohydrates with the conditions required for alkyne formation. There has been considerable interest in acetylenic carbohydrate derivatives, both in their own right,11 and as intermediates in synthesis of modified nucleosides.12,13 Alkyne-containing 2,2-dimethyl-1,3-dioxolanes have also found use in total synthesis.14,15 There is a single previous example of this approach in which a tetrahydropyran-containing ylide 1 was prepared and subjected to conventional pyrolysis at 200–250 °C (heating the solid above its mp) to afford alkyne 2 in 75% yield (Scheme 2).16 | ||
| Scheme 1 | ||
 | ||
| Scheme 2 | ||
Results and discussion
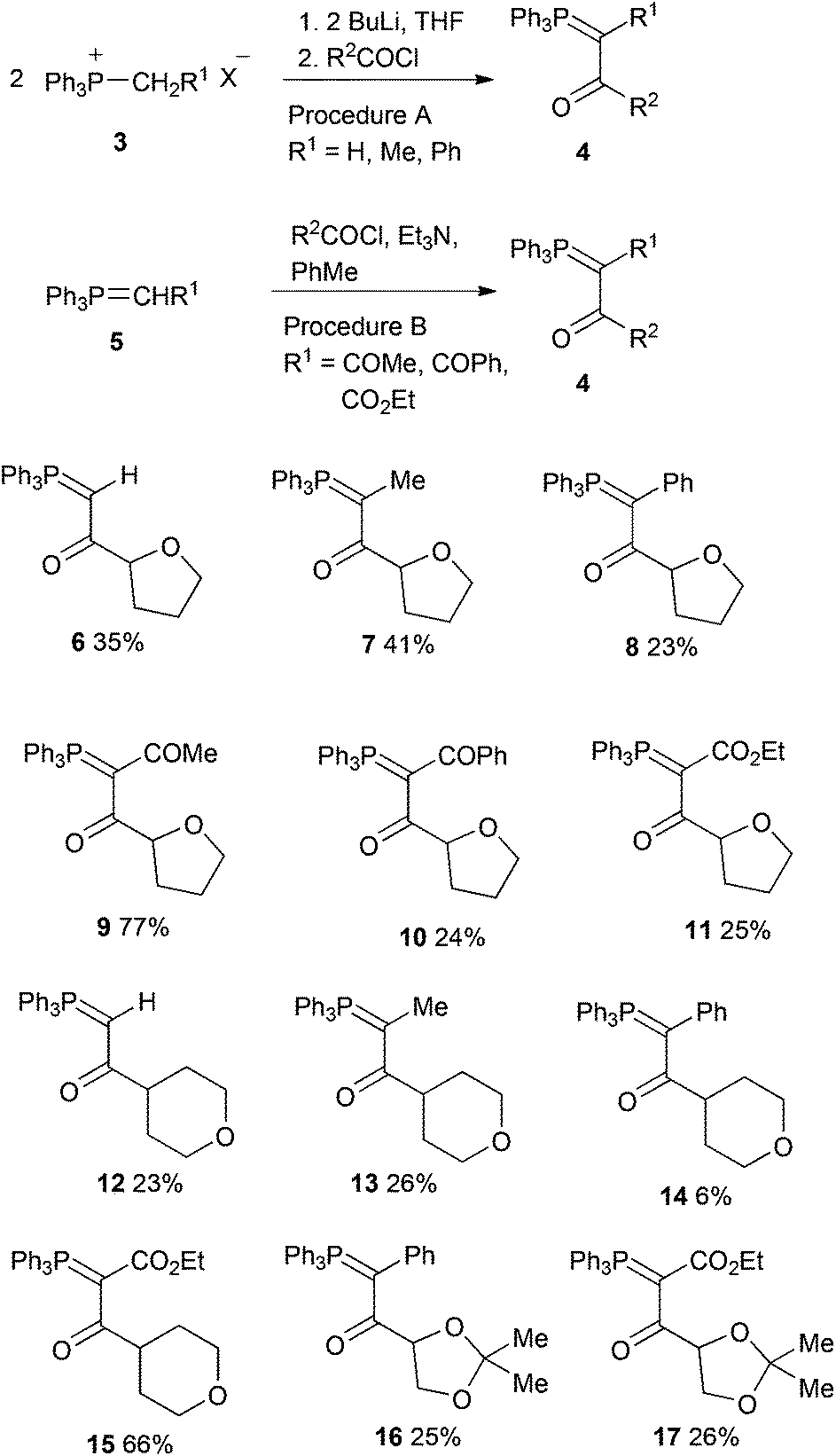
The ylides 4 were prepared by two different methods depending on the nature of R1 (Scheme 3). For monostabilised ylides (R1 = H, Me, Ph) reaction of the appropriate phosphonium salt 3 with butyllithium in THF followed by the acid chloride proceeded with “transylidation”17 requiring a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 reacting ratio, while for distabilised ylides (R1 = COMe, COPh, CO2Et) reaction of 5 with the acid chloride and triethylamine in toluene was used.18 Tetrahydrofuran-2-carbonyl chloride was prepared by reaction of the commercially available acid with oxalyl chloride, tetrahydropyran-4-carbonyl chloride was prepared by hydrolysis of methyl tetrahydropyran-4-carboxylate using sodium hydroxide followed by reaction with thionyl chloride, while 2,2-dimethyl-1,3-dioxolane-4-carbonyl chloride was prepared from mannitol diacetonide by sodium periodate cleavage19 followed by potassium permanganate oxidation of the resulting aldehyde and treatment of the potassium carboxylate with oxalyl chloride.20 The 12 ylides 6–17 were obtained in low to moderate yield as colourless or pale yellow crystals (Scheme 3). They all gave 31P NMR chemical shifts in the range δP +14.9–17.9 and the 13C NMR spectra showed a highly consistent and informative pattern of phosphorus coupling. X-Ray diffraction has previously been used to obtain valuable information on the structure of stabilised ylides, particularly in respect of the PC–CO torsion angle which is important for successful pyrolytic alkyne formation.21 Since no X-ray structures of ylides containing saturated oxygen heterocycles are known, we have determined the structure of one representative example from each of the three ring systems.
:1 reacting ratio, while for distabilised ylides (R1 = COMe, COPh, CO2Et) reaction of 5 with the acid chloride and triethylamine in toluene was used.18 Tetrahydrofuran-2-carbonyl chloride was prepared by reaction of the commercially available acid with oxalyl chloride, tetrahydropyran-4-carbonyl chloride was prepared by hydrolysis of methyl tetrahydropyran-4-carboxylate using sodium hydroxide followed by reaction with thionyl chloride, while 2,2-dimethyl-1,3-dioxolane-4-carbonyl chloride was prepared from mannitol diacetonide by sodium periodate cleavage19 followed by potassium permanganate oxidation of the resulting aldehyde and treatment of the potassium carboxylate with oxalyl chloride.20 The 12 ylides 6–17 were obtained in low to moderate yield as colourless or pale yellow crystals (Scheme 3). They all gave 31P NMR chemical shifts in the range δP +14.9–17.9 and the 13C NMR spectra showed a highly consistent and informative pattern of phosphorus coupling. X-Ray diffraction has previously been used to obtain valuable information on the structure of stabilised ylides, particularly in respect of the PC–CO torsion angle which is important for successful pyrolytic alkyne formation.21 Since no X-ray structures of ylides containing saturated oxygen heterocycles are known, we have determined the structure of one representative example from each of the three ring systems.
 | ||
| Scheme 3 Synthetic routes used and new ylides prepared. | ||
The structure of compound 10 (Fig. 1) shows an almost planar tetrahydrofuran ring and both carbonyl groups syn to the PC bond. However the degree of delocalisation in the sense P+–CC–O− as indicated by the CC and CO bond lengths is much greater for the CO–THF function with a torsion angle of 0.7° than for the COPh with a torsion angle of 34°.
 | ||
| Fig. 1 X-ray structure of 10 showing numbering scheme. Selected bond lengths and torsion angles: P(1)–C(7) 1.752(2), C(7)–C(8) 1.459(3), C(8)–O(8) 1.232(2), C(7)–C(6) 1.423(3), C(6)–O(6) 1.240(2) Å; P(1)–C(7)–C(8)–O(8) 34.2(2), P(1)–C(7)–C(6)–O(6) 0.7(2)°. | ||
In the tetrahydropyran series, the structure of the monostabilised ylide 13 was determined (Fig. 2) and this shows the tetrahydropyran in an almost perfect chair conformation with the oxo ylide function equatorial. The bond lengths again show the oxo ylide function to be substantially delocalised in the sense P+–CC–O− and PC and CO are syn.
 | ||
| Fig. 2 X-ray structure of 13 showing numbering scheme. Selected bond lengths and torsion angle: P(1)–C(19) 1.729(2), C(19)–C(20) 1.396(2), C(20)–O(20) 1.267(2) Å; P(1)–C(19)–C(20)–O(20) 2.4(2)°. | ||
The structure of 17 proved to be more complex with two separate and slightly different molecules in the unit cell (Fig. 3). In each case the ester carbonyl is anti to the PC bond while the dioxolanyl ketone is syn to it but, while the molecule containing P(41) has these functions almost coplanar (torsion angles 178.7 and 1.0°), the geometry for the other molecule containing P(1) is less ideal (torsion angles 156.8 and 10.6°).
 | ||
| Fig. 3 X-ray structure of 17 showing numbering scheme. Selected bond lengths and torsion angles: P(1)–C(19) 1.751(5), C(19)–C(24) 1.434(6), C(24)–O(24) 1.227(7), C(19)–C(20) 1.445(9), C(20)–O(20) 1.208(7), P(41)–C(59) 1.760(6), C(59)–C(60) 1.445(9), C(60)–O(60) 1.215(7), C(59)–C(64) 1.444(6), C(64)–O(64) 1.224(7) Å; P(1)–C(19)–C(24)–O(24) 10.6(7), P(1)–C(19)–C(20)–O(20) 156.8(4), P(41)–C(59)–C(60)–O(60) 178.7(4), P(41)–C(59)–C(64)–O(64) 1.0(7)°. | ||
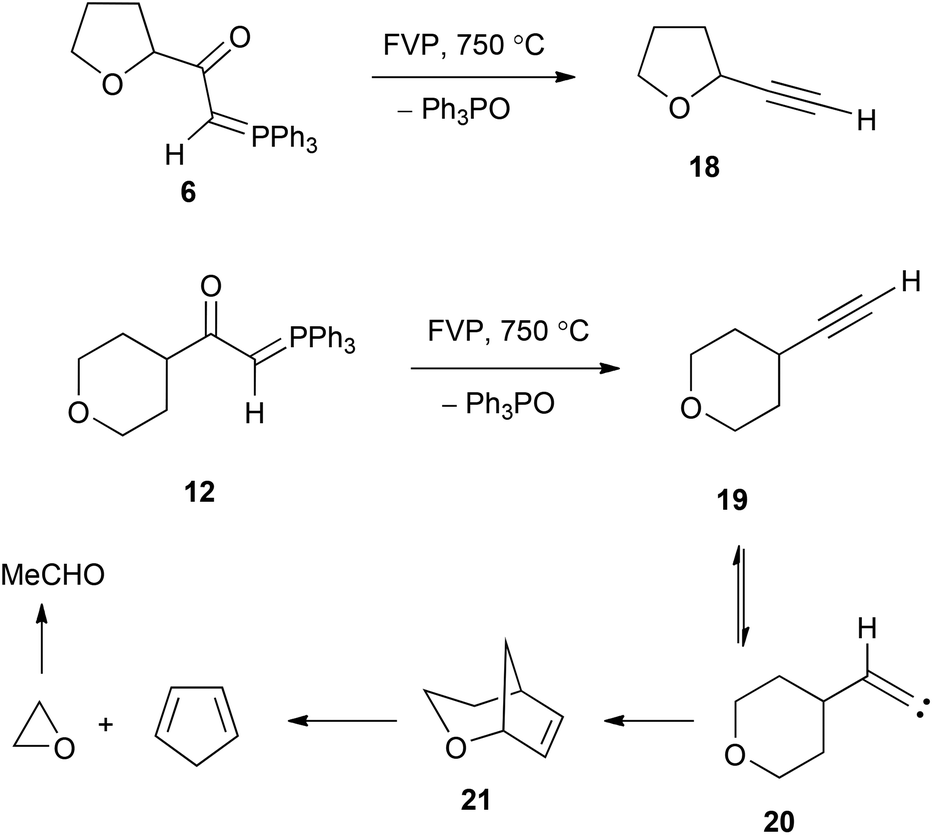
The behaviour of the compounds was now studied under flash vacuum pyrolysis conditions. The apparatus used has been described previously,22 and based on our extensive experience with oxo stabilised ylides, temperatures in the range 500–750 °C were used with a pressure of 1–5 × 10−2 Torr. The two compounds with a hydrogen atom on the ylidic bond, 6 and 12, reacted completely at 750 °C with clean extrusion of Ph3PO being observed in each case (Scheme 4). For 6 the other product was the expected terminal alkyne 18 (36%) together with a little furan and benzene from thermal degradation. In the case of the tetrahydropyran compound 12, the expected alkyne 19 (42%) was again formed but this was now accompanied by low yields of cyclopentadiene and acetaldehyde. We believe these to result from the mechanism shown in Scheme 4 in which the alkyne isomerises to the vinylidene 20 and this undergoes intramolecular CH insertion at the 2-position of the tetrahydropyran to form the bicyclic product 21. This is then set up to undergo a retro Diels–Alder reaction with formation of cyclopentadiene and oxirane, which isomerises to acetaldehyde. The formation of vinylidenes such as 20 by rearrangement of alkynes under FVP conditions is well known,23 and will be encountered again in the FVP of 9 and 10.
 | ||
| Scheme 4 | ||
The ylide 7 with a methyl group on the ylidic position reacted completely at 730 °C to give a 2:1 mixture of Ph3P and Ph3PO at the furnace exit. In the cold trap the expected alkyne 22 (20%) from loss of Ph3PO was accompanied by 2-propionyl-4,5-dihydrofuran 24 (30%) (Scheme 5). We propose that this product is formed by loss of Ph3P to give the (triplet) carbene 23, which then undergoes twofold intramolecular hydrogen atom abstraction to afford the product. In contrast the corresponding tetrahydropyran ylide 13 underwent exclusive elimination of Ph3PO at 710 °C to give the expected alkyne 25 in good yield as the only product. The extrusion of a phosphine as opposed to phosphine oxide from β-oxophosphonium ylides is very unusual, but has been observed in the FVP of a benzotriazolyl ylide,24,25 and also upon photolysis of Ph3PCHCOPh.26
 | ||
| Scheme 5 | ||
The two compounds 8 and 14 with a phenyl substituent on the ylidic carbon both reacted completely at 700 °C with exclusive elimination of Ph3PO. With the tetrahydrofuran compound 8 the product was the known alkyne 26 (56%) (Scheme 6), which showed good agreement with literature spectroscopic data,27,28 while for the tetrahydropyran compound 14, the expected alkyne product 27 (46%) was accompanied by the isomeric allene 28 (20%). The ylide 16 containing phenyl and 2,2-dimethyl-1,3-dioxolane groups was found to undergo loss of both Ph3PO and Ph3P to give separate products with the relative importance of the two competing pathways varying with temperature (Scheme 7). Thus at 650 °C, there was predominant loss of Ph3PO (Ph3PO/Ph3P 8:1) to afford the expected alkyne 29 in 48% yield readily identified by comparison with reported spectroscopic data.29 By increasing the pyrolysis temperature to 750 °C the importance of the Ph3P elimination route was increased (Ph3PO/Ph3P 3:1) and this allowed tentative identification of the resulting product as 2-phenyloxete 30. A correct HRMS measurement corresponding to 30 was obtained and, although peak overlap prevented full spectroscopic characterisation, the key signals in 1H and 13C NMR spectra were consistent with a highly shielded (enol ether) CH– [δH 6.16 (1 H, t, J 6.9); δC 93.9] adjacent to a –CH2O– [δH 5.14 (2 H, d, J 6.9); δC 78.7] function. The latter compares well with the value of δH 5.44 observed for the oxete CH2 in compound 34 (Scheme 8).30
 | ||
| Scheme 6 | ||
 | ||
| Scheme 7 | ||
 | ||
| Scheme 8 Previously reported oxetes. | ||
Simple oxetes such as this are almost unknown but there has been recent interest in more highly substituted examples such as the 4-methylene compounds 35 obtained by gold-catalysed cyclisation of α-hydroxybenzylallenes,31 and stable fully substituted compounds 36 prepared in high enantiomeric purity from ethyl trifluoropyruvate and alkynes.32 The parent compound has also been generated33 and was stable enough for a 1H NMR spectrum to be recorded at −25 °C [δH 6.70 (1 H, br s), 5.73 (1 H, br s) and 5.27 (2 H, br s)], but it isomerised to acrolein on warming to room temperature.
We rationalise the formation of 30, as shown in Scheme 7, by initial loss of Ph3P from ylide 16 to give the carbene 31 which undergoes intramolecular oxonium ylide formation to form 32. This can then rearrange as shown to give 33, which loses CO and acetone to afford the oxete. Further support for this route was provided by the observation of acetone among the pyrolysis products.
We next examined the two ylides 9 and 10 where there is potentially a choice between elimination of Ph3PO in two directions to give isomeric alkynyl ketones. In previous such cases there has been little selectivity with almost equal proportions of the two possible products being formed.34,35 This also proved to be the case here, with compound 9 undergoing complete extrusion of Ph3PO at 500 °C to afford a mixture of 37 (26%) and 39 (58%), while the benzoyl compound 10 gave a mixture of 38 (29%) and 40 (30%) under the same conditions (Scheme 9). All these alkynyl ketone products are previously unknown.
 | ||
| Scheme 9 | ||
When ylides 9 and 10 were pyrolysed at the higher temperature of 700 °C an interesting new process was observed. In each case the previously observed alkynyl ketones were still formed but these were accompanied by the 3-acylfurans 41 and 42, readily identified by comparison with literature spectroscopic data.36,37 Thus FVP of 9 at 700 °C gave 37 (18%), 39 (33%) and 41 (17%) while under the same conditions 10 gave 38 (12%), 40 (14%) and 42 (37%). As shown we propose that this involves isomerisation23 of the alkynyl ketones 39 and 40 to the vinylidene 43 which undergoes intramolecular insertion into the marked CH bond to give the oxabicyclic compounds 44 which are ideally set up to undergo retro-Diels Alder elimination of ethene to afford the observed acylfuran products.
Pyrolysis of ylides containing both adjacent ester and ketone carbonyl functions is well known to result in elimination of Ph3PO only from the latter to give acetylenic esters. The method was first developed by Märkl using conventional pyrolysis of methyl esters,3 but was later shown to also be amenable to FVP conditions using ethyl esters.38 An added advantage of using the latter technique is that, while efficient Ph3PO elimination to give the acetylenic esters occurs at 500 °C, simply increasing the temperature to 750 °C additionally leads to loss of the whole ester group to afford the alk-1-ynes. Mechanistic studies of this unusual process have been described.39 In agreement with this pattern, when the three ester-containing ylides 11, 15, and 17 were subjected to FVP, mixtures of the acetylenic esters and the alk-1-ynes were obtained with the ratio depending on the temperature (Scheme 10). Thus at 500 °C, FVP of 11 gave ester 45 (42%) together with the alkyne 18 (16%). At 750 °C, there was extensive decomposition with only products such as furan and cyclopentadiene isolated in low yield. FVP of the tetrahydropyan ylide 15 at 500 °C gave the ester 46 in 60% yield, while increasing the furnace temperature to 750 °C led to alkyne 19 (30%) together with the decomposition products cyclopentadiene (20%) and acetaldehyde (25%) formed from 19 as shown in Scheme 4.
 | ||
| Scheme 10 | ||
FVP of 17 at 600 °C gave mainly the ester 47 (68%) with a little of the alkyne 48 (9%) while at 650 °C the ratio of products had changed to 47 (39%) and 48 (30%). It therefore appears that lower temperature FVP of these three ylides provides an effective route to the acetylenic esters but for the alk-1-ynes FVP of ylides such as 6 and 12 is preferable.
Experimental
Instrumentation, general techniques and starting materials
Melting points were recorded on a Kofler hot-stage microscope and are uncorrected. Infra red spectra were recorded as Nujol mulls for solids and as thin films for liquids on a Perkin Elmer 1420 instrument. NMR spectra were obtained for 1H at 300, 400 or 500 MHz, for 13C at 75, 100 or 125 MHz and for 31P at 121 MHz all using Bruker instruments. All spectra were run on solutions in CDCl3 with internal Me4Si as reference for 1H and 13C and external H3PO4 for 31P. Chemical shifts are reported in ppm to high frequency of the reference and coupling constants J are in Hz. Mass spectra were obtained on a Micromass LCT spectrometer using electrospray ionisation.Tetrahydrofuran-2-carbonyl chloride was prepared (87%) by reaction of the commercially available acid with oxalyl chloride, tetrahydropyran-4-carbonyl chloride was prepared by hydrolysis of methyl tetrahydropyran-4-carboxylate using sodium hydroxide (quant.) followed by reaction with thionyl chloride (84%), while 2,2-dimethyl-1,3-dioxolane-4-carbonyl chloride was prepared from mannitol diacetonide by sodium periodate cleavage19 followed by potassium permanganate oxidation of the resulting aldehyde and treatment of the potassium carboxylate with oxalyl chloride.20
General procedure A for synthesis of monostabilised ylides
A suspension of the appropriate phosphonium salt (1 equiv.) in dry THF was stirred at rt under nitrogen while a solution of n-BuLi in hexanes (2.5 M, 1 equiv.) was added dropwise by syringe. After the addition the mixture was stirred for 30 min and then a solution of the acid chloride (0.5 equiv.) in dry THF was added dropwise. The mixture was stirred for 12 h and then partitioned between water and diethyl ether. The organic extract was dried (MgSO4) and evaporated to give the product. If necessary a little EtOAc was added to induce crystallisation and the products were recrystallised from EtOAc.General procedure B for synthesis of distabilised ylides
A solution of the appropriate stabilised ylide (1 equiv.) and triethylamine (1 equiv.) in dry toluene (15 cm3) was stirred at rt while a solution of the acid chloride (1 equiv.) in dry toluene (5 cm3) was added dropwise. After stirring for 12 h, the mixture was filtered to remove triethylamine hydrochloride and the filtrate was evaporated to give the product. If necessary a little EtOAc was added to induce crystallisation and the products were recrystallised from EtOAc.Tetrahydrofuran-containing ylides
P), 4.10–4.00 (1 H, m, 5-H), 3.90–3.80 (1 H, m, 5-H), 2.30–2.15 (1 H, m) and 2.10–1.80 (3 H, m); δC (100 MHz) 193.6 (d, J 3, CO), 133.0 (d, J 10, C-2 of Ph), 132.0 (d, J 1, C-4 of Ph), 128.8 (d, J 12, C-3 of Ph), 127.0 (d, J 90, C-1 of Ph), 83.0 (d, J 13, 2-CH), 68.9 (5-CH2), 48.5 (d, J 109, PCH), 30.9 (3-CH2) and 25.7 (4-CH2); δP (121 MHz) +16.2; m/z (ESI) 771.28 (2M + Na, 7%), 397.13 (M + Na, 12) and 375.15 (M + H, 100).
C), 29.3 (3-CH2), 25.9 (4-CH2) and 12.3 (d, J 12, Me); δP (121 MHz) +17.9; m/z (ESI) 389.16 (M + H, 100).
C), 69.2 (5-CH2), 30.4 (3-CH2) and 26.2 (4-CH2); δP (121 MHz) +15.5; m/z (ESI) 923.34 (2M + Na, 42%), 473.16 (M + Na, 6) and 451.18 (M + H, 100).
C), 80.9 (d, J 6, 2-CH), 68.8 (5-CH2), 30.1 (d, J 4, Me), 29.6 (3-CH2) and 25.3 (4-CH2); δP (121 MHz) +15.6; m/z (ESI) 855.30 (2M + Na, 12%), 439.14 (M + Na, 100) and 417.16 (M + H, 18).
C), 79.7 (d, J 9, 2-CH), 69.1 (5-CH2), 30.1 (3-CH2) and 25.4 (4-CH2); δP (121 MHz) +17.5; m/z (ESI) 979.33 (2M + Na, 62%), 501.16 (M + Na, 55) and 479.18 (M + H, 100).
C), 58.4 (OEt), 30.9 (3-CH2), 25.2 (4-CH2) and 13.7 (OEt); δP (121 MHz) +17.1; m/z (ESI) 915.32 (2M + Na, 20%) and 447.17 (M + H, 100).
Tetrahydropyran-containing ylides
P), 3.50–3.35 (2 H, m), 2.45–2.30 (1 H, m, 4-H) and 1.90–1.75 (4 H, m); δC (100 MHz) 195.1 (d, J 1, CO), 132.9 (d, J 10, C-2 of Ph), 131.9 (d, J 1, C-4 of Ph), 128.8 (d, J 12, C-3 of Ph), 127.2 (d, J 90, C-1 of Ph), 68.1 (2,6-CH2), 49.2 (d, J 107, PCH), 46.2 (d, J 14, 4-CH) and 30.8 (3,5-CH2); δP (121 MHz) +15.9; m/z (ESI) 389.17 (M + H, 100).
C), 40.7 (d, J 10, 4-CH), 29.2 (3,5-CH2) and 12.7 (d, J 14, Me); δP (121 MHz) +16.8; m/z (ESI) 425.16 (M + Na, 12%) and 403.18 (M + H, 100).
C), 67.8 (2,6-CH2), 41.1 (d, J 9, 4-CH) and 29.6 (3,5-CH2); δP (121 MHz) +14.9; m/z (ESI) 951.37 (2M + Na, 18%), 487.18 (M + Na, 27%) and 465.20 (M + H, 100).
C), 68.0 (2,6-CH2), 58.3 (OEt), 43.3 (d, J 7, 4-CH), 29.4 (3,5-CH2) and 13.7 (OEt); δP (121 MHz) +17.5; m/z (ESI) 943.35 (2M + Na, 32%), 483.17 (M + Na, 7%) and 461.19 (M + H, 100).
1,3-Dioxolane-containing ylides
C), 68.0 (dioxolane C-5), 26.2 (Me) and 26.0 (Me); δP (121 MHz) +16.1; m/z (ESI) 481.19 (M + H, 100).
C), 68.89 (dioxolane C-5), 58.4 (OEt), 26.1 (Me), 25.7 (Me) and 13.6 (OEt); δP (121 MHz) +16.9; m/z (ESI) 975.34 (2M + Na, 35%), 499.16 (M + Na, 60), 477.18 (M + H, 100).
X-ray structure determination
Compound 10, C31H27O3P, M 478.53, yellow prism. Monoclinic, space group P21/c, a 9.9232(13), b 21.836(2), c 11.4086(13) Å, β 96.272(4)°, V 2457.3(5) Å3, Z 4, Dc 1.293 Mg m−3, T 173 K, 26295 reflections, 4506 unique (Rint 0.048). R1 0.0484, wR2 0.1407, R indices based on 3994 data with I > 2σ(I), 316 parameters. Data were recorded using a Rigaku XtaLB P200, Mo Kα radiation (confocal optic, λ 0.71073 Å) and Saturn detector. The structure was solved by direct methods and refined using full-matrix least-squares methods.
Compound 13, C26H27O2P, M 402.47, colourless prism. Monoclinic, space group P21/c, a 11.829(3), b 11.011(3), c 16.248(4) Å, β 93.341(7)°, V 2112.7(9) Å3, Z 4, Dc 1.265 Mg m−3, T 93 K, 23466 reflections, 3850 unique (Rint 0.0592). R1 0.0369, wR2 0.1090, R indices based on 3336 data with I > 2σ(I), 263 parameters. Data were recorded using a Rigaku XtaLB P200, Mo Kα radiation (confocal optic, λ 0.71073 Å) and Saturn detector. The structure was solved by direct methods and refined using full-matrix least-squares methods.
Compound 17, C28H29O5P, M 476.51, colourless prism. Triclinic, space group P1, a 9.345(4), b 10.512(3), c 14.341(4) Å, α 98.497(2), β 106.816(11), γ 110.513(8)°, V 1213.3(7) Å3, Z 2, Dc 1.304 Mg m−3, T 125 K, 13591 reflections, 6259 unique (Rint 0.0836). R1 0.0570, wR2 0.1352, R indices based on 5802 data with I > 2σ(I), 619 parameters. Data were recorded using a Rigaku XtaLB P200, Cu Kα radiation (confocal optic, λ 1.54187 Å) and Saturn detector. The structure was solved by direct methods and refined using full-matrix least-squares methods.
Crystallographic data (excluding structure factors) for the structures included in this paper have been deposited at the Cambridge Crystallographic Data Centre as supplementary publication No. CCDC 1435297 (10), 1435298 (13) and 1435299 (17).
Flash vacuum pyrolysis (FVP)
This was carried out using the apparatus previously described.22 The contact time in the hot zone was estimated to be ∼10 ms. In each case a solid collected at the furnace exit which was found to be either Ph3PO or a mixture of Ph3PO and Ph3P. In the cold trap the other product(s) collected as a liquid or oil and this was dissolved out using either CDCl3 (small scale) for direct NMR analysis, or CH2Cl2 (large scale) for purification and characterisation by the usual methods.FVP of ylide 6 (150 mg) at 750 °C gave at the furnace exit Ph3PO; δP +29.2, and in the cold trap:
2-Ethynyltetrahydrofuran
18 (14 mg, 36%) as a colourless liquid (Found 95.0493. C6H7O (M − H) requires 95.0497); δH (300 MHz) 4.62–4.58 (1 H, m, 2-H), 4.00–3.92 (1 H, m, 5-H), 3.86–3.75 (1 H, m, 5-H), 2.43 (1 H, d, J 2.0, ![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH) and 2.24–1.85 (m, 4H, 3,4-H); δC (75 MHz) 83.8 (C, –C), 72.5 (CH, –C), 67.9 (CH2, C-5), 67.8 (CH, C-2), 33.2 (CH2) and 25.3 (CH2).
CH) and 2.24–1.85 (m, 4H, 3,4-H); δC (75 MHz) 83.8 (C, –C), 72.5 (CH, –C), 67.9 (CH2, C-5), 67.8 (CH, C-2), 33.2 (CH2) and 25.3 (CH2).
FVP of ylide 12 (110 mg) at 750 °C gave at the furnace exit Ph3PO; δP +29.2, and in the cold trap:
A mixture containing 4-ethynyltetrahydropyran19 (13 mg, 42%) as a colourless liquid (Found: 111.0812. C7H11O (M + H) requires 111.0810); νmax/cm−1 3293 (C–H), 2240 (CC), 1262, 1090, 1058 and 813; δH (400 MHz) 3.905 (2 H, ddd, J 11.6, 5.6, 3.6, 2,6-H), 3.505 (2 H, ddd, J 11.6, 8.4, 3.2, 2,6-H), 2.68–2.61 (1 H, m, 4-H), 2.11 (1 H, d, J 2.4, CH), 1.88–1.80 (2 H, m, 3,5-H) and 1.74–1.64 (2 H, m, 3,5-H); δC (100 MHz) 86.6 (C, –C), 69.1 (CH, –C), 66.2 (CH2), 32.0 (CH2) and 25.9 (CH), together with cyclopentadiene (4%); δH 6.57 (2 H, m), 6.47 (2 H, m) and 2.99 (2 H, m); δC 133.1 (CH), 132.2 (CH) and 41.6 (CH2) and acetaldehyde (3%); δH 9.79 (1 H, q, J 3) and 2.21 (3 H, d, J 3).
FVP of ylide
7 (200 mg) at 730 °C gave at the furnace exit a 2:1 mixture of Ph3P; δP −5.5 and Ph3PO; δP +29.2, and in the cold trap:
A colourless liquid consisting of a mixture of 2-(prop-1-ynyl)tetrahydrofuran22 (20%); δH (400 MHz) 4.58–4.50 (1 H, m, 2-H), 3.98–3.85 (2 H, m, 5-H), 1.90–1.80 (2 H, m), 1.75–1.70 (2 H, m) and 1.84 (3 H, d, J 1.8); δC (100 MHz) 79.0 (C, –C), 70.7 (C, –C), 68.3 (CH), 67.8 (CH2), 33.3 (CH2), 25.3 (CH2) and 3.5 (Me) [good agreement of δH with lit.40] and 2-(propionyl)-4,5-dihydrofuran24 (30%) (Found 127.0754. C7H11O2 (M + H) requires 127.0759); δH (300 MHz) 5.94 (1 H, t, J 3.0), 4.47 (2 H, t, J 9.8), 2.82 (2 H, td, 9.8, 3.0), 2.65 (2 H, q, J 7.2) and 1.12 (3 H, t, J 7.2); δC (100 MHz) 193.0 (CO), 156.0 (C–), 110.7 (CH–), 70.1 (OCH2), 32.0 (COCH2), 30.5 (4-CH2) and 8.0 (CH3). Preparative TLC allowed separation of the latter in pure form.
FVP of ylide 13 (70 mg) at 710 °C gave at the furnace exit Ph3PO; δP +29.2, and in the cold trap:
4-(Prop-1-ynyl)tetrahydropyran
25 (65%) (Found: 125.0969. C8H13O (M + H) requires 125.0966); νmax/cm−1 2233 (CC); δH (300 MHz) 3.89 (2 H, ddd, J 11.7, 5.1, 3.9, 2,6-H), 3.47 (2 H, ddd, J 11.7, 8.7, 2.8, 2,6-H), 2.60–2.50 (1 H, m, 4-H), 1.81 (3 H, d, J 2.0) 1.82–1.74 (2 H, m, 3,5-H) and 1.68–1.55 (2 H, m, 3,5-H); δC (75 MHz) 81.7 (C, –C), 76.4 (C, –C), 66.5 (CH2), 32.6 (CH2), 26.3 (CH) and 3.5 (Me).
FVP of ylide 8 (80 mg) at 700 °C gave at the furnace exit Ph3PO; δP +29.2, and in the cold trap:
2-(Phenylethynyl)tetrahydrofuran
26 (56%) as a colourless liquid (Found 195.0776. C12H12ONa (M + Na) requires 195.0786); νmax/cm−1 2231 (CC), 1727, 1599, 1490, 1054, 757 and 692; δH (300 MHz) 7.6–7.2 (5 H, m), 4.81 (1 H, dd, J 7.2, 5.0, 2-H), 4.05–3.98 (1 H, m, 5-H), 3.89–3.82 (1 H, m, 5-H), 2.25–2.15 (1 H, m), 2.15–2.00 (2 H, m) and 2.00–1.85 (1 H, m); δC (75 MHz) 131.7 (2 CH), 128.22 (CH), 128.17 (2 CH), 122.8 (C), 89.0 (C, –C), 84.4 (C, –C), 68.6 (CH2), 67.9 (CH), 33.4 (CH2) and 25.5 (CH2) [good agreement of δH27 and δC28 with lit.].
FVP of ylide 14 (70 mg) at 700 °C gave at the furnace exit Ph3PO; δP +29.2, and in the cold trap:
A mixture of 4-(phenylethynyl)tetrahydropyran27 (46%) (Found 186.1042. C13H14O (M) requires 186.1045); δH (300 MHz) 7.45–7.25 (5 H, m), 4.00–3.89 (2 H, m, 2,6-H), 3.60–3.50 (2 H, m, 2,6-H), 2.90–2.80 (1 H, m, 4-H), 1.95–1.85 (2 H, m, 3,5-H) and 1.82–1.70 (2 H, m, 3,5-H); δC (75 MHz) 131.6 (2 CH), 128.2 (2 CH), 126.6 (CH), 123.6 (C), 92.2 (C, –C), 81.5 (C, –C), 66.4 (CH2), 32.3 (CH2) and 26.8 (CH), and 4-(phenylvinylidene)tetrahydropyran28 (20%); δH (300 MHz) 7.45–7.25 (5 H, m), 6.10 (1 H, quintet, J 2.1), 3.89–3.75 (2 H, m, 2,6-H), 2.38–2.33 (2 H, m, 3,5-H); δC (75 MHz) 200.7 (C), 135.2 (C), 128.6 (2 CH), 127.7 (2 CH), 126.7 (CH), 101.9 (C<), 93.7 (CH), 68.8 (CH2) and 31.3 (CH2).
FVP of ylide
16 (65 mg) at 650 °C gave at the furnace exit an 8:1 mixture of Ph3PO; δP +29.2 and Ph3P; δP −5.5, and in the cold trap:
4-Phenylethynyl-2,2-dimethyl-1,3-dioxolane
29 (48%) (Found 225.0883. C13H14O2Na (M + Na) requires 225.0891); νmax/cm−1 2234, 2212 (CC), 1709, 1680, 1599, 1491, 1065, 758 and 692; δH (300 MHz) 7.48–7.40 (2 H, m), 7.35–7.25 (3 H, m), 4.95 (1 H, t, J 6.3, 4-H), 4.24 (1 H, dd, J 8.0, 6.3, 5-H), 4.01 (1 H, dd, J 8.0, 6.3, 5-H), 1.54 (3 H, q, J 0.6) and 1.43 (3 H, q, J 0.6); δC (75 MHz) 131.7 (2 C), 128.6 (C-4 of Ph), 128.2 (2 C), 122.2 (C-1 of Ph), 110.3 (dioxolane C-2), 86.2 (–C), 85.8 (–C), 70.0 (dioxolane C-5), 66.0 (dioxolane C-4), 26.2 and 26.0 (CMe2) [good agreement of δH and δC with lit.27]. Acetone (20%); δH 2.17; δC 207.0 and 30.9, was also present.
FVP of ylide
16 (107 mg) at 750 °C gave at the furnace exit a 3:1 mixture of Ph3PO; δP +29.2 and Ph3P; δP −5.5, and in the cold trap a mixture of 29 (data as above), acetone, and:
2-Phenyloxete
30 (Found 133.0649. C9H9O (M + H) requires 133.0653); δH (300 MHz) 6.16 (1 H, t, J 6.9) and 5.14 (2 H, d, J 6.9); δC (75 MHz) 93.9 (CH) and 78.7 (CH2). Due to peak overlap the phenyl and quaternary C– signals could not be observed with certainty.
FVP of ylide 9 (70 mg) at 500 °C gave at the furnace exit Ph3PO; δP +29.2 and in the cold trap a mixture of:
1-(Tetrahydrofuran-2-yl)but-2-yn-1-one
37 (26%); δH (400 MHz) 4.44 (1 H, dd, J 8.6, 5.8), 3.95–3.80 (2 H, m), 2.30–2.00 (4 H, m) and 2.07 (3 H, s); δC (75 MHz) 188.8 (CO), 93.7 (C, –C), 83.7 (CH), 78.3 (C, –C), 69.6 (CH2), 29.3 (CH2), 25.25 (CH2) and 4.3 (CH3) and 4-(tetrahydrofuran-2-yl)but-3-yn-2-one39 (58%); δH (400 MHz) 4.74 (1 H, dd, J 8.0, 4.8), 4.1–3.9 (2 H, m), 2.2–1.8 (4 H, m) and 2.35 (3 H, s); δC (75 MHz) 184.3 (CO), 91.0 (C, –C), 83.5 (C, –C), 68.3 (CH2), 67.6 (CH), 32.7 (CH2), 32.6 (CH3) and 25.34 (CH2).
FVP of ylide 9 (110 mg) at 700 °C gave at the furnace exit Ph3PO; δP +29.2 and in the cold trap a mixture of 37 (18%), 39 (33%) data as above, and:
3-Acetylfuran 41 (17%); δH (300 MHz) 8.03 (1 H, dd, J 1.5, 0.9), 7.45 (1 H, dd, J 2.0, 1.5), 6.77 (1 H, dd, J 2.0, 0.9) and 2.45 (3 H, s); δC (75 MHz) 192.6 (CO), 147.5 (CH), 144.2 (CH), 128.0 (C), 108.5 (CH) and 27.8 (CH3) [good agreement of δH and δC with lit.36].
FVP of ylide 10 (70 mg) at 500 °C gave at the furnace exit Ph3PO; δP +29.2 and in the cold trap:
A mixture of 3-phenyl-1-(tetrahydrofuran-2-yl)prop-2-yn-1-one38 (29%); δH (300 MHz) 7.65–7.35 (5 H, m), 4.58 (1 H, dd, J 8.7, 5.7), 4.14–3.85 (2 H, m) and 2.40–1.90 (4 H, m); δC (75 MHz) 83.9 (CH), 69.8 (CH2), 29.7 (CH2) and 25.4 (CH2) [due to low intensity and peak overlap, the signals for CO, –C and Ph could not be assigned with certainty] and 1-phenyl-3-(tetrahydrofuran-2-yl)prop-2-yn-1-one40 (30%); δH (300 MHz) 7.65–7.35 (5 H, m), 4.89 (1 H, dd, J 7.8, 4.8), 4.14–3.85 (2 H, m) and 2.40–1.90 (4 H, m); δC (75 MHz) 68.4 (CH2), 67.9 (CH), 32.9 (CH2) and 25.4 (CH2) [due to low intensity and peak overlap, the signals for CO, –C and Ph could not be assigned with certainty]. Aromatic CH signals for 38 and 40 were at 134.2, 133.2, 130.9, 129.6, 128.63 and 128 58.
FVP of ylide 10 (50 mg) at 700 °C gave at the furnace exit Ph3PO; δP +29.2 and in the cold trap a mixture of 38 (12%), 40 (14%) data as above, and:
3-Benzoylfuran 42 (37%); δH (300 MHz) 7.93 (1 H, dd, J 1.5, 0.9), 7.87–7.82 (2 H, m), 7.62–7.56 (1 H, m), 7.51 (1 H, dd, J 1.8, 1.5), 7.50–7.46 (2 H, m) and 6.92 (1 H, dd, J 1.8, 0.9) [good agreement of δH with lit.37].
FVP of ylide 11 (120 mg) at 500 °C gave at the furnace exit Ph3PO; δP +29.2 and in the cold trap a mixture of:
Ethyl 3-(tetrahydrofuran-2-yl)propynoate
45 (42%) (Found 167.0703. C9H11O3 (M − H) requires 167.0708); νmax/cm−1 2239 (CC), 1717, 1368, 1254, 1052, 1031, 860 and 752; δH (400 MHz) 4.73 (1 H, dd, J 8.0, 4.0, 2-H), 4.23 (2 H, q, J 7.0), 3.99–3.92 (1 H, m, 5-H), 3.89–3.80 (1 H, m, 5-H), 2.30–2.20 (1 H, m, 4-H), 2.20–2.00 (2 H, m), 2.00–1.85 (1 H, m) and 1.31 (3 H, t, J 7.0); δC (100 MHz) 153.3 (CO), 86.9 (C, –C), 76.1 (C, –C), 68.3 (CH2), 67.5 (CH), 62.0 (CH2), 32.7 (CH2), 25.3 (CH2) and 13.9 (CH3) and 2-ethynyltetrahydrofuran18 (16%), data as for FVP of 6.
FVP of ylide 11 (120 mg) at 750 °C gave at the furnace exit Ph3PO: δP +29.2 and in the cold trap a mixture containing low yields of furan, cyclopentadiene and acetaldehyde.
FVP of ylide 15 (100 mg) at 500 °C gave at the furnace exit Ph3PO; δP +29.2 and in the cold trap:
Ethyl 3-(tetrahydropyran-4-yl)propynoate
46 (60%) (Found 205.0830. C10H14O3Na (M + Na) requires 205.0841); νmax/cm−1 2239 (CC), 1712, 1260, 1250, 1095, 1021 and 747; δH (500 MHz) 4.23 (2 H, q, J 7.0), 3.90 (2 H, ddd, J 11.6, 5.6, 3.6, 2,6-H), 3.51 (2 H, ddd, J 11.8, 8.8, 3.0, 2,6-H), 2.81–2.75 (1 H, m, 4-H), 1.91–1.84 (2 H, m, 3,5-H), 1.79–1.71 (2 H, m, 3,5-H) and 1.32 (3 H, t, J 7.0); δC (125 MHz) 153.8 (CO), 90.3 (C, –C), 73.8 (C, –C), 66.1 (CH2), 62.0 (CH2), 31.0 (CH2), 26.1 (CH) and 14.0 (CH3).
FVP of ylide 15 (100 mg) at 750 °C gave at the furnace exit Ph3PO; δP +29.2 and in the cold trap 4-ethynyltetrahydropyran19 (30%) together with cyclopentadiene (20%) and acetaldehyde (25%); data as for FVP of 12.
FVP of ylide 17 (130 mg) at 600 °C gave at the furnace exit Ph3PO; δP +29.2 and in the cold trap:
A mixture of ethyl 3-(2,2-dimethyl-1,3-dioxolan-4-yl)propynoate47 (68%) (Found 221.0782. C10H14O4Na (M + Na) requires 221.0790); νmax/cm−1 2244 (CC), 1716 (CO), 1374, 1248, 1067 and 846; δH (300 MHz) 4.82 (1 H, dd, J 6.6, 5.6, 4-H), 4.24 (2 H, q, J 7.2), 4.21 (1 H, dd, J 8.4, 6.6, 5-H), 4.05 (1 H, dd, J 8.4, 5.6, 5-H), 1.50 (3 H, q, J 0.6), 1.39 (3 H, q, J 0.6) and 1.31 (3 H, t, J 7.2); δC (75 MHz) 153.0 (CO), 111.2 (dioxolane C-2), 84.2 (–C), 77.0 (–C), 69.2 (dioxolane C-5), 64.9 (dioxolane C-4), 62.2 (CH2CH3), 26.0 and 25.7 (CMe2) and 13.9 (CH2CH3) and 4-ethynyl-2,2-dimethyl-1,3-dioxolane48 (9%) (Found 125.0602. C7H9O2 (M − H) requires 125.0603); δH (300 MHz) 4.72 (1 H, td, J 6.3, 2.1, 4-H), 4.18 (1 H, dd, J 8.1, 6.3, 5-H), 3.95 (1 H, dd, J 8.1, 6.3, 5-H), 2.51 (1 H, d, J 2.1, CH), 1.50 (3 H, q, J 0.6) and 1.39 (3 H, q, J 0.6); δC (75 MHz) 110.5 (dioxolane C-2), 85.4 (–C), 73.9 (HC), 69.8 (dioxolane C-5), 65.2 (dioxolane C-4), 26.1 and 25.8 (CMe2).
FVP of ylide 17 (120 mg) at 650 °C gave at the furnace exit Ph3PO; δP +29.2 and in the cold trap a mixture of 47 (39%) and 48 (30%), data as above.
Conclusions
Our results show that the tetrahydrofuran, tetrahydropyran and 2,2-dimethyl-1,3-dioxolane rings are generally stable under the conditions required for alkyne formation using the FVP of stabilised phosphonium ylides. Increasing the pyrolysis temperature does lead to observation of new, largely carbene-based, processes in some cases but provided the temperature is carefully controlled the use of this method to construct acetylenic carbohydrate derivatives should be possible and further results on this will be reported shortly.Notes and references
- R. A. Aitken and A. W. Thomas, in Chemistry of the functional groups, Supplement A3, ed. S. Patai, Wiley, New York, 1997, pp. 473–536 Search PubMed.
- S. Trippett and D. M. Walker, J. Chem. Soc., 1959, 3874–3876 RSC.
- G. Märkl, Chem. Ber., 1961, 94, 3005–3010 CrossRef.
- A. W. Johnson, in Ylides and Imines of Phosphorus, Wiley, New York, 1993, pp. 129–151 Search PubMed.
- R. A. Aitken and J. I. Atherton, J. Chem. Soc., Chem. Commun., 1985, 1140–1141 RSC.
- R. A. Aitken and A. N. Garnett, New J. Chem., 2009, 33, 2402–2404 RSC.
- R. A. Aitken and L. Murray, J. Org. Chem., 2008, 73, 9781–9783 CrossRef CAS PubMed.
- R. A. Aitken, N. Karodia, T. Massil and R. J. Young, J. Chem. Soc., Perkin Trans. 1, 2002, 533–541 RSC.
- R. A. Aitken and Y. Boubalouta, in Advances in Heterocyclic Chemistry, ed. E. F. V. Scriven and C. A. Ramsden, Elsevier, Oxford, 2015, ch. 2, vol. 115, pp. 93–150 Search PubMed.
- E. J. Horn and J. Gervay-Hague, J. Org. Chem., 2009, 74, 4357–4359 CrossRef CAS PubMed.
- J. M. J. Tronchet, A. P. Bonenfant, F. Perret, A. Gonzalez, J.-B. Zumwald, E. M. Martinez and B. Bachler, Helv. Chim. Acta, 1980, 63, 1181–1189 CrossRef CAS.
- R. M. Adlington, J. E. Baldwin, G. J. Pritchard and K. C. Spencer, Tetrahedron Lett., 2000, 41, 575–578 CrossRef CAS.
- D. E. Bays, R. P. C. Cousins, H. J. Dyke, C. D. Eldred, B. D. Judkins, M. Pass and A. M. K. Pennell, US Pat, 6492348 B1, 2002 Search PubMed.
- P. Nakache, E. Ghera and A. Hassner, Tetrahedron Lett., 2000, 41, 5583–5587 CrossRef CAS.
- K. Fujiwara, Y. Hirose, D. Sato, H. Kawai and T. Suzuki, Tetrahedron Lett., 2010, 51, 4263–4266 CrossRef CAS.
- R. S. Vartanyan, Zh. V. Kazaryan and S. A. Vartanyan, Chem. Heterocycl. Compd. (Engl. Transl.), 1979, 253–254 CrossRef.
- H. J. Bestmann, Chem. Ber., 1962, 95, 58–63 CrossRef CAS.
- S. T. D. Gough and S. Trippett, J. Chem. Soc., 1964, 543–544 CAS.
- C. R. Schmid and J. D. Bryant, Org. Synth., 1995, 72, 6–9 CrossRef CAS; C. R. Schmid and J. D. Bryant, Org. Synth. Coll. Vol., 1998, 9, 450–453 Search PubMed.
- M. J. Earle, A. Abdur-Rashid and N. D. Priestley, J. Org. Chem., 1996, 61, 5697–5700 CrossRef CAS.
- R. A. Aitken, N. Karodia and P. Lightfoot, J. Chem. Soc., Perkin Trans. 2, 2000, 333–340 RSC.
- R. A. Aitken and J. I. Atherton, J. Chem. Soc., Perkin Trans. 1, 1994, 1281 RSC.
- R. F. C. Brown, Recl. Trav. Chim. Pays-Bas, 1988, 107, 655–661 CrossRef CAS.
- R. A. Aitken, I. M. Fairhurst, A. Ford, P. E. Y. Milne, D. W. Russell and M. Whittaker, J. Chem. Soc., Chem. Commun., 1993, 1517–1519 RSC.
- R. A. Aitken, I. M. Fairhurst, A. Ford, P. E. Y. Milne, D. W. Russell and M. Whittaker, J. Chem. Soc., Perkin Trans. 1, 1997, 3107–3112 RSC.
- R. R. Da Silva, V. G. Toscano and R. G. Weiss, J. Chem. Soc., Chem. Commun., 1973, 567–568 RSC.
- D. S. Brown, M. Bruno, R. J. Davenport and S. V. Ley, Tetrahedron, 1989, 45, 4293–4308 CrossRef CAS.
- A. R. Katritzky, S. Rachwal and B. Rachwal, J. Chem. Soc., Perkin Trans. 1, 1990, 1717–1725 RSC.
- W. Shen and L. Wang, J. Org. Chem., 1999, 64, 8873–8879 CrossRef CAS PubMed.
- T. Otsuki, Bull. Chem. Soc. Jpn., 1974, 47, 3089–3093 CrossRef.
- B. Alcaide, P. Almendros, T. Martínez del Campo and I. Fernández, Chem. Commun., 2011, 47, 9054–9056 RSC.
- K. Aikawa, Y. Hioki, N. Shimizu and K. Mikami, J. Am. Chem. Soc., 2011, 133, 20092–20095 CrossRef CAS PubMed.
- P. C. Martino and P. B. Shevlin, J. Am. Chem. Soc., 1980, 102, 5429–5430 CrossRef CAS.
- P. A. Chopard, R. J. G. Searle and F. H. Devitt, J. Org. Chem., 1965, 30, 1015–1019 CrossRef CAS.
- Y. Shen, W. Cen and Y. Huang, Synthesis, 1985, 159–160 CrossRef CAS.
- C. Gryparis, I. N. Lykakis, C. Efe, I.-P. Zaravinos, T. Vidali, E. Kladou and M. Stratakis, Org. Biomol. Chem., 2011, 9, 5655–5658 CAS.
- T. Satoh, T. Itaya, K. Okuro, M. Miura and M. Nomura, J. Org. Chem., 1995, 60, 7267–7271 CrossRef CAS.
- R. A. Aitken, C. E. R. Horsburgh, J. G. McCreadie and S. Seth, J. Chem. Soc., Perkin Trans. 1, 1994, 1727–1732 RSC.
- R. A. Aitken and J. J. Morrison, ARKIVOC, 2008,(x), 103–112 Search PubMed.
- M. Apparu and J. K. Crandall, J. Org. Chem., 1984, 49, 2125–2130 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Copies of 1H and 13C NMR spectra of all compounds. CCDC 1435297–1435299. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ob02467f |
| ‡ Present address: School of Biology, Chemistry and Forensic Science, University of Wolverhampton, Wulfruna Street, Wolverhampton, WV1 1LY, UK. E-mail: nazira.karodia@wlv.ac.uk |
| This journal is © The Royal Society of Chemistry 2016 |