
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licenceγ-Sultam-cored N,N-ligands in the ruthenium(II)-catalyzed asymmetric transfer hydrogenation of aryl ketones†
Slavko
Rast
a,
Barbara
Modec
b,
Michel
Stephan‡
a and
Barbara
Mohar
*a
aNational Institute of Chemistry, Hajdrihova 19, 1001 Ljubljana, Slovenia. E-mail: barbara.mohar@ki.si
bFaculty of Chemistry and Chemical Technology, University of Ljubljana, Večna pot 113, 1000 Ljubljana, Slovenia
First published on 8th January 2016
Abstract
The synthesis of new enantiopure syn- and anti-3-(α-aminobenzyl)-benzo-γ-sultam ligands 6 and their application in the ruthenium(II)-catalyzed asymmetric transfer hydrogenation (ATH) of ketones using formic acid/triethylamine is described. In particular, benzo-fused cyclic ketones afforded excellent enantioselectivities in reasonable time employing a low loading of the syn ligand-containing catalyst. A never-before-seen dynamic kinetic resolution (DKR) during reduction of a γ-keto carboxylic ester (S7) derivative of 1-indanone is realized leading as well to excellent induction.
Introduction
The efficient asymmetric transfer hydrogenation (ATH) of ketones employing a HCO2H/Et3N binary mixture can be currently achieved under mild conditions by three generations of RSO2-DPEN-based chiral Ru(II) complexes (available in both enantiomeric forms, DPEN = trans-1,2-diphenylethylenediamine) (Fig. 1).1–5 Noyori and co-workers’ chiral [RuCl(TsDPEN)(η6-arene)]-type complexes (1st generation) were the starting point of such catalyzed asymmetric transformation both on the applied and fundamental levels.2,3 Then, intracovalent tethering of the diamine and η6-arene ligand units (2nd and 3rd generations) led to an increased longevity of the catalytic species improving thus the turnover number.4,5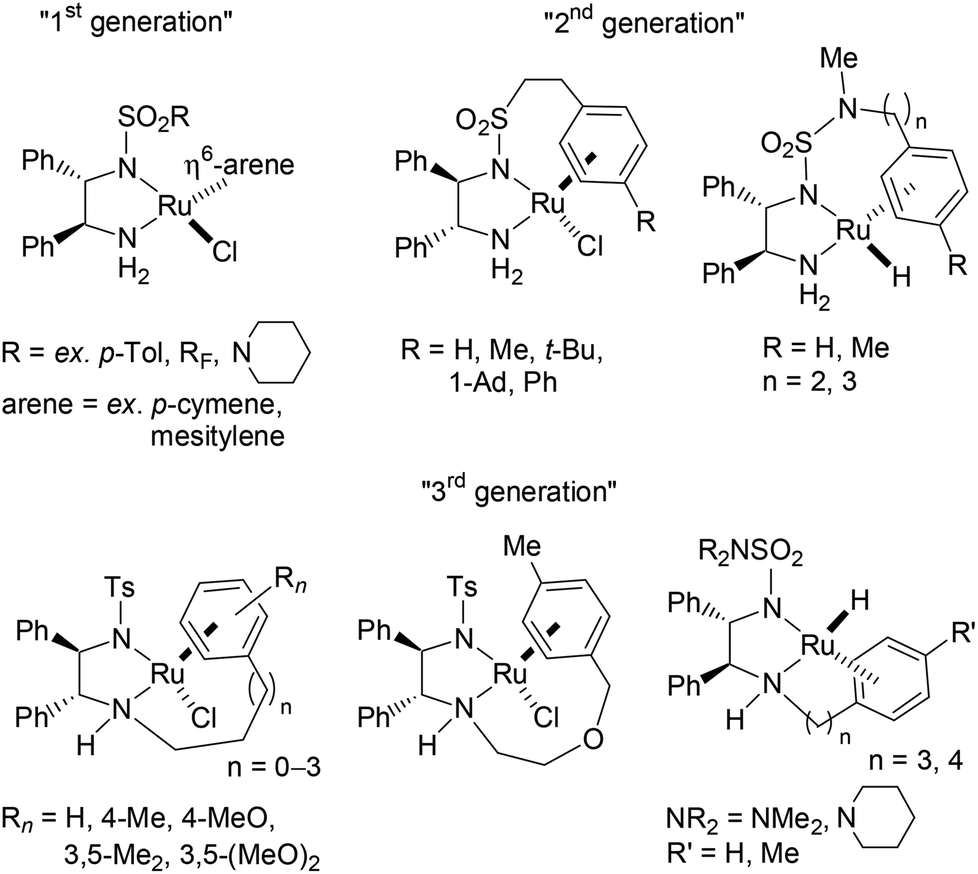 | ||
| Fig. 1 “SO2DPEN”-embedded ATH-efficient chiral Ru(II) complexes. | ||
Exploring the origin of the stereocontrol by the structural stereoarray of the 1st generation ligands and aiming to enhance the enantioselectivity and catalyst activity, empirical modifications of the chiral elements were undertaken (Fig. 2). In particular, TsDPEN skeletal alteration at the level of its ethylene-bridge substituents on position 1 or 2 revealed the critical importance of their aromatic nature (and inherent steric bulk) as well as the advantage of their anti disposition.2a,6
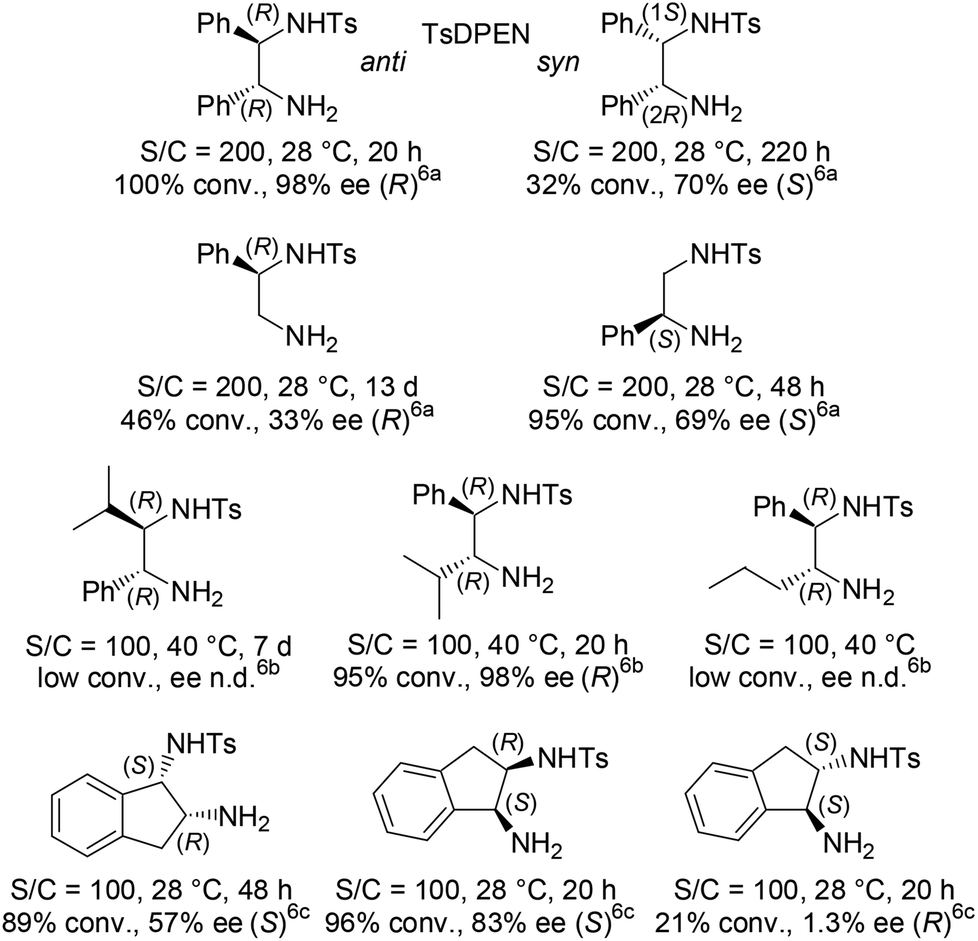 | ||
Fig. 2 Representative results of [RuCl(TsDPEN or “altered TsDPEN”)(p-cymene)]-catalyzed ATH of acetophenone using HCO2H/Et3N 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2. :2. | ||
With our ongoing research interest in this area, we present hereafter the synthesis of 5-membered cyclic minimalist TsDPEN analogs (Fig. 3) and their investigation in the ATH of various classes of ketones. This new design possesses a partial degree of stereochemical rigidity, maintaining however TsDPEN structural elements of the vic-diaryls disposition and the mono N-sulfonyl group. Such a structure gives rise to two possible pairs of syn and anti diastereomers of which preparation was targeted.
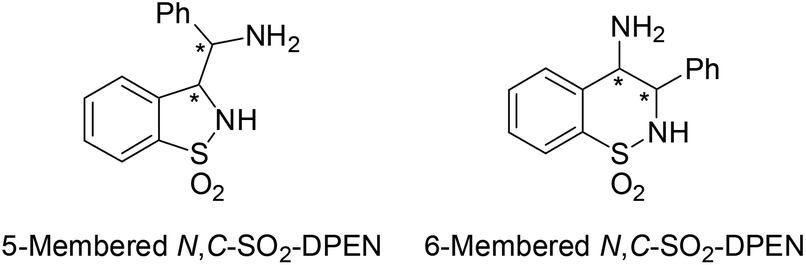 | ||
| Fig. 3 Possible heterocyclic regioisomers of “structurally-simplified TsDPEN”. | ||
Results and discussion
Synthesis of the new 5-membered cyclic N,C-SO2-DPEN ligands
In this study, we opted for a non-asymmetric synthetic strategy emphasizing on a convenient access to these ligands with a late-stage resolution. Thus, the NaOMe-mediated Wittig reaction of sodium ortho-formylbenzenesulfonate with benzyltriphenylphosphonium chloride afforded pure sodium (E)-β-styrylbenzenesulfonate [(E)-1] in 59% yield after recrystallization (Scheme 1). Conversion into the (E)-sulfonamide (E)-2 followed by epoxidation with m-CPBA and reaction with LiOMe in MeOH led to the regioselective formation of the 6-endo-tet-cyclized trans-δ-sultam trans-4 in high overall yield (82%). Such regioselectivity is supported by 1H, 13C-HMBC analysis from a correlation between the proximal aromatic hydrogen of the 1,1-dioxo-benzo-1,2-thiazinane core and the carbon atom bearing the hydroxyl group (see the ESI†).7 Tandem in situ O-mesylation-intra-N-alkylation furnished in 76% yield the trans-configured aziridine-cored product trans-5 (for its X-ray structure showing the aromatic rings in trans and a chiral angular N atom, see the ESI†).§ Alternatively to this circuitous approach, a straightforward single-step conversion of (E)-2 into trans-5via Rh2(OAc)4-catalyzed aziridination using PhI(OAc)28 was achieved in 57% yield. Further on, consecutive aziridine highly regioselective ring-opening with sodium azide in MeCN/H2O (4:1) and Pd/C-catalyzed hydrogenation gave the syn-3-(α-aminobenzyl)-benzo-γ-sultam syn-6. The (3S,1′R)- and (3R,1′S)-configured enantiomeric ligands 6 were separated by preparative chiral HPLC in >99% ee and 78% combined yields. The absolute configuration of the 1st eluting enantiomer was determined by X-ray analysis of its (S)-CSA salt (Fig. 4).9
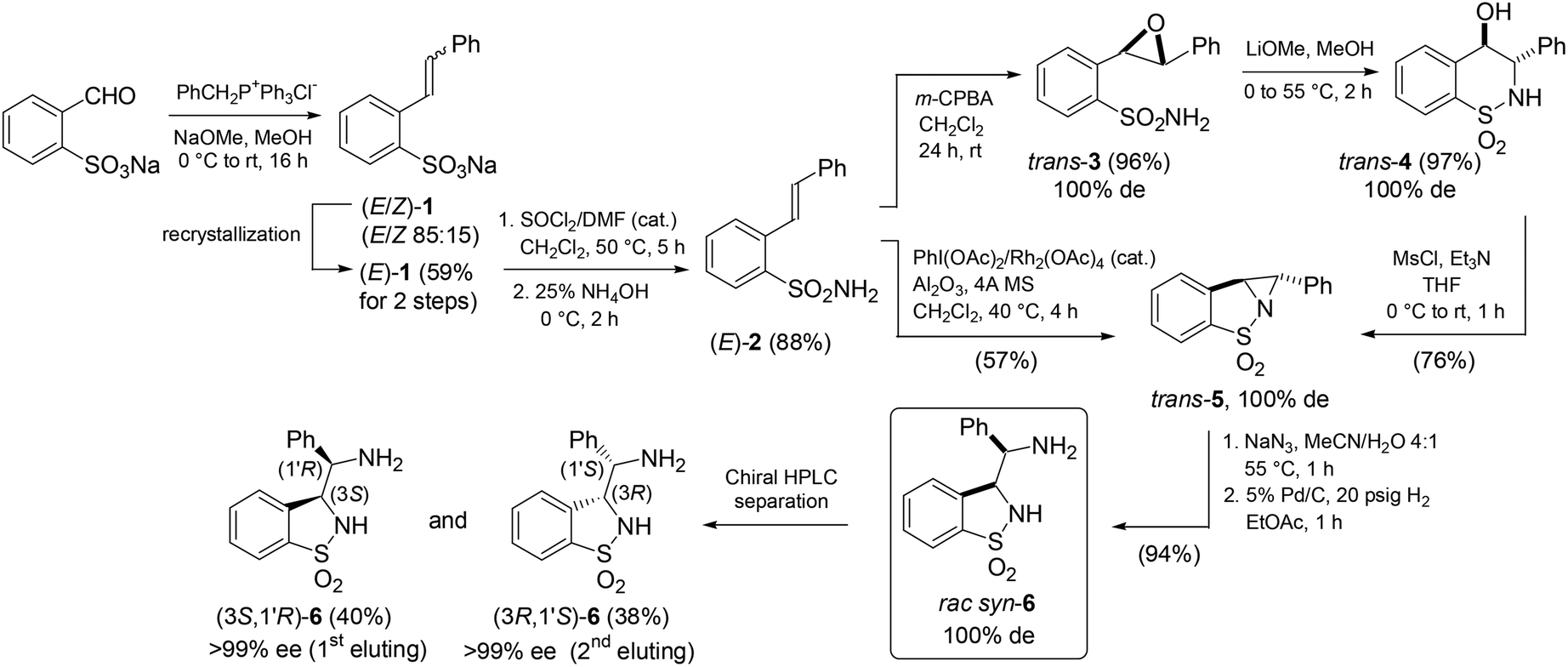 | ||
| Scheme 1 Preparation of the syn-3-(α-aminobenzyl)-benzo-γ-sultam ligand 6 enantiomers. | ||
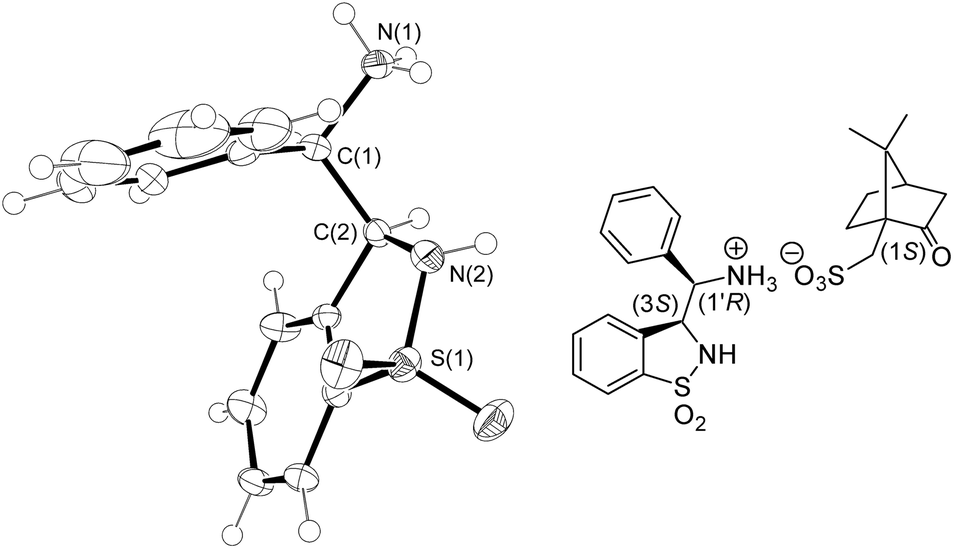 | ||
| Fig. 4 ORTEP drawing of the (1S)-camphor-10-sulfonic acid ((S)-CSA) salt of the HPLC 1st eluting syn-6 enantiomer at the 50% probability level ((S)-CSA was omitted for clarity; for full details, see the ESI†). | ||
Next, the complementary diastereomer anti-3-(α-aminobenzyl)-benzo-γ-sultam anti-6 was prepared analogously, however by resorting to NaHMDS as the base10 in the Wittig step (Scheme 2). The resulting ∼1:1 (E/Z)-isomeric mixture 1 was directly engaged in the further transformation (via the sulfonamide 2) into the aziridine 5 upon Rh2(OAc)4-catalyzed aziridination. The cis and trans diastereomers 5 were separated by silica gel chromatography at this stage of the sequence (28% yield for cis-5) and the former was converted as above into the corresponding racemic anti-3-(α-aminobenzyl)-benzo-γ-sultam anti-6. Its (3R,1′R)- and (3S,1′S)-configured enantiomers were separated by preparative chiral Supercritical Fluid Chromatography (SFC) affording the ligands in >99% ee and 44% combined yields. The absolute configuration of the 1st eluting enantiomer was equally determined by X-ray analysis of its (S)-CSA salt (Fig. 5).11
 | ||
| Scheme 2 Preparation of the anti-3-(α-aminobenzyl)-benzo-γ-sultam ligand 6 enantiomers. | ||
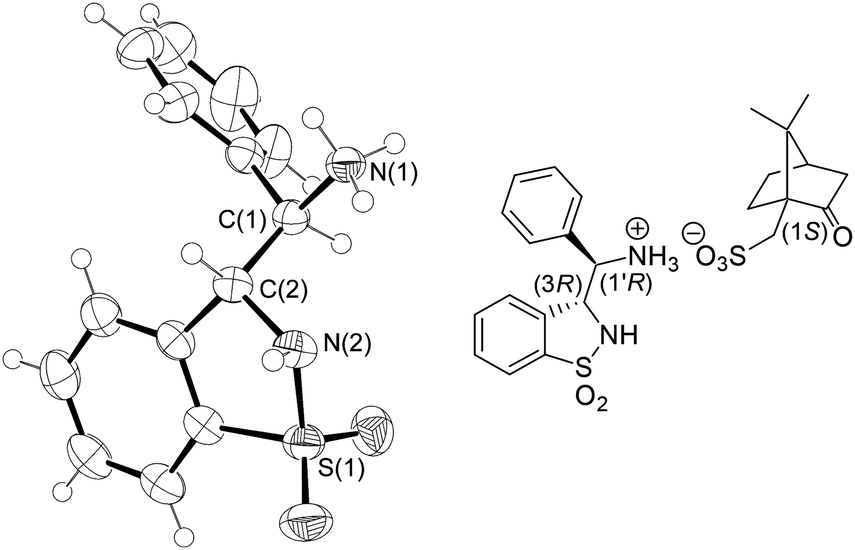 | ||
| Fig. 5 ORTEP drawing of the (1S)-camphor-10-sulfonic acid ((S)-CSA) salt of the SFC 1st eluting anti-6 enantiomer at the 50% probability level ((S)-CSA was omitted for clarity; for full details, see the ESI†). | ||
Evaluation of the new sulfonamido-amine ligands 6 in ATH
The assessment of the efficiency in ATH of the Ru(II) complexes incorporating the new 3-(α-aminobenzyl)-benzo-γ-sultam ligands was first conducted on the representative benchmark substrates acetophenone (S1), ethyl benzoylacetate (S2), 1-indanone (S3), and α-tetralone (S4) (Table 1). The Ru complexes were prepared from a [RuCl2(η6-arene)]2 precursor and the enantiopure sulfonamido-amine ligand 6 (syn or anti; 1.1 equiv. to Ru atom) at 40 °C (1 h) in 1,2-dichloroethane. The catalysts’ screening with an S/C = 200 was performed at 40 °C using HCO2H/Et3N 5:2.
|
|
|||||
|---|---|---|---|---|---|
| Ketone | Ligand | η6-Arene | t (h) | Conv. (%) | ee (%) |
|
a Reaction conditions: S/C = 200, ketone (1.0 mmol), 1,2-dichloroethane (1 mL), HCO2H/Et3N 5:2 (250 μL), 40 °C. Conversion was determined by 1H NMR of the extracted crude. Isolated yields were 96–98%. ees were determined by chiral GC or HPLC. (R)-Configured alcohols were obtained [(R,R)-TsDPEN used]. For further details, see the Experimental section.
b Reaction in neat HCO2H/Et3N 5:2 (500 μL).
|
|||||
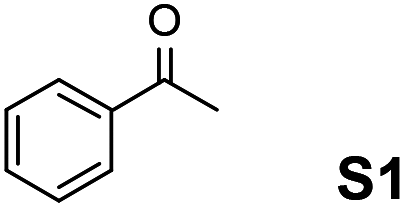
|
syn-6 | p-Cymene | 4 | >99 | 78 |
| 3b | 100 | 85 | |||
| syn-6 | Mesitylene | 7 | 80 | 72 | |
| anti-6 | p-Cymene | 4 | >99 | 58 | |
| anti-6 | Mesitylene | 7 | 90 | 72 | |
| TsDPEN | p-Cymene | 4 | 25 | 97 | |
| 4b | 25 | 97 | |||
| 20b | 99 | 97 | |||

|
syn-6 | p-Cymene | 3 | 100 | 92 |
| syn-6 | Mesitylene | 3 | 100 | 86 | |
| anti-6 | p-Cymene | 3 | 100 | 54 | |
| anti-6 | Mesitylene | 3 | 100 | 82 | |
| TsDPEN | p-Cymene | 7 | 85 | 98 | |
| 7b | >99 | 98 | |||
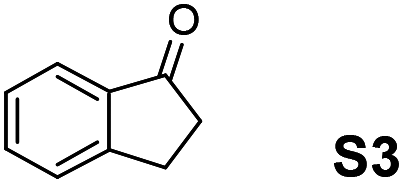
|
syn-6 | p-Cymene | 4 | 100 | 99 |
| anti-6 | p-Cymene | 5 | >99 | 95 | |
| anti-6 | Mesitylene | 7 | 90 | 95 | |
| TsDPEN | p-Cymene | 4b | 50 | - | |
| 20b | >99 | 99 | |||
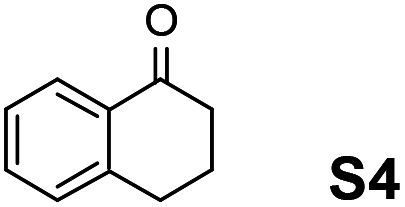
|
syn-6 | p-Cymene | 3 | 100 | 99 |
| syn-6 | Mesitylene | 7 | 80 | 98 | |
| anti-6 | p-Cymene | 7 | 95 | >99 | |
| anti-6 | Mesitylene | 7 | 85 | >99 | |
| TsDPEN | p-Cymene | 3 | 25 | 99 | |
| 3b | 40 | 99 | |||
| 20b | 98 | 98 | |||

The outcome of this exploratory profiling clearly revealed the faster reduction rate using the new sulfonamido-amine ligands 6versus the more flexible TsDPEN and “altered TsDPEN” ligands, or Wills’ conformationally locked indane-cored sulfonamido-amine ligands in Fig. 2. Also, it was noticed for TsDPEN that a supplemental amount of HCO2H/Et3N 5:2 was required in order to revitalize the reduction and drive it to completion. However, acetophenone (S1) and its α-ethoxycarbonyl-substituted derivative S2 afforded lower enantioselectivities (up to 85% ee and 92% ee, respectively) than the ones with the TsDPEN reference. Noteworthily, as (R)-1-phenylethanol was the major resulting enantiomer, the sense of induction on acetophenone (S1) of the syn-(3R,1′S)-6 and anti-(3R,1′R)-6 revealed to be in line with the one expected with the syn-(1R,2S)-TsDPEN (as its syn-(1S,2R)-enantiomer led to (S)-1-phenylethanol) or observed with anti-(R,R)-TsDPEN (Fig. 2).
Noticeably, the performance of syn-(3R,1′S)-6 was particularly good against the benzo-fused ketones, 1-indanone (S3) and α-tetralone (S4), as up to 99% ee with full conversion was obtained in reasonable times leading as well to (R)-configured alcohols.2a Also, (R)-configured products (95 to >99% ee) were formed employing the anti-(3R,1′R)-6 demonstrating hence that the chirality on the C(3) atom (bearing the sulfonamido group) predominantly determines the enantiofacial discrimination.
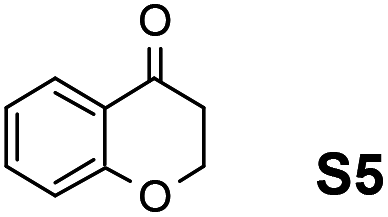
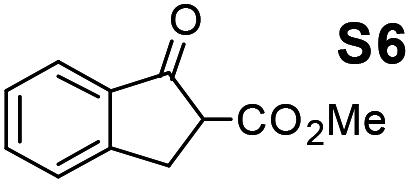
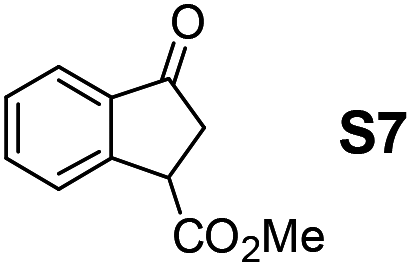
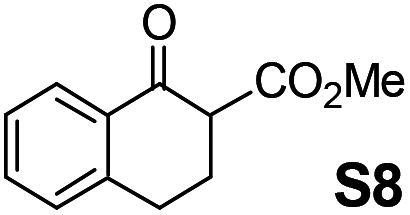
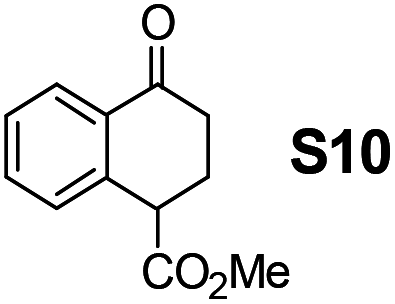
Therefore, the [RuCl2(p-cymene)]2/(3R,1′S)-6 complex was further screened on a series of methoxycarbonyl-substituted 1-indanones (S6–S7) and methoxycarbonyl-substituted α-tetralones (S8–S10) (Table 2).
| Ketone | S/C | t (h) | Conv. (%) | Product | |
|---|---|---|---|---|---|
| cis/trans | ee (%) | ||||
|
a Reaction conditions: [RuCl2(p-cymene)]2/(3R,1′S)-6, ketone (1.0 mmol), 1,2-dichloroethane (1 mL), HCO2H/Et3N 5:2 (250 μL), 40 °C. Conversion and cis/trans ratio (dr) were determined by 1H NMR of the extracted crude. Isolated yields were 94–99%. ees were determined by chiral GC or HPLC. (R)-Configured alcohols were obtained in all the cases. For further details, see the Experimental section.
b Determined by 19F NMR of the (R)-Mosher ester; trans-stereoisomers were not separated.
|
|||||
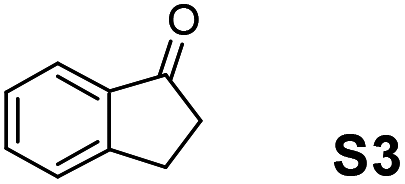
|
1000 | 16 | 95 | — | 99 |
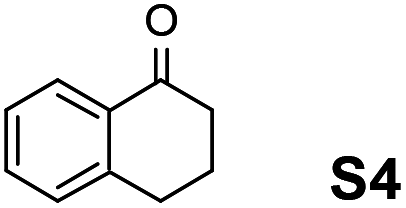
|
1000 | 16 | >99 | — | 99 |
|
1000 | 12 | 100 | — | 99 |
|
200 | 4 | 100 | 97:3 |
>99 (cis) |
|
200 | 6 | 100 | 95:5 |
99 (cis) |
| >99 (trans) | |||||
|
200 | 6 | >99 | 98:2 |
>99 (cis) |

|
100 | 6 | >99 | 50:50 |
99 (cis)b |
|
200 | 3 | 100 | 50:50 |
>99 (cis) |
| >99 (trans) | |||||
Employing an S/C = 1000, a 99% ee coupled with a high conversion was obtained within 16 h for 1-indanone (S3), α-tetralone (S4), and 4-chromanone (S5).
Shifting to functionalized variants, ATH of racemic 2- or 3-methoxycarbonyl-1-indanone (S6 and S7) using an S/C = 200 resulted in high diastereoselectivities (cis/trans 97:3 and 95:5, respectively) and 99% enantioselectivity indicating a dynamic kinetic resolution (DKR)12,13 occurring during their reduction. Regioisomeric methoxycarbonyl-α-tetralones S8–S10 displayed as well an excellent ATH outcome. A high enantioselectivity (>99% ee) was attained in all the cases, with S8 undergoing DKR12 in 98:2 cis/trans ratio. Noteworthily, S9 exhibited some kinetic resolution (KR) during ATH as, at ∼75% conversion, 1H NMR indicated a 60:40 cis/trans ratio.14
Most interestingly, in a related fashion to the reported DKR during ATH of readily enolizable β-keto esters (such as S6 and S8),12 a first-time DKR during reduction of a γ-keto ester as rac-3-methoxycarbonyl-1-indanone15 (S7) was encountered (Scheme 3). We hypothesize that this DKR occurs by keto–enol tautomerization of this 5-membered cyclic γ-keto ester via the racemization-prone dual benzylic and allylic carbon. This stereolability could be accentuated in the transition state (TS) influenced by the electrostatic interaction as shown in Scheme 3. We assume that the configuration of the GC-detected trans-product (5.3%) is as depicted supported by the 99% ee obtained with 1-indanone (S3). By contrast, the 6-membered δ-keto ester higher homolog S10 is unable to undergo racemization (under the test conditions) of its methoxycarbonyl-borne benzylic carbon precluding thus DKR.
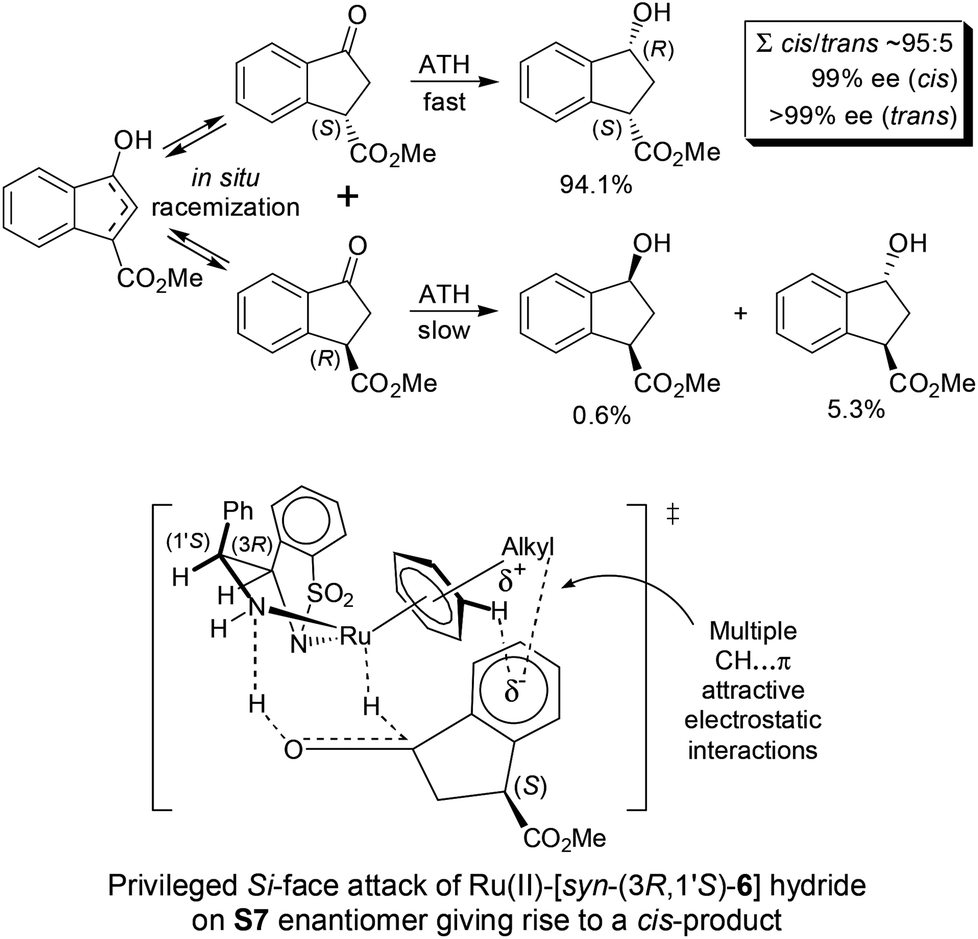 | ||
| Scheme 3 DKR of rac-3-methoxycarbonyl-1-indanone (S7) during ATH (Table 2). | ||
The [Ru(TsDPEN)(η6-arene)]-catalyzed ATH mechanism was established by Noyori, Ikariya and co-workers who considered multiple CH⋯π attractive electrostatic interactions in the Ru(II)-hydride-ketone TS.16 In the case of racemic ketone S7, a more facile reduction of the (S)-enantiomer (vs. the (R)-enantiomer) is attributable to the CO2Me group outwards orientation minimizing the steric interference in the TS.
Conclusions
We have successfully prepared diastereo- and enantiopure syn- and anti-(α-aminobenzyl)-benzo-γ-sultam ligands 6 and explored them in Ru(II)-catalyzed ATH of conventional ketones in the presence of formic acid/triethylamine 5:2. The ATH rate and enantioselectivity using the syn isomer ligand were better than with the anti isomer. High enantioselectivities (99 to >99% ee) were obtained for the benzo-fused cyclic ketones S3–S10 based on 1-indanone or α-tetralone. Namely, a first-time ever DKR of a γ-keto carboxylic ester occurred during reduction. In fact, ketone S7 derived from 1-indanone afforded the corresponding γ-hydroxy ester in a high cis/trans ratio (95:5) and excellent ee (99% ee for cis, >99% ee for trans).
Finally, this study is yet another example of how skeletal changes in a chiral ligand design can translate into unpredictable catalyst properties.
Experimental section
Materials and methods
The following non-commercial ketones were prepared according to literature procedures: 2-methoxycarbonyl-1-indanone17 and 2-methoxycarbonyl-1-tetralone18 from 1-indanone and α-tetralone, respectively, using dimethyl carbonate; 3-methoxycarbonyl-1-indanone19 by esterification of 3-oxo-1-indanecarboxylic acid; 3-methoxycarbonyl-1-tetralone20 starting by a Stobbe condensation of benzaldehyde with dimethyl succinate; 4-methoxycarbonyl-1-tetralone19 from 2-phenyl-glutaric anhydride.All reactions were conducted under an inert atmosphere (nitrogen or argon) using anhydrous solvents. HCO2H/Et3N 5:2 (azeotrope) was prepared by adding Et3N (280 mL, 2 mol) to HCO2H (190 mL, 5 mol) at 0 °C under a nitrogen atmosphere and used as such. Analytical thin-layer chromatography (TLC) was performed using Silica Gel 60 F254 pre-coated plates (0.25 mm thickness); Rf values are reported and visualization was accomplished by irradiation with an UV lamp (254 nm) and/or staining with KMnO4 solution. Silica gel 60 (40–63 μm) was used for flash column chromatography. 1H (299.9 MHz; internal Me4Si) and 13C (75.4 MHz; internal CDCl3, δ 77.00) spectra were recorded for solutions in CDCl3 if not stated otherwise. HRMS measurements were obtained on a Q-TOF instrument equipped with an orthogonal Z-spray ESI interface.
:15 ratio [by 1H NMR in DMSO-d6; δ 6.54 (d, J = 12.3 Hz) is the characteristic signal for the (Z)-isomer and δ 8.25 (d, J = 16.6 Hz) for the (E)-isomer]. Recrystallization from MeCN led to the title product as a white powder (8.14 g, 59% yield). 1H NMR (300 MHz, DMSO-d6): δ = 7.13 (d, 16.6 Hz, 1 H), 7.18–7.41 (m, 5 H), 7.50–7.53 (m, 2 H), 7.75–7.81 (m, 2 H), 8.25 (d, J = 16.6 Hz, 1 H). 13C NMR (76 MHz, DMSO-d6): δ = 125.3, 126.52, 126.58, 127.1, 127.5, 128.1, 128.3, 128.7, 128.9, 134.3, 137.8, 145.7. HRMS-ESI (m/z): [M]+ calcd for C14H1123NaO3S, 282.0327; found, 282.0326.
:2:1) and then EtOAc/MeOH (4:1). Fractions containing the product/triphenylphosphine oxide were concentrated, and the solid suspended in H2O (400 mL) and stirred at rt for 3 h. The insoluble matter was filtered off and the filtrate concentrated and dried under high vacuum at 60 °C affording the title product with a ∼1:1 E/Z ratio [by 1H NMR in DMSO-d6; δ 6.54 (d, J = 12.3 Hz) is the characteristic signal for the (Z)-isomer and δ 8.25 (d, J = 16.6 Hz) for the (E)-isomer] (7.70 g, 91% yield) as a pale yellow powder.
:1). After concentration and trituration with iPr2O, the title product (5.17 g, 88%) was obtained as colorless crystals; mp 145–146 °C. 1H NMR (300 MHz, CDCl3): δ = 4.72 (s, 2 H), 7.07 (d, J = 16 Hz, 1 H), 7.28–7.51 (m, 4 H), 7.50–7.65 (m, 3 H), 7.73 (d, J = 8 Hz, 1 H), 7.97 (d, J = 16 Hz, 1 H), 8.07 (dd, J = 8, 1 Hz, 1 H). 13C NMR (76 MHz, CDCl3): δ = 124.4, 127.1, 127.5, 128.0, 128.2, 128.7, 128.9, 133.0, 134.6, 136.3, 136.5, 138.9. HRMS-ESI (m/z): [M + H]+ calcd for C14H14NO2S, 260.0745; found, 260.0748.
:1) (4.00 g, 14.18 mmol) following a similar procedure to that for (E)-2. The title product (3.17 g, 86% yield; E/Z ∼1:1) was obtained as an amber-colored oil.
:1, 8:2, 7:3 then 6:4) affording the title product (1.07 g, 76% yield) as colorless crystals. Its structure was determined by X-ray analysis (see the ESI†). 1H NMR (300 MHz, CDCl3): δ = 3.48 (d, J = 3 Hz, 1 H), 4.18 (d, J = 3 Hz, 1 H), 7.39 (m, 5 H), 7.54–7.70 (m, 3 H), 7.74–7.81 (m, 1 H). 13C NMR (76 MHz, CDCl3): δ = 51.2, 57.5, 123.4, 125.5, 126.4, 128.7, 129.0, 130.3, 133.5, 133.6, 134.0, 136.0. HRMS-ESI (m/z): [M + H]+ calcd for C14H12NO2S, 258.0589; found, 258.0586.
:1) (4.00 g, 15.4 mmol) in CH2Cl2 (160 mL) were successively added at rt Al2O3 (3.9 g, 38.6 mmol), molecular sieves 4 Å (4 g), Rh2(OAc)4 (341 mg, 0.77 mmol) and PhI(OAc)2 (7.46 g, 23.17 mmol). The resulting mixture was stirred at 40 °C for 4 h, brought to rt and filtered through a plug of silica gel eluting with CH2Cl2. The crude was purified on silica gel eluting with toluene/EtOAc (24:1) affording trans-5 (0.97 g, 25% yield) and cis-5 (1.1 g, 28% yield) as an oil. cis-5: 1H NMR (300 MHz, CDCl3): δ = 4.31 (d, J = 5 Hz, 1 H), 4.62 (d, J = 5 Hz, 1 H), 6.92–7.02 (m, 2 H), 7.03–7.21 (m, 3 H), 7.35–7.52 (m, 2 H), 7.61–7.70 (m, 1 H), 7.71–7.82 (m, 1 H). 13C NMR (76 MHz, CDCl3): δ = 47.3, 55.8, 122.5, 126.4, 127.2, 127.8, 128.5, 130.3, 131.0, 133.2, 134.1, 137.1. HRMS-ESI (m/z): [M + H]+ calcd for C14H12NO2S, 258.0589; found, 258.0588.
:1, 30 mL) was heated at 55 °C for 1 h, then brought to rt and concentrated. The residue was partitioned between H2O (pH 4.5) and CH2Cl2, and the combined organic layers were dried (Na2SO4) and concentrated. To the resulting oil (1.3 g) in EtOAc (30 mL), 5% Pd/C (130 mg) was added and the mixture was hydrogenated using Parr apparatus at 20 psig H2 for 1 h. After filtration through Celite and concentration, the residue was purified on silica gel eluting successively with CH2Cl2, CH2Cl2/EtOAc (4:1 then 1:2) and EtOAc/EtOH (4:1), affording syn-6 (910 mg, 94% yield) as a yellowish syrup. 1H NMR (300 MHz, CDCl3): δ = 2.11 (br s, 3 H), 4.36 (d, J = 6 Hz, 1 H), 4.90 (d, J = 6 Hz, 1 H), 7.02–7.19 (m, 1 H), 7.28–7.62 (m, 7 H), 7.66–7.86 (m, 1 H). 13C NMR (76 MHz, CDCl3): δ = 59.0, 63.1, 121.4, 125.8, 127.1, 128.2, 128.9, 129.6, 132.5, 136.6, 137.0, 140.6. HRMS-ESI (m/z): [M + H]+ calcd for C14H15N2O2S, 275.0854; found, 275.0849. The syn-6 enantiomers were separated by preparative HPLC on ChiralPak IC (5 μm, 250 × 30 mm), mobile phase CH2Cl2/EtOH (98:2), flow rate 42.5 mL min−1, 25 °C, UV detection at 260 nm. Analytical HPLC conditions: ChiralPak IC (5 μm, 250 × 4.6 mm), mobile phase: CH2Cl2/EtOH (98:2), flow rate: 1 mL min−1, 25 °C, UV detection at 230 nm.21
General ATH procedure for ketones listed in Tables 1 and 2
:2.
:0.01 mmol Ru atom; with S/C = 200:0.005 mmol Ru atom; with S/C = 1000:0.001 mmol Ru atom) was added HCO2H/Et3N 5:2 (250 μL; 500 μL when no cosolvent was used) followed by the ketone (1.0 mmol). This mixture was stirred at 40 °C with continuous mild N2 sweeping. The reaction progress was monitored by 1H NMR. Workup: the reaction mixture was partitioned between Et2O (5 mL) and H2O (5 mL). The aq. layer was re-extracted with Et2O (5 mL) and the combined organic layers were filtered through a bed of silica gel/Na2SO4 and concentrated.
Characterization and ee determination of the ATH prepared chiral alcohols
:2), flow rate 1.0 mL min−1, UV detection at 254 nm; racemate: tR 22.6 min (S), 26.2 min (R).
:5), flow rate 1.0 mL min−1, UV detection at 220 nm; racemate: tR 10.0 min (S), 11.7 min (R).
:3 by 1H NMR; >99% ee (cis). 1H NMR (300 MHz, CDCl3): δ = 2.77 (d, J = 6.1 Hz, 1 H), 2.97–3.21 (m, 1 H), 3.34–3.53 (m, 2 H), 3.79 (s, 3 H), 5.35 (t, J = 5.9 Hz, 1 H), 7.23–7.32 (m, 3 H), 7.39–7.48 (m, 1 H). ee was determined by chiral GC analysis12,23a on a Chiralsil-DEX CB column (25 m × 0.25 mm), 130 °C (isothermal); racemates (cis/trans = 55:45): tR 33.7 min [cis-(R,R)], 34.8 min [cis-(S,S)], 45.0 min (trans-1), 50.0 min (trans-2).
:5 by 1H NMR; 98.8% ee (cis), >99% ee (trans); mp 105–108 °C; [α]20D –37.5 (c 1.15 in CHCl3). 1H NMR (CDCl3): δ = 2.20 (m, 0.05 H, trans), 2.33 (app t, J = 14.5, 8.0 and 6.8 Hz, 1 H), 2.59 (ddd, J = 14.5, 8.0 and 6.8 Hz, 1 H), 2.84 (m, 0.05 H, trans), 3.14 (br s, 1 H), 3.72 (s, 0.15 H, trans), 3.76 (s, 3 H), 4.02 (dd, J = 8.0 and 2.8 Hz, 1 H), 4.24 (dd, 0.05 H, trans), 5.16 (m, 1 H), 5.46 (t, 0.05 H, trans), 7.30–7.39 (m, 3 H), 7.50–7.53 (m, 1 H). 13C NMR (CDCl3): δ = 38.5, 48.4, 52.6, 75.2, 124.8, 125.4, 128.6, 128.9, 140.3, 145.4, 175.8. HRMS-ESI (m/z): [M + Na]+ calcd for C11H12O323Na, 215.0684; found, 215.0686. ees were determined by chiral GC analysis on a Chiralsil-DEX CB column (25 m × 0.25 mm), 130 °C (isothermal); racemates (cis/trans = 97:3): tR 16.8 min [cis-(1S,3R)], 18.6 min [cis-(1R,3S)], 24.1 min (trans-1), 25.2 min (trans-2). Recrystallization from hexane/CH2Cl2 afforded colorless needles (162 mg, 84% yield, >99.9% de, >99.9% ee) which were used for absolute configuration determination by X-ray analysis (see the ESI†).
:2.4 by 1H NMR; 99.4% ee (cis). 1H NMR (300 MHz, CDCl3): δ = 2.07–2.31 (m, 2 H), 2.75–2.99 (m, 4 H), 3.70 (s, 3 H), 4.80 (d, J = 9.0 Hz; CH, trans diastereomer), 5.05 (m, 1 H), 7.12–7.14 (m, 1 H), 7.19–7.25 (m, 2 H), 7.37–7.42 (m, 1 H). ee was determined by chiral HPLC analysis on a Chiralpak IB-3 column (25 cm). Eluent hexane/2-PrOH (97:3), flow rate 1.0 mL min−1, UV detection at 220 nm; racemates (a mixture prepared by weighing both the corresponding ATH products derived from enantiomeric catalysts): tR 15.4 min (trans-1), 17.4 min [cis-(R,R)], 19.7 min (trans-2), 32.7 min [cis-(S,S)].12
:50 by 1H NMR; 99% ee (cis). 1H NMR (300 MHz, CDCl3): δ = 1.70–1.71 (m, 1 H), 1.92–2.04 (m, 2 H), 2.22 (d, J = 8.1 Hz, 1 H), 2.35–2.42 (m, 1 H), 2.44–2.52 (m, 1 H), 2.85–3.19 (m, 6 H), 3.73 and 3.75 (2s, 2 × 3 H), 4.82–4.91 (m, 2 H), 7.10–7.28 (m, 6 H), 7.32–7.37 (m, 1 H), 7.54–7.56 (m, 1 H). HRMS-ESI (m/z): [M + Na]+ calcd for C12H14O323Na, 229.0841; found, 229.0846. Racemates (cis/trans = 96:4). ee was determined by 19F NMR (CDCl3) of the (R)-Mosher ester (prepared from (S)-Mosher's acid chloride): δ 71.95 [cis-(1R,3R)], 72.00 [cis-(1S,3S)], 72.24 (trans; stereoisomers not separated). The absolute configuration was assigned based on the general observed trend of enantioselectivity.
:50 by 1H NMR; >99% ee (cis), >99% ee (trans). 1H NMR (300 MHz, CDCl3): δ = 1.72–1.80 (m, 1 H), 2.31–1.88 (m, 7 H), 2.62 (br s, 2 H), 3.67 and 3.69 (2s, 2 × 3 H), 3.75–3.83 (m, 2 H), 4.65–4.73 (m, 2 H), 7.23–7.27 (m, 6 H), 7.39–7.47 (m, 2 H). HRMS-ESI (m/z): [M + Na]+ calcd for C12H14O323Na, 229.0841; found, 229.0845. ees were determined by chiral GC analysis on a Chiralsil-DEX CB column (25 m × 0.25 mm), 150 °C (isothermal); racemates (cis/trans = 72:28): tR 21.1 min [cis-(1R,4S)], 21.9 min [trans-(1R,4R)], 22.7 min [cis-(1S,4R)], 24.4 min [trans-(1S,4S)]. The absolute configuration was assigned based on the general observed trend of enantioselectivity.
Acknowledgements
This work was supported by the Ministry of Higher Education, Science, and Technology of the Republic of Slovenia (grant number P1-242).Notes and references
- For reviews, see: (a) S. Gladiali and E. Alberico, Chem. Soc. Rev., 2006, 35, 226–236 RSC; (b) T. Ikariya, K. Murata and R. Noyori, Org. Biomol. Chem., 2006, 4, 393–406 RSC; (c) T. Ikariya and A. J. Blacker, Acc. Chem. Res., 2007, 40, 1300–1308 CrossRef CAS; (d) C. Wang, X. Wu and J. Xiao, Chem. – Asian J., 2008, 3, 1750–1770 CrossRef CAS; (e) D. Wang and D. Astruc, Chem. Rev., 2015, 115, 6621–6686 CrossRef CAS; (f) F. Foubelo, C. Najera and M. Yus, Tetrahedron: Asymmetry, 2015, 15–16, 769–790 CrossRef.
- For various ATH ArSO2-DPEN ligands, see: (a) A. Fujii, S. Hashiguchi, N. Uematsu, T. Ikariya and R. Noyori, J. Am. Chem. Soc., 1996, 118, 2521–2522 CrossRef CAS; therein, {Ru[(S,S)-TsDPEN](mesitylene)} (S/C = 200, 28 °C, 48 h) led with 1-indanone (S3) or α-tetralone (S4) to 99% ee (S) (>99% conv.) but testing S4 at 60 °C led to 98% ee (>99% conv.) within 6 h. (b) Y. Ma, H. Liu, L. Chen, X. Cui, J. Zhu and J. Deng, Org. Lett., 2003, 5, 2103–2106 CrossRef CAS; (c) D. Šterk, M. Stephan and B. Mohar, Tetrahedron Lett., 2004, 45, 535–537 CrossRef; (d) P. N. Liu, P. M. Gu, F. Wang and Y. Q. Tu, Org. Lett., 2004, 6, 169–172 CrossRef CAS; (e) Z. Zhou, Y. Sun and A. Zhang, Cent. Eur. J. Chem., 2011, 9, 175–179 CrossRef CAS.
- (a) B. Mohar, A. Valleix, J.-R. Desmurs, M. Felemez, A. Wagner and C. Mioskowski, Chem. Commun., 2001, 2572–2573 RSC; (b) D. Šterk, M. Stephan and B. Mohar, Tetrahedron: Asymmetry, 2002, 13, 2605–2608 CrossRef; (c) Z. Luo, F. Qin, S. Yan and X. Li, Tetrahedron: Asymmetry, 2012, 23, 333–338 CrossRef CAS; (d) W.-W. Wang, Z.-M. Li, L. Su, Q.-R. Wang and Y.-L. Wu, J. Mol. Catal. A: Chem., 2014, 387, 92–102 CrossRef CAS; (e) X. Liu, C. Chen, Y. Xiu, A. Chen, L. Guo, R. Zhang, J. Chen and Z. Hou, Catal. Commun., 2015, 67, 90–94 CrossRef CAS.
- For 2nd generation Ru(II) complexes, see: (a) J. Hannedouche, G. J. Clarkson and M. Wills, J. Am. Chem. Soc., 2004, 126, 986–987 CrossRef CAS; (b) A. Kišić, M. Stephan and B. Mohar, Org. Lett., 2013, 15, 1614–1617 CrossRef; (c) A. Kišić, M. Stephan and B. Mohar, Adv. Synth. Catal., 2014, 356, 3193–3198 CrossRef.
- For 3rd generation Ru(II) complexes, see: (a) A. M. Hayes, D. J. Morris, G. J. Clarkson and M. Wills, J. Am. Chem. Soc., 2005, 127, 7318–7319 CrossRef CAS; (b) T. Touge, T. Hakamata, H. Nara, T. Kobayashi, N. Sayo, T. Saito, Y. Kayaki and T. Ikariya, J. Am. Chem. Soc., 2011, 133, 14960–14963 CrossRef CAS; (c) R. Soni, K. E. Jolley, G. J. Clarkson and M. Wills, Org. Lett., 2013, 15, 5110–5113 CrossRef CAS; (d) R. Soni, T. H. Hall, B. P. Mitchell, M. R. Owen and M. Wills, J. Org. Chem., 2015, 80, 6784–6793 CrossRef CAS; (e) A. Kišić, M. Stephan and B. Mohar, Adv. Synth. Catal., 2015, 357, 2540–2546 CrossRef.
- (a) A. Hayes, G. Clarkson and M. Wills, Tetrahedron: Asymmetry, 2004, 15, 2079–2084 CrossRef CAS; (b) B. Zhang, H. Wang, G.-Q. Lin and M.-H. Xu, Eur. J. Org. Chem., 2011, 4205–4211 CrossRef CAS; (c) M. J. Palmer, J. A. Kenny, T. Walsgrove, A. M. Kawamoto and M. Wills, J. Chem. Soc., Perkin Trans. 1, 2002, 416–427 RSC; (d) P. Roszkowski, J. K. Maurin and Z. Czarnocki, Tetrahedron: Asymmetry, 2013, 24, 643–650 CrossRef CAS.
- The preparation of (R)-3-hydroxymethyl-1,1-dioxo-benzo-1,2-thiazolidine was described by following a related synthetic strategy. Therein, in addition to a 5-exo-type cyclization, the formation of trans-(3R,4S)-4-hydroxy-3-hydroxymethyl-1,1-dioxo-benzo-1,2-thiazinane derived from 6-endo-type cyclization occurred as well. For this, see: K. H. Ahn, H. H. Baek, S. J. Lee and C.-W. Cho, J. Org. Chem., 2000, 65, 7690–7696 CrossRef CAS.
- J.-L. Liang, S.-X. Yuan, P. W. H. Chan and C.-M. Che, Org. Lett., 2002, 4, 4507–4510 CrossRef CAS.
- C24H30N2O6S2, FW 506.62, monoclinic, space group P21 (no. 4), a = 12.5236(3), b = 7.53800(17), c = 12.9539(3) Å, β = 92.676(2)°, V = 1221.55(5) Å3, Z = 2, T = 150(2) K, dcalcd = 1.377 g cm−3, μ = 0.261 mm−1, 11729 measured reflections, 5599 unique reflections (Rint = 0.0280), 324 refined parameters, R1 [I > 2σ(I)] = 0.0482, wR2 [all data] = 0.1230.
- P. A. Byrne and D. G. Gilheany, J. Am. Chem. Soc., 2012, 134, 9225–9239 CrossRef CAS.
- C24H30N2O6S2, FW 506.62, monoclinic, space group P21 (no. 4), a = 13.1356(2), b = 7.03520(10), c = 14.6189(2) Å, β = 112.8645(8)°, V = 1244.81(3) Å3, Z = 2, T = 293(2) K, dcalcd = 1.352 g cm−3, μ = 0.256 mm−1, 20767 measured reflections, 5599 unique reflections (Rint = 0.0280), 323 refined parameters, R1 [I > 2σ(I)] = 0.0474, wR2 [all data] = 0.1300.
- For DKR during Ru-TsDPEN-catalyzed (S/C = 200) ATH of 2-ethoxycarbonyl-1-indanone (S6 analog) (6 days, 81% yield, cis/trans >99:1, 99% ee for cis) and 2-ethoxycarbonyl-1-tetralone (S8 analog) (6 days, 90% yield, cis/trans >99:1, 99% ee for cis) using HCO2H/Et3N 5:2 at rt, see: A. Ros, A. Magriz, H. Dietrich, J. M. Lassaletta and R. Fernández, Tetrahedron, 2007, 63, 7532–7537 CrossRef CAS.
- For a review on DKR in ATH, see: (a) H. Pellisier, Tetrahedron, 2011, 67, 3769–3802 CrossRef. For selected recent DKR examples in ATH, see: (b) B. Seashore-Ludlow, F. Saint-Dizier and P. Somfai, Org. Lett., 2012, 14, 6334–6337 CrossRef CAS; (c) K. M. Steward, E. C. Gentry and J. S. Johnson, J. Am. Chem. Soc., 2012, 134, 7329–7332 CrossRef CAS; (d) S.-M. Son and H.-K. Lee, J. Org. Chem., 2014, 79, 2666–2681 CrossRef CAS; (e) F. Xu, M. J. Zacuto, Y. Kohmura, J. Rosen, A. Gibb, M. Alam, J. Scott and D. Tschaen, Org. Lett., 2014, 16, 5422–5425 CrossRef CAS; (f) P.-G. Echeverria, J. Cornil, C. Férard, A. Guérinot, J. Cossy, P. Phansavath and V. Ratovelomanana-Vidal, RSC Adv., 2015, 5, 56815–56819 RSC; (g) T. Cheng, Q. Ye, Q. Zhao and G. Liu, Org. Lett., 2015, 17, 4972–4975 CrossRef CAS.
- For a kinetic resolution of racemic cis-3-hydroxymethyl-indanol or -tetralol by oxidation in acetone using [Ru(TsDPEN)(p-cymene)], see: M. Caro, M. Torrado, C. F. Masaguer and E. Raviña, Tetrahedron: Asymmetry, 2003, 14, 3689–3696 CrossRef.
- A preparation of enantiomeric 3-ethoxycarbonyl-1-indanone (96% ee) by asymmetric hydroacylation of o-formyl-atropic acid ethyl ester using {Rh[(R)-BINAP]}ClO4 has been achieved. For this, see: K. Kundu, J. V. McCullagh and A. T. Morehead Jr., J. Am. Chem. Soc., 2005, 127, 16042–16043 CrossRef CAS.
- (a) P. A. Dub and T. Ikariya, J. Am. Chem. Soc., 2013, 135, 2604–2619 CrossRef CAS; (b) A. Matsuoka, C. A. Sandoval, M. Uchiyama, R. Noyori and H. Naka, Chem. – Asian J., 2015, 10, 112–115 CrossRef CAS.
- H. O. House and C. B. Hudson, J. Org. Chem., 1970, 35, 647–651 CrossRef CAS.
- L.-Q. Cui, Z.-L. Dong, K. Liu and C. Zhang, Org. Lett., 2011, 13, 6488–6491 CrossRef CAS.
- C. J. O'Donnell, R. A. Singer, J. D. Brubaker and J. D. McKinley, J. Org. Chem., 2004, 69, 5756–5759 CrossRef.
- H. Malik, F. P. J. T. Rutjes and B. Zwanenburg, Tetrahedron, 2010, 66, 7198–7203 CrossRef CAS.
- Separation of rac syn-6 enantiomers by preparative HPLC was outsourced to Chiral Technologies Europe (France).
- Separation of rac anti-6 enantiomers by preparative SFC was outsourced to WuXi AppTec Co., Ltd (China).
- For chiral GC analysis, see: (a) Ref. 5e; ; (b) Ref. 3b .
Footnotes |
| † Electronic supplementary information (ESI) available: X-ray crystallographic data for racemic trans-5, [syn-(3S,1′R)-6]·(S)-CSA, [anti-(3R,1′R)-6]·(S)-CSA, and the S7 ATH major reduction product, HPLC and GC chromatograms of ATH products, and 1H, 13C NMR and HMBC spectra. CCDC 1436151–1436154. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ob02352a |
| ‡ Present address: PhosPhoenix SARL, 115, rue de l′Abbé Groult, 75015 Paris, France. |
| § CCDC 1436151 [for syn-(3S,1′R)-6·(S)-CSA], CCDC 1436152 [for anti-(3R,1′R)-6·(S)-CSA], CCDC-1436153 [for trans-5], and CCDC 1436154 [for S7-reduced] contain the crystallographic data for this paper. |
| This journal is © The Royal Society of Chemistry 2016 |